

Background: The mechanisms by which bacterial ligands alter angiogenesis remain unknown.
Results: Lipopolysaccharide-mediated Angiopoietin-2-dependent autocrine angiogenesis in lung endothelial cells is regulated by NADPH oxidase 2.
Conclusion: Endothelial Nox2 regulates Angiopoietin-2-dependent angiogenesis.
Significance: This study presents new data regarding the regulation of proinflammatory angiogenesis.
Keywords: Angiogenesis, Endothelial Cell, LPS, Lung Injury, NADPH Oxidase, Angiopoeitin-2
Abstract
Sepsis-mediated endothelial Angiopoeitin-2 (Ang2) signaling may contribute to microvascular remodeling in the developing lung. The mechanisms by which bacterial cell wall components such as LPS mediate Ang2 signaling in human pulmonary microvascular endothelial cells (HPMECs) remain understudied. In HPMEC, LPS-induced Ang2, Tie2, and VEGF-A protein expression was preceded by increased superoxide formation. NADPH oxidase 2 (Nox2) inhibition, but not Nox4 or Nox1 inhibition, attenuated LPS-induced superoxide formation and Ang2, Tie2, and VEGF-A expression. Nox2 silencing, but not Nox4 or Nox1 silencing, inhibited LPS-mediated inhibitor of κ-B kinase β (IKKβ) and p38 phosphorylation and nuclear translocation of NF-κB and AP-1. In HPMECs, LPS increased the number of angiogenic tube and network formations in Matrigel by >3-fold. Conditioned media from LPS-treated cells also induced angiogenic tube and network formation in the presence of Toll-like receptor 4 blockade but not in the presence of Ang2 and VEGF blockade. Nox2 inhibition or conditioned media from Nox2-silenced cells attenuated LPS-induced tube and network formation. Ang2 and VEGF-A treatment rescued angiogenesis in Nox2-silenced cells. We propose that Nox2 regulates LPS-mediated Ang2-dependent autocrine angiogenesis in HPMECs through the IKKβ/NF-κB and MAPK/AP-1 pathways.
Introduction
Vascular remodeling in bronchopulmonary dysplasia (BPD),2 characterized histologically by a paucity of blood vessels and dysmorphic arborization in the distal lung, are hallmarks of the disease in the post-surfactant era (1, 2). With increasing use of non-invasive ventilation and controlled use of supplemental oxygen, the contribution of chorioamnionitis and bacterial sepsis to pulmonary vascular injury in premature infants has become more evident (3–5). Gram-negative bacterial cell wall components such as LPS released during sepsis induce a change in the endothelial phenotype from a quiescent phenotype to a “proinflammatory” phenotype (6, 7). This switch is associated with increased Angiopoietin-2 (Ang2) expression and disruption of homeostatic Angiopoietin 1 (Ang1)/Tie2 signaling, resulting in endothelial expression of cellular adhesion molecules and cytokines that facilitate lung inflammation and injury (1, 6–8). Furthermore, Ang2 and Ang1 expression is regulated temporally in the developing lung and facilitates development of the pulmonary vascular network (1, 9). Given the importance of Ang2 signaling in neonatal lung injury and pulmonary vascular development, elucidating the mechanisms underlying bacteria-mediated Ang2 expression in the pulmonary endothelium will contribute to a better understanding of the pathogenesis of BPD and assumes translational significance (9, 10).
In endothelial cells, Ang1 secreted from pericytes, smooth muscle cells, and fibroblasts binds to Tie2 (endothelial cell surface tyrosine kinase receptor), inducing tyrosine autophosphorylation (6, 7). Constitutive Ang1/Tie2 signaling promotes endothelial survival and migration and antagonizes the proinflammatory effects of cytokines and VEGF in the mature endothelium (7, 11). Ang2 competes with Ang1 for Tie2 and promotes the expression of cellular adhesion molecules such as vascular cell adhesion molecule 1 (VCAM-1), increases vascular permeability, and augments leukocyte transmigration (7, 10, 11). The effects of Ang2 on angiogenesis are modulated by the presence or absence of VEGF. In the presence of VEGF, Ang2 induces migration, proliferation, and the sprouting of new blood vessels, whereas, in the absence of VEGF, endothelial cell death and vessel regression occur (7, 11). Although systemic LPS administration has been shown to augment Ang2 expression in the liver, lung, and other tissues, the mechanisms involved in LPS-mediated Ang2 expression in lung endothelial cells remain unknown (12). Specifically, the involvement of redox signaling in the regulation of Ang2-dependent signaling and endothelial cell angiogenesis has not been studied.
