

Background: Polymorphisms within solute carrier family 22 member 3 (SLC22A3) affects the risk of cardiovascular disease.
Results: The polymorphism rs3088442 decreases SLC22A3 mRNA stability and inhibits lipopolysaccharide-induced inflammatory responses.
Conclusion: This polymorphism decreased CHD risk by controlling vascular inflammation.
Significance: Our findings will elucidate the relationship between SLC22A3 variants, inflammation, and CHD pathogenesis.
Keywords: Atherosclerosis, Gene Expression, Inflammation, MicroRNA (miRNA), Single-nucleotide Polymorphism (SNP), LPS-induced Inflammatory Response, SLC22A3, Histamine, miR-147, rs3088442
Abstract
Recent genome-wide association studies have identified single-nucleotide polymorphism (SNPs) within the SLC22A3 (solute carrier family 22 member 3) gene associated with coronary heart disease (CHD) in the Caucasian population. We performed molecular analysis to investigate the potential role of SLC22A3 variants in CHD. Our study showed that the common polymorphism rs3088442 G→A, which is localized in the 3′ UTR of the SLC22A3 gene, was associated with a decreased risk of CHD in the Chinese population by a case control study. In silico analysis indicated that G→A substitution of SNP rs3088442 created a putative binding site for miR-147 in the SLC22A3 mRNA. By overexpressing miR-147 or inhibiting endogenous miR-147, we demonstrated that SNP rs3088442 G→A recruited miR-147 to inhibit SLC22A3 expression. Moreover, SLC22A3 deficiency significantly decreased LPS-induced monocytic inflammatory response by interrupting NF-κB and MAPK signaling cascades in a histamine-dependent manner. Notably, the expression of SLC22A3A was also suppressed by LPS stimulus. Our findings might indicate a negative feedback mechanism against inflammatory response by which SLC22A3 polymorphisms decreased the risk of CHD.
Introduction
In recent years, there has been an increasing recognition of the link between inflammation and coronary heart disease (CHD).3 The pathogenic role of endotoxins (such as lipopolysaccharide (LPS)) in atherosclerosis initiation has attracted increased attention, because endotoxins are usually considered as a potentially important source of inflammation. Circulating monocytes, known to respond to extremely low levels of LPS, play a central role in endotoxin-induced atherosclerosis. Once activated by LPS, monocytes are recruited to the vessel walls, where they differentiate into macrophages and take up lipids, eventually forming foam cells, which are characteristic of atherosclerotic lesions. The impaired function or expression of key genes involved in monocytic inflammation has often led to a reduction of atherosclerosis in LPS-challenged apoE−/− mice (1, 2). Several studies showed that functional polymorphisms in the LPS signaling pathway influenced the risk of atherosclerosis by modulating the monocytic inflammatory response (3, 4). Therefore, an investigation of the genetic factors involved in endotoxin-induced inflammation might lead to new approaches in CHD treatment and prevention.
The SLC22A3 (solute carrier family 22 member 3) gene, which encodes a 62-kDa trans-membrane protein OCT3 (organic cation transporter3), is required for outward transport and clearance of bioamines and drugs, such as histamine, dopamine, and metformin (5, 6). Recently, increasing evidence has indicated that SLC22A3 was involved in the pathogenesis of cardiovascular diseases. A genomic haplotype association study identified the SLC22A3-LPAL2-LPA gene cluster on 6q26–27 as a risk locus for CHD, and an SNP rs2048327 within the SLC22A3 gene was associated with a decreased risk of CHD in a Caucasian population (7). Qi et al. (8) demonstrated that polymorphisms within this gene were associated with plasma lipoprotein(a) (an independent risk factor of atherosclerosis) levels among type 2 diabetic patients. Genetic variants in the SLC22A3-LPAL2-LPA gene cluster were associated with decreased early outgrowth colony-forming units and an increased risk of myocardial infarction (9).
Although there were several reports on the association between SLC22A3 polymorphisms and the risk of cardiovascular diseases, the relationship between SLC22A3 variations, monocytic inflammation, and CHD risk has not been systematically demonstrated. In this study, we reported a comprehensive investigation of a mechanistic link between the functional SLC22A3 polymorphism rs3088442 G→A, the inflammatory response, and the risk of atherosclerotic CHD. Our results provided direct evidence for a feedback inhibition against monocytic inflammatory response, indicating a potential mechanism for the athero-protective role of SNP rs3088442 G→A.
EXPERIMENTAL PROCEDURES
Cell Lines and Chemicals
SiHa (human cervical tumor cells), HEK293T (human embryonic kidney cells), and HepG2 (human hepatocellular liver carcinoma cells) were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS). Human acute monocytic leukemia cells (THP-1), human umbilical vein endothelial cells (HUVECs), and human epithelial carcinoma cells (HeLa) were cultured in RPMI 1640 medium (Invitrogen) supplemented with 10% FBS.
Actinomycin D and histamine dihydrochloride were purchased from Sigma. siRNAs used in this study were synthesized by GenePharma Co., Ltd. (Shanghai, China). miRNA mimics and antagomiR were provided by RioboBio Co., Ltd. (Guangzhou, Guangdong, China). Anti-OCT3 polyclonal antibody (SC-18515), anti-actin polyclonal antibody (SC-1616), and anti-p65 monoclonal antibody (SC-8008) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-JNK (AJ518), anti-phospho-JNK (AJ516) polyclonal antibody, anti-ERK (AM076), and anti-phospho-ERK (AM071) antibody were provided by Beyotime Co., Ltd. (Shanghai, China). Anti-p38 (21245-1) and anti-phospho-p38 (11252-1) antibodies were purchased from Signalway (Nanjing, Jiangsu, China).
Dual-Luciferase Reporter Assay
Cells were seeded into 96-well plates at 5000 cells/well and incubated at 37 °C for 24 h. Luciferase constructs were transfected using Lipofectamine 2000 (Invitrogen) following the manufacturer's protocol. At 24–36 h post-transfection, cells were lysed, and luciferase assays were performed using a Dual-Luciferase reporter assay system (Promega, Madison, WI) on a single automatic injection Mithras luminometer (Berthold Technologies, Bad Wildbad, Germany) following the protocol provided by manufacturer. Ratios of firefly luciferase readings to Renilla luciferase readings were calculated (10).
RNA Extraction and Analysis
Total cellular RNA was extracted using TRIzol (Invitrogen). cDNA was obtained with SuperScriptIII® first strand synthesis system (Invitrogen) for RT-PCR. Gene expression was monitored by quantitative real time PCR using SYBR Green PCR master mix (Invitrogen). mRNA levels were normalized to actin mRNA. The sequences of primers are listed in Table 1. The level of miR-147 was monitored by qRT-PCR using primer set provided by RioboBio Co., Ltd. (Guangzhou, Guangdong, China).
TABLE 1.
