

Abstract
Genome structure and gene expression depend on a multitude of chromatin-binding proteins. The binding properties of these proteins to native chromatin in intact cells are largely unknown. Here, we describe an approach based on combined in vivo photobleaching microscopy and kinetic modeling to analyze globally the dynamics of binding of chromatin-associated proteins in living cells. We have quantitatively determined basic biophysical properties, such as off rate constants, residence time, and bound fraction, of a wide range of chromatin proteins of diverse functions in vivo. We demonstrate that most chromatin proteins have a high turnover on chromatin with a residence time on the order of seconds, that the major fraction of each protein is bound to chromatin at steady state, and that transient binding is a common property of chromatin-associated proteins. Our results indicate that chromatin-binding proteins find their binding sites by three-dimensional scanning of the genome space and our data are consistent with a model in which chromatin-associated proteins form dynamic interaction networks in vivo. We suggest that these properties are crucial for generating high plasticity in genome expression.
Organization of DNA into higher-order chromatin structure serves to accommodate the genome within the spatial confines of the cell nucleus and acts as an important regulatory mechanism (22, 36, 46, 60). Establishment, maintenance, and alterations of global and local chromatin states are modulated by the combined action of a multitude of chromatin-binding proteins. The nucleosome, containing histone proteins, acts as a structural scaffold and as an entry point for regulatory mechanisms (60, 63). Nonhistone proteins, including the HMG proteins, further contribute to the structural maintenance and regulation of chromatin regions (6, 61). In heterochromatin, specific factors such as HP1 convey a transcriptionally repressed state, possibly by influencing higher-order chromatin structure (19, 27). Histone-modifying enzymes such as histone acetyl- and methyltransferases are instrumental in generating epigenetic marks on chromatin domains (60). Chromatin remodeling factors act on specific sites to facilitate access to regulatory DNA elements. Once accessible, transcriptional activators bind specific sequences on DNA and recruit the basal transcription machinery (37, 44, 46). All of these steps involve binding of proteins to chromatin.
Due to their functional significance, chromatin-associated proteins have been extensively characterized—mostly by biochemical extraction and in vitro binding assays. Little is known about the dynamics of how chromatin proteins bind to their target sites in native chromatin in living cells. In vivo microscopy techniques are providing novel tools to study chromatin proteins in living cells (32, 39, 41, 50). Qualitative analysis of photobleaching experiments has revealed a wide range of dynamic behavior for chromatin-associated proteins. The bulk of core histones is immobile on DNA, whereas the linker histone H1 and the TATA-binding protein are relatively stably, yet dynamically, associated (10, 16, 34, 38, 43). Replication and repair factors, on the other hand, are highly mobile in their unengaged state but become temporarily immobilized upon binding to their sites of action (31, 57). Several transcriptional activators and repressors have high mobility, implying transient binding to chromatin (3, 17, 30, 31, 40, 47, 49, 59). However, the majority of these studies are qualitative, and in only a few cases have quantitative parameters such as binding rates or residence times been extracted from in vivo microscopy data. Furthermore, it is not clear from the limited number of analyzed proteins how generally applicable the concept of transient binding of chromatin-associated proteins is.
In this report, we describe a computational microscopy approach to quantitatively determine in native chromatin of intact cells the binding properties of chromatin-associated proteins. By analysis of a wide range of chromatin proteins, including structural proteins, remodeling factors, and transcriptional coactivators, as well as basal and specific transcription factors, we demonstrate that transient binding is a general property of chromatin-associated proteins. Our results suggest that chromatin-binding proteins find their binding sites largely by three-dimensional scanning of the genome space, and we speculate that dynamic interaction networks play a critical role in the control of gene expression.
MATERIALS AND METHODS
Cell culture and growth rate.
BHK, HeLa, NIH 3T3, and CHO cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100-U/ml penicillin, 100-μg/ml streptomycin, and 2 mM l-glutamine at 37°C in an atmosphere enriched with 5% CO2. Hepa-1 cells were grown in alpha minimum essential medium (Gibco BRL) supplemented with 7% fetal bovine serum (HyClone Labs) to prevent nuclear translocation of AhR. Cells were routinely maintained in a 37°C incubator with 5% CO2. Cells were treated with a ligand, 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), at 10 nM for 1 h in all experiments. Cells were transfected by electroporation with a BTX square wave pulse at 650 V, 99 μs. Transiently transfected cells were observed 14 h after transfection. Stable cell lines expressing H10-green fluorescent protein (GFP) were generated and grown as previously described (28).
Fusion proteins.
Details of the fusion proteins used in this study are found in the references cited in Table 1. Myc, Mad, Max, BRG, PCAF, and NF1 were in the pEGFP-C1 vector.
TABLE 1.
Characterization of fusion proteins
| Protein | Function | Functionality of GFP fusiona | Cell typeb | Source or reference |
|---|---|---|---|---|
| GFP | Fluorescent marker | NA | a, b, c | 9 |
| a, b, c | ||||
| H2B | Core histone | In vivo nucleosome assembly | a, b | 33 |
| H1° | Linker histone | In vitro binding | c | 43 |
| H1°ΔC | Linker histone | In vitro binding | c | 43 |
| HMGB1 | Chromatin structure | In vivo binding | a, b | 54 |
| HMGN1 | Chromatin structure | In vitro binding | a, b, c | 51 |
| HMGN1-E22, -E24 | Chromatin structure | In vitro binding | a, b, c | 51 |
| HP1β | Heterochromatin formation | In vitro binding | a, d | 13 |
| In vivo complementation | 23 | |||
| BRG1 | Chromatin remodeling | ND | a | ND |
| PCAF | Histone modifier | ND | a | ND |
| AhR | Receptor | In vivo activation | e | 20 |
| In vivo complementation | ||||
| ARNT | Coactivator | In vivo activation | e | 20 |
| In vivo complementation | ||||
| C/EBP | Transcription factor | In vitro binding | a | 56 |
| NF1 | Transcription factor | ND | a | ND |
| Jun | Transcription factor | In vitro binding | a | T. Kerppolac |
| Fos | Transcription factor | In vitro binding | a | T. Kerppolac |
| Myc | Transcription factor | In vitro binding | a | 64 |
| Max | Transcription factor | In vitro binding | a | 64 |
| Mad | Transcription factor | In vitro binding | a | 64 |
| FBP | Transcription factor | In vitro binding | a | 29a; D. Levensc |
| XBP | Transcription factor | In vivo complementation | a | 30 |
| BRD4 | Chromatin binding | In vitro binding | a | 15 |
NA, not applicable; ND, not determined.
The following cell types were used: baby hamster kidney cells (a), HeLa (b), NIH 3T3 (c), Chinese hamster ovary (d), and Hepa-1 (e).
Personal communication.
Imaging.
