

Background: The unfolded protein response (UPR) has been implicated in retinal cell death but the mechanism is unknown.
Results: Cigarette smoke extract induces RPE cell apoptosis, which is alleviated by enhancing the UPR function.
Conclusion: UPR activation is required for RPE survival through up-regulation of Nrf2.
Significance: Enhancing Nrf2 and adaptive UPR protects the RPE against oxidative injury and apoptosis.
Keywords: apoptosis, cigarette smoke, nuclear factor 2 (erythroid-derived 2-like factor) (NFE2L2) (Nrf2), oxidative stress, unfolded protein response (UPR), RPE
Abstract
Recent studies have revealed a role of endoplasmic reticulum (ER) stress-induced unfolded protein response (UPR) in the regulation of RPE cell activity and survival. Herein, we examined the mechanisms by which the UPR modulates apoptotic signaling in human RPE cells challenged with cigarette smoking extract (CSE). Our results show that CSE exposure induced a dose- and time-dependent increase in ER stress markers, enhanced reactive oxygen species (ROS), mitochondrial fragmentation, and apoptosis of RPE cells. These changes were prevented by the anti-oxidant NAC or chemical chaperone TMAO, suggesting a close interaction between oxidative and ER stress in CSE-induced apoptosis. To decipher the role of the UPR, overexpression or down-regulation of XBP1 and CHOP genes was manipulated by adenovirus or siRNA. Overexpressing XBP1 protected against CSE-induced apoptosis by reducing CHOP, p-p38, and caspase-3 activation. In contrast, XBP1 knockdown sensitized the cells to CSE-induced apoptosis, which is likely through a CHOP-independent pathway. Surprisingly, knockdown of CHOP reduced p-eIF2α and Nrf2 resulting in a marked increase in caspase-3 activation and apoptosis. Furthermore, Nrf2 inhibition increased ER stress and exacerbated cell apoptosis, while Nrf2 overexpression reduced CHOP and protected RPE cells. Our data suggest that although CHOP may function as a pro-apoptotic gene during ER stress, it is also required for Nrf2 up-regulation and RPE cell survival. In addition, enhancing Nrf2 and XBP1 activity may help reduce oxidative and ER stress and protect RPE cells from cigarette smoke-induced damage.
Introduction
Age-related macular degeneration (AMD)2 is a leading cause of blindness in elderly people (1, 2). The early pathological changes of AMD include focal pigmentary irregularities of the posterior retina, increased lipofuscin in the retinal pigment epithelium (RPE), basal deposits, thickening of Bruch's membrane, and drusen formation (3). The disease can slowly progress to geographic atrophy or advanced “dry” AMD characterized by RPE atrophy accompanied by degeneration and loss of photoreceptors, or “wet” AMD characterized by abnormal new vessel formation from the choroid, both resulting in irreversible and severe vision loss (4). Studies to date have identified a number of risk factors, including genetic factors such as Y402H polymorphism of the complement factor H (CFH) gene (5, 6), and environmental factors. These factors impose significant influence on the development and progression of AMD. Cigarette smoking is considered the most important environmental risk factor for AMD (7–10). Cigarette smoke preferentially targets the RPE resulting in apoptosis of RPE cells, basement membrane thickening and subretinal deposits (11, 12). The mechanisms by which cigarette smoking enhances the RPE cell injury remain poorly understood.
The endoplasmic reticulum (ER) is a central hub for protein folding and maturation. Dysfunction of the ER results in ER stress, which activates the unfolded protein response (UPR), a sophisticated stress response program, to modulate cell activity and survival through regulation of protein synthesis, folding, and degradation (13). There are three key pathways of the UPR: 1) PERK-eIF2α-ATF4-CHOP pathway, 2) IRE1-XBP1 pathway, and 3) ATF6-chaporene pathway (14, 15). The latter two pathways primarily regulate ER chaperones, such as GRP78 and p58IPK, thereby increasing the capacity of protein folding (15–19). The early activation of PERK increasing the eIF2α phosphorylation reduces protein translation rate, which enables the ER to recover from its stressed condition. However, persistent ER stress activates the pro-apoptotic gene C/EBP homologous protein (CHOP), a downstream factor of the PERK pathway, resulting in apoptosis and cell death (14, 20, 21).
Aqueous whole-phase cigarette smoke extract (CSE) has been widely used to study the effects of cigarette smoke in cultured cells and disease models, such as chronic obstructive pulmonary disease, cancer, and cardiovascular diseases (22–28). The extract comprises water-soluble components of both the particulate and gas phases of cigarette smoke, which were believed to be present in the bloodstream of smokers (29). Importantly, it contains high levels of pro-oxidant compounds and free radicals that induce oxidative injury of various cells and tissues including RPE cells (30). In human bronchial epithelial and lung cells, CSE elicits ER stress likely through ROS resulting in CHOP-dependent apoptosis (31–33). In agreement with this finding, recent studies show that cigarette smoke induces ER stress in the RPE (34–36). In cultured RPE cells, hydroquinone, a potent oxidant in cigarette smoke, differentially regulates the UPR pathways by up-regulating CHOP expression but suppressing XBP1-mediated adaptive UPR signaling (36). Overexpressing XBP1 or decreasing CHOP level by reducing ER stress or inhibition of its upstream regulator, eIF-2a phosphorylation, protects cells from hydroquinone-induced apoptosis (36), suggesting a role of the UPR in regulating the RPE cell survival during oxidative injury.
In the present study, we examined the role of the UPR factors in RPE cells challenged with CSE and intended to understand the interactions between oxidative stress and ER stress and signaling pathways of Nrf2, XBP1s, and CHOP in CSE-induced apoptosis of RPE cells.
EXPERIMENTAL PROCEDURES
Preparation of CSE
Cigarette smoke extract (dissolved in DMSO, 40 mg/ml total particular matter, nicotine content = 6%) was purchased from Murty Pharmaceuticals (Lexington, KY) and was kept at −20 °C. Before each treatment, CSE was freshly prepared into working solutions and diluted with HEPES buffer as previously described (37).
Cell Culture
Human RPE (ARPE-19) cells (American Type Culture Collection, ATCC, Manassas, VA) were cultured with DMEM/F12 medium containing 10% fetal bovine serum (FBS) and 1% antibiotic/antimycotic. While growing to 70% confluence, cells were starved overnight with low-serum (1% FBS) DMEM/F12 medium to reduce antioxidant pool in the medium and make the cells quiescent and then subjected to desired treatment.
Construction and Transduction of Adenoviruses
Recombinant adenovirus expressing human Nrf2 was constructed using the AdEasy system ((Agilent Technologies) according to the manufacturer's instructions. Briefly, full-length human Nrf2 gene was cloned using primers: forward primer ACCGCCACCATGGATTTGATTGACATAC; and reverse primer CTCGAGCTAGTTTTTCTTAACATCTGGCT, and inserted into the KpnI-XhoI sites of the vector pShuttle-CMV. The resultant plasmid was co-transformed with pAdEasy-1 adenoviral vector into BJ5183 Escherichia coli-competent cells by electroporation. The recombinant adenoviral plasmids were then transfected into the packing cell line 293AD to generate recombinant adenoviruses. Construction of adenoviruses expressing human spliced XBP1 was described elsewhere (38). Large-scale preparation of adenoviruses was completed using 293AD cells. Harvested adenoviruses were tittered and purified using Adeno-X Maxi Purification kit (Clontech Laboratories, Mountain View, CA). ARPE-19 cells were transduced with adenoviruses as described previously (38). Adenovirus expressing LacZ was used as the control. After 24 h of transduction, cells were starved with 1% FBS DMEM/F12 medium followed by CSE treatment.
