

Background: Techniques to localize proteins in situ at high resolution are important but limited.
Results: Combining SNAP tag technology with cryo-electron tomography, we precisely localized proteins within the N-DRC that are important for ciliary motility.
Conclusion: The developed method was applied to localize proteins with ∼3 nm resolution without interfering with the complex function.
Significance: The method is a powerful tool for studies of proteins in situ.
Keywords: Chlamydomonas, Cilia, Cryo-electron Microscopy, Dynein, Electron Tomography, DRC3 and DRC4, FAP134, PF2, Clonable EM Tag, Nexin-Dynein Regulatory Complex
Abstract
Cryo-electron tomography (cryo-ET) has reached nanoscale resolution for in situ three-dimensional imaging of macromolecular complexes and organelles. Yet its current resolution is not sufficient to precisely localize or identify most proteins in situ; for example, the location and arrangement of components of the nexin-dynein regulatory complex (N-DRC), a key regulator of ciliary/flagellar motility that is conserved from algae to humans, have remained elusive despite many cryo-ET studies of cilia and flagella. Here, we developed an in situ localization method that combines cryo-ET/subtomogram averaging with the clonable SNAP tag, a widely used cell biological probe to visualize fusion proteins by fluorescence microscopy. Using this hybrid approach, we precisely determined the locations of the N and C termini of DRC3 and the C terminus of DRC4 within the three-dimensional structure of the N-DRC in Chlamydomonas flagella. Our data demonstrate that fusion of SNAP with target proteins allowed for protein localization with high efficiency and fidelity using SNAP-linked gold nanoparticles, without disrupting the native assembly, structure, or function of the flagella. After cryo-ET and subtomogram averaging, we localized DRC3 to the L1 projection of the nexin linker, which interacts directly with a dynein motor, whereas DRC4 was observed to stretch along the N-DRC base plate to the nexin linker. Application of the technique developed here to the N-DRC revealed new insights into the organization and regulatory mechanism of this complex, and provides a valuable tool for the structural dissection of macromolecular complexes in situ.
Introduction
Most cellular events rely on the functions of macromolecular machines, which in turn depend upon these macromolecules being properly assembled. A better understanding of the functions of these nanomachines and the cellular events that they drive requires information about the subunit organization of native macromolecular complexes in situ, i.e. the determination of the precise locations of the component proteins in relation to each other when the complexes are maintained in their physiological environment. Visualizing individual proteins in their functional context is a longstanding goal in biology, and although there are many labeling tools available for light microscopy, this technique lacks the overall structural resolution of electron microscopy (EM). Thus, for decades, EM has been instrumental for studying complexes in cells at up to ∼5 nm resolution (1). More recently, cryo-electron tomography (cryo-ET)3 in combination with subtomogram averaging has emerged as a new powerful imaging technique that can provide three-dimensional views of native complexes, organelles, and small cells to a resolution of ∼3 nm (2–4). However, even at this resolution, the low contrast images obtained by cryo-ET do not typically provide sufficient detail to directly identify and localize most proteins or their interactions.
Although clonable fluorescent reporters such as green fluorescent protein (GFP) have revolutionized imaging using light microscopy, determining the locations of proteins using EM-compatible probes has remained challenging (1). For example, immuno-gold labeling, the most widely used labeling method in EM, often suffers from low efficiency, fidelity, and precision (the distance between the target protein and gold is 20–30 nm).
Here, we developed an in situ localization method integrating SNAP tag technology with cryo-ET, which we tested using cilia and flagella as a model system. Motile cilia and flagella are important organelles for cell motility and for producing fluid flow, e.g. in the mammalian airway. The microtubule-based core structure of cilia and flagella, called the axoneme, contains thousands of dynein motors that require proper regulation to generate the waveforms typical for these organelles. The highly conserved nexin-dynein regulatory complex (N-DRC) is one of the key regulators of axonemal dyneins and is essential for proper motility of cilia and flagella (5). At least 11 proteins (DRC1–11) have been assigned to the N-DRC based on proteomic approaches (5–7). N-DRC mutations usually cause defective ciliary motility (8), and importantly, the lack of CCDC164 (DRC1) or CCDC65 (DRC2) has been identified as a cause of primary ciliary dyskinesia, a chronic human respiratory disease (9–11). Knowledge of the precise locations of the individual components of the N-DRC and of their arrangement within the larger N-DRC structure is not yet available, but it is essential for a better understanding of the regulatory mechanism of the N-DRC.
Several cryo-ET studies of cilia and flagella have compared wild type (WT) and mutants to visualize the structural defects caused by the mutation, and thus to infer the probable locations of specific subunits in axonemal complexes (12), including the N-DRC (13, 14). Such comparisons, however, are very limited because many proteins in a macromolecular complex are important for the assembly and/or stability of other components, i.e. the observed defects often extend beyond the mutated protein. For example, a null mutation of DRC4 (also known as pf2) causes the loss or reduction of the levels of most DRC subunits (loss of DRC3–7 and 11, and reduction of DRC8–10) in the complex (5–7). Moreover, the loss of a subunit might result in the rearrangement of the remaining proteins in the complex, shifting the apparent defect into a location different from the native site. Therefore, labeling the component of interest within an otherwise WT-like structure would allow a more direct and accurate determination of the locations of the components.
Labeling techniques that have been combined with cryo-EM imaging include immunogold labeling (15) and, more recently, clonable tags, which have the advantage of higher labeling specificity and resolution (16, 17). To avoid disruption of the assembly or architecture of macromolecular complexes, clonable tags are usually designed to be relatively small (e.g. GFP is 27 kDa), but many of them are too small for direct in situ visualization using cryo-ET. Therefore, for EM visualization, small clonable tags are often specifically linked to additional electron-dense probes, such as metal ion binding by metallothionein (7 kDa) (16, 18), or tags such as MiniSOG that catalyze the photooxidation of diaminobenzidine into a localized precipitate that can be visualized by OsO4 treatment (19, 20). Despite these recent advances, the techniques used are still limited and/or are not compatible with cryo-EM, because of low contrast, toxicity of metal ion solutions, and/or the requirement to chemically fix and post-stain the cells. Therefore, there is still a critical need for the development of a widely applicable clonable tag-based labeling technique that can precisely and accurately determine the locations of proteins of interest in situ.
The clonable SNAP tag (20 kDa), derived from the DNA repair protein O6-alkylguanine-DNA-alkyltransferase, forms a covalent bond specifically with O6-benzylguanine (BG) derivatives, which can be designed as fluorescent probes (21). The commercially available SNAP tag technology is widely applied in biochemical and cell biological studies, including live cell imaging and super-resolution fluorescence microscopy (22–24). Here, we extend the application of the SNAP tag technology by developing a protocol that is compatible with cryo-ET. We have also applied this new protocol to more precisely localize two subunits, DRC3 (FAP134) and DRC4 (PF2), within the structure of the intact N-DRC in the genetic model organism Chlamydomonas, a flagellated green algae. Both subunits were roughly localized to the linker region of the N-DRC in previous work by cryo-ET and WT mutant comparisons (14). Our new method allowed the precise localization of the N and C termini of DRC3 and the C terminus of DRC4 by ligating the fused SNAP tag with biotin and 1.4-nm-sized streptavidin-nanogold (strep-Au). After cryo-ET and subtomogram averaging, we found that DRC3 is located within the L1 projection of the nexin linker that is connected to an inner arm dynein. In addition to previous studies, we found that DRC4 is not only part of the nexin linker but also extends along the entire base plate of the N-DRC structure. Comparisons of these structures with wild type indicate that the overall native structure has not been disrupted by the introduction of the tags. Our precise and accurate localization of termini of N-DRC components has provided new insights into the mechanism of N-DRC assembly and regulation of flagellar motility, and it further demonstrates that our SNAP tag-based EM-labeling method is a powerful tool for localizing protein components of macromolecular complexes in situ.