For microscopy, transfected cells were plated and observed in LabTek II chambers (Nalgene). Live-cell microscopy was performed on a Zeiss 510 confocal microscope using the 488-nm laser line of an Ar laser (nominal output, 40 mW; beam width at specimen, 0.2 μm). All experiments were done at 37°C, and imaging was done with a ×100 objective, NA 1.3. Scanning was bidirectional at the highest possible rate using a ×5 zoom, with a pinhole of 1 Airy unit. Laser power for bleaching was maximal. For imaging, the laser power was attenuated to 0.1% of the bleach intensity. For fluorescence recovery after photobleaching (FRAP) experiments, five single-prebleach images were acquired, followed by two iterative bleach pulses of 223 ms each. Single-section images were then collected at 534-ms intervals for 60s. Recovery of signal in the bleached region and loss of signal in the unbleached region were measured as average intensity signals in a region comprising at least 50% of the bleached or unbleached area. The size of this measurement region was identical in all experiments. Signal loss during the recovery period was less than 5% of the initial fluorescence signal. Completeness of bleaching in three dimensions (3D) was confirmed by inspection of image stacks of fixed cells. All recovery and loss curves were generated from background-subtracted images. The fluorescence signal measured in a region of interest was singly normalized to the prebleach signal in the region of interest:
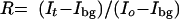 |
(1) |
where Io is the average intensity in the region of interest during prebleach, It is the average intensity in the region of interest at time point t, and Ibg is the background signal determined in a region outside of the cell nucleus.
Kinetic modeling.
Quantitative analysis of half-FRAP data was carried out as described in detail previously by a classical compartmental approach (48). Each photobleaching data set was fit to a sum of exponentials and showing that systematic deviations could not be eliminated with fewer binding sites and that addition of an additional site resulted in an unidentifiable system whose parameter values had unacceptable coefficients of variation. Most proteins analyzed showed biphasic behavior, and the kinetic modeling procedure is described for a model with two distinct types of binding events.
Using standard chemical kinetic principles, a system of ordinary differential equations with constant coefficients (rate constants) characterizing the processes of binding, unbinding, and (when active) photobleaching was generated (52). Since the absolute abundances of each protein and their binding sites are often unknown, second order association processes were converted to pseudo-first-order processes by combining the unknown steady-state binding site abundance with the corresponding second order rate constant to yield a first order association rate constant. This has the additional advantage that the nonlinear system of differential equations is converted to a linear system. The resulting equations are
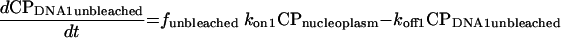 |
(2) |
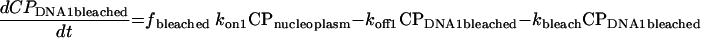 |
(3) |
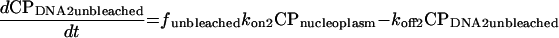 |
(4) |
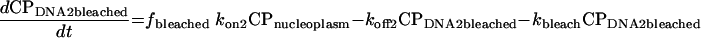 |
(5) |
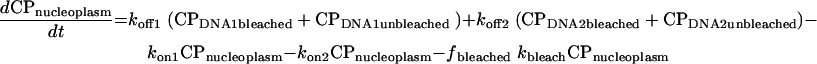 |
(6) |
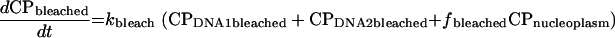 |
(7) |
In these equations, CP represents chromatin-associated protein; subscripts DNA1 and DNA2 indicate the two classes of binding sites; subscripts bleached and unbleached indicate the regions exposed to and not exposed to the bleaching laser pulse; fbleached and funbleached are the corresponding fractions of nuclear area; subscript nucleoplasm indicates the free pool of chromatin-associated protein in the nucleoplasm; kon1 and kon2 are the effective first order rate constants for chromatin-associated protein binding to sites 1 and 2, respectively; koff1 and koff2 are the rate constants for unbinding of chromatin-associated protein from sites 1 and 2, respectively; and kbleach is the rate constant characterizing the photobleaching process.
Rate constants for corresponding processes in the bleached and unbleached regions were constrained to be equal, and the proportion of the binding sites in the bleached versus unbleached parts of the nucleus was assumed to be the same as the proportion of the observed nuclear plane of focus occupied by the bleached and unbleached regions of interest. We further assumed that analysis of this single confocal slice was unaffected by the loss or gain of tagged molecules to or from nuclear regions above and below the slice.
Because it is difficult to resolve kon from half-FRAP data alone, we enforced an additional constraint. Based on our knowledge that diffusion is fast on this time scale, we assumed that in the unbleached region all of the free pool of protein and none of the bound pool was bleached by the time the first post-bleach image was collected. This allowed estimation of the fraction of the protein bound and, in turn, enforces a constraint on the values of kon1 and kon2 relative to koff1 and koff2.
Initial conditions were obtained by solving the model for an arbitrary synthetic rate of 10 molecules per s and a very slow degradation rate constant of 0.0005 s−1. As only normalized fluorescence data are analyzed, the absolute values of these steady-state abundances have no impact on the model solutions or on the dissociation rate constants and mean residence times reported here, but they do allow initial fluorescence to be correctly apportioned between fast and slow binding sites.
Data collected from the unbleached portion of the nucleus were fitted to the sum of the fast and slow compartments in the unbleached portion of the model. Data for the bleached portion of the nucleus were fitted to the sum of the fast and slow compartments in the bleached section of the model. For example, normalized recovery kinetics were fitted to
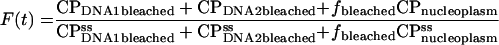 |
(8) |
where the superscript ss indicates the steady-state value. All CP values refer to relative amounts.
The key parameters of interest are the mean residence times, which were calculated from the corresponding dissociation rate constants:
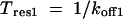 |
(9) |
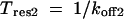 |
(10) |
Fractions of total binding associated with fast (1) or slow (2) binding sites are
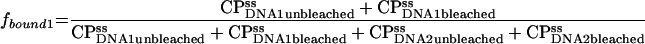 |
(11) |
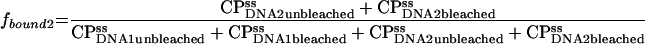 |
(12) |
The kinetic model was implemented by using SAAM II software (SAAM Institute, Inc., Seattle, Wash.). Generalized nonlinear least-squares fitting and parameter optimization were performed in SAAM II, and the coefficients of variation were obtained directly from the SAAM II Statistics window (4, 14). When fbleached, koff1, kbleach, kon2, and koff2 were allowed to adjust simultaneously, convergence was achieved in less than 20 iterations. Coefficients of variation for the reported parameter estimates were, respectively, on the order of 0.3, 7, 0.6, 30, and 9%.
RESULTS
Measurement of protein-chromatin interaction dynamics in living cells.
FRAP can be used to quantitatively measure binding kinetics of proteins to unperturbed chromatin in living cells. The rationale for this approach is based on the property that FRAP recovery kinetics reflect the overall mobility of a protein (39, 50). For proteins that do not interact with any cellular structures, FRAP kinetics are a direct reflection of their translational motion properties (41, 47, 62). In contrast, for proteins that bind to relatively immobile structures such as chromatin, binding events slow down the protein's overall mobility (41, 50, 62). Since diffusion over the micrometer range as measured in FRAP experiments occurs within 10 to 100 ms, even transient interactions of a protein with chromatin are rate limiting and severely slow down the overall recovery rate of a protein in a FRAP assay. Experimental FRAP recovery kinetics of chromatin-associated proteins are thus determined primarily by the protein's binding properties and allow extraction of information about protein-chromatin interactions in vivo (50).