Small-interfering RNAs
Cells were transfected with siRNAs against human Nrf2, XBP1 or CHOP (Santa Cruz Biotechnology, Santa Cruz, CA) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) following the manufacturer's instruction and previously described (39). Lipofectamine 2000 or a nonsilencing scrambled siRNA with oligonucleotide sequence that does not recognize any known homology to mammalian genes was used as a negative control. The knockdown efficiency was determined by target protein levels using Western blotting.
Western Blot Analysis
Cells were lysed using lysis buffer containing 150 mm NaCl, 1% Igepal, 50 mm Tris, 1 mm EDTA, and 10% protease inhibitor mixture. Protein quantification was performed with the bicinchoninic acid (BCA) method (Thermo Scientific, Rockford, IL). Ten micrograms of total cellular protein was fractionated on 10% SDS-PAGE gels, electroblotted onto an Immun-blot polyvinylidene difluoride membrane (Bio-Rad), and blocked with 5% nonfat dry milk in TBST buffer for 1 h. After blocking, membranes were blotted overnight at 4 °C with following primary antibodies: anti-GRP78 (1:1000, Abcam), anti-p-eIF2α (1:1000, Cell Signaling), anti-ATF4 (CREB2, 1:1000, Santa Cruz), anti-CHOP (1:1000, Cell Signaling), anti-XBP1 (1:1000, Santa Cruz), anti-Nrf2 (1:1000, Santa Cruz), anti-p58IPK(1:1000, Cell Signaling), anti-ATF6 (1:1000, ABCAM), anti-cleaved-caspase-3 (1:500, Cell Signaling), anti-p-p38(1:1000, Cell Signaling), and anti-eIF2α (1:1000, Novus Biologicals, Littleton, CO). After incubation with HRP-conjugated secondary antibodies, membranes were developed with chemiluminescence substrate (Thermo Fisher Scientific, Rockford, IL. 34076) using Vision Works LS image acquisition and analysis software (UVP, Upland, CA). Membranes were reblotted with anti-β-actin (1:20,000, Abcam) for normalization. The bands were semi-quantified by densitometry using the same software.
MTT (3-(4,5-Dimethylthiazolyl-2)-2,5-diphenyltetrazolium Bromide) Assay
MTT assay was performed to assess the viability of ARPE-19 cells as described previously (38). Cells were seeded into a 96-well plate for each assay. Culture medium was refreshed, and 10 μl of MTT reagent was added to 100 μl of medium in each well. Cells were incubated at 37 °C for 2 h. The precipitation was then dissolved by adding 100 μl of detergent reagent into each well and incubated at room temperature for 4 h. The absorbance was measured at 570 nm with a microplate reader (Perkin Elmer, Waltham, MA), and data were analyzed.
TUNEL (Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling) Assay
TUNEL assay was performed to detect apoptosis using the In Situ Cell Death Detection Kit, TMR red (Roche Diagnostics Corp., Indianapolis, IN) following the manufacturer's protocol (40). Briefly, cells on coverslips were fixed with 4% paraformaldehyde (PFA) for 1 h, permeabilized in 0.1% citrate buffer containing 0.1% Triton X-100 for 2 min on ice, then incubated in TUNEL reaction mix containing nucleotides and terminal deoxynucleotidyl transferase (TdT) at 37 °C for 1 h. Incubation without the TdT enzyme was conducted as negative control. After incubation, the coverslip was mounted onto a slice using mounting medium containing 4′-6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA) and observed under an Olympus AX70 microscope (Olympus, Japan).
In Situ Trypan Blue Staining
After treatment, ARPE-19 cells were stained in situ with 0.04% Trypan Blue in DMEM/F12 medium for 15 min (41). Trypan Blue-stained cells and total cells were counted per 10× field under an invert microscope (Zeiss, Germany). At least 5 fields were counted and averaged for each replicate, and results were obtained from three independent experiments.
Reverse Transcription Polymerase Chain Reaction (RT-PCR)
Total RNA from ARPE-19 cells was extracted using the E.Z.N.A. Total RNA Kit I (Omega Bio-Tek, Norcross, GA) according to the manufacturer's protocol. cDNA synthesis was performed using the Maxima First Strand cDNA Synthesis Kit (Fermentas, Glen Burnie, MD). PCR was performed using PCR Master Mix (Fermentas) as described (40). The primers for human XBP1 were 5′-TTA CGA GAG AAA ACT CAT GGC-3′ and 5′-GGG TCC AAG TTG TCC AGA ATG C-3′. PCR products were resolved and run on a 2.5% agarose/1× TAE gel (40, 42).
Intracellular ROS and Mitochondrial Morphology Analysis
Levels of intracellular reactive oxygen species (ROS) were assessed using CellROX (Fluorescence Probes, Invitrogen). Briefly, cells were incubated with CellROX Deep Red Reagent (5 μm) for 30 min (43) and then incubated with MitoTracker® Green FM (Invitrogen) at 500 nm for another 30 min to determine morphologic changes of the mitochondria and the distribution of ROS (44). After three washes with PBS, cells were observed and imaged under a Zeiss LSM confocal microscope. ROS levels were measured fluorescence density and quantified using Image-J software.
Statistical Analysis
All quantitative data are presented as mean ± S.D. Statistical analyses were performed using unpaired Student's t test for two group data and one-way analysis of variance (ANOVA) with Bonferroni's multiple comparison test for three groups or more. Differences were considered statistically significant at p < 0.05.
RESULTS
CSE Induces ER Stress and Apoptosis in ARPE-19 Cells
To determine if CSE is sufficient to induce ER stress, ARPE-19 cells were exposed to a broad range of doses (0.004–320 μg/ml) of CSE for 24 h. This dose range overlaps with the plasma levels of water-soluble components of cigarette smoke in smokers (37), and moreover, the concentrations of nicotine in the CSE solutions (0.24 ng/ml-19.2 μg/ml) overlap with plasma levels of nicotine found in smokers (45). Results showed that 80 μg/ml-320 μg/ml of CSE significantly increased expression of GRP78 and phosphorylation of eIF2α, while CSE increased ATF4 and CHOP expression only at 320 μg/ml (Fig. 1, A and B). To determine the time course of CSE's effects on ER stress, cells were exposed to 320 μg/ml of CSE for 6–48 h. Results showed that level of GRP78 and phosphorylation of eIF2α increased at 6 h and continued to increase until 24 h. The levels of ATF4 and CHOP increased from 6 h, peaked at 12 h, and slightly declined at 24 h. In addition, the level of activated ATF6, another major ER stress marker, was increased from 6 to 24 h. Similar changes were observed in Nrf2, a major transcription factor that up-regulates antioxidant genes, which may reflect increased oxidative stress upon CSE exposure (Fig. 1, C and D).
FIGURE 1.

Cigarette smoking extract induced ER stress and apoptosis in ARPE-19 cells. ARPE-19 cells were exposed to CSE (0.004 μg/ml-320 μg/ml) for up to 24 h. A, protein levels of ER markers or pro-apoptosis markers were determined by Western blotting in cells treated with CSE for 6 h (p-eIF2α, eIF2α and GRP78) or 12 h (ATF4 and CHOP). B, expression of p-eIF2α (normalized with eIF2α), GRP78, ATF4 and CHOP were quantified by densitometry of Western blots. C, protein levels of ER stress markers, Nrf2, p-p38, and cleaved caspase-3 were determined by Western blotting in cells treated with 320 μg/ml of CSE for 6–24 h. D, expression of p-eIF2α (normalized with eIF2α), GRP78, ATF4, CHOP, ATF6, Nrf2, p-p38, and cleaved caspase-3 were quantified by densitometry. E, apoptosis of ARPE-19 cells was examined by TUNEL assay after CSE treatment for 24 h. Scale Bar: 50 μm. F, quantification of TUNEL-positive cells. G, cell viability of ARPE-19 cells with or without 320 μg/ml of CSE treatment for 24 h. All data were expressed as mean ± S.D., from three independent experiments. *, p < 0.05; **, p < 0.01 versus control.