NADPH oxidases (Nox) have been reported to regulate endothelial responses to bacterial ligands and proinflammatory stimuli (13–15). Nox enzymes belong to a family of multimeric proteins that catalyze one electron reduction of oxygen to generate superoxide using NADPH as substrate (14). Nox-dependent redox signaling serves key physiologic processes in the endothelium but is also implicated in pathological processes such as ischemia-reperfusion, inflammation, and cell death (14, 16). Nox2, Nox4, and Nox1 have all been shown previously to mediate cytokine expression in response to LPS in endothelial cells of varied lineages (13, 15, 17). In this study, we investigated the hypothesis that Nox regulates LPS-mediated Ang2 signaling and angiogenesis in human pulmonary microvascular endothelial cells (HPMECs). Here we demonstrate that Nox2 regulates LPS-mediated Ang2 and VEGF-A expression in HPMECs through the IKKβ/NF-κB and p38/AP-1 pathways. We also show that Ang2- and VEGF-A-mediated autocrine angiogenesis is regulated by Nox2 in lung endothelial cells.
EXPERIMENTAL PROCEDURES
Cell Culture and Reagents
Fetal HPMECs (ScienCell, Carlsbad, CA) were used between passages 3–5 for all experiments. HPMECs were grown in endothelial cell medium (ECM) supplemented with fetal bovine serum, antibiotics, and endothelial cell growth serum as recommended by the manufacturer (ScienCell) in a humidified incubator containing 5% CO2 at 37 °C. Ultrapure LPS (100 ng/ml) and human TLR4 neutralizing antibody (Ab-TLR4, 5 μm) were purchased from Invivogen (San Diego, CA). Tiron, potassium phosphate, EGTA, sucrose, lucigenin, and NADPH were purchased from Sigma. Collagenase, FBS, DMEM, and PEG-superoxide dismutase were purchased from Sigma (400 units/ml). VAS2870 (3-benzyl-7-(2-benzoxazolyl) thio-1,2,3,-triazolo (4,5-d) pyrimidine, 10 μm), a reversible Nox inhibitor, was obtained from Vasopharm (a gift from Dr. Reinhard Schinzel, Würzburg, Germany). Recombinant VEGF-A (rhVEGF-A, 25 ng/ml) and Ang2 (rhAng2, 25 ng/ml) were purchased from R&D Systems (Minneapolis, MN). A combined neutralizing antibody against Ang2 and VEGF (Ab-Ang2/VEGF, 500 ng/ml) was a gift from the Roche Innovation Center (Pharma Research and Early Development, Penzberg, Germany).
Mice
Care of mice before and during the experimental procedures was conducted in accordance with the policies of the Biomedical Resource Center, Medical College of Wisconsin, and the National Institutes of Health guidelines for the care and use of laboratory animals. All protocols had prior approval from the Medical College of Wisconsin Institutional Animal Care and Use Committee. C57BL/6 mice were obtained from Charles River Laboratories (Franklin, CT).
Isolation of Endothelial Cells from Murine Lungs
For endothelial cell isolation, cells from two to three neonatal C57BL/6 pups (7 days old) were pooled per condition. Mice were injected intraperitoneally with 1 mg/kg LPS or saline, and lungs were harvested after 18 h following sacrifice of the animals. Harvested lungs were minced with sterile scissors in ice-cold DMEM and then transferred to 15 ml of prewarmed 1 mg/ml collagenase solution in DMEM. The mixture was allowed to rotate for 45 min at 37 °C. The digested tissue was then passed through a 14-gauge cannula attached to a 20-ml syringe several times, followed by passage through a 70-μm cell strainer, and washed with 20% FBS plus DMEM. Cells were then centrifuged at 400 × g for 5 min, and the supernatant was aspirated. The cell pellet was resuspended with 0.1% BSA in PBS. The suspension was incubated with anti-PECAM-1 antibody-conjugated DynabeadsTM (Invitrogen) prepared according to the protocol of the manufacturer in a rocker for 15 min at room temperature. Upon completion, the cells were washed with PBS three times, and the proteins were extracted with radioimmune precipitation assay buffer following standard protocol.