The FRAP approach was experimentally validated by direct comparison of well-characterized corresponding pairs of wild-type and binding-impaired mutants of chromatin-associated proteins (Fig. 1A and B). To this end, half of the nucleus of a cell expressing a functional GFP-tagged protein of interest was bleached with a 400-ms bleach pulse. The recovery of the fluorescence signal in the bleached region and the loss of the signal in the unbleached regions were monitored simultaneously by time-lapse microscopy to provide complementary data sets (Fig. 1A and B). As previously reported, a stably expressed functional fusion protein between GFP and the linker histone H10 showed relatively slow fluorescence recovery and loss (Fig . 1A and B). The t50, defined as the time required to reach half-maximal recovery, for H10-GFP was 127 s, and complete recovery and loss were reached within 420 s (38, 43). In contrast, H10-Δ7-GFP, which contains seven point mutations within the globular domain and only binds weakly to cruciform DNA in vitro (26), showed dramatically faster recovery and faster loss than the corresponding wild-type protein (Fig. 1A and B). t50 was 7.3 s, and full recovery was reached within 57 s (Fig. 1B). Note that the experimental data are only normalized to the prebleach signal, and since about 50% of the initial signal is bleached, complete recovery is indicated by recovery of the signal to ∼50% of the prebleach value (Fig. 1B).
FIG. 1.

FRAP for the analysis of chromatin-binding proteins. (A) FRAP of pairs of wild-type and DNA binding-deficient chromatin proteins H10 and HMGN1. The H10 mutant contains seven point mutations within the globular domain previously demonstrated to reduce chromatin binding. The HMGN1 mutant contains mutations S20E and S24E, which abolish nucleosome binding. Fusion proteins were imaged before and during recovery after bleaching of about half the nuclear area. Images were taken at the indicated times after end of the bleach pulse. As shown in panel B, the average fluorescence intensities in the bleached (open symbols) and unbleached (solid symbols) regions were measured over time. Interference with DNA binding results in significantly faster recovery of the mutants. Bar, 5 μm. (B) Quantitative analysis of FRAP recovery of wild type and mutant H10, HMGN1 or GFP-NLS as a nonbinding control. Convergence of recovery and loss curves indicates the absence of an immobile fraction. Values are averages ± standard deviation from at least 10 cells from three experiments.
The sensitivity of FRAP to chromatin binding events was further confirmed by analysis of HMGN1-GFP and its double point mutant, HMGN1-EE-GFP, containing mutations S20E and S24E in the nucleosome binding domain in transiently transfected HeLa cells (51). The functionality of HMGN1-GFP and the loss of binding of HMGN1-EE-GFP have previously been demonstrated in vitro and in vivo (51). The recovery kinetics for the wild type, HMGN1-GFP-wt, were significantly slower than that of the mutant (Fig. 1A and B). t50 for the wild type was 17.2 s, but t50 for the nonbinding mutant was only 4.3 s, and total recovery was reached after 52 s for the wild type but within 14.8 s for the mutant (Fig. 1B). Similar results were obtained for wild-type and mutant pairs of HP1, UBF, Brd4, and FBP (data not shown) (13). As expected, recovery of GFP-NLS, which does not bind chromatin specifically, was very rapid and was completed within ∼ 2 s after bleaching (Fig. 1B). No recovery or loss was observed in chemically fixed cells. Differences in recovery kinetics between wild-type and mutant pairs cannot be explained by differences in molecular weight, since all mutants contain substitutions rather than deletions. The observed differences between wild-type and mutant proteins also exclude the possibility that the recovery kinetics of the wild type reflect nonspecific binding due to saturation of binding sites caused by overexpression. Taken together, these observations demonstrate that photobleaching is a sensitive tool to probe the binding interactions of proteins with native chromatin in intact cells.
Transient binding is a general feature of chromatin-associated proteins.
To gain an impression of the interaction dynamics of a wide range of proteins with chromatin, we examined factors involved in various aspects of chromatin function, including structure, remodeling, histone modification, and transcriptional activation (Fig. 2 and 3). The functionality of most GFP fusions has been previously tested by in vivo and/or in vitro assays (Table 1). Most tested fusion proteins localized homogeneously throughout the nucleus, were excluded from the nucleolus, and did not accumulate to any significant extent in nuclear foci (Fig. 2A). Cells expressing low levels of the fusion proteins were used in all experiments, although no significant differences in recovery rates were observed in cells expressing higher levels.
FIG. 2.

Localization and FRAP recovery and loss curves for chromatin-associated proteins. (A) Most fusion proteins were homogeneously distributed within the nucleus and excluded from the nucleolus. (B) The indicated proteins were imaged before and during recovery after bleaching of about half the nuclear area. Average fluorescence intensities in the bleached (open squares) and the unbleached region (solid squares) were measured over time. The fluorescence signal equilibrated in most cases within about 30 to 60 s, suggesting rapid exchange of proteins on chromatin. Convergence of the two curves indicates the absence of any substantial immobile, stably bound fraction. Values are averages ± standard deviation from at least 10 cells from three experiments.
FIG. 3.

Kinetic characteristics of FRAP curves for chromatin proteins. The time required to reach half-maximal recovery (t50) (A) and the time required to reach a stable plateau for recovery and loss (B) were determined for all chromatin proteins. For most chromatin proteins, t50 was between 3 and 8 s and recovery was complete within 30 to 45 s. Exceptions are the core and linker histones. Values are averages ± standard deviation from at least 10 cells from three experiments.
Qualitative analysis of FRAP experiments revealed that the vast majority of fusion proteins exhibited rapid recovery kinetics (Fig. 2B). With the exception of the positive control H10 and the FUSE-binding protein FBP, half-times of recovery and loss were typically within 3 to 8 s after the bleach pulse and recovery was completed within about 30 to 45 s for most proteins (Fig. 3). Inspection of corresponding pairs of recovery and loss curves showed that the two measurements for all proteins converged and reached a common plateau (Fig. 2B). Convergence represents complete equilibration of the fluorescence signal between the unbleached and the bleached region. Such convergence can only be achieved when the entire protein population is dynamically exchanged and no immobile, statically bound population is present. Comparable results were obtained when the same protein was analyzed in different cell types (Table 1).
We conclude from these observations that a wide variety of chromatin-associated proteins, irrespective of function or structural features, bind only transiently to chromatin in the nucleus of living cells and that virtually the entire population is dynamically exchanged. Transient binding of proteins to chromatin thus appears to be a common feature of many chromatin-associated proteins.
Kinetic modeling of protein-chromatin interactions in vivo.
To extract specific quantitative information about binding rates and residence times, we applied computational kinetic modeling methods (48, 50). We generated a mathematical model based on standard principles of chemical kinetics to describe the kinetic behavior of chromatin-binding proteins in the cell nucleus (Fig. 4A). We assumed that a chromatin-associated protein moves randomly through the nucleoplasm and binds at random intervals to chromatin. On average, the protein resides on chromatin for a certain period of time, which we refer to as mean residence time, before it dissociates and moves to another binding site. The overall movement of a chromatin-associated protein through nuclear space is thus determined by two factors: translational mobility between binding sites and the binding reactions themselves. Because the translational, diffusional mobility is much faster than the rate-limiting binding events, the movement of a protein through nuclear space can be described in a first approximation as a simple association/dissociation reaction (see Materials and Methods). Since the absolute abundances of each protein and their binding sites are unknown, second order association processes were converted to pseudo-first-order processes by combining the unknown steady-state binding site abundance with the corresponding second order rate constant to yield a first order association rate constant. This has the additional advantage that the nonlinear system of differential equations is converted to a linear system (see Materials and Methods for details).
FIG. 4.