EXPERIMENTAL PROCEDURES
Strains
Chlamydomonas reinhardtii strains used in this study are listed in Table 1. A pUC57 plasmid containing a Chlamydomonas codon-optimized sequence encoding SNAP was provided by W. S. Sale (Emory University, Atlanta, GA). A SNAP-tagged version of DRC4 (PF2) was constructed by insertion of the codon-optimized SNAP sequence into a 6.9-kb XmaI subclone containing the complete WT DRC4 gene (25). The SNAP tag was amplified using primers with BstEII restriction sites, ligated into pGEM-T Easy (Promega, Inc., Madison, WI), and then digested and re-ligated into a unique BstEII site located within the DRC4 plasmid. Sequence analysis confirmed that the SNAP tag was inserted into the 12th exon of the DRC4 gene in the proper orientation and reading frame. DRC4-SNAP rescued strains were generated by transforming the null mutant pf2-4 with the DRC4-SNAP plasmid and the selectable marker pSI103 containing the aphVIII gene (26) and plating them on medium containing 10 μg/ml paromomycin. Positive transformants were screened for recovery of wild type motility and expression of the SNAP tag as described below. To generate a plasmid encoding N-terminally SNAP-tagged DRC3, a fragment containing the first exon of DRC3 was excised at the MfeI sites from a plasmid containing full-length DRC3 (pDRC3 (27)) and cloned into pBluescript II SK at the EcoRI site (pDRC3-MfeI). To make a blunt end after the start codon, full-length pDRC3-MfeI was amplified by PCR with the following set of primers: DRC3ATGup (5′-CATTTTGGCTTGCTACGCGGC-3′) and DRC3ATGdown (5′-CCGAGTCTCGAGCGTCTAATC-3′). A cassette of cDNA encoding SNAP followed by a linker of serine and glycine were amplified from the plasmid containing the codon-optimized SNAP by PCR with the following set of primers: SNAP5 (5′-GACAAGGACTGCGAGATGAAGCGC-3′) and SNAP3+SG (5′-GCCCGAGGAGCCCGGGGCGCCGAT-3′), and ligated into pDRC3-MfeI at the blunt end site (pDRC3-MfeI+SNAP). A fragment of pDRC3-MfeI+SNAP containing the MfeI sites was amplified by PCR with the following set of primers: DRC3MfeN (5′-GGTTCAATTGGTCACCGGCTCGGC-3′) and DRC3MfeC (5′-CCTTCAATTGCAGCTTGGTCAGGGTG-3′). The PCR product was digested with MfeI and inserted into pDRC3 at the MfeI sites (pSNAP-DRC3). To generate SNAP-DRC3 cells, about 106 of the drc3 cells were transformed with linearized 1.4 μg of pSNAP-DRC3 containing the marker gene aph 7″ gene (28) as described previously.4 Colonies on Tris acetate/phosphate plates containing hygromycin B were randomly picked and screened using immunofluorescence microscopy with anti-DRC3. Expression of SNAP-DRC3 in the selected strain was confirmed by Western blot with anti-DRC3 and anti-SNAP. Cells were grown in liquid Tris acetate/phosphate medium (29).
TABLE 1.
Strains used in this study
| Name | Strain | Phenotype |
|---|---|---|
| WT | CC-125, 137c mt+ | WT waveform, fast swimming |
| pf2 | CC-4483, pf2-4 | Altered waveform, slow swimming |
| DRC4-SNAP | pf2-4::DRC4-SNAP | WT waveform, fast swimming |
| drc3 | drc3 | Altered waveform, slow swimming |
| DRC3-SNAP | drc3::DRC3-SNAP | WT waveform, fast swimming |
| SNAP-DRC3 | drc3::SNAP-DRC3 | WT waveform, fast swimming |
Analysis of Motility Phenotypes
Motility phenotypes were assessed by phase contrast microscopy using a Zeiss Axioskop with a ×40 objective and a ×1.6 eyepiece. Cells were recorded with a halogen light source, a red filter, a ×20 objective and a Rolera-MGI camera (QImaging, Surray, British Columbia, Canada) using 50-ms exposures for several seconds. The forward swimming velocities of individual cells were measured using the Metamorph software package, version 7.1.7.0 (Molecular Devices, Downington, PA), calibrated with a stage micrometer. 70–90 cells were measured per strain.
Axoneme Preparation
Axonemes were isolated using the pH shock method as described previously (30). Briefly, Chlamydomonas cells were collected by centrifugation and washed with fresh minimal medium (M-N/5 medium) (31). The pelleted cells were resuspended in pH shock buffer containing 10 mm HEPES, pH 7.4, 1 mm SrCl2, 4% sucrose, and 1 mm dithiothreitol (DTT). Flagella were detached from cells by adding 0.5 m acetic acid to the solution to reduce the pH to 4.3. After 80 s, the pH was increased to 7.2 by adding 1 m KOH. Then the solution was supplemented with 5 mm MgSO4, 1 mm EGTA, 0.1 mm EDTA, and 100 μg/ml aprotinin, pepstatin, and leupeptin. The flagella were separated from the cell bodies by centrifuging at 1800 × g for 10 min at 4 °C and purified by two additional centrifugation steps at 2400 × g for 10 min at 4 °C with a 20% sucrose cushion. Purified flagella were demembranated with 1% IGEPAL CA-630 (Sigma) for 20 min at 4 °C. After centrifugation at 10,000 × g for 10 min, the pelleted axonemes were resuspended in HMEEK buffer (30 mm HEPES, pH 7.4, 5 mm MgSO4, 1 mm DTT, 1 mm EGTA, 1 mm EDTA, and 25 mm KCl).
Fluorescence Light Microscopy
Cover glasses (Fisher Scientific) were treated with 0.1% polyethyleneimine at room temperature for 10 min and air-dried after removing the polyethyleneimine solution. 100 μl of isolated axonemes were then applied to the treated cover glasses. After 10 min, unattached axonemes were washed away with HMEEK buffer. The cover glasses were incubated with a droplet of 3 μm SNAP tag BG-substrate (which forms a covalent bond with the SNAP tag) SNAP-Cell® TMR Star (New England Biolabs, Ipswich, MA) in HMEEK buffer with 1% BSA for 2 h at room temperature. Excess SNAP-Cell® TMR Star was removed by three washing steps with HMEEK buffer. In the first washing step, the cover glasses were incubated with HMEEK buffer for 30 min to improve the washing. The cover glasses were mounted on glass slides (Fisher Scientific) using PermaFluorTM (Thermo Scientific) mounting medium, and the samples were observed using a spinning disk confocal microscope (Marianas; Intelligent Imaging Innovations 3i Inc.) with a ×60 Plan Apochromat objective (N.A. 1.4). Image acquisition and analysis were performed using SlideBook 5.0 (3i Inc.).
Gel Electrophoresis and Western Blotting
Samples of WT, pf2-4, and DRC4-SNAP axonemes were separated by SDS-polyacrylamide gel and transferred to Western blots as described previously (7). Antibodies were diluted in Tropix I-Block (Applied Biosystems, Bedford, MA) at the following dilutions: DRC4 peptide (1:1000 (7)), RSP16 (1:10,000 (32)), and SNAP (1:10:000, New England Biolabs). Immunoreactive bands were detected using alkaline phosphatase-conjugated secondary rabbit antibodies and the Tropix chemiluminescent detection system according to the manufacturer's instructions.
For analysis of the DRC3-SNAP axonemes, 1 μl of 1 mm SNAP tag substrate BG-biotin (SNAP-Biotin; New England Biolabs) was added to 200 μl of the purified axonemes in HMEEK buffer and incubated overnight at 4 °C. Unbound BG-biotin was removed by centrifugation at 10,000 × g for 10 min at 4 °C and another two wash steps with HMEEK buffer (without DTT). The axoneme pellet was dissolved in lysis buffer (7 m urea, 2 m thiourea, 4% CHAPS, 65 mm DTT, and 2% IPG buffer (pH 3–10NL; GE Healthcare)). 20 μg of axonemal proteins in lysis buffer were separated on SDS-polyacrylamide minigels and then transferred to nitrocellulose membranes. After blocking with nonfat milk for 1 h at room temperature, the membranes were probed either with polyclonal primary antibodies (DRC2 at 1:3000 and DRC3 at 1:5000) overnight at 4 °C or with streptavidin-HRP (1:1000) for 1 h at room temperature. After three wash steps with TBST buffer (20 mm Tris-base, 150 mm NaCl, 0.1% Tween 20, pH 7.4) for 10 min, the membranes that were probed with primary antibodies were then incubated with secondary antibodies (anti-rabbit HRP at 1:10,000) for 1 h at room temperature. All membranes were then washed three times with TBST buffer for 10 min, and the HRP signals were detected with the enhanced chemiluminescence (ECL) detection system (Promega).
Streptavidin Nanogold Labeling and Cryo-sample Preparation
One microliter of 1 mm BG-biotin or BG-(PEG)12-biotin (New England Biolabs; PEG linker available on request) was added to 200 μl of the purified axonemes in HMEEK buffer and incubated overnight at 4 °C. Unbound BG-substrate was removed by three wash steps with HMEEK buffer and centrifugation at 10,000 × g for 10 min at 4 °C. The axoneme pellet was resuspended in 200 μl of HMEEK buffer (without DTT). After adding 5 μl of 80 μg/ml 1.4-nm-sized streptavidin nanogold particles (strep-Au, Nanoprobes, Inc.), the suspension was incubated at 4 °C for 4 h. After adding 800 μl of HMEEK buffer (without DTT), the labeled axonemes were pelleted by centrifugation at 10,000 × g for 10 min at 4 °C and resuspended in 200 μl of HMEEK buffer (without DTT).
An aliquot of labeled axonemes was used for cryo-sample preparation, and the remaining labeled axonemes were stained with a silver enhancement kit to verify the labeling efficiency (see below). The cryo-samples were prepared as reported previously (34). Briefly, 3 μl of isolated axonemes (labeled or unlabeled for control) in HMEEK buffer and 1 μl of 5-fold concentrated and BSA-treated 10 nm colloidal gold (Sigma) (35) were applied to Quantifoil holey carbon grids (copper R2/2, Quantifoil Micro Tools GmbH, Jena, Germany) that were previously coated with 10 nm colloidal gold. Excess fluid was blotted from the grid for ∼2 s with Whatman filter paper and then plunge frozen in liquid ethane using a homemade plunge freezer to achieve sample vitrification. Frozen samples were stored in liquid nitrogen until EM imaging.