List of primers and probes used in the study
| Gene | Sequence |
|---|---|
| IL-6 | 5′-CTTCTCCACAAGCGCCTTCG-3′ |
| 5′-TACTCTTGTTACATGTCTCC-3′ | |
| IL-8 | 5′-GTGCAGTTTTGCCAAGGAGT-3′ |
| 5′-CTCTGCACCCAGTTTTCCTT-3′ | |
| MCP-1 | 5′-CAGCCAGATGCAATCAATGC-3′ |
| 5′-GTGGTCCATGGAATCCTGAA-3′ | |
| HDC | 5′-GGGGGTTTCTGAAGGGGATTGAGT-3′ |
| 5′-ATAGTGGCCGGGGATGAGGAAGAGAC-3′ | |
| SLC22A3 | 5′-CACAGCCCTTCCTGGGTTT-3′ |
| 5′-CAAGTCAAGGCTACCACGG-3′ | |
| TLR4 | 5′-AGATGGGGCATATCAGAGC-3′ |
| 5′-GTCCATCGTTTGGTTCTGG-3′ | |
| β-Actin | 5′-CCTGGCACCCAGCACAAT-3′ |
| 5′-GCCGATCCACACGGAGTACT-3′ | |
| Overlap PCR | 5′-GACCTGTTCACATTGCGAAGGTGTGTGGAAACCAAGGTGAG-3′ |
| 5′-CACACCTTCGCAATGTGAACAGGTC-3′ |
UTR Cloning and Point Mutant Construction
The 3′UTR fragment of SLC22A3 was amplified with primers (CGTCTAGAGGCCCCCGACAAAGACAGAA and GAGCCCAGGGGACACGGAGCAGTCTGAGGC). A point mutation (G→A) was introduced by overlap PCR with primers (sequences are listed in the Table 1). The fragments were digested with XbaI and inserted into the luciferase reporter vector pGL3-control (Promega, Madison, WI).
Protein Extraction and Western Blot Analysis
Cells were harvested in RIPA lysis buffer (Beyotime, Shanghai, China) containing 1 μm PMSF (Sigma) and a Protease Inhibitor Mixture (Roche Diagnostics). Proteins were separated by SDS-PAGE, transferred to nitrocellulose membranes, and immunoblotted with the indicated antibodies.
Monocyte Adhesion Assay
SLC22A3 or TLR4 expression in HUVECs or THP-1 cells was inhibited by knockdown with specific siRNA before performing the adhesion assay. HUVECs were cultured overnight to confluence in 6-well plates before initiating the adhesion assay. THP-1 cells were stimulated with LPS, prelabeled with calcein-AM (Sigma), added to the HUVECs layer, and allowed to adhere for 1 h at 37 °C. Nonadherent cells were removed by washing with PBS. Five different fields were randomly selected from each sample, and adherent cells were counted using a fluorescence microscope (11).
Transendothelial Migration Assay
SLC22A3 expression in THP-1 cells and HUVECs was inhibited by siRNA transfection. HUVECs (2 × 106 per well) were plated onto Costar Transwells (8-μm pore size and 6.5-mm diameter) that were coated with Matrigel (BD Biosciences). After 24 h, 1 × 106 THP-1 cells were stimulated with LPS, suspended in RPMI 1640 medium (containing 1% FBS), and added to the upper chamber. RPMI 1640 medium containing 10% FBS in the lower chamber served as the chemoattractant. After incubation for 24 h at 37 °C, THP-1 cells were collected from the bottom of the filter and stained with Hoechst 33342. Five different fields were randomly selected from each sample, and the migrated cells were counted using a fluorescence microscope (12, 13).
Enzyme-linked Immunosorbent Assay (ELISA)
Levels of IL-6, IL-8, or MCP-1 were determined by ELISA. The cell culture media were collected at the indicated times after LPS stimulation, and the secretion of pro-inflammatory mediators was detected according to the manufacturer's protocol (NeoBioscience, Shenzhen, Guangdong, China). Three independent experiments were performed.
mRNA Stability Assay
The fragment containing the coding sequencing and 3′ UTR of SLC22A3 was inserted into pEGFP-C1. Point mutation was introduced using bridged PCR. Two constructs pEGFP-SLC22A3G and pEGFP- SLC22A3A were transfected into HepG2 cells, respectively. Actinomycin D was added at the indicated hours just before harvest. Cells were lysed with TRIzol to isolate total RNA. SLC22A3 expression was analyzed by quantitative real time PCR using SYBR Green PCR master mix (Invitrogen). The mRNA levels were normalized to actin mRNA. The sequences of primers were listed as follows: forward primer 5′-GGACGACGGCAACTACAAGA-3′ and reverse primer 5′-TTGTACTCCAGCTTGTGCCC-3′.
Correlation Analysis of Genotype and Expression
The fresh blood samples were collected from healthy subjects and divided into two vials, one for mononuclear cell isolation and the other for genomic DNA extraction. Human peripheral blood mononuclear cells were isolated by Ficoll gradient centrifugation and maintained in RPMI 1640 medium containing 20% FBS. After a 4-day culture, 100 ng/ml LPS was added to induce the production of pro-inflammatory mediators. At 6 h post-LPS stimulation, cells were harvested and lysed with TRIzol. Total RNA was extracted to monitor the expression of SLC22A3 and mediators. The genomic DNA was extracted from whole blood with TIANamp Blood DNA kit (Tiangen, Beijing, China). A TaqMan assay was performed to test the genotypes for rs3088442 in all the healthy donors. The correlation of gene expression and genotypes was analyzed by independent t test with SPSS 5.0 (14). The study was conducted conforming to the Declaration of Helsinki for the use of human tissue and subjects, approved by the Ethics Committee of Tongji Medical College, and all participants provided written informed consent.
Intracellular Histamine Detection
THP-1 or HUVECs were transfected with siRNAs or miRNA mimics as indicated and stimulated with 100 ng/ml LPS at 48 h post-transfection. The cells were then harvested and washed twice with ice-cold PBS. Cell pellets were resuspended with RPMI 1640 medium (Invitrogen) and lysed by rapid freezing and thawing. Cell supernatants were collected by centrifugation at 12,000 rpm for 10 min to determine histamine level using enzyme immunoassay according the protocol provided by the manufacturer (Cayman, Ann Arbor, Michigan).
Correlation Analysis of Genotype and Monocyte Adhesion
The fresh blood samples were collected from healthy subjects and divided into two vials, one for mononuclear cell isolation and the other for genomic DNA extraction. The genomic DNA was extracted from whole blood with TIANamp Blood DNA kit (Tiangen, Beijing, China). A TaqMan assay was performed to test the genotypes for rs3088442 in all the healthy donors. The genomic DNA was extracted from whole blood with TIANamp Blood DNA kit (Tiangen, Beijing, China). A TaqMan assay was performed to test the genotypes for rs3088442 in all the healthy donors. Human peripheral blood mononuclear cells were isolated by Ficoll gradient centrifugation and maintained in RPMI 1640 containing 15% FBS, which was supplemented with nonessential amino acids. HUVECs were seeded into a 24-well plate and cultured overnight to confluence before initiating the adhesion assay. After a 7-day culture in vitro, LPS-challenged PBMCs (homozygotes) were stained with calcein AM, added to the HUVECs layer, and allowed to adhere for 1 h at 37 °C. Nonadherent cells were removed by washing with PBS 5 times. Three different fields were randomly selected from each sample, and adherent cells were counted using a fluorescence microscope. The correlation of gene expression and genotypes was analyzed by Mann-Whitney test with SPSS 5.0. The study was conducted conforming to the Declaration of Helsinki for use of human tissue and subjects, approved by the Ethics Committee of Tongji Medical College, and all participants provided written informed consent.