Quantification of Ang2, VEGF-A, Tie2, Nox1, Nox2, and Nox4 mRNA Expression Using Real-time PCR
Total RNA was extracted from HPMECs using the RNeasy mini kit from Qiagen (Valencia, CA), and cDNA was synthesized from 1 μg of RNA using the iScript cDNA synthesis kit (Bio-Rad) according to the instructions of the manufacturer. The transcripts were amplified, and gene expression data were collected on a Bio-Rad IQ5 with SYBR Green Mastermix. In experiments using PEG-superoxide dismutase and VAS2870, the chemicals were incubated with the cells for 1 h prior to LPS treatment. The primers for Ang2, VEGF-A, and Tie2 were obtained from Operon (Huntsville, AL). They consisted of Ang2 (sense, GAGGAACTGTCTCGAACT; antisense, GTGGAAGAGGACACAGTG), VEGF-A (sense, GGGCAGAATCATCACGAAGT; antisense, ATCTGCATGGTGATGTTGGA), and Tie2 (sense, TACTAATGAAGAAATGACCCTGG; antisense, GGAGTGTGTAATGTTGGAAATCT). The primers for Nox1, Nox2, and Nox4 were purchased commercially from Santa Cruz Biotechnology (Santa Cruz, CA), and the primers for 18 S were purchased commercially from MCLabs (San Francisco, CA). 18 S was used as the housekeeping gene. Relative gene expression of Nox1, Nox2, Nox4, Ang2, VEGF-A, and Tie2 was calculated with the Pflaffl method (18).
Immunoblotting for Quantifying Changes in Protein Expression and Phosphorylation
Whole cell lysates were prepared from HPMECs by lysing cells with a modified radioimmune precipitation assay buffer containing commercially available protease and phosphatase inhibitors (Sigma). Protein quantification was done using a BCA protein assay from Thermo Fisher (Rockford, IL) according to the instructions of the manufacturer using BSA as a standard. Immunoblotting was done following standard protocol. The primary antibodies for angiogenic expression were as follows: mouse anti-Tie2 (Cell Signaling Technology, Danvers, MA, 1:1000), rabbit anti-Ang2 (Abcam, Cambridge, MA, 1:1000), rabbit anti-VEGF-A (Santa Cruz Biotechnology, 1:500), and mouse anti-β-Actin (Sigma, 1:5000). For phosphorylation, the primary antibodies were as follows: rabbit anti-(p)IKKβ, rabbit anti-(p)p38 MAPK, rabbit anti-(p)SAPK/JNK, rabbit anti-p38 MAPK, rabbit anti-SAPK/JNK (Cell Signaling Technology, 1:1000), and mouse anti-IKKβ (Santa Cruz Biotechnology, 1:500). The antibodies used on Nox2 components were as follows: rabbit anti-Rac2 (Santa Cruz Biotechnology, 1:500), rabbit anti-p22phox (Santa Cruz Biotechnology, 1:500), rabbit anti-p47phox (Santa Cruz Biotechnology, 1:500), mouse anti-p67phox (BD Biosciences, 1:500), and goat anti-gp91phox (Santa Cruz Biotechnology, 1:500). Blots were developed using ECL, and blots were stripped for 15 min, if needed, with Restore Plus stripping buffer (Thermo Fisher). β-Actin or the corresponding non-phosphorylated antibody was used for normalization, and densitometry was performed using ImageJ Software (National Institutes of Health).
Detection of NADPH-dependent Superoxide Formation
NADPH-dependent production of superoxide was quantified using a Tiron-inhibitable lucigenin chemiluminescence assay as described previously (19, 20). Briefly, 1–2 × 104 cells were seeded in a white 96-well plate (Bio-Rad) and grown for 24 h. Cells were treated with LPS (100 ng/ml) for 7–45 min. Each condition was run in quintuplicate, the Tiron-inhibition (5 μm) wells were run in duplicate, and a blank well was run with only lucigenin. After LPS incubation, cells were washed briefly with Hepes-buffered Saline twice. After the last wash, 100 μl of 50 μm potassium phosphate buffer with 1 mm EGTA and 1:1000 protease inhibitor mixture (Sigma) was added to each well. Immediately after addition of the phosphate buffer, a lucigenin reaction mixture was added to each well, containing the following reaction components (final concentration): NADPH (100 μm), sucrose (150 mm), and lucigenin (5 μm). After the lucigenin mixture, the chemiluminescent signals were collected every 1 min for 30 min in a 96-well plate, with the total relative light units summated for each well at the end of 10 min (luminometer from Turner Scientific, Madison, WI). The summated signal inhibited by Tiron was calculated and subtracted from each corresponding sample. Superoxide anion results are expressed as Tiron-inhibited NADPH oxidase activity detected by chemiluminescent signals (relative light units).