Staining with Silver Enhancement Kit
Axonemes labeled with strep-Au were concentrated by centrifugation at 10,000 × g for 10 min and resuspended in 30 μl of HMEEK buffer (without DTT). The axoneme solution was then dissolved in 30 μl of lysis buffer (without DTT). After mixing with SDS-polyacrylamide gel loading buffer (without DTT and without heat treatment), the proteins were separated on a 0.75-mm-thick SDS-polyacrylamide minigel. After two washes with Milli-Q water for 5 min, the nanogold-labeled DRC proteins on the gel were visualized by staining using a silver enhancement kit (Nanoprobes, Inc.) (33). Following image acquisition, the gel was post-stained with Coomassie Blue for visualization of all proteins.
Cryo-electron Tomography
Vitrified samples were imaged using a Tecnai F30 transmission EM (FEI, Eindhoven, The Netherlands) operated at 300 keV and equipped with a field emission gun and a postcolumn energy filter (Gatan, Pleasanton, CA). EM images were acquired at ×13,500 magnification (pixel size ∼1 nm), and at −8 μm defocus using a 2k × 2k charge-coupled device camera (Gatan, Pleasanton, CA). Tilt series of axonemes were automatically recorded from −65 to +65° with 1.5–2.5 angular increments using SerialEM software (36), under low dose conditions and in the zero-loss mode of the energy filter (20-eV slit width). The cumulative electron dose for each tilt series was limited to ∼100 e/Å2.
Image Processing
Three-dimensional tomograms were reconstructed using the IMOD software package (37) with alignment using fiducial markers and weighted back projection. For each strain, 3–10 tomograms of noncompressed or mildly compressed axonemes were chosen for further image processing (Tables 1 and 2). Subtomograms, each containing a 96-nm axonemal repeat, were extracted, aligned, and averaged with missing-wedge compensation using PEET software (3). An unsupervised classification approach incorporated in PEET (38) was used to analyze the presence or absence of strep-Au label in the axonemal repeats of WT, DRC4-SNAP, DRC3-SNAP, and SNAP-DRC3. During the classification analysis, the examined three-dimensional volume was limited to the targeted region with a mask. Axonemal repeats with consistent features (e.g. with strep-Au) were combined into class averages. The averaged structures were visualized in three-dimensions by isosurface rendering using the UCSF Chimera package (39), or in two-dimensions as tomographic slices using the IMOD software. To highlight electron densities that were missing in mutant axonemes compared with WT or rescue strains, we overlaid the respective three-dimensional structures using the Chimera package, identified and segmented the structural differences, and then colored the WT density that was missing in the mutant N-DRC in red (see Figs. 2–4). The densities of the strep-Au labels (only present in the labeled axonemes of rescued strains) were similarly identified and gold-colored in the N-DRC structures of the rescued strains (Figs. 3, 4, and 7). Information about the number of tomograms and 96-nm axonemal repeats included in the three-dimensional averaged structures, as well as the resolutions of the resulting images using the 0.5 Fourier shell correlation criterion (40), are summarized in Table 2.
TABLE 2.
Image processing information for strains used in this study
| Name | Strain | Tomograms included | Averaged repeats | Resolutiona |
|
|---|---|---|---|---|---|
| DMT | DRC | ||||
| nm | |||||
| WTb | CC-125, 137c mt+ | 9 | 1225 | 3.1 | 3.4 |
| pf2 | CC-4483, pf2-4 | 7 | 983 | 3.8 | 4.2 |
| DRC4-SNAP | pf2-4::DRC4-SNAP | 3 | 459 | 4.5 | 4.8 |
| Labeled DRC4-SNAP | pf2-4::DRC4-SNAP | 10 | 1514 | 3.0 | 3.5 |
| drc3c | drc3 | 6 | 940 | 3.2 | 3.9 |
| DRC3-SNAPc | drc3::DRC3-SNAP | 8 | 1180 | 3.3 | 3.8 |
| Labeled DRC3-SNAP | drc3::DRC3-SNAP | 7 | 1212 | 3.4 | 3.9 |
| SNAP-DRC3 | drc3::SNAP-DRC3 | 6 | 993 | 3.4 | 3.9 |
| Labeled SNAP-DRC3 | drc3::SNAP-DRC3 | 9 | 1479 | 3.5 | 3.9 |
a The 0.5 Fourier shell correlation criterion was used to estimate the resolution at two different locations as follows: the doublet microtubule (DMT; which usually has the highest resolution in the axonemal averages) and the N-DRC (which is usually at lower resolution, typical for associated complexes).
b WT was used as the control and its structure was refined using data originally published by Awata et al. (see Footnote 4).
c The drc3 and DRC3-SNAP structures were refined using data originally published by Awata et al. (see Footnote 4).
FIGURE 2.

SNAP-tagged DRC4 and DRC3 rescue the structural defects in pf2 and drc3. A, schematic of a Chlamydomonas axoneme in cross-sectional view, seen from the flagellar tip. ODA, IDA, and the N-DRC connect neighboring microtubule doublets, whereas the RS connect to the central pair complex (CPC). B–K, isosurface renderings of the three-dimensional structures of the 96-nm axonemal repeats in WT (B and C), pf2 (D and E), DRC4-SNAP (C-terminally SNAP-tagged DRC4, F and G), drc3 (H and I), and DRC3-SNAP (C-terminally SNAP-tagged DRC3, J and K) after cryo-ET and subtomogram averaging; cross-sectional views from distal (B, D, F, H, and J) and bottom views (C, E, G, I, and K) of the N-DRC (blue or red/blue). Note that the N-DRC defects are more severe in pf2 (D and E) than drc3 (H and I), but both the C-terminally SNAP-tagged DRC4 and DRC3 rescue strains, respectively, show recovery (colored red) of the N-DRC to WT morphology in DRC4-SNAP (F and G) and DRC3-SNAP (J and K). The hole in the A-B inner junction is indicated by an arrowhead (C). Other labels used are as follows: At, A-tubule; Bt, B-tubule; IDA e/g, inner dynein arms e and g; N-DRC, nexin-dynein regulatory complex; ODA, outer dynein arms; RS2, radial spoke 2; L1, L1 projection.
FIGURE 3.

Precise localization of the N terminus of PF2 (DRC4), and the N and C termini of DRC3 by SNAP tag labeling and cryo-ET. Isosurface renderings show the three-dimensional structures of the 96-nm axonemal repeats in WT (A and B), labeled DRC4-SNAP (C and D), labeled SNAP-DRC3 (N-terminally SNAP-tagged DRC3; E and F), and labeled DRC3-SNAP (G and H) after cryo-ET and subtomogram averaging. Purified axonemes with SNAP-tagged DRC4 (C and D) or DRC3 (E–H) were ligated with BG-(PEG)12-biotin and then with strep-Au (1.4 nm nanogold); cross-sectional views from distal (C, E, and G) and bottom views (D, F, and H) of the N-DRC (blue and red) and the associated SNAP-biotin-strep-Au label (marked with arrows and gold-colored in the SNAP-tagged strains; C–H) are shown. In the labeled DRC4-SNAP, SNAP-DRC3, and DRC3-SNAP strains, all structures (except for the N-DRC-associated label), including the ODA, IDA, and RS, have the same three-dimensional organization as in the untreated WT axonemes (A and B), suggesting that the conditions used for the SNAP tag labeling technique preserve the pristine structure of the axonemes. The additional density of SNAP-biotin-strep-Au (gold, arrows) is present as follows: in labeled DRC4-SNAP axonemes (C and D) at the end of the N-DRC base plate close to the B-tubule hole (black arrowhead in B), in the N terminus labeled DRC3 (E and F) at the transition region between the linker and base plate of the N-DRC, and in the C terminus labeled DRC3 at the edge of the L1 projection (G and H). Other labels used are as follows: At, A-tubule; Bt, B-tubule; IDA e/g, inner dynein arms e and g; N-DRC, nexin-dynein regulatory complex; ODA, outer dynein arms; RS2, radial spoke 2; L1, L1 projection.
FIGURE 4.

Classification analysis shows high labeling efficiency of DRC4-SNAP. Tomographic slices (A, B, and D–F) and isosurface renderings (C) of the averaged 96-nm axonemal repeats in cross-sectional (from distal; left image) and bottom view orientation (right image) show the presence of the SNAP-biotin-strep-Au density at the end of the N-DRC base plate in labeled DRC4-SNAP axonemes (golden arrows in B); this density does not appear in the unlabeled averaged WT (white arrows in A). The subtomogram average of all axonemal repeats from labeled DRC4-SNAP (B and C) was classified (D–F) focusing on the region with the label density; the results revealed three distinct classes 1–3 (D–F) that show the additional density of the biotin-strep-Au label (golden arrows) in slightly different positions, suggesting that the label is somewhat flexible and can pivot with the (PEG)12-linker around the SNAP tag of the fusion protein, i.e. at the C terminus of DRC4. The biotin-strep-Au density was nevertheless observed in all three classes, indicating nearly 100% labeling efficiency. Other labels are as follows: At, A-tubule; Bt, B-tubule; IDA e/g, inner dynein arms e and g; N-DRC, nexin-dynein regulatory complex; ODA, outer dynein arms; RS2, radial spoke 2. Scale bar, 20 nm.
FIGURE 7.