To determine whether CSE exposure induces apoptosis in RPE cells, activation of caspase-3, a key mediator of apoptosis, was examined by Western blot analysis of cleaved caspase-3. Results show that the level of cleaved caspase-3 significantly increased only after CSE (320 μg/ml) treatment for 24 h (Fig. 1, C and D). Activation of p38 MAPK (P38), another potential signaling molecule involved in apoptosis, was observed CSE-challenged cells. TUNEL assay showed that the number of apoptotic cells was significantly increased after CSE treatment (Fig. 1, E and F). Cell viability measured by MTT assay was markedly decreased by CSE exposure (Fig. 1G).
Antioxidant or Chemical Chaperone Ameliorates CSE-induced ER Stress and Apoptosis
N-Acetyl-cysteine (NAC) is an antioxidant that reduces oxidative stress induced by CSE (30). To determine whether ER stress induced by CSE is mediated by oxidative stress, we pre-treated ARPE-19 cells with 1 mm NAC and then exposed the cells with 320 μg/ml of CSE for 6–24 h. Activation of the UPR and apoptosis was determined by Western blot analysis. Results show that NAC reduced the expression of p-eIF2α, ATF4, CHOP, and alleviated activation of caspase-3 compared with CSE treatment only (Fig. 2, A and B), suggesting a potential role of oxidative stress in CSE-induction of ER stress. As expected, NAC also reduced the induction of Nrf2 by CSE (Fig. 2C). TUNEL assay and MTT demonstrated that pre-treatment with NAC protected ARPE-19 cells from apoptosis (Fig. 2, D and E) and alleviated the reduction of cell viability and mitigated cell death caused by CSE (Fig. 2, F and G).
FIGURE 2.

Antioxidant attenuated the CSE-induced ER stress and protected RPE cells from apoptosis. ARPE-19 cells were pretreated with 1 mm of NAC for 6 h and then incubated with 320 μg/ml of CSE for 6–24 h. A, representative Western blots of p-eIF2α and eIF2α in cells treated with CSE for 6 h, ATF4 and CHOP (CSE-treated for 12 h), and cleaved-caspase-3 (CSE-treated for 24 h). B, densitometry analysis of p-eIF2α (normalized with eIF2α), ATF4, CHOP, and cleaved-caspase-3 (normalized with β-actin). C, Nrf2 levels were examined by Western blotting. D, apoptosis was examined by TUNEL assay after CSE treatment for 24 h. Scale Bar: 50 μm. E, quantification of TUNEL-positive cells. F, cell viability result of ARPE-19 cells after CSE 24 h treatment with or without NAC pretreatment. G, cell death was detected using in situ Trypan Blue staining after CSE treatment for 24 h. All data were expressed as mean ± S.D., from three independent experiments. *, p < 0.05; **, p < 0.01 versus control; †, p < 0.05; ‡, p < 0.01 vs. CSE.
To decipher the importance of ER stress in CSE-induced apoptosis, TMAO, a chemical chaperone that facilitates protein folding thereby reducing ER stress, was applied in our study. ARPE-19 cells were pre-treated with TMAO and then challenged with CSE. In TMAO-pretreated cells, CSE induced much lower expression of ATF4, CHOP, and cleaved caspase-3 (Fig. 3, A and B). TUNEL assay confirmed the anti-apoptotic effect of TMAO (Fig. 3, C and D). These results indicate that ER stress plays a role in CSE-induced apoptosis in RPE cells.
FIGURE 3.

Chemical chaperone decreased CHOP expression and mitigated CSE-induced apoptosis. ARPE-19 cells were pretreated with TMAO for 6 h, followed by CSE (320 μg/ml) treatment for 6–24 h. A, representative Western blots of p-eIF2α and eIF2α (CSE- treated for 6 h), ATF4 and CHOP (CSE-treated for 12 h), and cleaved-caspase-3 (CSE-treated for 24 h). B, densitometry analysis of p-eIF2α (normalized with eIF2α), ATF4, CHOP, and cleaved-caspase-3 (normalized with β-actin). C, apoptosis of ARPE-19 cells was examined by TUNEL assay after CSE 320 μg/ml 24 h treatment with or without TMAO (200 mm, 6 h) pretreatment. Scale Bar: 50 μm. D, quantification of TUNEL-positive cells. All data were expressed as mean ± S.D., from three independent experiments. *, p < 0.05; **, p < 0.01 versus Ctrl; †, p < 0.05; ‡, p < 0.01 versus CSE.
Chemical Chaperone Decreases ROS Levels and Attenuates Mitochondrial Changes Caused by CSE
Evidence shows that CSE, which contains potent oxidants, leads to depletion of GSH and subsequent mitochondrial damage in ARPE-19 cells (30). To determine reducing ER stress by chemical chaperone affects ROS generation and mitochondria, ROS levels and mitochondrial morphology were measured in ARPE-19 cells after CSE treatment for 6 h, with or without pretreatment of NAC or TMAO. After 6 h of CSE treatment, APRE-19 cells showed significantly increased ROS, most of which co-localized with mitochondria, suggesting that mitochondria is a major source of ROS generation in CSE-treated cells (Fig. 4A). Significant lower levels of ROS were observed in cells pre-treated with TMAO or NAC, which indicates an inhibitory effect of chemical chaperone on oxidative stress (Fig. 4B). Additionally, using MitoTracker® staining of live cells, we observed changes in mitochondrial morphology after CSE treatment. As shown in Fig. 4A, mitochondria became fragmented and more diffusely distributed in CSE-treated cells. These changes were reduced in cells with NAC or TMAO treatment, indicating that inhibiting ER stress may protect mitochondria and alleviate ROS generation.
FIGURE 4.

Antioxidant and chemical chaperone reduced ROS level and alleviated mitochondrial changes induced by CSE. ARPE-19 cells were exposed to CSE (320 μg/ml) for 6 h, with or without NAC (1 mm) or TMAO (100 mm) pretreatment for 6 h. Increased ROS level and morphological changes of mitochondria in ARPE-19 cells was induced by CSE and attenuated by antioxidant NAC or chemical chaperone TMAO. A, representative images of ROS and mitochondrial changes under confocal microscope. Red: ROS. Green: MitoTracker®. Scale Bar: 20 μm. B, quantification of intracellular ROS level by measuring florescence density. All data were expressed as mean ± S.D., from three independent experiments. **, p < 0.01 versus Ctrl; ‡, p < 0.01 versus CSE.
Activation of XBP1 Is Important for Cell Survival after CSE Treatment
As a master coordinator of the adaptive UPR, XBP1 plays a vital role in maintaining the ER function by inducing ER chaperone and ERAD protein expression. RT-PCR and Western blotting results showed that 320 μg/ml or 400 μg/ml of CSE treatment for 24 h stimulated XBP1 mRNA splicing (Fig. 5A) and increased its protein expression (Fig. 5B). To determine the role of XBP1 in CSE-induced cell apoptosis, ARPE-19 cells were transduced with adenovirus expressing spliced (active form) XBP1 (Ad-XBP1) or LacZ (Ad-LacZ) as control. Adenoviral transduced cells were then treated with 320 μg/ml of CSE for 24 h. Western blotting showed that overexpressing XBP1 reduced the expression of CHOP, p-p38, and cleaved caspase-3 (Fig. 5, C and D). TUNEL assay (Fig. 5, E and F) and in situ Trypan Blue staining (Fig. 5, G and H) showed that CSE induced less apoptosis and cell death in Ad-XBP1-treated cells than in control cells. These results indicate that XBP1 overexpression attenuates ER stress and protects cells from apoptosis.