RESULTS
CHD-associated SNP rs3088442 G→A Is a Functional Regulatory Site
In this study, we first conducted a case control study that included 2660 CHD patients and 2674 controls to confirm the association of rs3088442 with the risk of CHD. Results indicated that compared with the GG genotype at rs3088442, the AA genotype significantly decreased the risk of CHD after adjustment for age, sex, smoking habits, body mass index, blood pressure, glucose levels, and lipid levels (odds ratio = 0.83, 95% confidence interval = 0.70–0.99, p = 0.038) (data not shown).
Bioinformatics assays with SNPinfo and microSniper suggested that G to A substitution of rs3088442 created a putative miR-147-binding site (Fig. 1A). As microRNAs often functioned as negative regulators for gene expression, we hypothesized that this SNP might decrease the risk of CHD because of SLC22A3 gene silencing.
FIGURE 1.

Allele-specific regulation of SLC22A3 by miRNA-147. A, in silico prediction of miR-147 and SLC22A3 mRNA interactions showed differences in binding within the seed region. B, SLC22A3A mRNA level decreased more sharply than SLC22A3G after actinomycin D treatment. The fragment containing the CDS and 3′UTR of SLC22A3 was inserted into pEGFP-C1. Point mutation was introduced by bridged PCR. HepG2 cells, which transfected with these constructs, were treated with actinomycin D for several hours as indicated. mRNA levels of SLC22A3 were quantified using quantitative RT-PCR. The SLC22A3/actin ratio was expressed as a percent of the value obtained for 0 h post-treatment and then plotted as the semi-log to reflect the difference of mRNA decay rate. C, rs3088442 G→A significantly down-regulated the relative luciferase activity of SLC22A3A 3′UTR fusion construct. Top, luciferase reporter constructs contained the 3′UTR of SLC22A3 gene. Gray boxes represented the 3′ UTR fragment used in the reporter constructs. Blue star represented the A allele of rs3088442. Bottom, luciferase activity of each construct in HeLa, HepG2, and HEK293T cells. The relative luciferase activity was analyzed by a dual-luciferase reporter system. Data were represented as mean ± S.D. Triplicate repeats were conducted showing similar results. D, rs3088442 G→A facilitated the affinity of SLC22A3 mRNA to has-miR-147. The miRNA mimics or antagomiR was co-transfected with reporter gene constructions into HepG2 cells. Left, miR-147 level increased 48 h at post-transfection with miR-147 mimics. Meanwhile, miR-147 antagomiR (anti-miR-147) significantly decreased the level of endogenous miR-147. miRNA level was monitored using qRT-PCR, and the small nuclear RNA U6 was used as a loading control. Negative control (NC) mimics and negative control antagomiR (NC2) was provided by RiboBio. Middle, relative luciferase activity of SLC22A3A UTR fusion construct dropped significantly after miR-147 transfection (n = 3). Right, miR-147 antagomiR abrogated the difference in luciferase activity of two constructs containing the A or G allele (n = 3). E, miRNA-147 mimics significantly decreased the SLC22A3A mRNA level. The miRNA mimics were transfected into HepG2 and SiHa cells respectively. At 48 h post-transfection, qRT-PCR was applied to detect the fluctuation of SLC22A3 mRNA levels (n = 3). The result was represented as th mean ± S.D. of three independent assays. F, miR-147 decreased SLC22A3A protein level sharply. The miRNA mimics were transfected into HepG2, THP-1, HUVECs, and SiHa cells, respectively. A slight fluctuation of SLC22A3G protein level was detected. The SLC22A3 protein level was analyzed by Western blot with anti-OCT3 specific antibody. SLC22A3A protein levels were normalized to actin. G, quantitative results of immunoblots in F. NS, not significant. Quantification of immunoblots is shown in F using ImageJ software. *, p value <0.05.
To verify this hypothesis, we first assessed the effects of rs3088442 G→A polymorphism on SLC22A3 mRNA stability by actinomycin D (an inhibitor of RNA synthesis) treatment. We transfected two constructs pEGFP-SLC22A3G and pEGFP-SLC22A3A into HepG2, and the levels of remnant fusion transcripts were analyzed by RT-PCR. As shown in Fig. 1B, the levels of SLC22A3A (containing the A allele) mRNA dropped significantly after treatment, compared with SLC22A3G (containing the G allele) in HepG2. We next constructed two reporter vectors encompassing the 3′ UTR fragment of SLC22A3 and performed functional analyses to compare the activities of the G or A allele in HEK293T, HeLa, and HepG2 cells by luciferase assay (Fig. 1C, top). As Fig. 1C, bottom, shows, the relative luciferase activity of the A allele was significantly lower than the G allele. Furthermore, we found that the miR-147 mimics reduced the relative luciferase activity of fusion transcript carrying the A allele more significantly, whereas depletion of miR-147 by antagomiR abrogated the difference in luciferase activity between the two alleles (Fig. 1D). Finally, the effects of miR-147 on the expression of endogenous SLC22A3A were assessed by real time PCR assay and Western blot. Fig. 1, E and F, showed that miR-147 mimics transfection of down-regulated SLC22A3A mRNA and protein expression in SiHa and human umbilical vein endothelial cells but not SLC22A3G in THP-1 and HepG2 cells. Together, these results indicated that the CHD-associated risk (the G allele) and protective (the A allele) haplotypes might result in a difference in SLC22A3 expression under the control of miR-147.
SLC22A3 Silencing Inhibits LPS-induced Inflammatory Response
Considering that monocytic inflammation was critical for CHD pathogenesis (6), the role of SLC22A3 in endotoxin-induced inflammatory responses was examined. THP-1 cells were transfected with specific siRNA against SLC22A3 (SLC22A3i) to inhibit its expression. Fig. 2A showed that SLC22A3i significantly reduced SLC22A3 protein levels. Then, we investigated the effect of SLC22A3 deficiency on NF-κB and MAPKs activation (two important downstream effectors of LPS signaling). As expected, LPS induced nuclear translocation of NF-κB and phosphorylation of MAPKs (JNK and p38) were both inhibited after SLC22A3i (specific small interfering RNA (siRNA) against SLC22A3) transfection (Fig. 2, B and C). We next examined the production of pro-inflammatory mediators whose expression was tightly controlled by the NF-κB and MAPKs pathways. Fig. 2, D and E, showed that sharp reductions in the mRNA and protein levels of interleukin 6 (IL-6), interleukin 8 (IL-8), and MCP-1 were detected in SLC22A3-deficient THP-1 cells. Consistently, we observed that mRNA expression of SLC22A3, IL-6, IL-8, and MCP-1 decreased in PBMCs from healthy subjects with an AA genotype, compared with subjects with one or both copies of G allele (Fig. 2F). Fig. 3 showed that PBMC-endothelial interactions significantly decreased in subjects with AA genotype, compared with those with GG genotype. To further evaluate the physiological consequences of SLC22A3 deficiency, we assessed monocyte adhesion and infiltration, key processes during vascular inflammation, which were known to be dependent on pro-inflammatory mediators (e.g. cytokines, chemokines, and adhesion molecules). As shown in Fig. 2G, lack of TLR4 and SLC22A3 both impaired the binding of THP-1 cells to confluent HUVECs layer induced by LPS. Fig. 2H indicated that SLC22A3i reduced transmigration of THP-1 monocytic cells through a HUVEC monolayer along a fetal bovine serum (FBS) gradient. Thus, SLC22A3 played a critical role in modulating monocytic inflammatory responses.