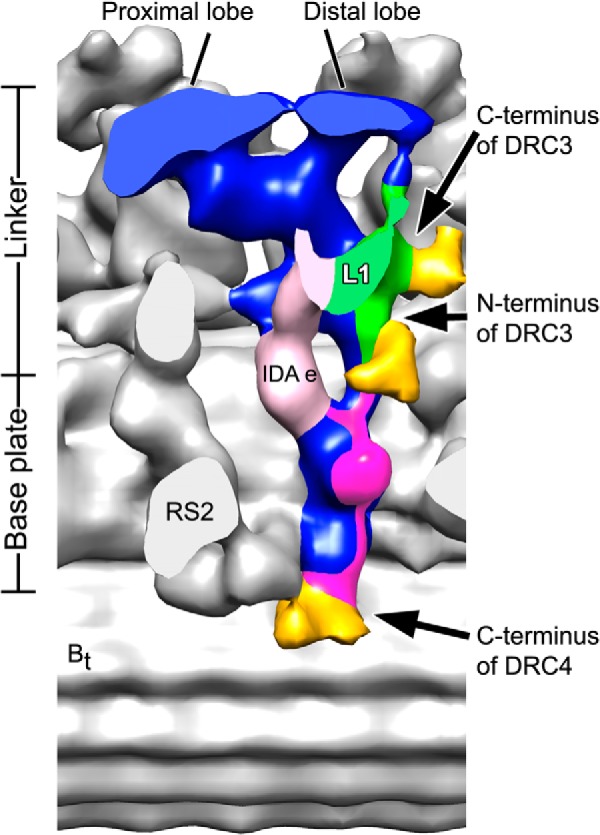
Precise localization of N and C termini of DRC3 and the C terminus of DRC4 in the N-DRC of Chlamydomonas axonemes. A modified three-dimensional isosurface image shows the locations of the SNAP-biotin-strep-Au labels (gold) at the N and C termini of DRC3 and the C terminus of DRC4 in Chlamydomonas axoneme. Note that the densities of the predicted locations of DRC3 and DRC4 are colored in green and magenta, respectively. The image was generated by merging the information from the averages of labeled SNAP-DRC3, DRC3-SNAP, and DRC4-SNAP axonemes. The modified image shows that the N terminus of DRC3 is close to the basal portion of the linker and to DRC4, whereas the C terminus of DRC3 (green) is close to the central portion of the linker and located at the edge of the L1 projection that is connected to dynein g. The DRC4 extends along the entire base plate of the N-DRC (magenta) with its C terminus located close to the hole in the A-B inner junction. The N terminus of DRC4 has not been localized; therefore, it cannot be excluded that DRC4 also locates along the nexin linker.
RESULTS
SNAP-tagged N-DRC Components Are Functional and Assemble Properly into Chlamydomonas Flagella
To study the proper assembly, distribution, and function of SNAP-tagged axonemal components in Chlamydomonas flagella, we transformed the SNAP-tagged DRC3 and DRC4 genes into the corresponding null mutants and screened for rescue of the motility defects. The swimming speed and waveform of the SNAP-tagged rescue strains were similar to those of WT (Fig. 1C; Table 1),4 demonstrating that the 20-kDa SNAP tag neither affected the regulatory function of the N-DRC nor its flagellar motility. After purifying axonemes from the DRC3-SNAP (C-terminally tagged) rescue strain, we ligated the SNAP with the live cell fluorescent probe SNAP-Cell® TMR Star. The fluorescence signal was observed along the entire lengths of the axonemes, indicating that DRC3-SNAP is properly distributed along the axonemes (Fig. 1E). We did not observe a fluorescence signal in axonemes of the WT control (Fig. 1E). Therefore, the SNAP tag of the fusion proteins exhibited high enzymatic activity, allowing covalent and highly specific binding of the fluorescent BG-substrate.
FIGURE 1.

SNAP-tagged Chlamydomonas axonemal proteins assemble in the flagella, rescue the defects seen in the corresponding null mutants to WT phenotype, and can be ligated with BG-substrates with high specificity and efficiency. A, schematic workflow of the SNAP tag labeling technique for cryo-ET studies. B, diagrams showing predicted polypeptide domains in the SNAP-tagged DRC4 and DRC3, respectively. The SNAP tag is shown in magenta. Coiled coil domains (CC) and leucine-rich repeats (LRR) are shown in gray. C, measurements of forward swimming velocity indicate that the transformed DRC4-SNAP rescued the motility defects of the pf2 mutant. D, Western blots indicate that the transformed DRC4-SNAP assembled into the axoneme. Western blots of purified axonemes from WT, pf2, and DRC4-SNAP strains were probed with antibodies against SNAP, RSP16, and DRC4. The endogenous DRC4 protein (55 kDa) was only detected in WT axonemes, whereas a higher molecular mass band (75 kDa) corresponding to SNAP-tagged DRC4 protein was detected by both SNAP and DRC4 antibodies in the DRC4-SNAP rescued axonemes. The RSP16 antibody shows similar loading amounts of axonemal proteins. E, purified axonemes from WT (negative control) and DRC3-SNAP were probed with fluorescent SNAP-Cell® TMR-Star, a SNAP tag substrate (red), which showed the assembly of SNAP-tagged axonemal proteins along the full-length of Chlamydomonas axonemes. Scale bar, 10 μm. F, Western blots of purified axonemes from WT, DRC3R (a DRC3 rescue strain without a SNAP tag), and two DRC3-SNAP strains were probed with streptavidin-horseradish peroxidase (HRP) and with antibodies against DRC3 and DRC2, which showed that the SNAP tags in the rescue strains react with BG-biotin. An expected DRC3 (60 kDa) band was only detected in WT and DRC3R axonemes, and an expected DRC3-SNAP (80 kDa) band was only detected in DRC3-SNAP axonemes by both streptavidin-HRP and the anti-DRC3 antibody. This anti-DRC3 antibody also detected another band at 70 kDa in the DRC3-SNAP axonemes that did not respond to streptavidin-HRP. The DRC2 bands show similar loading amounts of axonemal proteins. G, SNAP-tagged DRC3 can be labeled with 1.4-nm-sized strep-Au with high specificity and efficiency. Purified axonemes from WT and DRC3-SNAP were labeled with the SNAP substrate BG-biotin or BG-(PEG)12-biotin, and then with strep-Au, which was visualized by silver enhancement kit (Ag-kit, top panel). A 130-kDa band was detected by silver enhancement (top) and in a duplicated gel by DRC3 antibodies (bottom panel) only in DRC3-SNAP axonemes. Post-staining of the same gel with Coomassie Blue (middle panel) confirmed equivalent loading amounts of axonemal proteins. Untagged DRC3 (60 kDa) was only detected in WT axonemes.
SNAP-tagged N-DRC Components Can Be Labeled with Gold Nanoparticles in Intact Chlamydomonas Axonemes with High Efficiency and Specificity
To evaluate the binding of ligands to the SNAP-tagged N-DRC components, purified axonemes from WT, DRC3R (drc3 mutant rescued with an untagged DRC3 gene), and two DRC3-SNAP strains were conjugated with BG-biotin or the extended BG-(PEG)12-biotin. Western blots of the axonemes were probed with a biotin-binding complex of streptavidin and horseradish peroxidase and showed only a positive band for the two DRC3-SNAP axonemes at ∼80 kDa (Fig. 1F, top panel), which is consistent with the molecular weight of SNAP-tagged DRC3. There were no bands observed in the WT and DRC3R control axonemes (Fig. 1F, top panel), indicating that BG-biotin binds specifically to DRC3-SNAP. To confirm that the 80-kDa biotinylated protein is DRC3-SNAP, we also probed the Western blots with DRC3 antibody and observed labeling of the corresponding ∼80-kDa band in both DRC3-SNAP strains (Fig. 1F, middle panel). Unexpectedly, another band was observed at ∼70 kDa in both DRC3-SNAP strains that was not biotin-positive and could be a product of protein degradation. As expected, the anti-DRC3 antibodies also labeled bands at ∼60 kDa in both the WT and DRC3R axonemes (Fig. 1F, middle panel). As a loading control, the Western blots were also probed with an anti-DRC2 antibody, showing that the loading amounts were similar (Fig. 1F, bottom panel). These results suggest that the SNAP-tagged N-DRC components can be specifically ligated with BG-biotin in intact axonemes.