FIGURE 5.

Overexpression of XBP1s reduced CHOP expression and enhanced cell viability of ARPE cells. A–B, ARPE-19 cells were treated with CSE for 24 h. Splicing of XBP1 mRNA was visualized by RT-PCR (A). Protein level of XBP1s was determined by Western blotting and semi-quantified by densitometry (B). **, p < 0.01 versus control. C–H, ARPE-19 cells were transduced with Ad-XBP1s or Ad-LacZ for 24 h, followed by CSE treatment for additional 24 h. C, protein level of XBP1s, CHOP, p-p38, and cleaved-caspase-3 were determined by Western blotting. D, densitometry analysis of Western blots for XBP1s, CHOP, p-p38, and cleaved-caspase-3 (normalized with β-actin). E, apoptosis of ARPE-19 cells was examined by TUNEL assay. Scale Bar: 100 μm; F, quantification of apoptotic cells number (TUNEL-positive cells) from E. G, dead cell number was detected using in situ Trypan Blue staining after CSE treatment for 24 h. Arrows show Trypan Blue-stained cells. Scale Bar: 100 μm. H, quantification of dead cells (Trypan Blue-stained cells). All data were expressed as mean ± S.D., from three independent experiments. *, p < 0.05; **, p < 0.01 versus Ad-LacZ; #, p < 0.05; ##, p < 0.01 versus Ad-LacZ+ CSE.
To elucidate the role of endogenous XBP1, siRNA was used to knock down XBP1 gene in ARPE-19 cells and the knockdown efficiency was examined by Western blotting. Due to the short half-life of XBP1 proteins (22 min for XBP1s and 11 min for XBP1u), MG132, a proteasome inhibitor, was used to inhibit protein degradation. Specifically, cells were treated with XBP1 siRNA or control, then exposed to CSE for 24 h. During the last 4 h of the treatment, 10 μm of MG132 was added to culture medium. Our results show that protein level of XBP1s was reduced by 60% (Fig. 6, A and B). In XBP1 siRNA-treated cells, CSE induced less eIF2α phosphorylation, more p-p38 expression, and strongly increased caspase-3 activation (Fig. 6, C and D). However, down-regulation of XBP1 did not affect CSE-induced CHOP expression (Fig. 6, C and D). CSE also induced more morphologic changes and shrunken cells (Fig. 6E), and more reduction in cell viability in the group of XBP1-knockdown (Fig. 6F). TUNEL assay (Fig. 6, G and H) and Trypan Blue staining (Fig. 6, I and J) showed that CSE induced more apoptosis and cell death in the cells treated with XBP1 siRNA, suggesting endogenous XBP1 has a protective role in RPE cells challenged with CSE.
FIGURE 6.

Knockdown of XBP1 exacerbated CSE-induced cell apoptosis. ARPE-19 cells were transfected with XBP1 siRNA to knockdown XBP1 expression and then treated with CSE for 24 h. A, to determine the XBP1 siRNA knockdown efficiency, 10 μm of MG132, a proteasome inhibitor, was added to culture medium during the last 4 h of the CSE treatment. Western blotting shows down-regulation of XBP1s after XBP1 siRNA. B, quantification of XBP1 siRNA efficiency by densitometry. C, protein level of p-eIF2α, CHOP, p-p38 and cleaved-caspase-3 were determined by Western blotting. D, densitometry analysis of Western blots for p-eIF2α, CHOP, p-p38, and cleaved-caspase-3 (normalized with β-actin). E, representative images of siRNA-transfected ARPE-19 cells after CSE treatment for 24 h. Scale Bar: 200 μm. F, cell viability was quantified by MTT assay. G, apoptosis examined by TUNEL assay. Scale Bar: 100 μm. H, quantification of apoptotic cells number (TUNEL-positive cells) from G). I, cell death was detected using in situ Trypan Blue staining. Scale Bar: 200 μm. J, quantification of dead cells (Trypan Blue-stained cells). All data were expressed as mean ± S.D., from three independent experiments. *, p < 0.05; **, p < 0.01 versus mock; #, p < 0.05; ##, p < 0.01, versus mock + CSE.
CHOP Down-regulation Reduced Nrf2 Level and Exacerbated CSE-induced Cell Death
Compelling evidence shows that activation of CHOP is a key step in the execution of ER stress-associated apoptosis (14). Our study found that CSE induced CHOP expression in APRE-19 cells. To determine the role of CHOP in CSE-induced apoptosis, siRNA was applied to knock down CHOP expression. In CHOP siRNA-treated cells, CSE-induced CHOP expression was dramatically decreased compared with control cells (Fig. 7, A and B), and the expressions of CSE-induced Nrf2, p-eIF2α, and p-p38 were also reduced (Fig. 7, A and B). Although we had expected that CHOP inhibition protects ARPE-19 cells form CSE induced cell apoptosis and cell death, in contrast to our expectation, CSE induced more caspase-3 activation (Fig. 7, A and B), more morphologic changes and shrunken cells (Fig. 7C), more reduction of cell viability (Fig. 8D), more apoptosis (Fig. 7, E and F), and more cell death (Fig. 7, G and H) in CHOP-deficient cells compared with controls. These data indicate that although CHOP was generally considered to be a transcription factor involved in apoptosis, knockdown of CHOP may have an aggravating effect rather than a protective effect on CSE-induced apoptosis in RPE cells.
FIGURE 7.

Knockdown of CHOP attenuated CSE-induced ER stress but increased cell apoptosis. CHOP-siRNA was applied to knockdown CHOP expression in ARPE-19 cells, followed by CSE treatment for 24 h. A, Western blotting shows the down-regulation of CHOP after CHOP siRNA, and protein level of Nrf2, p-eIF2α, p-p38, and cleaved-caspase-3. B, quantification of CHOP, Nrf2, p-eIF2α, p-p38, and cleaved-caspase-3 by densitometry. C, representative phase contrast images of ARPE-19 cells. Scale Bar: 200 μm. D, cell viability of ARPE-19 cells measured by MTT assay after CSE 24 h treatment. E, apoptosis was examined by TUNEL assay after CSE treatment for 24 h. Scale Bar: 100 μm. F, quantification of apoptotic cells number (TUNEL-positive cells) from E). G, dead cell number was detected using in situ Trypan Blue staining after CSE treatment for 24h. Scale Bar: 200 μm. H, quantification of dead cells (Trypan Blue-stained cells). All data were expressed as mean ± S.D., from three independent experiments. *, p < 0.05; **, p < 0.01 versus mock; #, p < 0.05; ##, p < 0.01, versus mock + CSE.
FIGURE 8.

Knockdown of Nrf2 exacerbates CSE induced ER stress and cell death. ARPE-19 cells were transfected with Nrf2 siRNA to knockdown Nrf2 expression and then treated with CSE for 24 h. A, protein level of Nrf2, CHOP, p-p38, and cleaved-caspase-3 were determined by Western blotting analysis. B, semi-quantification of Nrf2, CHOP, p-p38, and cleaved-caspase-3 (normalized with β-actin) by densitometry. C, apoptosis was examined by TUNEL assay after CSE treatment for 24 h. Scale Bar: 100 μm. D, quantification of TUNEL-positive cells from C). E, cell death was detected using in situ Trypan Blue staining after CSE treatment for 24 h. Arrows show Trypan Blue-stained cells. Scale Bar: 100 μm. F, quantification of dead cells (Trypan Blue-stained cells). All data were expressed as mean ± S.D., from three independent experiments. *, p < 0.05; **, p < 0.01 versus mock; #, p < 0.05; ##, p < 0.01, versus mock + CSE.