FIGURE 2.

SLC22A3 gene silencing suppressed LPS-induced inflammatory responses. A, transfection with SLC22A3i decreased the SLC22A3 protein level. THP-1 cells transfected with SLC22A3i were challenged with LPS at 48 h post-transfection. The SLC22A3 protein level was monitored by Western blot at 6 h post-LPS stimulation. Quantitative results of immunoblots in A are shown in lower panel. B, knockdown of SLC22A3 significantly inhibited the translocation of NF-κB into nuclei induced by LPS stimulus. THP-1 cells transfected with SLC22A3i were challenged with LPS at 48 h post-transfection. The nuclear proteins and cytosolic proteins were extracted using NE-PER nuclear protein extraction kit. The protein levels of p65 in nuclei and cytoplasm were analyzed by Western blot. Actin was used as a loading control. Triplicate repeats were conducted showing similar results. Quantitative results of immunoblots in B are shown in the lower panel. C, knockdown of SLC22A3 significantly decreased phosphorylated protein levels of JNK and p38. THP-1 cells transfected with SLC22A3i were challenged with LPS at 48 h post-transfection. The protein levels of JNK and phosphorylated JNK, p38, phospho-p38, ERK, and phospho-ERK were analyzed by Western blot. Triplicate repeats were conducted showing similar results. Quantitative results of immunoblots in C are shown in the right panel. D, knockdown of SLC22A3 significantly suppressed the expression of pro-inflammatory mediators IL-6, IL-8, MCP-1, and histamine synthase (HDC). THP-1 cells transfected with SLC22A3i were challenged with LPS at 48 h post-transfection. The production of IL-6, IL-8, MCP-1, and HDC was monitored by qRT-PCR and normalized to the amount of actin (n = 3). E, knockdown of SLC22A3 inhibited the secretion of pro-inflammatory mediators IL-6, IL-8, and MCP-1. THP-1 cells transfected with SLC22A3i were challenged with LPS at 48 h post-transfection. Culture media were collected, and pro-inflammatory mediator levels were measured by ELISA at 24 h post-LPS stimulation (n = 3). F, correlation of pro-inflammatory mediator mRNA expression and the rs3088442 polymorphism. PBMCs were isolated from 37 healthy subjects, and the genotype was determined by TaqMan assay. PBMCs were cultured in vivo for 6 days and then stimulated with LPS for 24 h. The mRNA levels of pro-inflammatory mediators by quantitative real time PCR. The relative expression of SLC22A3, IL-6, IL-8, and MCP-1 was normalized to α-actin. The box plot showed the distribution of mRNA expression clustered according to different genotypes. The expression of SLC22A3 and pro-inflammatory mediators induced by LPS stimulation decreased significantly in subjects carrying AA alleles (n = 9) compared with other genotypes (GG, n = 6; GA, n = 22), as compared by the Student's t test. G, knockdown of SLC22A3 inhibited monocyte (THP-1) adhesion. LPS-challenged THP-1 monocytic cells were stained with calcein AM (green) and allowed to interact for 1 h with confluent HUVEC monolayers. Untreated cells were used as a negative control, and TLR4-deficient cells (TLR4i) were used as a positive control. Three independent repeats were performed, and similar results were obtained. Adherent cells were quantified in the right panel. Data represented three repeated experiments and are shown as mean ± S.D. H, SLC22A3i blocked transendothelial migration of THP-1 monocytic cells. LPS-challenged THP-1 monocytic cells were allowed to transmigrate across confluent HUVECs along an FBS gradient during 24 h. Cells in the lower chamber were collected and stained with Hoechst 33342 (blue). Untreated cells were used as a negative control. The experiments were repeated three times with similar results. Migrated cells were quantified in right panel. Data represent three repeated experiments and are shown as mean ± S.D.
FIGURE 3.
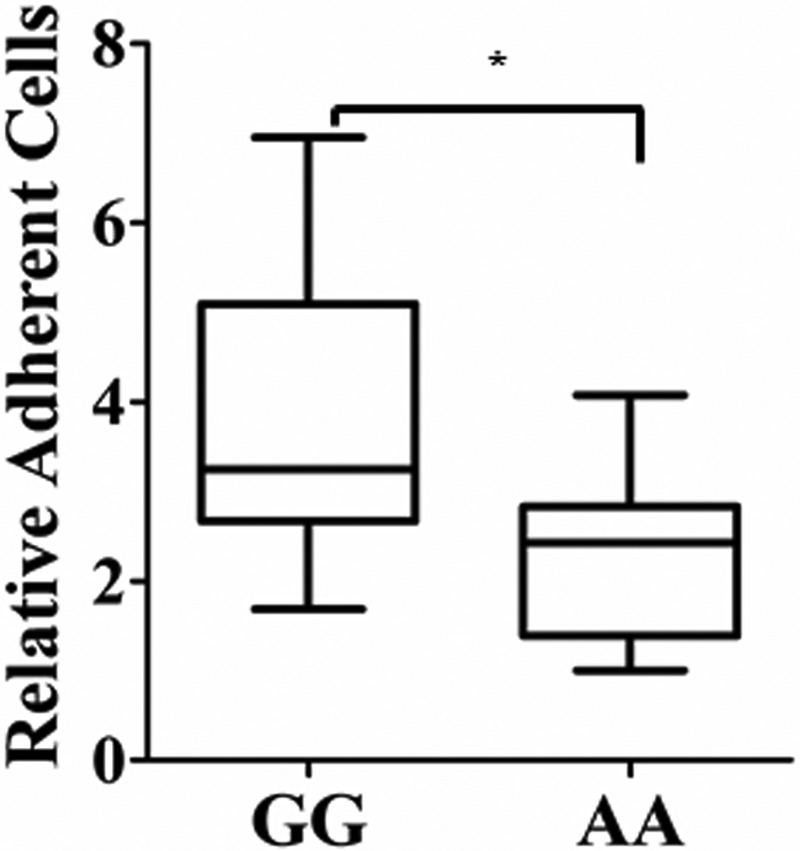
Correlation of adherent PBMCs and the rs3088442 polymorphism. PBMCs were collected from 50 healthy subjects, and the genotype was determined using TaqMan assay. After a 7-day incubation in vitro, homozygotes were challenged with LPS, labeled with calcein AM, and allowed to interact for 1 h with confluent HUVEC monolayers. The box plot showed the distribution of adhesion efficiency clustered according to a different genotype. PBMC-endothelial interactions decreased significantly in subjects carrying the AA genotype (n = 7) compared with the GG genotype (n = 16), as compared by Mann-Whitney test (*, p < 0.05).