To achieve unambiguous visibility of a tag by (cryo-)EM requires the binding of an electron-dense probe. Therefore, we tested whether gold nanoparticles, which scatter electrons stronger than biological molecules like proteins and thus provide enhanced contrast, could be linked to the SNAP protein using the strong and highly specific biotin-streptavidin interaction. We purified axonemes of the WT and DRC3-SNAP strains and treated the intact axonemes first with BG-biotin or BG-(PEG)12-biotin and then with 1.4-nm-sized strep-Au, before removing unbound strep-Au by pelleting and washing. To test nanogold binding biochemically, labeled axonemes were dissolved in diluted lysis buffer under mild conditions, i.e. without DTT and heat treatment. This condition is critical to preserve the biotin-streptavidin interactions and to demonstrate nanogold binding on an SDS-polyacrylamide gel. After silver enhancement of the SDS-polyacrylamide gel, which is known to enlarge gold nanoparticles, a specific band could be observed at ∼130 kDa for the labeled DRC3-SNAP axonemes but not for the WT negative control (Fig. 1G, top panel). Coomassie Blue staining of the SDS-polyacrylamide gel showed that the loading amounts of axonemal proteins were similar (Fig. 1G, middle panel). Western blots of the same axonemal samples probed with anti-DRC3 antibody showed the presence of DRC3 in the 130-kDa nanogold-binding band in labeled DRC3-SNAP axonemes (Fig. 1G, bottom panel). Only a weak DRC3 signal was found at 80 and 70 kDa (Fig. 1G, bottom panel), demonstrating that DRC3-SNAP-biotin can be labeled with strep-Au with more than 90% efficiency. Comparison of the 130-kDa band of BG-biotin-ligated SNAP with that of BG-(PEG)12-biotin-ligated SNAP reveals that the (PEG)12 extension increased the overall labeling efficiency (Fig. 1G, top panel). Therefore, BG-(PEG)12-biotin was used in all cryo-ET studies. As expected, only a 60-kDa band, corresponding to the endogenous DRC3, was detected in Western blots of treated WT axonemes (Fig. 1G, bottom panel).
Structure of SNAP-tagged and Nanogold-ligated Axonemes Is Well Preserved and Resembles the Overall Structure of WT Axonemes
For a cryo-EM/ET labeling technique to be most beneficial, it is critical that the processing steps allow pristine preservation of the native structure of the specimen. To test how well the structure is preserved when using our labeling approach, we imaged axonemes from WT, pf2 null mutant, DRC4-SNAP rescue of pf2, drc3 null mutant, and DRC3-SNAP rescue of drc3 both with and without treatment with BG-(PEG)12-biotin and strep-Au using cryo-ET and subtomogram averaging (Figs. 2 and 3 and supplemental Videos 1–3). Comparison of cryo-tomograms and 96-nm axonemal averages between all studied strains reveals that all axonemal structures, except for the expected mutation defects and N-DRC labels, resembled each other and the WT structure, demonstrating that the developed labeling procedure preserves the pristine structure required for cryo-ET.
The axonemes of the Chlamydomonas pf2 mutant have previously been investigated by cryo-ET, but at relatively low resolution (14). Here, we reinvestigated pf2 axonemes and confirm that most of the N-DRC is missing, including the major part of the nexin link and central linker region (Fig. 2, D and E, and supplemental Video 1). In contrast to the previous study, however, we found that not only the base plate protrusion but the entire base plate density were considerably reduced (Fig. 2, D and E). The structural defects in pf2 are completely rescued in the DRC4-SNAP (C-terminally SNAP-tagged) strain (Fig. 2, F and G). The mutant-WT and mutant-rescue comparisons clearly show that DRC4 is required for other N-DRC components to properly assemble into the axoneme. The structural similarity between the 96-nm axonemal repeat of WT (Fig. 2, B and C) and DRC4-SNAP (with or without ligand treatment) (Fig. 2, F and G), including the N-DRC, inner and outer dynein arms (IDA and ODA), and radial spokes (RS), confirms that the SNAP tag fusion allows proper assembly and organization of the axoneme. However, the subtomogram averages of untreated DRC4-SNAP axonemes also confirm that the 3-nm resolution is insufficient to directly visualize the addition of the 20-kDa SNAP tag, i.e. we cannot localize DRC4 in the N-DRC by simply comparing the axonemal structures of WT and untreated DRC4-SNAP.
Likewise, we imaged the three-dimensional axonemal structures of the drc3 mutant and DRC3-SNAP rescue strain by cryo-ET and subtomogram averaging (Fig. 2, H–K and supplemental Video 2). In drc3 axonemes we observed that the L1 projection, part of the distal lobe of the nexin link, and the connection between these structures were missing (Fig. 2, H and I). The estimated molecular weight of the densities missing in drc3 is ∼140 kDa,4 which clearly exceeds the 60 kDa of DRC3, suggesting that DRC3 is required for other N-DRC components to assemble into the N-DRC. The structural defects seen in drc3 (Fig. 2, H and I) are fully rescued to a WT-like structure in DRC3-SNAP (Fig. 2, J and K). As in the case of DRC4-SNAP rescue, no additional mass corresponding to the SNAP tag was visible in untreated DRC3-SNAP averages (Fig. 2, J and K) when compared with the WT structure (Fig. 2, B and C). These images demonstrated the proper assembly of the N-DRC structure despite the presence of the SNAP tag but also that the resolution of cryo-ET and subtomogram averaging is insufficient to visualize the SNAP tag.
Precise Localization of N and C Termini of DRC3 and C Terminus of DRC4 in Chlamydomonas Axonemes by Cryo-ET Imaging of the Gold-labeled SNAP Tag Fusion Proteins
Our biochemical results demonstrate that the SNAP-tagged N-DRC components can be labeled with strep-Au (Fig. 1G). We then tested whether the streptavidin-conjugated gold nanoparticles (1.4 nm in size) can be resolved in subtomogram averages of the axonemal repeats of Chlamydomonas flagella. We purified axonemes from DRC3-SNAP (C-terminally SNAP-tagged), SNAP-DRC3 (N-terminally SNAP-tagged), and DRC4-SNAP, and we treated the intact axonemes with BG-(PEG)12-biotin, followed by strep-Au. The labeled and washed axonemes were then imaged using cryo-ET, and the axonemal repeats were averaged. Except for the N-DRC, other axonemal structures, including IDAs, ODAs, and RSs, in all three SNAP-tagged and labeled strains resembled the WT structure (compare images of DRC4-SNAP (Fig. 3, C and D), SNAP-DRC3 (Fig. 3, E and F), and DRC3-SNAP (Fig. 3, G and H) to WT (Fig. 3, A and B)).
In the averaged 96-nm repeat of labeled DRC4-SNAP axonemes, additional density corresponding to strep-Au was found at the end of the N-DRC base plate close to the hole in the inner junction between the A- and B-tubule (gold-colored in Fig. 3, C and D, and supplemental Video 3), indicating the location of the C terminus of DRC4. Strep-Au density was also clearly present in tomographic slices of the DRC4-SNAP axonemes (Fig. 4B, golden arrow), and absent in the corresponding position in WT axonemes (Fig. 4A, white arrow). Although the location of the DRC4 C terminus in this region was unexpected based on previous interpretations of mutant images that suggested that DRC4 was limited to the central nexin-linker region (14), our results are consistent with the defects in pf2 observed here, specifically with the reduced N-DRC base plate (Fig. 2, D and E) and the reduction in IA4 (dynein e) that attaches to doublet microtubules through the N-DRC base plate (supplemental Video 1). Thus, our data demonstrate that DRC4 stretches from the central region and microtubule attachment site of the nexin linker, all along the entire N-DRC base plate to the inner junction between the A- and B-tubule.
The three-dimensional averages of the axonemal repeats of labeled SNAP-DRC3 and DRC3-SNAP also clearly show the additional densities of strep-Au (gold-colored in Fig. 3, E–H, and supplemental Video 3). The strep-Au-labeled C terminus of DRC3 is seen to be localized at the edge of the L1 projection close to the microtubule surface (Fig. 3, G and H), whereas the strep-Au-labeled N terminus of DRC3 is found to be slightly distant from the L1 projection in the transition region between the linker and base plate of the N-DRC (Fig. 3, E and F). This location suggests that DRC3 could be important for transducing regulatory signals from the radial spokes (RS2 and RS3) through the base plate of the N-DRC to the motor IA5 (dynein g).
As described above, our biochemical results suggest that the SNAP fusion protein was labeled with strep-Au with an efficiency of more than 90% (Fig. 1G). The electron density of the strep-Au probes appeared somewhat blurred-out in the three-dimensional averages of labeled DRC4-SNAP, SNAP-DRC3, and DRC3-SNAP (Figs. 4B, 5B), which may be due to some flexibility in the position of strep-Au around the 1–2-nm-long PEG linker between the BG and biotin groups. To analyze label flexibility and labeling efficiency, we performed classification analyses (see “Experimental Procedures” for details) (Figs. 4 and 5). Classification of strep-Au in the labeled DRC4-SNAP axoneme resulted in three classes, all of which appeared to contain additional density for strep-Au but in slightly different positions (Fig. 4, D–F). This analysis suggests a nearly 100% labeling efficiency. The classification analysis also revealed four classes of strep-Au labeling for DRC3-SNAP and three classes for SNAP-DRC3. The additional density of the strep-Au label was visible in all classes (Fig. 5C), suggesting that both the N and C termini of DRC3 were labeled with nearly 100% efficiency. In summary, our results with DRC3 and DRC4 indicate that the SNAP tag can be labeled with high efficiency (nearly 100%) and then readily be visualized by cryo-ET and subtomogram averaging.
FIGURE 5.