siRNA-mediated Nox1, Nox2, and Nox4 Gene Silencing
siRNA sequences targeting Nox1, Nox2, and Nox4 were purchased from Santa Cruz Biotechnology, and transfections were performed as before (13). Briefly, cells were cultured with antibiotic-free ECM until 60–80% confluent. The media was then aspirated, and cells were washed twice with siRNA transfection medium (Santa Cruz Biotechnology). The plates were then incubated with either the control or siRNA strand (1 μg) in transfection medium and incubated for 16 h. Subsequently, the reagents were aspirated, and normal ECM was gently put on the plates. The optimal period of silencing was determined as 36 h for Nox1 and 48 h for Nox2 and Nox4 using mRNA and protein studies. The silencing efficiency was determined by PCR (primers mentioned above) and by immunoblotting using goat anti-Nox1 (1:500), goat anti-Nox2 (1:500), and rabbit anti-Nox4 (1:500) antibodies purchased from Santa Cruz Biotechnology. β-Actin was used for normalization.
Immunoprecipitation of Nox2
Cells grown to 90% confluence in 50-mm dishes were treated with LPS for 15 or 30 min or left untreated. The beads were prepared with goat anti-gp91phox antibody (Santa Cruz Biotechnology) according to the protocol of the manufacturer using SureBeadsTM protein G magnetic beads for immunoprecipitation (Bio-Rad). A sample (500 μg) of the protein was incubated with the magnetic antibody beads overnight at 4 °C. Upon completion, the beads were magnetized and washed twice with ice-cold TBS, after which 50 μl of 2× Laemmli buffer was incubated with the beads and heated at 95 °C prior to immunoblotting. Blots were incubated overnight at 4 °C with goat anti-gp91phox (Santa Cruz Biotechnology, 1:500) and rabbit anti-p47phox (Santa Cruz Biotechnology, 1:500). Blots were developed using ECL, and densitometry was performed using ImageJ software (National Institutes of Health).
Cellular Fractions and Transcription Factor Assays
Nuclear and cytoplasmic fractions were obtained with the use of a commercially available nuclear extraction kit (Cayman Chemicals, Ann Arbor, MI). The nuclear fractions obtained were quantified using a standard BCA assay (Thermo Fisher) following the protocol of the manufacturer. 5 μg of nuclear extract/well was used with the NF-κB (p65) transcription factor ELISA (Cayman Chemicals) and TRANSAMTM AP-1 c-JUN (Active Motif, Carlsbad, CA) according to the protocols provided by the manufacturer, and all samples were run in duplicate. The nuclear fractions were immunoblotted with rabbit anti-histone (Epitomics, Burlingame, CA, 1:5000) to normalize for protein loading.
Angiogenic Tube and Network Formation Assay in Matrigel
HPMECs grown to ∼80% confluence in 6-well culture plates were either silenced or left unsilenced. After 36 h of silencing, media was replaced with serum-free media, followed by treatment with LPS for 10 h. Cells were then detached with Tryple Express (Invitrogen), resuspended in basal ECM, and 6 × 104 cells in 300 μl of media were plated on to a 24-well Matrigel matrix-coated plate (BD Biosciences). Angiogenesis was assessed 12 h after cells were seeded on Matrigel. For experiments using the human TLR4-neutralizing antibody (Ab-TLR4, 5 μm), the cells were treated with the inhibitor for 1 h prior to LPS incubation. For rescue experiments with rhAng2 and rhVEGF-A, cells were treated for 10 h prior to seeding in Matrigel. Calcein AM (BD Biosciences) fluorescent dye was used to enhance the visibility of tube and network formation in Matrigel. Angiogenesis was evaluated by counting the number of tube and network formations in one quadrant (the same one for each condition) and multiplying by four. Only tubular structures connecting two cell clusters were considered for measurements, whereas cell clusters with at least three emanating tubular structures were considered to be a network. Representative images were taken using a Zeiss Observer Z1 fluorescence microscope with an attached camera at ×5 magnification.
For experiments with conditioned media (CM), HPMEC supernatants were collected 24 h after LPS treatment from control, LPS-treated, and LPS plus siNox2-treated cells grown in serum free media. HPMECs grown to 75% confluence in a 6-well culture plate were treated with conditioned media from different conditions for 10 h. A TLR4-neutralizing antibody (Ab-TLR4, 5 μm) was used to block LPS signaling in all experiments with conditioned media. Cells were then detached with Tryple Express, and the Matrigel assay was performed as above. For rescue experiments with rhAng2 and rhVEGF-A, cells were treated for 10 h prior to seeding in Matrigel. For experiments with Ab-Ang2/VEGF (blocking antibody), HPMEC were treated with 500 ng/ml of antibody for 30 min before addition of CM.