SLC22A3 Controls Monocytic Inflammatory Response by Modulating Histamine Synthesis
As previous research indicated that SLC22A3 played an essential role in controlling histamine synthesis (a key mediator of vascular inflammation) (6), we determined whether SLC22A3 regulated the inflammatory response by modulating histamine synthesis. First, we examined whether LPS/NF-κB (MAPKs) signaling could be suppressed by exogenous histamine. Fig. 4A indicated that LPS-induced nuclear translocation of NF-κB was inhibited by histamine. Similarly, the protein level of phosphorylated JNK and p38 also decreased after histamine application (Fig. 4B). Then, we monitored the production of pro-inflammatory mediators. As expected, the expression and secretion of pro-inflammatory mediators IL-6, IL-8, and MCP-1 induced by LPS also were inhibited by histamine (Fig. 4, C and D). As a consequence, LPS-induced monocyte adhesion and trans-endothelial migration (TEM) were inhibited after treatment with exogenous histamine (Fig. 4, E and F). Interestingly, histamine treatment or knockdown of SLC22A3 both caused a reduction in the mRNA level of HDC (histidine decarboxylase, a synthase of histamine) (Figs. 2D and 4C). Fig. 5 showed similar results that SLC22A3i increased intracellular histamine levels significantly. It suggested that SLC22A3 might play a role in LPS-induced inflammatory response via controlling the histamine level. Moreover, we examined whether HDC gene knockdown could reverse the inhibition of inflammatory responses. Our results showed that silencing of the HDC gene significantly up-regulated the production of pro-inflammatory mediators (IL-6, IL-8, and MCP-1) in SLC22A3-deficient cells (Fig. 6A) and reversed SLC22A3-deficient THP-1 cell adhesion and TEM (Fig. 6, B and C). Therefore, our findings suggested that SLC22A3 played a role in an LPS-stimulated inflammatory response in a histamine-dependent manner.
FIGURE 4.

Exogenous histamine inhibited LPS-induced inflammatory response. A, exogenous histamine suppressed the activation of NF-κB. THP-1 cells were treated with 1 mm histamine for 4 h and stimulated with LPS for a further 2 h. Then challenged or unchallenged cells were collected, and nuclear and cytoplasmic protein was isolated to detect the protein level of NF-κB, respectively. The experiments were repeated three times with similar results. Quantitative results of immunoblots in Fig. 3A are shown in the lower panel. B, histamine inhibited phosphorylation of p38 and JNK induced by LPS stimulation. THP-1 cells were pretreated with histamine and stimulated with LPS. Cells were harvested to analyze the protein level of total and phospho-p38, JNK, and ERK by Western blot. Three independent repeats were performed, and similar results were obtained. Quantitative results of immunoblots in Fig. 3B are shown in the lower panel. C, exogenous histamine significantly decreased the expression of pro-inflammatory mediators. THP-1 cells were treated with 1 mm histamine for 4 h and stimulated with LPS for a further 6 h. Total RNA was extracted with TRIzol, and the expression of pro-inflammatory mediators was analyzed by qRT-PCR. Data were expressed as mean ± S.D. (n = 3). D, exogenous histamine suppressed the secretion of IL-6, IL-8, and MCP-1 induced by LPS treatment. THP-1 cells were treated with 1 mm histamine for 4 h and stimulated with LPS for a further 6 h. Cell media were collected to measure the level of mediators by ELISA. Data were expressed as mean ± S.D. (n = 3). E, exogenous histamine suppressed monocyte (THP-1) adhesion. LPS-challenged THP-1 cells were pretreated with 1 mm histamine dihydrochloride for 4 h and applied to confluent HUVEC monolayers. Three independent repeats were performed, and similar results were obtained. Quantitative analysis of Fig. 3E is shown in the right panel. Data represent three repeated experiments and are shown as mean ± S.D. F, exogenous histamine suppressed monocyte (THP-1) TEM. LPS-challenged THP-1 cells were pretreated with 1 mm histamine dihydrochloride for 4 h and applied to confluent HUVEC monolayers. Monocytic cells were allowed to trans-migrate across confluent HUVECs along an FBS gradient during 24 h. Cells in the lower chamber were collected and stained with Hoechst 33342 (blue). Quantitative analyses of Fig. 3F are shown in the right panel. Data represented three repeated experiments and are shown as mean ± S.D.
FIGURE 5.
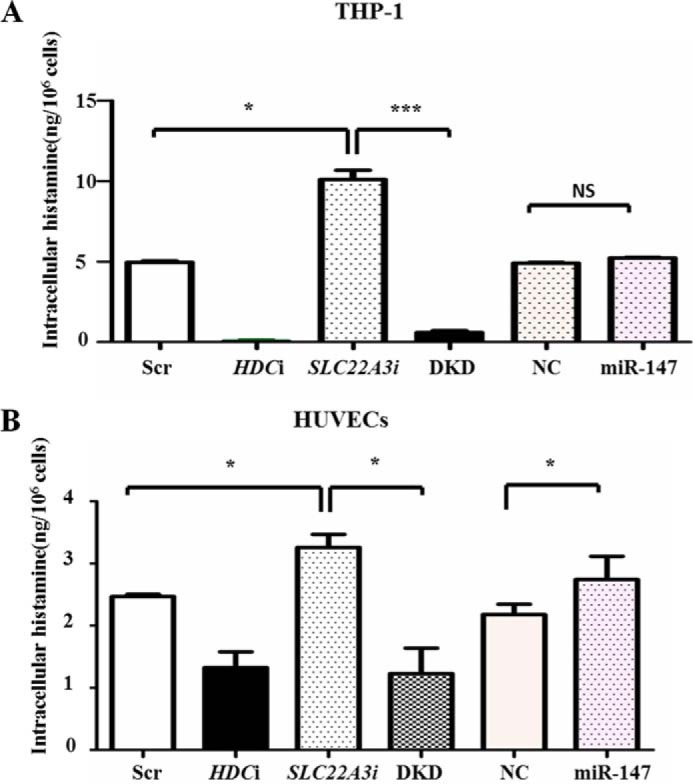
SLC22A3 deficiency increases intracellular histamine level. THP-1 (A) or HUVECs (B) were transfected with Scrambled control (Scr), HDCi, SLC22A3i, DKD (HDCi + SLC22A3i), negative control mimics and miR-147 mimics respectively. After LPS stimulation, cells were harvested to determine intracellular histamine level by enzyme immunoassay. *, p ≤ 0.05; ***, p ≤ 0.001.
FIGURE 6.

SLC22A3 deficiency inhibited LPS-induced inflammatory response in a histamine-dependent way. A, knockdown of HDC with siRNA reversed the inhibition of inflammatory mediator production induced by SLC22A3 deficiency. siRNA against HDC (HDCi) significantly decreased the mRNA level of the HDC gene. THP-1 cells were transfected with HDCi or SLC22a3i together or respectively and were stimulated with LPS at 48 h post-transfection. Total RNA was extracted to analyze the expression of IL-6, IL-8, and MCP-1 by RT-PCR. Data are represented as mean ± S.D., *, p ≤ 0.05; **, p ≤ 0.01. The experiments were repeated three times with similar results. B and C, knockdown of HDC with siRNA reversed the inhibitory effects on monocyte adhesion and TEM induced by SLC22A3 deficiency. THP-1 cells transfected with SLC22A3i or HDCi were stimulated with LPS and added to confluent HUVEC monolayers. Untreated cells were used as a negative control. Monocyte adhesion and TEM were observed under the fluoroscope. Three independent repeats were performed, and similar results were obtained. Quantitative analysis of adherent cells and migrated cells is shown in the right panel. Data represented three repeated experiments and are shown as mean ± S.D.