Kinetic modeling of chromatin binding. (A) Compartmental model for analysis of half-FRAP photobleaching data. The model consists of an unbound nucleoplasmic pool of protein that exchanges with two classes of nuclear binding sites, a rapidly exchanging pool and a slowly exchanging pool. These pools exist in both the unbleached and bleached regions of the nucleus and are replicated in order to accurately simulate the experimental bleaching protocols. Arrows represent processes of binding and unbinding; they are labeled with the corresponding rate constants. Protein synthesis and degradation were assumed to be negligible on the time scale of these photobleaching experiments. Diffusion events are neglected since they are much faster than the binding events and are not rate limiting. (B) Least-square best fit of experimental recovery and loss data for Mad and FBP. For Mad, a two-site binding model gives a significantly better fit than a one-site binding model. For FBP, a one-site binding model is sufficient. Symbols represent experimental data, and lines represent best fits.
The kinetic model contains as parameters the on and off rate constants and the abundances of bound and unbound molecules (Fig. 4A) (Materials and Methods). The size of the total bound fraction was experimentally determined by taking advantage of the fact that the diffusion time of a protein is much shorter than the bleach time used (Table 2). For each protein, the fluorescence intensity in the unbleached region before bleaching was compared to the intensity immediately after the bleach pulse. This difference is an indication of the bound fraction, since a bleach pulse of 446 ms completely abolishes the fluorescence signal from freely diffusing, unbound molecules in the unbleached region (data not shown). For all chromatin-associated proteins, the bound fraction was around 90% or higher (Table 2). This observation suggests that the vast majority of molecules are at any give time bound to chromatin. The size of the bound fraction was used as a constraint for the fitting of the experimental data to the modeling data.
TABLE 2.
Kinetic properties of chromatin proteins
| Protein | Fast fraction
|
Slow fraction
|
Total bound fraction (%) | ||||
|---|---|---|---|---|---|---|---|
| koff1 | Mean residence time (s) | Fraction size (%) | koff2 | Residence time (s) | Fraction size (%) | ||
| GFP | <1 | <1 | |||||
| H2B | >3,600 | >98 | >98 | ||||
| H1° | <0.004 | 183 | >99 | >99 | |||
| H1°ΔC | 0.082 | 14.4 | 99 | 99.0 | |||
| HMGB1 | 0.209 | 4.7 | 95.9 | 95.9 | |||
| HMGN1 | 0.241 | 4.1 | 19.9 | 0.040 | 24.8 | 79.5 | 99.5 |
| HMGN1-E20, E24 | 0.294 | 3.4 | 96.5 | 96.5 | |||
| HP1β | 0.089 | 11.3 | 88.1 | 0.013 | 73.0 | 12.0 | 99 |
| BRG1 | 0.326 | 3.1 | 24.9 | 0.51 | 19.4 | 66.2 | 91.1 |
| PCAF | 0.209 | 4.7 | 81.0 | 0.058 | 17.1 | 12.7 | 93.7 |
| AhR | 0.253 | 4.0 | 42.9 | 0.039 | 25.6 | 56.1 | 98.9 |
| ARNT | 0.137 | 7.3 | 90.1 | 0.032 | 30.9 | 7.8 | 97.9 |
| C/EBP | 0.15 | 6.7 | 64.5 | 0.053 | 18.8 | 35.5 | 99.0 |
| NF1 | 0.216 | 4.6 | 34.4 | 0.061 | 16.2 | 58.1 | 92.5 |
| Jun | 0.169 | 5.9 | 19.5 | 0.037 | 27.3 | 75.2 | 94.7 |
| Fos | 0.417 | 2.4 | 32.4 | 0.068 | 14.6 | 54.2 | 86.6 |
| Myc | 0.180 | 5.5 | 45.1 | 0.061 | 16.3 | 54.1 | 99.1 |
| Max | 0.197 | 5.1 | 64.3 | 0.071 | 13.9 | 34.5 | 98.7 |
| Mad | 0.270 | 3.7 | 76.7 | 0.051 | 19.5 | 21.2 | 97.9 |
| FBP | 0.016 | 63.6 | 99.7 | 99.7 | |||
| XBP | 0.343 | 2.9 | 78.6 | 0.043 | 23.1 | 18.6 | 97.3 |
| BRD4 | 0.127 | 7.9 | 27.6 | 0.026 | 38.1 | 62.4 | 90.0 |
In preliminary modeling analyses, we found that the recovery kinetics for most proteins could be most accurately fit by two exponentials (data not shown) (Tables 3 and 4), indicating that most proteins were present in the nucleus in at least two distinguishable populations with distinct binding kinetics. The complete kinetic model for a protein with two kinetic populations is shown in Fig. 4A, and the set of differential equations describing the model is shown in Materials and Methods. For simplicity, we refer to the two populations as “fast” and “slow.”
TABLE 3.
Statistical significance of parameter fitsa
| Protein | No. of phases | Fast fraction
|
Slow fraction
|
Total bound fraction (%) | ||
|---|---|---|---|---|---|---|
| koff | Fraction size (%) | koff | Fraction size (%) | |||
| H2B | 1 | 2.2 | <0.1 | 0.2 | ||
| H1° | 2 | 3.75 | 0.3 | 0.3 | ||
| H1°ΔC | 1 | 4.2 | 7.1 | <0.1 | ||
| HMGB1 | 1 | 1.7 | <0.1 | <0.1 | ||
| HMGN1 | 2 | 10.6 | 7.0 | 17.6 | 17.6 | <0.1 |
| HMGN1-E20, -E24 | 1 | 3.4 | 9.6 | <0.1 | ||
| HP1β | 2 | 11.3 | 4.7 | 8.9 | 12.0 | <0.1 |
| BRG1 | 2 | 11.2 | 5.2 | 2.1 | 1.9 | 0.1 |
| PCAF | 2 | 5.3 | 5.5 | 23.6 | 3.5 | 0.8 |
| AhR | 2 | 11.3 | 7.5 | 5.8 | 5.7 | <0.1 |
| ARNT | 2 | 0.9 | 1.2 | 4.7 | 7.2 | 0.7 |
| C/EBP | 2 | 6.7 | 8.5 | 6.2 | 4.8 | 0.4 |
| NF1 | 2 | 10.7 | 11.7 | 4.1 | 6.9 | 0.2 |
| Jun | 2 | 6.5 | 4.4 | 1.2 | 1.1 | 0.4 |
| Fos | 2 | 1.0 | 7.9 | 4.5 | 4.7 | 0.5 |
| Myc | 2 | 16.7 | 23.4 | 10.1 | 19.5 | <0.1 |
| Max | 2 | 9.3 | 13.8 | 12.4 | 25.8 | <0.1 |
| Mad | 2 | 3.9 | 10.4 | 8.2 | 10.4 | <0.1 |
| FBP | 1 | 0.3 | 0.13 | <0.1 | ||
| XBP | 2 | 3.8 | 1.9 | 7.9 | 7.9 | <0.1 |
| BRD4 | 2 | 8.4 | 8.2 | 3.3 | 3.6 | 0.6 |
Statistical significance is expressed as the coefficient of variation (CV [percentage error relative to average value]). Note that the CV values for Tres are identical to those of koff, since Tres = 1/koff.
TABLE 4.