Nrf2 Regulates CHOP Expression and Apoptosis in RPE Cells
Nrf2 is a central regulator of the anti-oxidant system, inducing expression of genes encoding phase II detoxification enzymes and antioxidant proteins (46). To determine whether down-regulation of Nrf2 expression by CHOP deletion is responsible for enhanced caspase-3 activation and apoptosis, we used siRNA to knock down the Nrf2 gene in ARPE-19 cells. We found that in the cells pre-treated with Nrf2 siRNA, CSE caused more robust CHOP and p-p38 expression and caspase-3 activation compared with mock controls (Fig. 8, A and B). In line with these changes, TUNEL and Trypan Blue staining assays showed more apoptotic cells (Fig. 8, C and D) and more dead cells (Fig. 8, E and F) in Nrf2 siRNA group after CSE treatment. These results suggest that Nrf2 down-regulation may contribute to aggravated apoptosis caused by CHOP inhibition.
To further validate the role of Nrf2 on CHOP expression and apoptosis induced by CSE, we overexpressed Nrf2 using adenovirus in ARPE-19 cells. Western blotting showed that Nrf2 overexpression remarkably reduced expression CHOP, p-p38, cleaved caspase-3 induced by CSE when compared with control adenovirus (Fig. 9, A and B). TUNEL assay showed that overexpression of Nrf2-protected cells from CSE-induced apoptosis (Fig. 9, C and D). In situ Trypan Blue staining showed decreased number of dead cells in Nrf2 overexpression group after CSE treatment (Fig. 9, E and F). Together, these data revealed a potent effect of Nrf2 on reducing CHOP expression and apoptosis in CSE-challenged RPE cells.
FIGURE 9.
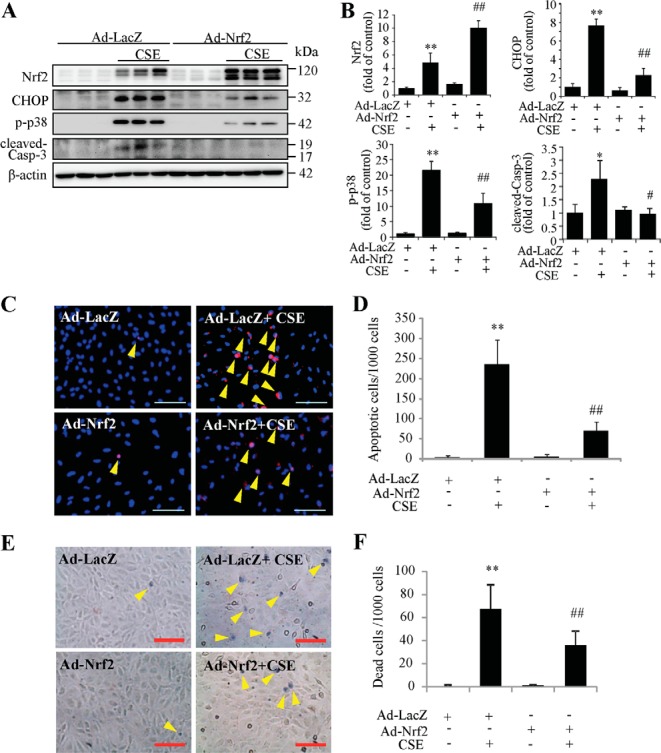
Overexpression of Nrf2 reduces CSE induced ER stress and protects RPE cells from apoptosis. ARPE-19 cells were transduced with Ad-Nrf2 or Ad-LacZ for 24 h, followed by CSE treatment for additional 24 h. A, protein level of Nrf2, CHOP, p-p38, and cleaved-caspase-3 were determined by Western blotting. B, semiquantification of Nrf2, CHOP, p-p38, and cleaved-caspase-3 (normalized with β-actin) by densitometry. C, apoptosis was examined by TUNEL assay after CSE treatment for 24 h. Scale Bar: 100 μm. D, quantification of TUNEL-positive cells from C). E, cell death was examined using in situ Trypan Blue staining after CSE treatment for 24 h. Arrow shows Trypan Blue- stained cells. Scale Bar: 100 μm. F, quantification of dead cells (Trypan Blue-stained cells). All data were expressed as mean ± S.D., from three independent experiments. *, p < 0.05; **, p < 0.01 versus Ad-lacZ; #, p < 0.05; ##, p < 0.01, versus Ad-LacZ + CSE.
DISCUSSION
Cigarette smoking is the most important environmental risk factor for AMD, and has been confirmed to induce damage in RPE cells (7–10). Studies have shown that CSE induces ER stress markers such as phosphorylation of eIF2α and the nuclear form of ATF6 in lung lysates from mice (47), GRP78 and CHOP in human bronchial epithelial cells (hBEC) and in the lungs of mice exposed to cigarette smoke (32). Recent work by our group and others also suggests induction of ER stress by cigarette smoke in the RPE (34–36), however, the mechanisms by which ER stress regulates signaling pathways implicated in stress response and apoptosis are not well understood. In the present study, we found that CSE increased the expression of GRP78 and p-eIF2α, and induced ATF4 and CHOP expression in a time- and dose-dependent manner in cultured human RPE cells. The increased p-eIF2α reflects the activation of PERK in UPR, which indirectly inactivates eIF2α and inhibits mRNA translation (14). At the same time, p-eIF2α activates transcription factor ATF4. ATF4 induces its downstream transcription factor CHOP, which is considered to control genes involved in apoptosis (14). Our study shows that CSE induced apoptosis and reduced the cell viability in ARPE-19 cells, indicating that CSE induces cell apoptosis when persistent ER stress exits. We also found that CSE induced ATF6 and Nrf2, two protective factors expressed during ER stress, in ARPE-19 cells at 6 h of CSE exposure. This result may indicate an auto-protective mechanism of ARPE-19 cells in early stages of ER stress.
CSE contains a large amount of free radicals that can induce oxidative stress (29, 48–51). As described above, oxidative stress can induce ER stress. However, how oxidative stress affects CSE-induced ER stress in ARPE-19 cells is not clear. In our study, antioxidant NAC alleviated CSE-induced ER stress, protected RPE cells from apoptosis, and increased cell viability in ARPE-19 cells, suggesting that oxidative stress is involved in the induction of CSE-induced ER stress. We also found that chemical chaperone TMAO significantly attenuated CSE-induced ER stress, and protected ARPE-19 cells from CSE-induced apoptosis, indicating that ER stress is important to CSE-induced cell apoptosis. Previous studies have shown that CSE disturbs oxidative balance, including increased lipid peroxidation (4-HNE) and mitochondrial superoxide production, as well as decreased intracellular glutathione (GSH) in RPE cells (30). In the present study, we found that CSE increased ROS accumulation in ARPE-19 cells, confirming the CSE induced oxidative stress. The accumulated ROS partly co-located with mitochondria, causing mitochondria shrank and fragmented. Pre-treatment of cells with the antioxidant NAC or the chemical chaperone TMAO alleviated CSE-induced ER stress and ROS accumulation, and protected cells from apoptosis. Interestingly, our preliminary study indicates that inducing mitochondrial ROS generation was sufficient to activate the UPR signaling (data not shown). In addition, CSE induced a reduction in mitochondrial potential, which was partially prevented by NAC but not TMAO (data not shown). These results indicate that in RPE cells ER stress and mitochondria-associated oxidative stress are closely associated and interdependently regulate each other, both contributing to apoptosis induced by CSE.