Statistical Analysis
Statistical analysis was done using STATA 12 (StataCorp LP, Dallas, TX). Data are presented as mean ± S.D. p < 0.05 was considered significant for experiments. -Fold changes in protein levels relative to the untreated control cells were quantified by densitometry and compared between various treatments using analysis of variance. Changes in superoxide levels, transcription factor levels, and angiogenic formations relative to the untreated, control cells were compared between using analysis of variance. The Bonferroni test was used in conjunction with analysis of variance to perform pairwise comparisons between groups. For mRNA studies, changes in gene expression with various treatments were calculated relative to expression in control cells and compared between different treatment groups using analysis of variance.
RESULTS
LPS-induced Ang2, Tie2, and VEGF-A Expression in HPMEC Is Associated with Increased Oxidative Stress
The effect of LPS (100 ng/ml) on Ang2, Tie2, and VEGF-A RNA and protein expression were studied in HPMECs. LPS-induced Ang2, Tie2, and VEGF-A RNA expression was evident at 3 h, peaked by 7 h, and decreased by 24 h (Fig. 1A). Correspondingly, LPS-mediated expression of Ang2, Tie2, and VEGF-A protein was detectable by 12 h, peaked at 24 h, and decreased by 36 h (Fig. 1, B and C). TLR4 (an LPS recognition receptor) blockade using a specific antibody resulted in a dose-dependent inhibition of Ang2 and VEGF-A expression (Fig. 1, D and E). These data show that LPS mediates TLR4-dependent expression of angiogenic markers in HPMECs.
FIGURE 1.

LPS induces the expression of angiogenic markers and superoxide in HPMECs. A, Ang2, VEGF-A, and Tie2 mRNA expression quantified by real-time PCR 3, 7, and 24 h after LPS treatment (n = 4). *, p < 0.02 (Ang2, control versus 3, 7, and 24 h LPS); **, p < 0.02 (VEGF-A, control versus 3, 7, and 24 h LPS); ***, p < 0.01 (Tie2, control versus 3 and 7 h LPS). B, cell lysates obtained from LPS-treated or control HPMECs at 12, 24, and 36 h were immunoblotted for Tie2, Ang2, and VEGF-A. C, densitometry quantification of angiogenic markers. *, p = 0.007 (12 h Tie2, control versus LPS); #, p = 0.003 (12 h Ang2, control versus LPS); $, p < 0.01 (the rest of the comparisons are between control versus LPS for the respective proteins) (n = 4). D, Ang2 and VEGF-A protein expression was quantified in cell lysates by immunoblotting 24 h after treatment with LPS or LPS + Ab-TLR4. E, densitometric quantification of Ang2 and VEGF-A following treatment with LPS and Ab-TLR4. *, p < 0.01 (Ang2, control versus LPS, LPS versus 2.5 μm, and LPS versus 5 μm); $, p < 0.03 (VEGF, control versus LPS, LPS versus 2.5 μm, and LPS versus 5 μm) (n = 3). F, superoxide formation in HPMECs was quantified at various time points after LPS treatment using a lucigenin-derived chemiluminescence assay. *, p = 0.02 (control versus 7 min LPS); **, p < 0.001 (control versus 15 min LPS); ***, p < 0.02 (control versus 30 min LPS); $, p = 0.04 (control versus 45 min LPS) (n = 3). G, lucigenin chemiluminescence was quantified at 1-min intervals in control and LPS-treated (15 min) HPMECs as before. *, p < 0.05 (control versus LPS for all comparisons, n = 3). H, Ang2, VEGF-A, and Tie2 RNA expression quantified by real-time PCR 3 h after LPS treatment. *, p < 0.02 (Ang2, control versus 3 h LPS, LPS versus LPS + VAS2870, and LPS versus LPS + PEG-superoxide dismutase); **, p < 0.02 (VEGF-A, control versus 3 h LPS, LPS versus LPS + VAS2870, and LPS versus LPS + PEG-superoxide dismutase); ***, p < 0.01 (Tie2, control versus 3 h LPS, LPS versus LPS + VAS2870, and LPS versus LPS + PEG-superoxide dismutase) (n = 3). Cells were pretreated with VAS2870 and PEG-superoxide dismutase for 1 h before LPS.