Evaluation of a one-site versus a two-site model
| Protein | Akaike information criteriona
|
|
|---|---|---|
| 2-site model | 1-site model | |
| H2B | NA | −3.82 |
| H10 | −2.8 | −2.74 |
| HMGB1 | NA | −2.94 |
| HMGN1 | −3.23 | −3.02 |
| HP1β | −4.1 | −3.28 |
| BRG1 | −3.16 | −2.85 |
| PCAF | −3.04 | −2.74 |
| AhR | −2.56 | −2.30 |
| ARNT | −3.56 | −3.27 |
| C/EBP | −3.54 | −3.08 |
| NF1 | −3.11 | −2.84 |
| Jun | −3.87 | −3.29 |
| Fos | −2.77 | −2.55 |
| Myc | −2.71 | −2.54 |
| Max | −3.20 | −2.95 |
| Mad | −3.17 | −2.54 |
| FBP | NA | −3.31 |
| XBP | −3.09 | −2.43 |
| BRD4 | −3.44 | −2.89 |
Experimental data were fitted to either a one or a two-site binding model. A variant of the Akaike information criterion (AIC) was used to identify the best model for each protein (2). The AIC is a function of the objective function used by the parameter estimation routine in SAAM II, the number of adjustable parameters, and the number of data points. This function penalizes an improvement in the residual sum of squares whenever additional parameters are added to the model. The model with the minimum AIC is always preferred. The lower value is in boldface. With the exception of H2B, HMGB1, and FBP, all experimental data fit more accurately a two-site model than a one-site model. NA, not applicable.
Quantitative binding properties of chromatin-associated proteins in vivo.
Using this kinetic model, we simulated in silico the FRAP experiments to obtain best fits between the experimental data and the simulation (Fig. 4B). Best-fit parameters for each protein's off rates, mean residence times, and the sizes of the fast and slow fractions were obtained by using a generalized least-squares method (Fig. 4B and Table 2). Error margins for all best-fit parameters were typically around 10% of the measured values, and coefficients of variance are given in Table 3. The obtained values are based on the assumption that the total of available specific and nonspecific binding sites is in excess of the number of molecules of the observed protein. To ensure that a kinetic model containing two distinct types of binding sites was more accurate than a simple single-site model, we determined best parameter fits for all proteins for a one-site or two-site model and assessed the goodness of fit according to the Akaike information criterion (Table 4). With the exception of H2B, HMGB1, and FBP, all proteins fit more accurately to a two-site model (Fig. 4B and Table 4).
The two koff rates obtained from the two-site binding models typically differed by roughly an order of magnitude (Table 2): A fast population with a koff between 0.15 and 0.3 and a slow population with a koff between 0.03 and 0.07 (Table 2). These values correspond to residence times (defined as 1/koff) of the fast population of 3 to 6 s and 15 to 30 s for the slow population (Table 2). The shortest mean residence times of ∼2 s were found for Jun and XBP, whereas FBP and HP1β had the longest mean residence times of ∼ 70 s. As a control, GFP had a nominal mean residence time of less than 1 s, which is within our experimental error (Table 2). We confirmed that core histones had mean residence times on the order of hours (Table 2). Furthermore, in agreement with earlier qualitative reports, we determined the mean residence time of H10 to be ∼3 min and a H10-ΔC mutant, which lacks the entire C terminus and has severely reduced DNA binding activity, had a mean residence time on the order of 14 s (Table 2) (43). When fits were forced to a single binding site model, mean residence times on the order of 5 to 30s were obtained for most proteins (data not shown). It is likely that populations with faster binding dynamics exist that are below our temporal resolution limit. However, computer simulations show that the experimentally observed FRAP recovery kinetics are inconsistent with large populations of mean residence times in the subsecond range (data not shown).
We next determined the relative sizes of the fast and slow fractions for each protein by using best-fit analysis. As might be expected based on the diverse functions of the analyzed proteins, most proteins showed distinct combinations of residence time and the relative proportions of slow and fast binding fractions (Table 2). For some proteins, the major fraction showed rapid binding, whereas for other proteins the rapid fraction only represented a minor population (Table 2). For example, only about 20% of BRG1 or Jun molecules were in the fast fraction, whereas more than 80% of PCAF or ARNT molecules made up the fast fraction (Table 2). As a control, we find that impairment of nucleosome binding activity of HMGN1 results as predicted in complete loss of the slow fraction and increases the fast fraction to more than 96%, compared to less than 20% for the wild-type protein (Table 2).
Two proteins, HMGB1 and FBP, fit more accurately to a single-site model, although their mean residence times were dramatically different (Tables 2 and 4). While the DNA binding protein HMGB1 is a rapid binder, with its entire population having a mean residence time of ∼4 s, the entire population of the TFIIH interacting protein FBP had a mean residence time of more than 60 s (Table 2). The short mean residence time of HMGB1 is consistent with its proposed role as a stimulator of chromatin remodeling via transient interaction with chromatin (5).
Structural proteins had an overall tendency for slower turnover. The slow fraction of HP1β with a mean residence time of 72 s had the longest mean residence time of all proteins, and although more than 80% of the protein turns over within 11 s, even this mean residence time is by far the longest among the fast fractions (Table 2). This long residence time is inconsistent with a suggested role of FBP in RNA binding. Similarly, the relatively long mean residence time of 24.8 s in combination with its large slow fraction of almost 80% makes the HMGN1 one of the more stably associated proteins (Table 2). Thus, although even structural proteins interact in a very transient fashion with chromatin, their somewhat lower turnover might be critical for the establishment and maintenance of stable chromatin domains (12).
The two enzymatic chromatin-associated proteins BRG1 and the histone deacetylase PCAF showed very distinct kinetic profiles (Table 2). BRG1 was predominantly present in a relatively slow fraction with a mean residence time of about 20 s; in contrast, PCAF was highly enriched in a rapid fraction with a mean residence time of less than 5 s. The differences might reflect the protein's different functions as PCAF modifies histone tails in a presumably very rapid enzymatic reaction, whereas BRG1 might be required for longer periods of time to bring about the structural changes involved in chromatin remodeling.
The transcriptional activator ARNT was predominantly (90%) found in a rapidly exchanging fraction, and only about 8% of the total was bound for longer periods (30 s). The size of this slow fraction is similar to estimates based on localization data of the fraction of ARNT present in transcriptionally engaged AhR-ARNT complexes (20). Furthermore, the estimated fast fraction of AhR of about 42% is in the same range as the fraction estimated to be associated with cellular transcription sites. These data are consistent with a model in which ARNT scans potential target genes and then recruits AhR temporarily to these genes, resulting in their activation (20).
The sum of these results demonstrates that transient binding is a common feature of many chromatin-associated proteins in vivo, that chromatin-associated proteins exist in several, kinetically distinct populations, and that at steady state the major population of each protein is bound to chromatin rather than present in a soluble form in the nucleoplasm.