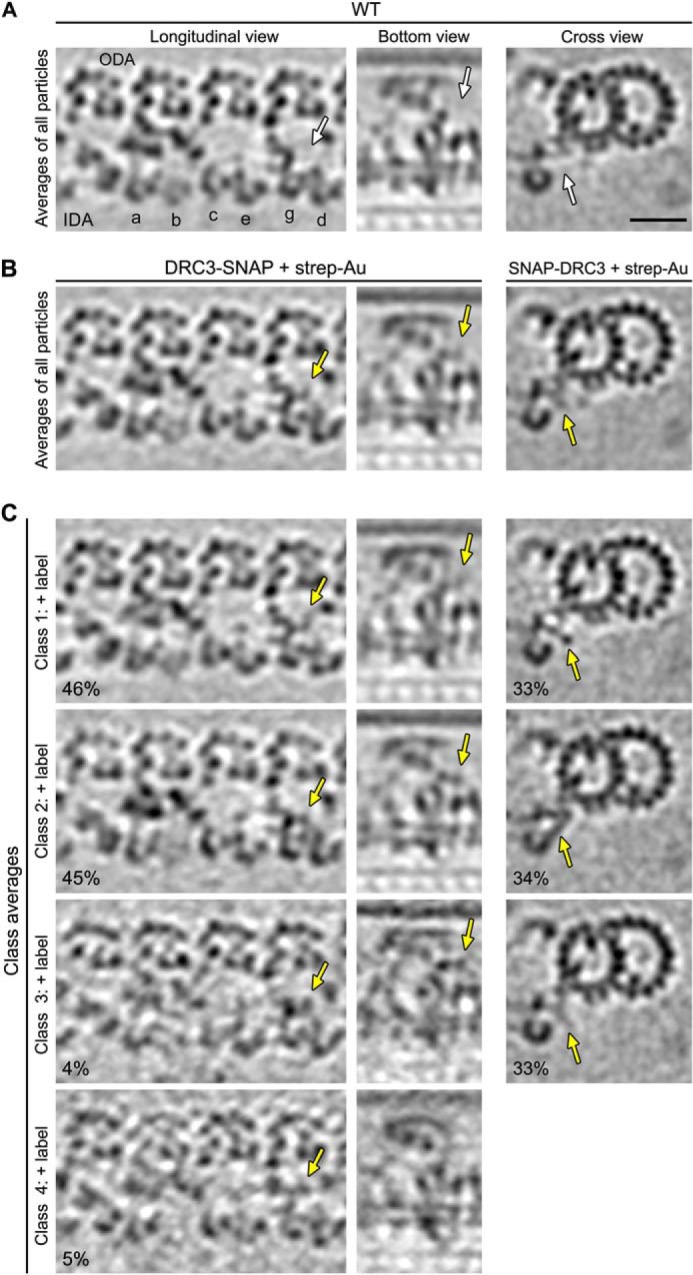
Classification analyses show high labeling efficiency for the N and C termini of SNAP-tagged DRC3. Tomographic slices of the averaged 96-nm axonemal repeats in longitudinal front view (left column), bottom view (middle column), and cross-section (right column) show the presence of SNAP-biotin-strep-Au density (golden arrows in B) in labeled DRC3-SNAP (B, left and middle columns) and SNAP-DRC3 (B, right column) axonemes compared with the unlabeled WT average (white arrows in A). The subtomogram averages of all axonemal repeats from both labeled SNAP-DRC3 and DRC3-SNAP axonemes (B) were classified focusing on the region with the label density; the results revealed distinct classes for each strain (four and three classes for DRC3-SNAP and SNAP-DRC3, respectively) that show the additional density of the biotin-strep-Au label (golden arrows in C) in slightly different positions, suggesting that the label is somewhat flexible and can pivot with the (PEG)12-linker around the SNAP tag of the fusion protein, i.e. at the N and C termini of DRC3. The biotin-strep-Au density was nevertheless observed in all classes, indicating nearly 100% labeling efficiency. Scale bar, 20 nm.
Density of the N-DRC Base Plate Is Not Completely Missing in pf3 Mutant Axonemes
Our previous proteomic study revealed that a small amount of DRC4 protein was still present in axonemes of the Chlamydomonas pf3 mutant (6). However, in a previous cryo-ET study that averaged all repeat units of pf3 axonemes, the N-DRC base plate could not be detected (14). Here, we performed a classification analysis of the N-DRC base plate regions in WT and pf3 axonemes, and we found that the N-DRC base plate was not only present in all WT repeats, but also in 17% of the pf3 repeats (Fig. 6, A and C). The lack of the N-DRC base plate in the remaining 83% of the pf3 axonemal repeats (Fig. 6D) dominates the subtomographic average, including all repeats, explaining why the remnant N-DRC base plate in pf3 axonemes was not observed in the previous study (see also Fig. 6B). These results are consistent with our previous proteomic and structural study and support that at least some part of DRC4 localizes to the N-DRC base plate.
FIGURE 6.
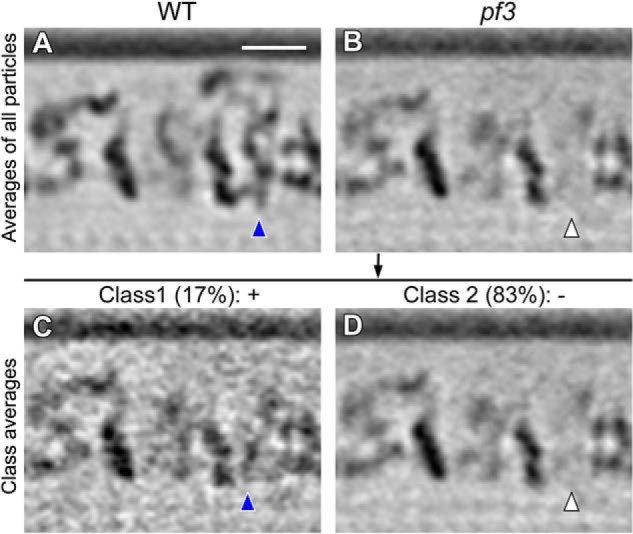
Classification analysis of pf3 axonemes reveals that the N-DRC base plate is not completely missing. Tomographic slices of the averaged 96-nm axonemal repeats in longitudinal bottom view (orientation similar to the right image in each panel of Fig. 4) show the presence of the N-DRC base plate in all WT (A, blue arrowhead) and in 17% of the pf3 axonemal repeats (C, blue arrowhead). As the density of the N-DRC base plate is missing from 83% of the pf3 axonemal repeats (D, white arrowhead), it is hardly visible in the average of all pf3 axonemal repeats (B, white arrowhead). Classification analysis was performed focusing on the base plate region of the N-DRC. Scale bar, 20 nm.
DISCUSSION
With the development and increasing application of cryo-ET to in situ studies, labeling techniques with high efficiency and specificity are in demand, yet currently the available labeling methods are often limited in their suitability for cryo-ET studies. Recently, biotin carboxyl carrier protein that can bind biotin covalently has been used successfully to tag and localize axonemal proteins in Chlamydomonas axonemes using cryo-ET (41–43). Here, we developed a SNAP tag-based labeling approach for cryo-ET studies. The SNAP tag technology has the advantage that it is a commercially available and already widely used label for cell biological studies, offering a variety of probes for live cell fluorescence studies. Here, we expand the SNAP tag applications to localize proteins with high precision in their cellular context using cryo-ET imaging and subtomogram averaging. The distance between the target protein and the ligated strep-Au is ∼5 nm, which is considerably smaller and more precise than the 20–25 nm of immunogold labels with primary and secondary antibodies. In addition, the results of our Western blots, silver-enhanced SDS gels, and EM classification analyses demonstrate that the strep-Au bound to SNAP-tagged proteins in intact axonemes with high specificity and close to 100% efficiency.
Visualization using SNAP tag technology requires access of the BG group of the substrate/probe to the catalytic site of the SNAP domain to form a covalent bond. The three-dimensional structure and architecture of a macromolecular complex are often unknown before the labeling experiments and therefore a possible problem is the accessibility of the SNAP tag by the BG ligand. Although (PEG)12 adds 1–2 nm of a possibly flexible linker between the protein of interest and the strep-Au, we chose BG-(PEG)12-biotin as a ligand for our cryo-ET studies because it labeled the target protein with even higher efficiency than did BG-biotin. This increase in labeling efficiency might be due to decreased steric hindrance as the small BG group on an extended linker might have easier access to the SNAP. We found that possible structural flexibility between the target protein and strep-Au due to the (PEG)12-linker can be addressed using a classification analysis and class averaging, which at the same time provides feedback about labeling efficiency. Similar to other clonable tags, the active site of the SNAP tag might be structurally buried inside a protein or complex, completely hindering access of the BG substrate and strep-Au. Such a problem could be addressed by introducing the SNAP tag in different sites of the target protein.
The quality of the three-dimensional reconstructions of axonemes after they were exposed to the conditions used for SNAP tag labeling, i.e. treatment with BG ligand and nanogold, clearly shows that the overall native structure is preserved during the labeling procedure and that this labeling procedure is thus compatible with cryo-ET imaging. These results with axonemes suggest that this new SNAP tag labeling approach for cryo-ET could in general be a powerful tool for determining the locations of proteins in situ with high precision.