To assess the effect of LPS on oxidative stress in HPMECs, we measured NADPH-dependent superoxide formation. Superoxide formation represents a summation of data collected every minute over 10 min after timed LPS treatments. LPS caused a temporal increase in superoxide formation, which was detectable by 7 min, peaked at 15 min, and persisted at 45 min (Fig. 1F). The time course of superoxide formation after LPS treatment for 15 min is shown in Fig. 1G. To demonstrate the relevance of LPS-induced superoxide to the expression of angiogenic markers, we conducted experiments with VAS2870 (a NOX inhibitor) and PEG-superoxide dismutase (a superoxide anion scavenger) (21, 22). Pretreatment with VAS2870 (10 μm) or PEG-superoxide dismutase (400 units/ml) for 1 h attenuated LPS-mediated induction of Ang2, VEGF-A, and Tie2 RNA at 3 h (Fig. 1H). These data suggest that LPS-mediated superoxide formation is important for induction of angiogenic markers.
LPS Induces Lung Endothelial Expression of Tie2 and Ang2 in Vivo
To investigate whether changes in Tie2 expression observed in vitro with LPS in HPMEC are observed in vivo, we isolated mouse lung endothelial cells 18 h after intraperitoneal LPS (1 mg/kg) injection in 7-day-old mice. Mouse lung endothelial cells showed a >2.2-fold increase in Tie2 and Ang2 expression after systemic LPS (Fig. 2, A and B). These data demonstrate that LPS induces Tie2 protein in lung endothelial cells in neonatal mice.
FIGURE 2.
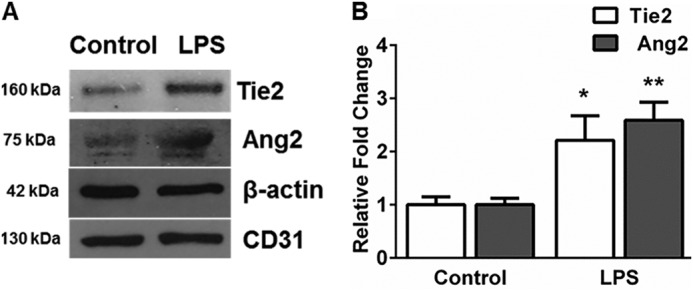
Systemic LPS induces Tie2 and Ang2 in mouse lung endothelial cells. A, protein from mouse lung endothelial lysates harvested 18 h after intraperitoneal LPS were immunoblotted for Ang2 and Tie2. B, quantification by densitometry is shown for Tie2 and Ang2 from endothelial cells. *, p = 0.03 (Tie2, control versus LPS); **, p = 0.01 (Ang2, control versus LPS) (n = 3).
LPS Stimulates Angiogenic Tube and Network Formation in HPMECs in a VEGF-A/Ang2-dependent Manner
We then examined the effect of LPS on in vitro HPMEC angiogenesis in a Matrigel-based assay used by other investigators (23). LPS increased angiogenic tube and network formation in HPMECs >2-fold in a TLR4-dependent manner at 12 h (Fig. 3, A and B). We then performed experiments with conditioned media to determine whether LPS-mediated VEGF-A and Ang2 expression stimulates autocrine angiogenesis. CM from LPS-treated cells or addition of recombinant Ang2 and VEGF-A (both 25 ng/ml) to CM from control cells induced a >3-fold increase in tube and networks (Fig. 3, C and D). The induction of angiogenesis with conditioned media derived from LPS-treated cells was not altered with TLR4 blockade (Ab-TLR4) but was strongly inhibited with a neutralizing antibody against Ang2/VEGF (Ab-Ang2/VEGF) (Fig. 3, C and D). These data demonstrate that LPS stimulates Ang2 and VEGF-A-dependent autocrine angiogenesis in lung microvascular endothelial cells in vitro.
FIGURE 3.