Expression of SLC22A3A Is Controlled by LPS Stimulation
As murine miR-147 functioned as a negative feedback regulator for LPS/TLR4-stimulated inflammatory responses (15), we suggested that its human counterpart could play a similar role. To verify this hypothesis, we treated HUVECs and THP-1 cells with LPS for the indicated time and harvested them to detect the expression of SLC22A3 and miR-147. As Fig. 7 shows, the level of miR-147 increased steadily, accompanied by a gradual reduction of SLC22A3A expression in HUVECs. In contrast, no obvious fluctuation of SLC22A3G protein level could be detected in THP-1 cells, although LPS stimulation up-regulated the level of miR-147 sharply. The quantitative result of SLC22A3 protein expression was shown in Fig. 7B (left panel). Moreover, we examined whether miR-147 suppressed inflammatory responses by controlling SLC22A3A expression. miR-147 mimics reduced the level of SLC22A3A (Fig. 1F) and pro-inflammatory mediators (IL-6, IL-8, and MCP-1) in HUVECs (Fig. 8A). The antagomiR-147 treatment increased the mRNA level of SLC22A3A and up-regulated the expression of pro-inflammatory mediators (Fig. 8B). However, the expression of SLC22A3 and the pro-inflammatory mediators did not alter obviously after miR-147 or antagomiR-147 transfection in THP-1 cells (Fig. 8, C and D). Moreover, miR-147 mimics significantly decreased monocyte adhesion, and the reduction could be reversed by HDCi (Fig. 9). Taken together, our results suggest that LPS stimulation up-regulated the miR-147 expression to suppress the inflammatory response (Fig. 8) via suppression of SLC22A3 expression (Fig. 1, E and F).
FIGURE 7.

Expression of SLC22A3A is suppressed by LPS stimulation. LPS induced up-regulation of miR-147 and, in turn, decreased the expression of SLC22A3A protein. THP-1 cells and HUVECs were seeded into 6-well plates and incubated with RPMI 1640 or DMEM medium containing 10% FBS. 100 ng/ml LPS was added at the indicated times (0, 12, and 24 h) before harvest. After stimulation, we collected cells to analyze the expression of miR-147 and SLC22A3. A, LPS treatment up-regulated the expression of miR-147 in a time-dependent manner. miR-147 level was examined using the miR-147-specific qRT-PCR primer set. The results were normalized against the amount of U6 miRNA. B, protein level of SLC22A3A decreased sharply after LPS stimulation as time elapsed in HUVECs. In contrast, no sharp fluctuation of SLC22A3G protein level was observed in THP-1 cells. Left, whole cell lysates from cells treated with LPS for indicated times were subjected to Western blot analyses using antibodies against OCT3 and actin protein. The results of a representative experiment are shown. Right, densitometric quantification of the data shown in left panel. The results are presented as arbitrary units representing ratios of normalized densitometric values of SLC22A3 protein detected in samples. All experiments (A and B) were repeated three times with similar results. NS, not significant.
FIGURE 8.

Inhibition of endogenous miR-147 expression increased pro-inflammatory mediator expression in HUVECs. HUVECs (A) or THP-1 (C) cells were transfected with miR-147 mimics or antagomiR and then stimulated with LPS at 48 h post-transfection. Negative control mimics (NC) and negative control antagomiR (antagomiR-NC) were used as negative controls. Total RNA was extracted with TRIzol reagent. miR-147 antagomiR increased the expression of SLC22A3, IL-6, IL-8, and MCP-1. A and C, expression of SLC22A3, IL-6, IL-8, and MCP-1 was monitored by one-step RT-PCR. B and D, quantification of RT-PCR results shown in left panel using ImageJ software. *, p value < 0.05; **, p value < 0.01; ***, p value < 0.001, n = 3.
FIGURE 9.

Overexpression of miR-147 decreased THP-1 adhesion, which could be partially reversed by knockdown of HDC. HUVECs were transfected with miR-147 mimics or co-transfected with miR-147 and HDCi. THP-1 cells were stimulated with LPS and applied to confluent HUVEC monolayers. A, adherent cells were calculated under a fluoroscope. B, quantification of monocyte adhesion assay. *, p value <0.05; ***, p value <0.001, n = 3. C, SLC22A3 expression in S4A.
DISCUSSION
In this study, we proposed a novel protective mechanism against the inflammatory response mediated by SNP rs3088442 G→A, which may contribute to a decreased risk of CHD (Fig 10). Here, we obtained several lines of evidence to support this hypothesis. First, we confirmed that the A allele of rs3088442 could enhance the suppression of SLC22A3 expression by miR-147 (Fig. 1). Second, our results suggested that LPS stimulation led to up-regulation of miR-147 and down-regulation SLC22A3A expression (Fig. 7). Third, functional studies showed that SLC22A3 silencing significantly inhibited pro-inflammatory mediator (IL-6, IL-8, and MCP-1) production, impaired leukocyte-endothelial interaction, and suppressed monocyte infiltration (Fig. 2, D, E, G, and H). Fourth, pro-inflammatory mediator production decreased significantly in PBMCs carrying the AA allele of rs3088442 compared with other genotypes (Fig. 2F). Moreover, the adhesion of PBMCs carrying the AA allele of rs3088442 decreased significantly, compared with the GG genotype (Fig. 3). Taken together, it might suggest LPS stimulation up-regulated miR-147 levels to suppress SLC22A3A expression and, ultimately, led to a feedback inhibition of inflammation induced by itself (as Fig 10 shows).
FIGURE 10.

Schematic diagrams of LPS-induced signaling cascade in cells carrying different alleles of rs3088442. LPS stimulation initiated the secretion of several inflammatory mediators, including histamine, pro-inflammatory cytokines, and chemokines. Human miR-147 could also be up-regulated by LPS engagement by an unknown mechanism. A, in HepG2 cells (the G allele), miR-147 failed to inhibit the expression of the SLC22A3G gene. Newly synthesized histamine could be outwardly transported by trans-membrane transporter SLC22A3 to augment LPS-induced inflammatory responses successfully. B, miR-147 bound to the UTR of SLC22A3A and suppressed its expression. SLC22A3 deficiency inhibited outward transport of newly synthesized histamine, led to accumulation of intercellular histamine, and suppressed the signaling cascade for LPS in a negative feedback manner. Interruption of LPS signaling pathway often decreased the inflammatory response.
MicroRNAs are a class of 22-nucleotide-long endogenous noncoding RNAs that participate in regulating the expression of target genes by binding to the 3′ UTR. Previous reports indicated that over 30% of human genes could be regulated by miRNAs. Not surprisingly, microRNAs were key regulators in the pathogenic and physiological processes of CHD, including vascular cell differentiation, contraction, migration, proliferation, and inflammation (16–18). Therefore, SNPs, located within or near the target sites of miRNAs, often influenced the function of miRNAs and were associated with vascular diseases (19–21). Murine miR-147 has been identified as an inhibitor for TLR-mediated inflammation (15). However, the physiological role of its human counterpart in CHD pathogenesis remained largely unknown. Hoekstra et al. (22) reported that the level of human miR-147 dropped significantly in peripheral blood mononuclear cells from patients with CHD compared with those from healthy donors, indicating that miR-147 might act as a protective factor for CHD. Here, we reported a stronger binding of miR-147 to the A allele of rs3088442 (Fig. 1A). Consistent with the work of Nies et al. (23) that the SLC22A3 variant (rs3088442) was correlated with decreased SLC22A3 expression in hepatocytes, our study showed that SNP rs3088442 recruited miR-147 to suppress SLC22A3A expression (Fig. 1, D and F) and, ultimately, decreased LPS-induced inflammation (Figs. 2F and 8). Moreover, the case control study indicated that the A allele of rs3088442 was associated with a decreased risk of CHD (data not shown). Our findings suggested a potential mechanism by which miRNA-147 plays a role in atheroprotection.