The application of this technique to axonemes has improved our understanding of the structures and functions of cilia and flagella. Motile cilia/flagella are found almost ubiquitously in eukaryotic cells, from single-celled algae to humans. Their motility mechanism is complicated and controlled by several regulatory elements, including the ODA, IDA, RS, central pair complex, and N-DRC. The N-DRC is a 2-MDa large regulatory hub with at least 11 subunits that is thought to maintain outer doublet alignment, limit microtubule sliding, and regulate dynein activity (5, 7). In a previous cryo-ET study, we predicted the approximate locations of DRC1–7 within the N-DRC by comparing various drc mutants with the WT structure (14). DRC1 and DRC2 were predicted to form the N-DRC base plate; DRC3, DRC4, and DRC7 were localized to the central linker region; and DRC5 and DRC6 were localized to the part of the nexin linker that connects to the neighboring B-tubule. Recent studies found at least four new N-DRC subunits (DRC 8–11) by coimmunoprecipitation or iTRAQ labeling combined with mass spectroscopy (6, 7). Previous biochemical and structural studies (6, 7, 14) reported that DRC4 is crucial for the assembly of the nexin linker, but the molecular mechanisms underlying the assembly and function of the N-DRC are still unknown. Dissection of the precise three-dimensional architecture and subunit organization of the N-DRC would be an important step toward a better understanding of the inner workings of the N-DRC.
In addition to having successfully developed a SNAP tag labeling technique for cryo-ET as described above, we have also applied this technology to precisely determine the locations of the termini of DRC3 and DRC4 within the N-DRC in Chlamydomonas axonemes. Our three-dimensional reconstructions of the axonemal repeats with tagged N-DRC components revealed that the N terminus of DRC3 is close to the basal portion of the nexin linker and to DRC4, whereas the C terminus of DRC3 is close to the central portion of the linker and located at the edge of the L1 projection that connects to dynein g (Fig. 7). Together with the structural defects found in the drc3 null mutant axonemes, i.e. the missing L1 projection, our results suggest that DRC3 forms the L1 projection with both termini located away from the interface with the motor domain of dynein g.
Given the previous observations, it was somewhat surprising that the C terminus of DRC4 was localized to the end of the N-DRC base plate close to the hole in the A-B inner junction. However, our new three-dimensional averages of the pf2 (DRC4 null) mutant showed structural defects along the length of the N-DRC base plate, i.e. the base plate in this mutant is markedly thinner than in WT axonemes, which is consistent with the nanogold labeling of the DRC4 C terminus. These observations, together with the central role of DRC4 in the assembly of the nexin linker and its previously shown direct interactions with DRC3 (7, 25), suggest that DRC4 has an elongated morphology along the entire base plate extending to the nexin linker of the N-DRC (Fig. 7). Previous studies reported that DRC4 can bind to microtubules through its microtubule-binding domain and, in addition, interacts with other N-DRC subunits, such as DRC1 and DRC2 (7, 11). Even a mutated DRC4 lacking amino acids 96–151 (sup-pf3 strain) still binds to microtubules (7). Although DRC4 is required for proper assembly of the nexin link and its subunits (DRC3 and DRC5–7), DRC4 does not seem to be required for proper assembly of DRC1 and DRC2 into the base plate, as seen by the remaining base plate density in the pf2 mutant. In contrast, DRC1 and DRC2 seem to be important for DRC4 assembly into the N-DRC, because DRC4 is reduced and more easily extracted in the Chlamydomonas DRC1 null mutant pf3 (6, 11) and is markedly reduced in the DRC2 (CCDC65) mutant human patient (10). Both DRC1 and DRC2 are missing in pf3, and no base plate density was detected by previous cryo-ET studies (14). Here we performed a classification analysis (see under “Experimental Procedures”) of the base plate region of the pf3 mutant. We observed a small base plate density remaining in about a quarter of the axonemal repeats of pf3 (Fig. 6), which is consistent with the biochemical data that DRC4 is reduced but not completely missing from pf3 axonemes (6). The location of parts of DRC4 to the N-DRC base plate is also consistent with previous observations that dynein e is reduced in pf2 axonemes (7, 8, 14). In our structural study, most of the nexin linker is missing in the pf2 mutant, suggesting that the other missing subunits of the N-DRC are recruited or stabilized by DRC4 directly or indirectly.
Electron density corresponding to the distal lobe and the L1 projection of the nexin linker as well as the connection between these two structures that is observed in the cryo-ET structures of the WT axonemes are missing from the corresponding images of drc3 N-DRC (Fig. 2).4 These structures are not likely formed by DRCs 1, 2, 4–7, 8, and 11 because these DRCs are found with normal abundances in drc3 axonemes according to biochemical analysis.4 The estimated molecular mass of the total missing density is about 140 kDa larger than DRC3 itself, suggesting that DRC3 is required for the assembly or stability of other N-DRC subunits localized in the distal lobe and connector part of the nexin linker. Our data also showed that the assembly of dynein g was not affected in drc3. Therefore, the observed defective motility in drc3 cells is likely due to failure of the proper regulation of dynein g by DRC3, which forms the L1 projection.
Like the previously reported biotin carboxyl carrier protein tag and many other EM labeling methods, one remaining limitation of the SNAP tag/strep-Au approach is the requirement that EM-visible probes (here BG-biotin and strep-Au) are able to access the cloned tag. Cilia and flagella can be demembranated, and the isolated axonemes are sufficient to generate beating upon addition of ATP and at the same time allow delivery of the required labeling compounds to the intact axonemes. Delivery of such probes through an intact plasma membrane in live cells, however, has not (yet) been shown to be feasible. Nanogold particles have nevertheless been shown to cross cellular membranes (44, 45). Therefore, future development of a BG-nanogold probe that can reversibly cross membranes could considerably increase the applications of the SNAP labeling technique for cryo-ET studies of intact cells.
In summary, our labeling and precise localization results have provided new insights into the assembly and organization of the N-DRC, opening a new window to explore the communication between the N-DRC and other axonemal structures. The SNAP tag labeling technique for cryo-ET/subtomogram averaging developed here is a powerful tool for localizing proteins in macromolecular complexes in situ.
Acknowledgments
We are grateful to Maureen Wirschell (University of Mississippi) and Win Sale (Emory University) for providing the pUC57 plasmid containing a Chlamydomonas codon-optimized sequence encoding SNAP and to Ivan Correa (New England Biolabs) for providing the BG-(PEG)12-biotin. We thank Chen Xu for training and management of the Brandeis EM facility.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 GM083122 (to D. N.), R01 GM055667 (to M. E. P.), and R37 GM030626 (to G. B. W.). This work was also supported by Robert W. Booth Endowment at University of Massachusetts Medical School (to G. B. W.) and by the March of Dimes Foundation (to D. N.).

This article contains supplemental Videos 1–3.
J. Awata, K. Song, J. Lin., S. M. King, D. Nicastro, and G. B. Witman, submitted for publication.
- cryo-ET
- cryo-electron tomography
- EM
- electron microscopy
- N-DRC
- nexin-dynein regulatory complex
- strep-Au
- streptavidin-conjugated nanogold particle
- IDA
- inner dynein arm
- ODA
- outer dynein arm
- RS
- radial spoke
- BG
- O6-benzylguanine
- PEG
- polyethylene glycol.