DISCUSSION
We describe and apply here a combined microscopy-computation approach to analyze the binding dynamics of a wide range of proteins with chromatin in the nucleus of living cells. We find by quantitative analysis that transient binding is a common property of many chromatin-associated proteins. Mean residence times are typically on the order of 2 to 20 s, and for most proteins, no significant population of immobile, statically associated molecules was detected. Although we cannot strictly rule out the possibility that detection of more stable interactions were obscured due to overexpression of the fluorescently tagged protein, several lines of evidence argue against this scenario. First, transient binding with similar residence times was observed for a range of proteins representing a spectrum of endogenous proteins, whose abundances and numbers of endogenous binding sites differ. If factors such as endogenous abundance and available binding sites critically and artifactually determined our results, a larger range of residence times would be expected. Second, the dynamic behavior of a given protein was similar among several cell types, but differences between proteins were observed within a given cell type. Third, relatively small protein fractions can be detected by our method. We estimate the abundance of the overexpressed protein to be typically between 10,000 to 100,000 molecules per nucleus (18). Since the sensitivity in detecting kinetic fractions is about 5% of the total protein (Table 3), kinetically distinct populations of 500 to 5,000 molecules can be detected. Thus, even relatively low-abundance binding events would be detected against a pool of overexpressed protein. Fourth, comparison of pairs of wild-type and mutant proteins in all cases showed a dramatic reduction in the residence time of the mutant protein, suggesting that the observed slower binding of the wild-type protein was not due to nonphysiological binding. Fifth, our results are consistent with qualitative observations in numerous well-characterized experimental systems (3, 8, 13, 16, 17, 30, 31, 35, 38, 40, 51, 57, 59). For example, in the case of HP1, similar results in FRAP experiments have been obtained in transient and stable expression systems as well as in a gene replacement system (13, 23). Importantly, the dynamic properties of HP1 as well as its yeast homologue, swi6, were independent of the expression level of the GFP-fusion protein (12). Since HP1 behaves similarly to several of the proteins observed here, it is likely that these binding events are also representative of the physiological behavior of these proteins. Furthermore, although we cannot strictly exclude the possibility that some of the retardation in FRAP kinetics is due to nonchromatin-mediated binding, for example, to a nuclear matrix or to storage compartments, the sensitivity of the FRAP curves of six chromatin-associated proteins to mutations that affect chromatin binding suggests that our measurements indeed primarily reflect binding to chromatin.
While we can accurately fit the experimental data for most proteins to a kinetic model containing two distinct binding types, the biological significance of the two kinetic fractions is unclear and might be different for each protein. It is tempting to suggest that the fast and slow fractions represent nonspecific and specific binding events for each protein. However, the inability to accurately determine critical parameters such as the number of endogenous molecules and binding sites and the ratio of exogenous and endogenous protein for most of the analyzed proteins makes it difficult to generalize this interpretation. The biological meaning of each kinetic fraction should be determined on a protein-to-protein basis. Regardless, the most likely cause for generating distinct kinetic fractions is the temporary immobilization of transcription factors upon their functional binding to distinct types of target sequences. Well-documented examples of modulation of the dynamic properties of proteins due to their functional interaction with chromatin include the steroid receptors, which become temporarily immobilized on target genes upon stimulation with ligand (59), RNA polymerases I and II upon transcriptional engagement (3, 17, 35), and DNA repair and replication factors upon association with repair and replication sites (30, 31, 57).
Although our measurements provide an estimate of residence times, they do not address the underlying cause for the exchange of a protein on chromatin. Mean residence times on the order of tens of seconds or minutes are unlikely to simply reflect spontaneous exchange events but might point to an involvement of regulatory activities. Recruitment of members of the chaperone family to specific genes during hormone activation and an effect of molecular chaperone activity on the mobility of the glucocorticoid receptor (GR) and the progesterone nuclear receptors have been reported (21, 25). Similarly, proteosome activity influences the exchange dynamics of glucocorticoid and estrogen receptors (55, 58, 59). Furthermore, a role for chromatin remodeling in the active exchange of the GR has been proposed, since it appears that GR is actively displaced from the template during the remodeling process (24, 29, 45). It will be important to determine whether these active processes are limited to controlling the binding of steroid receptors or are generally involved in the rapid exchange of proteins on chromatin.
A 3D genome-scanning model for chromatin-associated proteins.
Our observations of transient binding and the large bound fraction of all analyzed chromatin-associated proteins support a model in which a single molecule of a chromatin-associated protein resides on chromatin for a few seconds and then dissociates and diffuses for a relatively short period of time before it associates with a new site, most likely on a different chromatin fiber (53). While we have not systematically determined the time a molecule spends diffusing through the nucleoplasm between binding events, preliminary observations on the linker histone H1 indicate that this time is on the order of 200 to 400 ms (T. Misteli, unpublished observation). Regardless, the dynamic exchange of chromatin-associated proteins combined with a large population of bound molecules generates a steady state in which a predominantly bound population of molecules continuously samples the genome for appropriate binding sites by diffusional hopping between chromatin fibers. This mode of action is an effective way of ensuring availability of chromatin-associated proteins throughout the nucleus and is a simple means to efficiently target proteins to their binding sites (42).
Dynamic protein interaction networks on chromatin.
Dynamic exchange of chromatin-associated proteins has implications for chromatin function and gene regulation. The characteristic transient binding of proteins to a site in the genome results in a high flux of molecules at a particular binding site. As a consequence, the maintenance of a protein's occupancy, or of a protein complex on chromatin, requires the continuous supply of the involved factors. A requirement for continuous supply of repressor has been demonstrated in yeast, where silencing of mating-type genes requires the continuous presence of its silencers (11). Similarly, in mammalian cells, the continuous maintenance of active ribosomal genes and the formation of preinitiation and transcription complexes require the uninterrupted supply of polymerase components (17).
As a consequence of the lability of protein-chromatin interactions, simple competition among potential binding partners for any given site in the genome emerges as a major determinant of chromatin states (42, 53). Upon dissociation of any protein, the available binding site is open for competition by multiple binding factors. Since the outcome of this competition is largely dependent on the local concentration and the affinity of the competing factors, dynamic competition is a simple, but effective, means to modulate chromatin states. In heterochromatin of yeast and mammals, for example, the mandatory structural protein HP1, which only binds transiently to chromatin, can be displaced from pericentromeric heterochromatin within minutes upon alterations of the chromatin compaction status (13). Similarly, expression of the GAL4 activator in a reporter system in Drosophila can overcome heterochromatic silencing, most likely by affecting the binding site occupancy of silencing factors (1). While these examples consist of two known competitors, more complex dynamic interaction networks acting on chromatin are likely commonplace. One such network is the dynamic competitive interactions between the linker histone H1 and multiple HMG proteins as recently described in living cells (7, 8). The interplay of multiple proteins in this manner in the form of dynamic interaction networks can account for some of the most general properties of gene expression systems, such as rapid response, plasticity, diversity, robustness, and integration of signals. Since the transient binding of proteins to chromatin is a critical component of such interaction networks, we suggest that the dynamic nature of chromatin-associated proteins is a key feature of gene regulation processes (42, 53).
Acknowledgments
We thank T. Kerppola, D. Levens, and E. Prochownik for reagents.
J.V. was partially funded by grant IAA5039103 from the Academy of Sciences of the Czech Republic and MSM111100003 from the Ministry of Education, Youth and Sports. D.T.B. was supported by NSF grant MCB-0235800. J.V. acknowledges receipt of a Journal of Cell Science Traveling Fellowship. T.M. is a Fellow of the Keith R. Porter Endowment for Cell Biology.