The early stage of UPR is the predominant adaptive cytoprotective response against ER stress. Studies have shown that the transcription factor XBP1s activates UPR genes, such as chaperon p58IPK, to relieve ER stress (18, 19). Although hydroquinone, a potent oxidant in cigarette smoke, suppressed XBP1-mediated adaptive UPR signaling (36), we found that CSE increased the expression of UPR factor XBP1s. In our study we found that overexpression of XBP1s reduced the CSE-induced CHOP and p-p38, protecting ARPE-19 cell from CSE-induced apoptosis. In contrast, knockdown of XBP1 decreased the baseline level of p-eIF2α and CSE induced eIF2α phosphorylation, up-regulated P38 activation, and increased the sensitivity of ARPE-19 cell to CSE-induced cell apoptosis. However, knockdown of XBP1 did not induce more CHOP expression. Our results suggest that knockdown of XBP1 enhances the sensitivity of ARPE-19 cells to CSE-induced apoptosis, which may act through an alternative pathway to CHOP. These results confirmed the protective effect of UPR factor XBP1s in cultured human RPE cells during CSE exposure.
CHOP is considered a major pro-apoptotic gene induced by ER stress. Evidence demonstrates that activation of CHOP induces a variety of downstream genes involved in apoptosis, while deficiency of CHOP protects cells from ER stress-induced apoptosis (52–56). However, some recent studies have shown discrepant results. For instance, Esposito et al. showed that knock-out of CHOP exaggerated LPS-induced inflammation and kidney injury in mice, suggesting that CHOP inhibits the inflammatory response in renal cells and at the same time provides a protective effect against kidney injury (57). Chen et al. found that CHOP deficiency enhanced apoptosis in hippocampal cells and impaired memory-related behavioral performances in mice with tunicamycin treatment (58). Recently, Nashine et al. observed that CHOP deficiency did not protect but exacerbate rod photoreceptor death in retina degeneration mice, with up-regulation of p-eIF2a and decreased spliced XBP1, which indicated that CHOP may be a survival factor for rod photoreceptors in severe retinitis pigmentosa mice (59). Furthermore, Cano et al. showed that CHOP knockdown decreased cell viability in ARPE-19 cells when exposed to 500 μg/ml CSE (35). Likewise, we found that CSE induced more apoptosis and cell injury in CHOP-deficient cells. These observations suggest that despite the pro-apoptotic effect of CHOP, a baseline level of CHOP seems to be required for photoreceptor and RPE cell survival. However, it remains unclear why loss of CHOP results in more cell death in stressed retina and RPE.
To address this question, we evaluated the expression of Nrf2, p-eIF2α, and p-p38 in cells treated with siRNA against CHOP after exposure to CSE. Nrf2 is a central regulator of the anti-oxidant system, inducing expression of genes encoding phase II detoxification enzymes and antioxidant proteins to attenuate oxidative stress and protect cell survival (46). Recently, Lei Wang et al. found that Nrf2 was altered in human AMD specimens, and Nrf2 deficiency promoted cellular oxidative damage and a pro-inflammatory environment in CSE exposed RPE cells (60). In our study, we found that in CHOP knockdown cells, p-p38, a MAPK factor involved in cell death, was decreased, which may indicate that knockdown of CHOP to some extend alleviates cell damage through MAPK pathway; meanwhile, Nrf2 and p-eIF2α were significantly decreased. Overexpression of Nrf2 significantly reduced CHOP-dependent apoptosis in RPE cells; in contrast, knockdown of Nrf2 exacerbated CSE induced cell apoptosis, suggesting that CHOP knockdown exacerbated cell death at least partially through the down-regulation of Nrf2. In addition, the decreased p-eIF2α level may lead to a decreased capacity to slow down protein formation, which would increase the unfolded protein accumulation, thereby exacerbating ER stress and cell damage.
In summary, our study showed that CSE induces ER stress associated with oxidative stress and mitochondrial damage, playing an important role in apoptosis of RPE cells death. The CSE induced UPR and CHOP activation is essential for induction of Nrf2, which suppresses ER stress and protects against apoptosis. Activation of XBP1, the central regulator of the adaptive UPR, is also important for RPE survival during ER and oxidative stress. Thus, harnessing the protective factors such as XBP1 and Nrf2 may provide novel therapeutic targets to protect RPE cells from cigarette smoke-associated damage of RPE cells.
Acknowledgments
We thank Dr. Laurie Glimcher (Weill Cornell Medical College, Cornell University) for XBP1 adenovirus, Dr. Junhua Li and Maulasri Bhatta for technical help, and Dr. Steven J. Fliesler (SUNY-Buffalo and VAWNYHS) for helpful discussions and suggestions.
This work was supported, in part, by National Institutes of Health/NEI Grants EY019949, EY025061, by grants from American Diabetes Association and American Health Assistance Foundation, and by an Unrestricted Grant to the Dept. of Ophthalmology, SUNY-Buffalo, from Research to Prevent Blindness (all to S. X. Z.).
- AMD
- age-related macular degeneration
- ATF
- activating transcription factor
- CHOP
- C/EBP homologous protein
- CSE
- cigarette smoke extract
- eIF2α
- eukaryotic translation initiation factor 2α
- ER
- endoplasmic reticulum
- IRE1
- inositol-requiring trans-membrane kinase/endonuclease 1
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- Nrf2
- NF-E2-related factor 2
- p-eIF2α
- phospho-eIF2α
- p-p38
- phospho-P38
- PERK
- protein kinase RNA-like endoplasmic reticulum kinase
- qPCR
- quantitative real-time reverse-transcriptase polymerase chain reaction
- RPE
- retinal pigment epithelium
- siRNA
- small interfering RNA
- TUNEL
- terminal deoxynucleotidyl transferase dUTP nick end labeling
- UPR
- unfolded protein response
- XBP1
- x-box-binding protein 1.