Histamine was usually considered as a vasoactive autacoid, which stimulated immune and inflammatory responses directly or synergistically with endotoxins (24–26). Genetic removal of the HDC gene significantly decreased inflammatory responses in apoE knock-out atherosclerotic mice (27). However, there is increasing evidence to indicate the protective role of histamine in vascular inflammation. Several studies found histamine injection diminished induction of endothelial adhesion molecules, impaired platelet adhesion, and inhibited thrombosis (28, 29). Schneider et al. (6, 30) suggested exposure to high levels of histamine inhibited Th2-type immune responses by suppressing its own synthesis. Consistently, our results also showed LPS induced the expression of histamine synthase, and pro-inflammatory mediators could be suppressed by treatment with high levels of histamine (Fig. 4, C and D). Taken together, these data suggested histamine synthesis might be regulated in a negative feedback manner and that disruption of its synthesis significantly inhibited immune and inflammatory responses.
SLC22A3 was responsible for the outward transport of newly synthesized histamine. Schneider et al. (6, 30) showed that OCT3 ligands or SLC22A3 knock-out significantly inhibited histamine synthesis. We also found impaired expression of the HDC gene was detected in SLC22A3-deficient monocytes (Fig. 2D), suggesting that SLC22A3 might be required for histamine synthesis. Our findings also showed that miR-147 or SLC22A3i inhibited SLC22A3A expression (Fig. 1F) and increased the intracellular level of histamine (Fig. 5). Both high histamine levels and SLC22A3 deficiency suppressed LPS-induced inflammatory response (Fig. 4), suggesting that histamine suppressed its own synthesis and inflammatory response in a feedback manner. Furthermore, SLC22A3 deficiency inhibited LPS-induced inflammatory responses (Fig. 2, D–H), which could be reversed by knockdown of HDC (Fig. 6). Consistently, Schneider et al. (6) indicated that SLC22A3 deficiency or histamine treatment also inhibited IL-6 and IL-4 production in basophils, which could be abrogated by an HDC inhibitor or knockdown of the HDC gene (6). Thus, our results revealed that SLC22A3 played a role in monocytic inflammation via controlling intracellular histamine levels in a feedback manner.
However, some limitations still need further investigation. First, histologic evidence collected from a murine model of atherosclerosis will be required to verify our hypothesis that SLC22A3 polymorphism exerted a protective effect against CHD by controlling monocytic inflammation. Second, the miR-147 level and SLC22A3 expression in PBMCs from CHD patients should be examined to evaluate the role of miR-147 in the pathogenesis of CHD.
In summary, this study revealed a protective mechanism mediated by SLC22A3A protein against inflammatory responses. Our findings may offer a better understanding of the mechanism by which the SLC22A3 polymorphism decreases CHD susceptibility. In addition, our research identified the role of SLC22A3 in the control of monocytic inflammation.
This study was supported by National Basic Research Program Grant 2011CB503806 and Natural National Scientific Foundation of China Grant 81230069.
- CHD
- coronary heart disease
- HDC
- histidine decarboxylase
- HDCi
- siRNA against HDC
- HUVEC
- human umbilical vein endothelial cell
- MCP
- monocyte chemoattractant protein
- OCT3
- organic cation transporter 3
- SLC22A3
- solute carrier family 22 member 3
- SLC22A3i
- siRNA against SLC22A3
- TEM
- trans-endothelial migration
- TLR4i
- siRNA against TLR4
- PBMC
- peripheral blood mononuclear cell
- SNP
- single-nucleotide polymorphism.
REFERENCES
- 1. Gu L., Okada Y., Clinton S. K., Gerard C., Sukhova G. K., Libby P., Rollins B. J. (1998) Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol. Cell 2, 275–281 [DOI] [PubMed] [Google Scholar]
- 2. Ishibashi M., Egashira K., Zhao Q., Hiasa K., Ohtani K., Ihara Y., Charo I. F., Kura S., Tsuzuki T., Takeshita A., Sunagawa K. (2004) Bone marrow-derived monocyte chemoattractant protein-1 receptor CCR2 is critical in angiotensin II-induced acceleration of atherosclerosis and aneurysm formation in hypercholesterolemic mice. Arterioscler. Thromb. Vasc. Biol. 24, e174–e178 [DOI] [PubMed] [Google Scholar]
- 3. den Dekker W. K., Cheng C., Pasterkamp G., Duckers H. J. (2010) Toll-like receptor 4 in atherosclerosis and plaque destabilization. Atherosclerosis 209, 314–320 [DOI] [PubMed] [Google Scholar]
- 4. Lavergne E., Labreuche J., Daoudi M., Debré P., Cambien F., Deterre P., Amarenco P., Combadière C., and GENIC Investigators. (2005) Adverse associations between CX3CR1 polymorphisms and risk of cardiovascular or cerebrovascular disease. Arterioscler. Thromb. Vasc. Biol. 25, 847–853 [DOI] [PubMed] [Google Scholar]
- 5. Chen L., Pawlikowski B., Schlessinger A., More S. S., Stryke D., Johns S. J., Portman M. A., Chen E., Ferrin T. E., Sali A., Giacomini K. M. (2010) Role of organic cation transporter 3 (SLC22A3) and its missense variants in the pharmacologic action of metformin. Pharmacogenet. Genomics 20, 687–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schneider E., Machavoine F., Pléau J. M., Bertron A. F., Thurmond R. L., Ohtsu H., Watanabe T., Schinkel A. H., Dy M. (2005) Organic cation transporter 3 modulates murine basophil functions by controlling intracellular histamine levels. J. Exp. Med. 202, 387–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Trégouët D. A., König I. R., Erdmann J., Munteanu A., Braund P. S., Hall A. S., Grosshennig A., Linsel-Nitschke P., Perret C., DeSuremain M., Meitinger T., Wright B. J., Preuss M., Balmforth A. J., Ball S. G., Meisinger C., Germain C., Evans A., Arveiler D., Luc G., Ruidavets J. B., Morrison C., van der Harst P., Schreiber S., Neureuther K., Schäfer A., Bugert P., El Mokhtari N. E., Schrezenmeir J., Stark K., Rubin D., Wichmann H. E., Hengstenberg C., Ouwehand W., Wellcome Trust Case Control Consortium, Cardiogenics Consortium, Ziegler A., Tiret L., Thompson J. R., Cambien F., Schunkert H., Samani N. J. (2009) Genome-wide haplotype association study identifies the SLC22A3-LPAL2-LPA gene cluster as a risk locus for coronary artery disease. Nat. Genet. 41, 283–285 [DOI] [PubMed] [Google Scholar]