REFERENCES
- 1.Ahmad, K., and S. Henikoff. 2001. Modulation of a transcription factor counteracts heterochromatic gene silencing in Drosophila. Cell 104:839-847. [DOI] [PubMed] [Google Scholar]
- 2.Akaike, H. 1974. A new look at statistical model identification. IEEE Trans. Auto. Control 19:716-723. [Google Scholar]
- 3.Becker, M., C. Baumann, S. John, D. A. Walker, M. Vigneron, J. G. McNally, and G. L. Hager. 2002. Dynamic behavior of transcription factors on a natural promoter in living cells. EMBO Rep. 3:1188-1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bell, B. M. 2001. Approximating the marginal likelihood estimate for models with random parameters. Appl. Math. Comput. 119:57-75. [Google Scholar]
- 5.Bonaldi, T., G. Langst, R. Strohner, P. B. Becker, and M. E. Bianchi. 2002. The DNA chaperone HMGB1 facilitates ACF/CHRAC-dependent nucleosome sliding. EMBO J. 21:6865-6873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bustin, M. 1999. Regulation of DNA-dependent activities by the functional motifs of the high-mobility-group chromosomal proteins. Mol. Cell. Biol. 19:5237-5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Catez, F., D. T. Brown, T. Misteli, and M. Bustin. 2002. Competition between histone H1 and HMGN proteins for chromatin binding sites. EMBO Rep. 3:760-766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Catez, F., Y. Huan, K. J. Tracey, R. Reeves, T. Misteli, and M. Bustin. 2004. A network of dynamic interations between histone H1 and HMG proteins in chromatin. Mol. Cell. Biol. 24:4321-4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chalfie, M., Y. Tu, G. Euskirchen, W. Ward, and D. Prasher. 1994. Green fluorescent protein as a marker for gene expression. Science 263:802-805. [DOI] [PubMed] [Google Scholar]
- 10.Chen, D., C. S. Hinkley, R. W. Henry, and S. Huang. 2002. TBP dynamics in living human cells: constitutive association of TBP with mitotic chromosomes. Mol. Biol. Cell 13:276-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng, T. H., and M. R. Gartenberg. 2000. Yeast heterochromatin is a dynamic structure that requires silencers continuously. Genes Dev. 14:452-463. [PMC free article] [PubMed] [Google Scholar]
- 12.Cheutin, T., S. A. Gorski, K. M. May, P. B. Singh, and T. Misteli. 2004. In vivo dynamics of Swi6 in yeast: evidence for a stochastic model of heterochromatin. Mol. Cell. Biol. 24:3157-3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheutin, T., A. J. McNairn, T. Jenuwein, D. M. Gilbert, P. B. Singh, and T. Misteli. 2003. Maintenance of stable heterochromatin domains by dynamic HP1 binding. Science 299:721-725. [DOI] [PubMed] [Google Scholar]
- 14.Cobelli, C., and D. M. Foster. 1998. Compartmental models: theory and practice using the SAAM II software system. Adv. Exp. Med. Biol. 445:79-101. [DOI] [PubMed] [Google Scholar]
- 15.Dey, A., F. Chitsaz, A. Abbasi, T. Misteli, and K. Ozato. 2003. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Natl. Acad. Sci. USA 100:8758-8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dou, Y., J. Bowen, Y. Liu, and M. A. Gorovsky. 2002. Phosphorylation and an ATP-dependent process increase the dynamic exchange of H1 in chromatin. J. Cell Biol. 158:1161-1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dundr, M., U. Hoffmann-Rohrer, Q. Hu, I. Grummt, L. I. Rothblum, R. D. Phair, and T. Misteli. 2002. A kinetic framework for a mammalian RNA polymerase in vivo. Science 298:1623-1626. [DOI] [PubMed] [Google Scholar]
- 18.Dundr, M., J. G. McNally, J. Cohen, and T. Misteli. 2002. Quantitation of GFP-fusion proteins in single living cells. J. Struct. Biol. 140:92-99. [DOI] [PubMed] [Google Scholar]
- 19.Eissenberg, J. C., and S. C. R. Elgin. 2000. The HP1 protein family: getting a grip on chromatin. Curr. Opin. Gen. Dev. 10:204-210. [DOI] [PubMed] [Google Scholar]
- 20.Elbi, C., T. Misteli, and G. L. Hager. 2002. Recruitment of dioxin receptor to active transcription sites. Mol. Biol. Cell 13:2001-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elbi, C. C., D. A. Walker, G. Romero, W. P. Sullivan, D. O. Toft, G. L. Hager, and D. B. DeFranco. 2004. Molecular chaperones function as steroid receptor nuclear mobility factors. Proc. Natl. Acad. Sci. USA 2876-2881. 101: [DOI] [PMC free article] [PubMed]
- 22.Felsenfeld, G., and M. Groudine. 2003. Controlling the double helix. Nature 421:448-453. [DOI] [PubMed] [Google Scholar]
- 23.Festenstein, R., S. N. Pagakis, K. Hiragami, D. Lyon, A. Verreault, B. Sekkali, and D. Kioussis. 2003. Modulation of heterochromatin protein 1 dynamics in primary mammalian cells. Science 299:719-721. [DOI] [PubMed] [Google Scholar]
- 24.Fletcher, T. M., N. Xiao, G. Mautino, C. T. Baumann, R. Wolford, B. S. Warren, and G. L. Hager. 2002. ATP-dependent mobilization of the glucocorticoid receptor during chromatin remodeling. Mol. Cell. Biol. 22:3255-3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Freeman, B. C., and K. R. Yamamoto. 2002. Disassembly of transcriptional regulatory complexes by molecular chaperones. Science 296:2232-2235. [DOI] [PubMed] [Google Scholar]
- 26.Goytisolo, F. A., S. E. Gerchman, X. Yu, C. Rees, V. Graziano, V. Ramakrishnan, and J. O. Thomas. 1996. Identification of two DNA-binding sites on the globular domain of histone H5. EMBO J. 15:3421-3429. [PMC free article] [PubMed] [Google Scholar]
- 27.Grewal, S. I., and S. C. Elgin. 2002. Heterochromatin: new possibilities for the inheritance of structure. Curr. Opin. Genet. Dev. 12:178-187. [DOI] [PubMed] [Google Scholar]
- 28.Gunjan, A., B. T. Alexander, D. B. Sittman, and D. T. Brown. 1999. Effects of H1 histone variant overexpression on chromatin structure. J. Biol. Chem. 274:37950-37956. [DOI] [PubMed] [Google Scholar]
- 29.Hager, G. L., C. Elbi, and M. Becker. 2002. Protein dynamics in the nuclear compartment. Curr. Opin. Genet. Dev. 12:137-141. [DOI] [PubMed] [Google Scholar]
- 29a.He, L., A. Weber, and D. Levens. 2000. Nuclear targeting determinants of the far upstream element binding protein, a c-myc transcription factor. Nucleic Acids Res. 28:4558-4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoogstraten, D., A. L. Nigg, H. Heath, L. H. Mullenders, R. van Driel, J. H. Hoeijmakers, W. Vermeulen, and A. B. Houtsmuller. 2002. Rapid switching of TFIIH between RNA polymerase I and II transcription and DNA repair in vivo. Mol. Cell 10:1163-1174. [DOI] [PubMed] [Google Scholar]