REFERENCES
- 1. Klein R., Klein B. E., Jensen S. C., Meuer S. M. (1997) The five-year incidence and progression of age-related maculopathy: the Beaver Dam Eye Study. Ophthalmology 104, 7–21 [DOI] [PubMed] [Google Scholar]
- 2. Fine S. L., Berger J. W., Maguire M. G., Ho A. C. (2000) Age-related macular degeneration. NEJM 342, 483–492 [DOI] [PubMed] [Google Scholar]
- 3. Pieramici D. J., Bressler S. B. (1998) Age-related macular degeneration and risk factors for the development of choroidal neovascularization in the fellow eye. Curr. Opin. Ophthalmol. 9, 38–46 [DOI] [PubMed] [Google Scholar]
- 4. Ramkumar H. L., Zhang J., Chan C. C. Retinal ultrastructure of murine models of dry age-related macular degeneration (AMD). Prog. Retin. Eye Res. 29, 169–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Losonczy G., Fekete Á., Vokó Z., Takács L., Káldi I., Ajzner É., Kasza M., Vajas A., Berta A., Balogh I. (2011) Analysis of complement factor H Y402H, LOC387715, HTRA1 polymorphisms and ApoE alleles with susceptibility to age-related macular degeneration in Hungarian patients. Acta Ophthalmologica 89, 255–262 [DOI] [PubMed] [Google Scholar]
- 6. Souied E. H., Leveziel N., Richard F., Dragon-Durey M. A., Coscas G., Soubrane G., Benlian P., Fremeaux-Bacchi V. (2005) Y402H complement factor H polymorphism associated with exudative age-related macular degeneration in the French population. Molecular Vision 11, 1135–1140 [PubMed] [Google Scholar]
- 7. Age-Related Eye Disease Study Research, G. (2000) Risk factors associated with age-related macular degeneration. A case-control study in the age-related eye disease study: Age-Related Eye Disease Study Report Number 3. Ophthalmology 107, 2224–2232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Khan J. C., Thurlby D. A., Shahid H., Clayton D. G., Yates J. R., Bradley M., Moore A. T., Bird A. C., and Genetic Factors in AMD Study (2006) Smoking and age related macular degeneration: the number of pack years of cigarette smoking is a major determinant of risk for both geographic atrophy and choroidal neovascularisation. Br. J. Ophthalmol. 90, 75–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tamakoshi A., Yuzawa M., Matsui M., Uyama M., Fujiwara N. K., Ohno Y. (1997) Smoking and neovascular form of age related macular degeneration in late middle aged males: findings from a case-control study in Japan. Research Committee on Chorioretinal Degenerations. Br. J. Ophthalmol. 81, 901–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wilson G. A., Field A. P., Wilson N. (2001) Smoke gets in your eyes: smoking and visual impairment in New Zealand. New Zealand Med. J. 114, 471–474 [PubMed] [Google Scholar]
- 11. Fujihara M., Nagai N., Sussan T. E., Biswal S., Handa J. T. (2008) Chronic cigarette smoke causes oxidative damage and apoptosis to retinal pigmented epithelial cells in mice. PloS one 3, e3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Espinosa-Heidmann D. G., Suner I. J., Catanuto P., Hernandez E. P., Marin-Castano M. E., Cousins S. W. (2006) Cigarette smoke-related oxidants and the development of sub-RPE deposits in an experimental animal model of dry AMD. Invest. Ophthalmol. Vis. Sci. 47, 729–737 [DOI] [PubMed] [Google Scholar]
- 13. Kaufman R. J. (1999) Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 13, 1211–1233 [DOI] [PubMed] [Google Scholar]
- 14. Walter P., Ron D. (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086 [DOI] [PubMed] [Google Scholar]
- 15. Eizirik D. L., Cardozo A. K., Cnop M. (2008) The role for endoplasmic reticulum stress in diabetes mellitus. Endocr. Rev. 29, 42–61 [DOI] [PubMed] [Google Scholar]
- 16. Shuda M., Kondoh N., Imazeki N., Tanaka K., Okada T., Mori K., Hada A., Arai M., Wakatsuki T., Matsubara O., Yamamoto N., Yamamoto M. (2003) Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: a possible involvement of the ER stress pathway in hepatocarcinogenesis. J. Hepatol. 38, 605–614 [DOI] [PubMed] [Google Scholar]
- 17. Rutkowski D. T., Kang S. W., Goodman A. G., Garrison J. L., Taunton J., Katze M. G., Kaufman R. J., Hegde R. S. (2007) The role of p58IPK in protecting the stressed endoplasmic reticulum. Mol. Biol. Cell 18, 3681–3691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. van Huizen R., Martindale J. L., Gorospe M., Holbrook N. J. (2003) P58IPK, a novel endoplasmic reticulum stress-inducible protein and potential negative regulator of eIF2α signaling. J. Biol. Chem. 278, 15558–15564 [DOI] [PubMed] [Google Scholar]
- 19. Lee A. H., Iwakoshi N. N., Glimcher L. H. (2003) XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 23, 7448–7459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Harding H. P., Zhang Y., Bertolotti A., Zeng H., Ron D. (2000) Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 5, 897–904 [DOI] [PubMed] [Google Scholar]
- 21. Zinszner H., Kuroda M., Wang X., Batchvarova N., Lightfoot R. T., Remotti H., Stevens J. L., Ron D. (1998) CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 12, 982–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zeng D. X., Xu Y. J., Liu X. S., Wang R., Xiang M. (2011) Cigarette smoke extract induced rat pulmonary artery smooth muscle cells proliferation via PKCalpha-mediated cyclin D1 expression. J. Cell. Biochem. 112, 2082–2088 [DOI] [PubMed] [Google Scholar]
- 23. Wu W., Patel K. B., Booth J. L., Zhang W., Metcalf J. P. (2011) Cigarette smoke extract suppresses the RIG-I-initiated innate immune response to influenza virus in the human lung. AJP Lung Cell Molecular Physiology 300, L821–L830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim Y. J., Kim J. Y., Yoon J. Y., Kyung S. Y., Lee S. P., Jeong S. H., Moon C., Park J. W. (2011) Protective effect of aminophylline against cigarette smoke extract-induced apoptosis in human lung fibroblasts (MRC-5 cells). Basic Clin. Pharmacol. Toxicol. 109, 17–22 [DOI] [PubMed] [Google Scholar]
- 25. Chen H. W., Chien M. L., Chaung Y. H., Lii C. K., Wang T. S. (2004) Extracts from cigarette smoke induce DNA damage and cell adhesion molecule expression through different pathways. Chem. Biol. Interact. 150, 233–241 [DOI] [PubMed] [Google Scholar]
- 26. Yang W., Cui S., Ma J., Lu Q., Kong C., Liu T., Sun Z. (2012) Cigarette smoking extract causes hypermethylation and inactivation of WWOX gene in T-24 human bladder cancer cells. Neoplasma 59, 216–223 [DOI] [PubMed] [Google Scholar]
- 27. Xu L., Mao X. Y., Fan C. F., Zheng H. C. (2011) MTA1 expression correlates significantly with cigarette smoke in non-small cell lung cancer. Virchows Arch. 459, 415–422 [DOI] [PubMed] [Google Scholar]
- 28. Chen H. W., Lii C. K., Ku H. J., Wang T. S. (2009) Cigarette smoke extract induces expression of cell adhesion molecules in HUVEC via actin filament reorganization. Environ. Mol. Mutagen 50, 96–104 [DOI] [PubMed] [Google Scholar]
- 29. Orosz Z., Csiszar A., Labinskyy N., Smith K., Kaminski P. M., Ferdinandy P., Wolin M. S., Rivera A., Ungvari Z. (2007) Cigarette smoke-induced proinflammatory alterations in the endothelial phenotype: role of NAD(P)H oxidase activation. AJP Heart and Circulatory Physiology 292, H130–H139 [DOI] [PubMed] [Google Scholar]
- 30. Bertram K. M., Baglole C. J., Phipps R. P., Libby R. T. (2009) Molecular regulation of cigarette smoke induced-oxidative stress in human retinal pigment epithelial cells: implications for age-related macular degeneration. AJP Cell Physiol. 297, C1200–C1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Geraghty P., Wallace A., D'Armiento J. M. (2011) Induction of the unfolded protein response by cigarette smoke is primarily an activating transcription factor 4-C/EBP homologous protein mediated process. Int. J. Chron. Obstruct. Pulmon. Dis. 6, 309–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tagawa Y., Hiramatsu N., Kasai A., Hayakawa K., Okamura M., Yao J., Kitamura M. (2008) Induction of apoptosis by cigarette smoke via ROS-dependent endoplasmic reticulum stress and CCAAT/enhancer-binding protein-homologous protein (CHOP). Free Rad. Biol. Med. 45, 50–59 [DOI] [PubMed] [Google Scholar]