REFERENCES
- 1. McIntosh R., Nicastro D., Mastronarde D. (2005) New views of cells in 3D: an introduction to electron tomography. Trends Cell Biol. 15, 43–51 [DOI] [PubMed] [Google Scholar]
- 2. Hoenger A., Nicastro D. (2007) Electron microscopy of microtubule-based cytoskeletal machinery. Methods Cell Biol. 79, 437–462 [DOI] [PubMed] [Google Scholar]
- 3. Nicastro D., Schwartz C., Pierson J., Gaudette R., Porter M. E., McIntosh J. R. (2006) The molecular architecture of axonemes revealed by cryoelectron tomography. Science 313, 944–948 [DOI] [PubMed] [Google Scholar]
- 4. Gan L., Jensen G. J. (2012) Electron tomography of cells. Q. Rev. Biophys. 45, 27–56 [DOI] [PubMed] [Google Scholar]
- 5. Huang B., Ramanis Z., Luck D. J. (1982) Suppressor mutations in Chlamydomonas reveal a regulatory mechanism for flagellar function. Cell 28, 115–124 [DOI] [PubMed] [Google Scholar]
- 6. Lin J., Tritschler D., Song K., Barber C. F., Cobb J. S., Porter M. E., Nicastro D. (2011) Building blocks of the nexin-dynein regulatory complex in Chlamydomonas flagella. J. Biol. Chem. 286, 29175–29191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bower R., Tritschler D., Vanderwaal K., Perrone C. A., Mueller J., Fox L., Sale W. S., Porter M. E. (2013) The N-DRC forms a conserved biochemical complex that maintains outer doublet alignment and limits microtubule sliding in motile axonemes. Mol. Biol. Cell 24, 1134–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gardner L. C., O'Toole E., Perrone C. A., Giddings T., Porter M. E. (1994) Components of a “dynein regulatory complex” are located at the junction between the radial spokes and the dynein arms in Chlamydomonas flagella. J. Cell Biol. 127, 1311–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Austin-Tse C., Halbritter J., Zariwala M. A., Gilberti R. M., Gee H. Y., Hellman N., Pathak N., Liu Y., Panizzi J. R., Patel-King R. S., Tritschler D., Bower R., O'Toole E., Porath J. D., Hurd T. W., Chaki M., Diaz K. A., Kohl S., Lovric S., Hwang D. Y., Braun D. A., Schueler M., Airik R., Otto E. A., Leigh M. W., Noone P. G., Carson J. L., Davis S. D., Pittman J. E., Ferkol T. W., Atkinson J. J., Olivier K. N., Sagel S. D., Dell S. D., Rosenfeld M., Milla C. E., Loges N. T., Omran H., Porter M. E., King S. M., Knowles M. R., Drummond I. A., Hildebrandt F. (2013) Zebrafish ciliopathy screen plus human mutational analysis identifies C21orf59 and CCDC65 defects as causing primary ciliary dyskinesia. Am. J. Hum. Genet. 93, 672–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Horani A., Brody S. L., Ferkol T. W., Shoseyov D., Wasserman M. G., Ta-shma A., Wilson K. S., Bayly P. V., Amirav I., Cohen-Cymberknoh M., Dutcher S. K., Elpeleg O., Kerem E. (2013) CCDC65 mutation causes primary ciliary dyskinesia with normal ultrastructure and hyperkinetic cilia. PLoS One 8, e72299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wirschell M., Olbrich H., Werner C., Tritschler D., Bower R., Sale W. S., Loges N. T., Pennekamp P., Lindberg S., Stenram U., Carlén B., Horak E., Köhler G., Nürnberg P., Nürnberg G., Porter M. E., Omran H. (2013) The nexin-dynein regulatory complex subunit DRC1 is essential for motile cilia function in algae and humans. Nat. Genet. 45, 262–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mizuno N., Taschner M., Engel B. D., Lorentzen E. (2012) Structural studies of ciliary components. J. Mol. Biol. 422, 163–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bui K. H., Yagi T., Yamamoto R., Kamiya R., Ishikawa T. (2012) Polarity and asymmetry in the arrangement of dynein and related structures in the Chlamydomonas axoneme. J. Cell Biol. 198, 913–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Heuser T., Raytchev M., Krell J., Porter M. E., Nicastro D. (2009) The dynein regulatory complex is the nexin link and a major regulatory node in cilia and flagella. J. Cell Biol. 187, 921–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Linck R., Fu X., Lin J., Ouch C., Schefter A., Steffen W., Warren P., Nicastro D. (2014) Insights into the structure and function of ciliary and flagellar doublet microtubules: tektins, Ca2+-binding proteins, and stable protofilaments. J. Biol. Chem. 289, 17427–17444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mercogliano C. P., DeRosier D. J. (2007) Concatenated metallothionein as a clonable gold label for electron microscopy. J. Struct. Biol. 160, 70–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Oda T., Kikkawa M. (2013) Novel structural labeling method using cryo-electron tomography and biotin-streptavidin system. J. Struct. Biol. 183, 305–311 [DOI] [PubMed] [Google Scholar]
- 18. Diestra E., Fontana J., Guichard P., Marco S., Risco C. (2009) Visualization of proteins in intact cells with a clonable tag for electron microscopy. J. Struct. Biol. 165, 157–168 [DOI] [PubMed] [Google Scholar]
- 19. Ludwig A., Howard G., Mendoza-Topaz C., Deerinck T., Mackey M., Sandin S., Ellisman M. H., Nichols B. J. (2013) Molecular composition and ultrastructure of the caveolar coat complex. PLoS Biol. 11, e1001640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shu X., Lev-Ram V., Deerinck T. J., Qi Y., Ramko E. B., Davidson M. W., Jin Y., Ellisman M. H., Tsien R. Y. (2011) A genetically encoded tag for correlated light and electron microscopy of intact cells, tissues, and organisms. PLoS Biol. 9, e1001041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Keppler A., Gendreizig S., Gronemeyer T., Pick H., Vogel H., Johnsson K. (2003) A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol. 21, 86–89 [DOI] [PubMed] [Google Scholar]
- 22. Bosch P. J., Corrêa I. R., Jr., Sonntag M. H., Ibach J., Brunsveld L., Kanger J. S., Subramaniam V. (2014) Evaluation of fluorophores to label SNAP-tag fused proteins for multicolor single-molecule tracking microscopy in live cells. Biophys. J. 107, 803–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Corrêa I. R., Jr. (2014) Live-cell reporters for fluorescence imaging. Curr. Opin. Chem. Biol. 20, 36–45 [DOI] [PubMed] [Google Scholar]
- 24. Liu T. K., Hsieh P. Y., Zhuang Y. D., Hsia C. Y., Huang C. L., Lai H. P., Lin H. S., Chen I. C., Hsu H. Y., Tan K. T. (2014) A rapid SNAP-tag fluorogenic probe based on an environment-sensitive fluorophore for no-wash live cell imaging. ACS Chem. Biol. 9, 2359–2365 [DOI] [PubMed] [Google Scholar]
- 25. Rupp G., Porter M. E. (2003) A subunit of the dynein regulatory complex in Chlamydomonas is a homologue of a growth arrest-specific gene product. J. Cell Biol. 162, 47–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sizova I., Fuhrmann M., Hegemann P. (2001) A Streptomyces rimosus aphVIII gene coding for a new type phosphotransferase provides stable antibiotic resistance to Chlamydomonas reinhardtii. Gene 277, 221–229 [DOI] [PubMed] [Google Scholar]
- 27. Awata J., Takada S., Standley C., Lechtreck K. F., Bellvé K. D., Pazour G. J., Fogarty K. E., Witman G. B. (2014) NPHP4 controls ciliary trafficking of membrane and large soluble proteins at the transition zone. J. Cell Sci. 127, 4714–4727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Berthold P., Schmitt R., Mages W. (2002) An engineered Streptomyces hygroscopicus aph 7" gene mediates dominant resistance against hygromycin B in Chlamydomonas reinhardtii. Protist 153, 401–412 [DOI] [PubMed] [Google Scholar]
- 29. Gorman D. S., Levine R. P. (1965) Cytochrome f and plastocyanin: their sequence in the photosynthetic electron transport chain of Chlamydomonas reinhardtii. Proc. Natl. Acad. Sci. 54, 1665–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Witman G. B. (1986) Isolation of Chlamydomonas flagella and flagellar axonemes. Methods Enzymol. 134, 280–290 [DOI] [PubMed] [Google Scholar]
- 31. Iomini C., Till J. E., Dutcher S. K. (2009) Genetic and phenotypic analysis of flagellar assembly mutants in Chlamydomonas reinhardtii. Methods Cell Biol. 93, 121–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang C., Compton M. M., Yang P. (2005) Dimeric novel HSP40 is incorporated into the radial spoke complex during the assembly process in flagella. Mol. Biol. Cell 16, 637–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gregori L., Hainfeld J. F., Simon M. N., Goldgaber D. (1997) Binding of amyloid beta protein to the 20 S proteasome. J. Biol. Chem. 272, 58–62 [DOI] [PubMed] [Google Scholar]
- 34. Nicastro D. (2009) Cryo-electron microscope tomography to study axonemal organization. Methods Cell Biol. 91, 1–39 [DOI] [PubMed] [Google Scholar]
- 35. Iancu C. V., Tivol W. F., Schooler J. B., Dias D. P., Henderson G. P., Murphy G. E., Wright E. R., Li Z., Yu Z., Briegel A., Gan L., He Y., Jensen G. J. (2006) Electron cryotomography sample preparation using the Vitrobot. Nat. Protoc. 1, 2813–2819 [DOI] [PubMed] [Google Scholar]
- 36. Mastronarde D. N. (2005) Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 [DOI] [PubMed] [Google Scholar]
- 37. Kremer J. R., Mastronarde D. N., McIntosh J. R. (1996) Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 116, 71–76 [DOI] [PubMed] [Google Scholar]
- 38. Heumann J. M., Hoenger A., Mastronarde D. N. (2011) Clustering and variance maps for cryo-electron tomography using wedge-masked differences. J. Struct. Biol. 175, 288–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 40. Harauz G., Van Heel M. (1986) Exact filters for general geometry three dimensional reconstruction. Optik 73, 146–156 [Google Scholar]
- 41. Oda T., Yanagisawa H., Yagi T., Kikkawa M. (2014) Mechanosignaling between central apparatus and radial spokes controls axonemal dynein activity. J. Cell Biol. 204, 807–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Oda T., Yanagisawa H., Kamiya R., Kikkawa M. (2014) Cilia and flagella. A molecular ruler determines the repeat length in eukaryotic cilia and flagella. Science 346, 857–860 [DOI] [PubMed] [Google Scholar]
- 43. Oda T., Yanagisawa H., Kikkawa M. (2014) Detailed structural and biochemical characterization of the nexin-dynein regulatory complex. Mol. Biol. Cell mbc.E14-09-1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shah N., Zhang S., Harada S., Smith R. M., Jarett L. (1995) Electron microscopic visualization of insulin translocation into the cytoplasm and nuclei of intact H35 hepatoma cells using covalently linked Nanogold-insulin. Endocrinology 136, 2825–2835 [DOI] [PubMed] [Google Scholar]
- 45. Onelli E., Prescianotto-Baschong C., Caccianiga M., Moscatelli A. (2008) Clathrin-dependent and independent endocytic pathways in tobacco protoplasts revealed by labelling with charged nanogold. J. Exp. Bot. 59, 3051–3068 [DOI] [PMC free article] [PubMed] [Google Scholar]