- 8. Qi Q., Workalemahu T., Zhang C., Hu F. B., Qi L. (2012) Genetic variants, plasma lipoprotein(a) levels, and risk of cardiovascular morbidity and mortality among two prospective cohorts of type 2 diabetes. Eur. Heart J. 33, 325–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shaw S. Y., Cheng S., Cupples L. A., Larson M. G., McCabe E. L., Ngwa J. S., Wang Y. A., Martin R. P., Klein R. J., Hashmi B., Ajijola O. A., Lau E., O'Donnell C. J., Vasan R. S., Cohen K. S., Wang T. J. (2011) Genetic and clinical correlates of early-outgrowth colony-forming units. Circ. Cardiovasc. Genet. 4, 296–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guo H., Bai Y., Xu P., Hu Z., Liu L., Wang F., Jin G., Wang F., Deng Q., Tu Y., Feng M., Lu D., Shen H., Wu T. (2010) Functional promoter-1271G→C variant of HSPB1 predicts lung cancer risk and survival. J. Clin. Oncol. 28, 1928–1935 [DOI] [PubMed] [Google Scholar]
- 11. Yan T., Li Q., Zhou H., Zhao Y., Yu S., Xu G., Yin Z., Li Z., Zhao Z. (2012) Gu-4 suppresses affinity and avidity modulation of CD11b and improves the outcome of mice with endotoxemia and sepsis. PLoS One 7, e30110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Voura E. B., Ramjeesingh R. A., Montgomery A. M., Siu C. H. (2001) Involvement of integrin α(v)β(3) and cell adhesion molecule L1 in transendothelial migration of melanoma cells. Mol. Biol. Cell 12, 2699–2710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yu L., Fan Y., Chen B., Hu Y., Gao Y., Wei D. (2013) Suppression of metastasis of human pancreatic cancer cells to the liver by small RNA-mediated targeting of the midkine gene. Oncol. Lett. 6, 1338–1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kondo T., Ohno M., Shimokata K., Iino S., Inden Y., Murohara T., Hirai M. (2003) CD14 promoter polymorphism is associated with acute myocardial infarction resulting from insignificant coronary artery stenosis. Heart 89, 931–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu G., Friggeri A., Yang Y., Park Y. J., Tsuruta Y., Abraham E. (2009) miR-147, a microRNA that is induced upon Toll-like receptor stimulation, regulates murine macrophage inflammatory responses. Proc. Natl. Acad. Sci. U.S.A. 106, 15819–15824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ramírez C. M., Rotllan N., Vlassov A. V., Dávalos A., Li M., Goedeke L., Aranda J. F., Cirera-Salinas D., Araldi E., Salerno A., Wanschel A., Zavadil J., Castrillo A., Kim J., Suárez Y., Fernández-Hernando C. (2013) Control of cholesterol metabolism and plasma HDL levels by miRNA-144. Circ. Res. 112, 1592–1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang L., Jia X. J., Jiang H. J., Du Y., Yang F., Si S. Y., Hong B. (2013) MicroRNAs 185, 96, and 223 repress selective high-density lipoprotein cholesterol uptake through posttranscriptional inhibition. Mol. Cell. Biol. 33, 1956–1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wei Y., Nazari-Jahantigh M., Neth P., Weber C., Schober A. (2013) MicroRNA-126, -145, and -155: a therapeutic triad in atherosclerosis? Arterioscler. Thromb. Vasc. Biol. 33, 449–454 [DOI] [PubMed] [Google Scholar]
- 19. Li P., Liu Y., Yi B., Wang G., You X., Zhao X., Summer R., Qin Y., Sun J. (2013) MicroRNA-638 is highly expressed in human vascular smooth muscle cells and inhibits PDGF-BB-induced cell proliferation and migration through targeting orphan nuclear receptor NOR1. Cardiovasc. Res. 99, 185–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Martin M. M., Buckenberger J. A., Jiang J., Malana G. E., Nuovo G. J., Chotani M., Feldman D. S., Schmittgen T. D., Elton T. S. (2007) The human angiotensin II type 1 receptor +1166 A/C polymorphism attenuates microRNA-155 binding. J. Biol. Chem. 282, 24262–24269 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21. Wu C., Gong Y., Sun A., Zhang Y., Zhang C., Zhang W., Zhao G., Zou Y., Ge J. (2013) The human MTHFR rs4846049 polymorphism increases coronary heart disease risk through modifying miRNA binding. Nutr. Metab. Cardiovasc. Dis. 23, 693–698 [DOI] [PubMed] [Google Scholar]
- 22. Hoekstra M., van der Lans C. A., Halvorsen B., Gullestad L., Kuiper J., Aukrust P., van Berkel T. J., Biessen E. A. (2010) The peripheral blood mononuclear cell microRNA signature of coronary artery disease. Biochem. Biophys. Res. Commun. 394, 792–797 [DOI] [PubMed] [Google Scholar]
- 23. Nies A. T., Koepsell H., Winter S., Burk O., Klein K., Kerb R., Zanger U. M., Keppler D., Schwab M., Schaeffeler E. (2009) Expression of organic cation transporters OCT1 (SLC22A1) and OCT3 (SLC22A3) is affected by genetic factors and cholestasis in human liver. Hepatology 50, 1227–1240 [DOI] [PubMed] [Google Scholar]
- 24. Su E., Yu D., Cringle S. (2005) Histamine induces opposing vasoactive effects at different levels of the ocular vasculature. Curr. Eye Res. 30, 205–212 [DOI] [PubMed] [Google Scholar]
- 25. Delneste Y., Lassalle P., Jeannin P., Joseph M., Tonnel A. B., Gosset P. (1994) Histamine induces IL-6 production by human endothelial cells. Clin. Exp. Immunol. 98, 344–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tan X., Essengue S., Talreja J., Reese J., Stechschulte D. J., Dileepan K. N. (2007) Histamine directly and synergistically with lipopolysaccharide stimulates cyclooxygenase-2 expression and prostaglandin I2 and E2 production in human coronary artery endothelial cells. J. Immunol. 179, 7899–7906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang K. Y., Tanimoto A., Guo X., Yamada S., Shimajiri S., Murata Y., Ding Y., Tsutsui M., Kato S., Watanabe T., Ohtsu H., Hirano K., Kohno K., Sasaguri Y. (2011) Histamine deficiency decreases atherosclerosis and inflammatory response in apolipoprotein E knockout mice independently of serum cholesterol level. Arterioscler. Thromb. Vasc. Biol. 31, 800–807 [DOI] [PubMed] [Google Scholar]
- 28. Wang J., Al-Lamki R. S., Zhang H., Kirkiles-Smith N., Gaeta M. L., Thiru S., Pober J. S., Bradley J. R. (2003) Histamine antagonizes tumor necrosis factor (TNF) signaling by stimulating TNF receptor shedding from the cell surface and Golgi storage pool. J. Biol. Chem. 278, 21751–21760 [DOI] [PubMed] [Google Scholar]
- 29. Brown T. P., Forouzan O., Shevkoplyas S. S., Khismatullin D. B. (2013) Histamine reduces GPIbα-mediated adhesion of platelets to TNF-α-activated vascular endothelium. Thromb. Res. 131, 150–157 [DOI] [PubMed] [Google Scholar]
- 30. Schneider E., Machavoine F., Bricard-Rignault R., Levasseur M., Petit-Bertron A. F., Gautron S., Ribeil J. A., Launay J. M., Mecheri S., Côté F., Dy M. (2011) Downregulation of basophil-derived IL-4 and in vivo T(H)2 IgE responses by serotonin and other organic cation transporter 3 ligands. J. Allergy Clin. Immunol. 128, 864–871 [DOI] [PubMed] [Google Scholar]