- 31.Houtsmuller, A. B., S. Rademakers, A. L. Nigg, D. Hoogstraten, J. H. Hoeijmakers, and W. Vermeulen. 1999. Action of DNA repair endonuclease ERCC1/XPF in living cells. Science 284:958-961. [DOI] [PubMed] [Google Scholar]
- 32.Houtsmuller, A. B., and W. Vermeulen. 2001. Macromolecular dynamics in living cell nuclei revealed by fluorescence redistribution after photobleaching. Histochem. Cell Biol. 115:13-21. [DOI] [PubMed] [Google Scholar]
- 33.Kanda, T., K. F. Sullivan, and G. M. Wahl. 1998. Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr. Biol. 26:377-385. [DOI] [PubMed] [Google Scholar]
- 34.Kimura, H., and P. R. Cook. 2001. Kinetics of core histones in living human cells: little exchange of H3 and H4 and some rapid exchange of H2B. J. Cell Biol. 153:1341-1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kimura, H., K. Sugaya, and P. R. Cook. 2002. The transcription cycle of RNA polymerase II in living cells. J. Cell Biol. 159:777-782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Labrador, M., and V. G. Corces. 2002. Setting the boundaries of chromatin domains and nuclear organization. Cell 111:151-154. [DOI] [PubMed] [Google Scholar]
- 37.Lemon, B., and R. Tjian. 2000. Orchestrated response: a symphony of transcription factors for gene control. Genes Dev. 14:2551-2569. [DOI] [PubMed] [Google Scholar]
- 38.Lever, M. A., J. P. H. Th'ng, X. Sun, and M. J. Hendzel. 2000. Rapid exchange of histone H1.1 on chromatin in living cells. Nature 408:873-876. [DOI] [PubMed] [Google Scholar]
- 39.Lippincott-Schwartz, J., E. Snapp, and A. Kenworthy. 2001. Studying protein dynamics in living cells. Nat. Rev. Mol. Cell Biol. 2:444-456. [DOI] [PubMed] [Google Scholar]
- 40.McNally, J. G., W. G. Muller, D. Walker, R. Wolford, and G. L. Hager. 2000. The glucocorticoid receptor: rapid exchange with regulatory sites in living cells. Science 287:1262-1265. [DOI] [PubMed] [Google Scholar]
- 41.Meyvis, T. K., S. C. De Smedt, P. Van Oostveldt, and J. Demeester. 1999. Fluorescence recovery after photobleaching: a versatile tool for mobility and interaction measurements in pharmaceutical research. Pharm. Res. 16:1153-1162. [DOI] [PubMed] [Google Scholar]
- 42.Misteli, T. 2001. Protein dynamics: implications for nuclear architecture and gene expression. Science 291:843-847. [DOI] [PubMed] [Google Scholar]
- 43.Misteli, T., A. Gunjan, R. Hock, M. Bustin, and D. T. Brown. 2000. Dynamic binding of histone H1 to chromatin in living cells. Nature 408:877-881. [DOI] [PubMed] [Google Scholar]
- 44.Naar, A. M., B. D. Lemon, and R. Tjian. 2001. Transcriptional coactivator complexes. Annu. Rev. Biochem. 70:475-501. [DOI] [PubMed] [Google Scholar]
- 45.Nagaich, A. K., D. A. Walker, R. Wolford, and G. L. Hager. 2004. Rapid periodic binding and displacement of the glucocorticoid receptor during chromatin remodeling. Mol. Cell 14:163-174. [DOI] [PubMed] [Google Scholar]
- 46.Orphanides, G., and D. Reinberg. 2002. A unified theory of gene expression. Cell 108:439-451. [DOI] [PubMed] [Google Scholar]
- 47.Pederson, T. 2001. Protein mobility within the nucleus—what are the right moves? Cell 104:635-638. [DOI] [PubMed] [Google Scholar]
- 48.Phair, R. D., S. Gorski, and T. Misteli. 2004. Measurement of dynamic protein binding to chromatin in vivo using photobleaching microscopy. Methods Enzymol. 375:393-414. [DOI] [PubMed] [Google Scholar]
- 49.Phair, R. D., and T. Misteli. 2000. High mobility of proteins in the mammalian cell nucleus. Nature 404:604-609. [DOI] [PubMed] [Google Scholar]
- 50.Phair, R. D., and T. Misteli. 2001. Kinetic modelling approaches to in vivo imaging. Nat. Rev. Mol. Cell Biol. 2:898-907. [DOI] [PubMed] [Google Scholar]
- 51.Prymakowsa-Bosak, M., T. Misteli, J. E. Herrera, H. Shirakawa, Y. Birger, S. Garfield, and M. Bustin. 2001. Mitotic phosphorylation prevents the binding of HMGN proteins to chromatin. Mol. Cell. Biol. 21:5169-5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Purich, D. L., and R. D. Allison. 2000. Handbook of biochemical kinetics. Academic Press, San Diego, Calif.
- 53.Roix, J., and T. Misteli. 2002. Genomes, proteomes, and dynamic networks in the cell nucleus. Histochem. Cell Biol. 118:105-116. [DOI] [PubMed] [Google Scholar]
- 54.Scaffidi, P., T. Misteli, and M. E. Bianchi. 2002. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418:191-195. [DOI] [PubMed] [Google Scholar]
- 55.Schaaf, M. J., and J. A. Cidlowski. 2003. Molecular determinants of glucocorticoid receptor mobility in living cells: the importance of ligand affinity. Mol. Cell. Biol. 23:1922-1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schaufele, F., J. F. Enwright III, X. Wang, C. Teoh, R. Srihari, R. Erickson, O. A. MacDougald, and R. N. Day. 2001. CCAAT/enhancer binding protein alpha assembles essential cooperating factors in common subnuclear domains. Mol. Endocrinol. 15:1665-1676. [DOI] [PubMed] [Google Scholar]
- 57.Sporbert, A., A. Gahl, R. Ankerhold, H. Leonhardt, and M. C. Cardoso. 2002. DNA polymerase clamp shows little turnover at established replication sites but sequential de novo assembly at adjacent origin clusters. Mol. Cell 10:1355-1365. [DOI] [PubMed] [Google Scholar]
- 58.Stavreva, D. A., W. G. Muller, G. L. Hager, C. L. Smith, and J. G. McNally. 2004. Rapid glucocorticoid receptor exchange at a promoter is coupled to transcription and regulated by chaperones and proteasomes. Mol. Cell. Biol. 24:2682-2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stenoien, D. L., K. Patel, M. G. Mancini, M. Dutertre, C. L. Smith, B. W. O'Malley, and M. A. Mancini. 2001. FRAP reveals that mobility of oestrogen receptor-α is ligand and proteasome-dependent. Nat. Cell Biol. 3:15-23. [DOI] [PubMed] [Google Scholar]
- 60.Strahl, B. D., and C. D. Allis. 2000. The language of covalent histone modifications. Nature 403:41-45. [DOI] [PubMed] [Google Scholar]
- 61.Thomas, J. O., and A. A. Travers. 2001. HMG1 and 2, and related 'architectural' DNA-binding proteins. Trends Biochem. Sci. 26:167-174. [DOI] [PubMed] [Google Scholar]
- 62.Verkman, A. S. 2002. Solute and macromolecule diffusion in cellular aqueous compartments. Trends Biochem. Sci. 27:27-33. [DOI] [PubMed] [Google Scholar]
- 63.Woodcock, C. L., and S. Dimitrov. 2001. Higher-order structure of chromatin and chromosomes. Curr. Opin. Genet. Dev. 11:130-135. [DOI] [PubMed] [Google Scholar]
- 64.Yin, X., M. F. Landay, W. Han, E. S. Levitan, S. C. Watkins, R. M. Levenson, D. L. Farkas, and E. V. Prochownik. 2001. Dynamic in vivo interactions among Myc network members. Oncogene 20:4650-4664. [DOI] [PubMed] [Google Scholar]