LPS stimulates angiogenic tube and network formation in HPMECs in an autocrine manner. A, fluorescent microscope images depicting LPS-mediated tube and network formation on Matrigel. HPMECs were treated with LPS with or without a neutralizing TLR4 antibody (Ab-TLR4) for 10 h, followed by seeding on Matrigel-coated plates. Tube and network formation was assessed after a further 12 h. A tube is a connection between two junctions, and a network is more than two tubes branching from a junction. Images were taken at 5× magnification. B, graphical representation summarizing data from four different experiments in HPMECs (n = 4). *, p < 0.001 (control versus LPS tube and network formation); **, p = 0.001 (LPS versus Ab-TLR4 + LPS tube and network formation). C, HPMECs were treated with conditioned media from control (CM-control) or LPS-treated cells (CM-LPS) in the presence of Ang2/VEGF-A blockade (CM-LPS + Ab-Ang2/VEGF), TLR4 blockade (CM-LPS + Ab-TLR4), or recombinant VEGF-A and Ang2 (CM-control + rhAng2 + rhVEGF-A) for 10 h. Cells were then plated onto Matrigel, and angiogenic responses were assessed after 12 h as above. D, graphical representation summarizing data from three different experiments in HPMECs (n = 3). *, p < 0.001 (CM-Control versus CM-LPS tube and network formation); **, p < 0.003 (CM-LPS versus CM-LPS + Ab-Ang2/VEGF tube and network formation); ***, p < 0.001 (CM-Control versus CM-Control + rhAng2 + rhVEGF-A tube and network formation).
Nox2 Inhibition Attenuates LPS-mediated Oxidative Stress and Ang2 and VEGF-A Expression in HPMECs
To determine the source of oxidative stress in LPS-treated cells, Nox2, Nox4, and Nox1 were inhibited in HPMECs using siRNA. Experiments conducted to determine the appropriate dose and duration of silencing revealed that ∼60% silencing efficiency for Nox2 and Nox4 was achieved at 48 h and for Nox1 at 36 h (Fig. 4, A and B). We measured superoxide formation in HPMECs after inhibiting Nox2, Nox1, and Nox4. LPS-induced superoxide formation at 15 min was inhibited by >60% in Nox2-silenced cells but did not change significantly in Nox1- and Nox4-silenced cells (Fig. 4C).
FIGURE 4.

Effect of Nox1, Nox2, and Nox4 silencing on LPS-induced superoxide production in HPMECs. A, mRNA expression of Nox1, Nox2, and Nox4 quantified 48 h after silencing by real-time PCR. 18 S was used as a housekeeping gene. Expression is relative to untreated controls. *, p = 0.003 (control versus Nox1); **, p < 0.001 (control versus Nox2); ***, p < 0.001 (control versus Nox4) (n = 4). B, Nox1, Nox2, and Nox4 protein expression was quantified in cell lysates by immunoblotting after silencing with the respective siRNA. C, superoxide formation was quantified in HPMECs 15 min after LPS treatment under various test conditions using a lucigenin-derived chemiluminescence assay. *, p < 0.001 (control versus 15 min LPS); **, p < 0.005 (LPS versus siNox2 + LPS) (n = 3). D and E, Tie2, Ang2, and VEGF-A protein expression was examined in cell lysates 24 h after LPS treatment by immunoblotting (D), and blots were quantified by densitometry (E). *, p < 0.001 (control versus LPS); #, p < 0.001 (control versus LPS); $, p < 0.001 (control versus LPS); **, p < 0.001 (control versus siNox2 + LPS); ##, p = 0.008 (control versus siNox2 + LPS); $$, p = 0.008 (control versus siNox2 + LPS) (n = 4).
To identify the Nox isoform involved in LPS-mediated expression of angiogenic markers in HPMECs, we quantified Ang2, VEGF-A, and Tie2 after inhibiting Nox isoforms. Nox2 silencing, but not Nox1 or Nox4 silencing, suppressed LPS-induced Ang2, VEGF-A, and Tie2 expression at 24 h by >50% (Fig. 4, D and E). These data demonstrate that Nox2 regulates LPS-induced superoxide and Ang2, Tie2, and VEGF-A expression in HPMECs.
LPS Stimulates Assembly of the Nox2 Complex in HPMECs
The active Nox2 complex requires both the membrane-bound gp91phox/p22phox subunits along with the cytoplasmic p47phox, p67phox, and RAC2 subunits (14, 16). To demonstrate that all components of the Nox2 complex are present in HPMECs, we quantified the expression of subunits in control and LPS-treated HPMECs (15 min) by immunoblotting (Fig. 5A). Furthermore, to show that LPS activates Nox2 in HPMECs, we investigated whether p47phox coimmunoprecipitated with gp91phox after LPS treatment. Compared with control cells, we found that p47phox was bound to gp91phox in LPS-treated cells at 15 and 30 min (Fig. 5, B and C). These data suggest that components of the Nox2 complex are present in HPMECs and that LPS treatment results in coimmunoprecipitation of p47phox with gp91phox.