- 33. Min T., Bodas M., Mazur S., Vij N. (2011) Critical role of proteostasis-imbalance in pathogenesis of COPD and severe emphysema. J. Mol. Med. 89, 577–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kunchithapautham K., Atkinson C., Rohrer B. (2014) Smoke Exposure Causes Endoplasmic Reticulum Stress and Lipid Accumulation in Retinal Pigment Epithelium through Oxidative Stress and Complement Activation. J. Biol. Chem. 289, 14534–14546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cano M., Wang L., Wan J., Barnett B. P., Ebrahimi K., Qian J., Handa J. T. (2014) Oxidative stress induces mitochondrial dysfunction and a protective unfolded protein response in RPE cells. Free Rad. Biol. Med. 69, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen C., Cano M., Wang J. J., Li J., Huang C., Yu Q., Herbert T. P., Handa J., Zhang S. X. (2013) Role of UPR dysregulation in oxidative injury of retinal pigment epithelial cells. Antioxidants Redox Signal. 20, 2091–2106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Csiszar A., Labinskyy N., Podlutsky A., Kaminski P. M., Wolin M. S., Zhang C., Mukhopadhyay P., Pacher P., Hu F., de Cabo R., Ballabh P., Ungvari Z. (2008) Vasoprotective effects of resveratrol and SIRT1: attenuation of cigarette smoke-induced oxidative stress and proinflammatory phenotypic alterations. AJP Heart and Circulatory Physiology 294, H2721–H2735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhong Y., Li J., Wang J. J., Chen C., Tran J. T., Saadi A., Yu Q., Le Y. Z., Mandal M. N., Anderson R. E., Zhang S. X. (2012) X-box binding protein 1 is essential for the anti-oxidant defense and cell survival in the retinal pigment epithelium. PloS one 7, e38616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li J., Wang J. J., Zhang S. X. (2011) Preconditioning with endoplasmic reticulum stress mitigates retinal endothelial inflammation via activation of X-box binding protein 1. J. Biol. Chem. 286, 4912–4921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen C., Wang J. J., Li J., Yu Q., Zhang S. X. (2013) Quinotrierixin inhibits proliferation of human retinal pigment epithelial cells. Mol. Vis. 19, 39–46 [PMC free article] [PubMed] [Google Scholar]
- 41. Chang Y. S., Wu C. L., Tseng S. H., Kuo P. Y., Tseng S. Y. (2007) Cytotoxicity of triamcinolone acetonide on human retinal pigment epithelial cells. Invest. Ophthalmol. Vis. Sci. 48, 2792–2798 [DOI] [PubMed] [Google Scholar]
- 42. Lin J. H., Li H., Yasumura D., Cohen H. R., Zhang C., Panning B., Shokat K. M., Lavail M. M., Walter P. (2007) IRE1 signaling affects cell fate during the unfolded protein response. Science 318, 944–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Beazley K. E., Deasey S., Lima F., Nurminskaya M. V. (2012) Transglutaminase 2-mediated activation of beta-catenin signaling has a critical role in warfarin-induced vascular calcification. Arterioscl. Thromb. Vasc. Biol. 32, 123–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dou G., Sreekumar P. G., Spee C., He S., Ryan S. J., Kannan R., Hinton D. R. (2012) Deficiency of αB crystallin augments ER stress-induced apoptosis by enhancing mitochondrial dysfunction. Free Rad. Biol. Med. 53, 1111–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Benowitz N., Hukkanen J., Jacob P., III. (2009) Nicotine Chemistry, Metabolism, Kinetics and Biomarkers in Nicotine Psychopharmacology (Henningfield J., London E., Pogun S., eds), pp. 29–60, Springer; Berlin Heidelberg: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cano M., Thimmalappula R., Fujihara M., Nagai N., Sporn M., Wang A. L., Neufeld A. H., Biswal S., Handa J. T. (2010) Cigarette smoking, oxidative stress, the anti-oxidant response through Nrf2 signaling, and Age-related Macular Degeneration. Vis. Res. 50, 652–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kenche H., Baty C. J., Vedagiri K., Shapiro S. D., Blumental-Perry A. (2012) Cigarette smoking affects oxidative protein folding in endoplasmic reticulum by modifying protein disulfide isomerase. FASEB J. 27, 965–977 [DOI] [PubMed] [Google Scholar]
- 48. Kroening P. R., Barnes T. W., Pease L., Limper A., Kita H., Vassallo R. (2008) Cigarette smoke-induced oxidative stress suppresses generation of dendritic cell IL-12 and IL-23 through ERK-dependent pathways. J. Immunol. 181, 1536–1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cheng S. E., Luo S. F., Jou M. J., Lin C. C., Kou Y. R., Lee I. T., Hsieh H. L., Yang C. M. (2009) Cigarette smoke extract induces cytosolic phospholipase A2 expression via NADPH oxidase, MAPKs, AP-1, and NF-κB in human tracheal smooth muscle cells. Free Rad. Biol. Med. 46, 948–960 [DOI] [PubMed] [Google Scholar]
- 50. Edirisinghe I., Arunachalam G., Wong C., Yao H., Rahman A., Phipps R. P., Jin Z. G., Rahman I. (2010) Cigarette-smoke-induced oxidative/nitrosative stress impairs VEGF- and fluid-shear-stress-mediated signaling in endothelial cells. Antioxidants Redox Signaling 12, 1355–1369 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 51. Yoon J. S., Lee H. J., Chae M. K., Lee S. Y., Lee E. J. (2013) Cigarette smoke extract-induced adipogenesis in Graves' orbital fibroblasts is inhibited by quercetin via reduction in oxidative stress. J. Endocrinol. 216, 145–156 [DOI] [PubMed] [Google Scholar]
- 52. Friedman A. D. (1996) GADD153/CHOP, a DNA damage-inducible protein, reduced CAAT/enhancer binding protein activities and increased apoptosis in 32D c13 myeloid cells. Cancer Res. 56, 3250–3256 [PubMed] [Google Scholar]
- 53. Yamaguchi H., Wang H. G. (2004) CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem. 279, 45495–45502 [DOI] [PubMed] [Google Scholar]
- 54. Ariyama Y., Tanaka Y., Shimizu H., Shimomura K., Okada S., Saito T., Yamada E., Oyadomari S., Mori M., Mori M. (2008) The role of CHOP messenger RNA expression in the link between oxidative stress and apoptosis. Metab. Clin. Exp. 57, 1625–1635 [DOI] [PubMed] [Google Scholar]
- 55. Tamaki N., Hatano E., Taura K., Tada M., Kodama Y., Nitta T., Iwaisako K., Seo S., Nakajima A., Ikai I., Uemoto S. (2008) CHOP deficiency attenuates cholestasis-induced liver fibrosis by reduction of hepatocyte injury. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G498–505 [DOI] [PubMed] [Google Scholar]
- 56. Song B., Scheuner D., Ron D., Pennathur S., Kaufman R. J. (2008) Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J. Clin. Investig. 118, 3378–3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Esposito V., Grosjean F., Tan J., Huang L., Zhu L., Chen J., Xiong H., Striker G. E., Zheng F. (2013) CHOP deficiency results in elevated lipopolysaccharide-induced inflammation and kidney injury. Am. J. Physiol. Renal Physiol. 304, F440–F450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chen C. M., Wu C. T., Chiang C. K., Liao B. W., Liu S. H. (2012) C/EBP homologous protein (CHOP) deficiency aggravates hippocampal cell apoptosis and impairs memory performance. PloS one 7, e40801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nashine S., Bhootada Y., Lewin A. S., Gorbatyuk M. (2013) Ablation of C/EBP Homologous Protein Does Not Protect T17M RHO Mice from Retinal Degeneration. PLoS ONE 8, e63205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang L., Kondo N., Cano M., Ebrahimi K., Yoshida T., Barnett B. P., Biswal S., Handa J. T. (2014) Nrf2 signaling modulates cigarette smoke-induced complement activation in retinal pigmented epithelial cells. Free. Radic. Biol. Med. 70, 155–166 [DOI] [PMC free article] [PubMed] [Google Scholar]