FIGURE 5.

Subunits of Nox2 complex are present in HPMECs. A, Nox2 subunits were quantified in control and LPS-treated (15 min) HPMEC lysates by immunoblotting (n = 3). B and C, gp91phox was immunoprecipitated from control and LPS-treated (15 and 30 min) HPMECs, followed by immunoblotting for p47phox (B) with densitometry for the blots shown (C). *, p = 0.009 (control versus 15 min LPS); **, p < 0.01 (control versus 30 min LPS) (n = 3).
Effect of Nox2 Inhibition on LPS-mediated IKKβ, p38, and JNK Phosphorylation in HPMECs
To investigate the mechanisms underlying Nox-mediated regulation of Ang2, VEGF-A, and Tie2 expression, we examined the effect of LPS on phosphorylation events in the canonical Toll-like receptor signaling pathway (24). LPS-induced IKKβ phosphorylation (Ser177/181) was evident by 5 min, peaked at 10 min, and waned by 30 min (Fig. 6, A and C). Similarly, LPS mediated a temporal increase in p38 (Thr180/Tyr182) and JNK (Thr183/Tyr185) phosphorylation, with a peak at 60 min (Fig. 6, B and D). In Nox2-silenced cells, but not Nox4 or Nox1-silenced cells, LPS-induced IKKβ phosphorylation (10 min) and p38 phosphorylation (60 min) were attenuated significantly (Fig. 7, A–C). The effect of Nox isoform silencing on JNK phosphorylation was not selective and showed a marginal reduction. These data show that Nox2 inhibition attenuates IKKβ and p38 phosphorylation in HPMECs.
FIGURE 6.

Temporal changes in LPS-induced IKKβ, p38, and JNK phosphorylation in HPMECs. A and C, IKKβ phosphorylation was assessed in cell lysates after LPS treatment by Western blotting (A), and blots were quantified by densitometry. The mean –fold change in IKKβ phosphorylation relative to untreated controls from four experiments is shown (C). *, p < 0.001 (control versus 5 min LPS); **, p < 0.001 (control versus 10 min LPS); ***, p < 0.001 (control versus 15 min LPS); #, p = 0.003 (control versus 30 min LPS) (n = 4). B and D, Western blot (B) showing changes in p38 and JNK (MAPK) phosphorylation with LPS treatment at 15, 30, 60, and 120 min. The mean -fold-change in p38 and JNK phosphorylation relative to controls was quantified by densitometry (D). *, p = 0.001 (control versus 15 min LPS); **, p < 0.001 (control versus 30 min LPS); ***, p < 0.001 (control versus 60 min LPS); $, p = 0.001 (control versus 60 min LPS) (n = 4).
FIGURE 7.
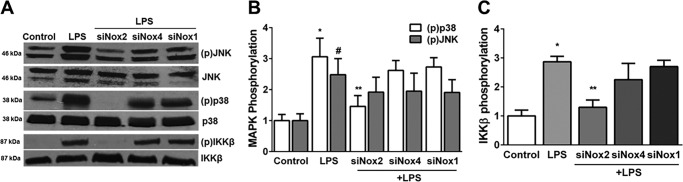
Effect of Nox inhibition on LPS-induced phosphorylation of p38, JNK, and IKKβ in HPMECs. A, immunoblot showing changes in phosphorylation of p38, JNK, and IKKβ following LPS treatment for 60 min (p38 and JNK) or 10 min (IKKβ) in control, LPS-treated, siNox2 + LPS, siNox4 + LPS, and siNox1 + LPS-treated cells. B, densitometric quantification of changes in p38 and JNK (MAPK) phosphorylation relative to controls. *, p < 0.001 (p38, control versus LPS); #, p < 0.01 (JNK, control versus LPS); **, p < 0.01 (p38, LPS versus LPS + siNox2) (n = 5). C, changes in IKKβ phosphorylation relative to untreated controls were quantified using densitometry. *, p < 0.001 (control versus LPS); **, p < 0.001 (LPS versus siNox2 + LPS) (n = 5).
NF-κB and AP-1 Activation Induced by LPS Is Modulated by Nox2 Silencing in HPMECs
The promoters of Ang2, VEGF-A, and Tie2 have transcription factor binding sites for NF-κB and AP-1 (25–28). To examine the role of Nox isoforms in the activation of transcription factors that regulate expression of angiogenesis genes in HPMECs, we quantified NF-κB and AP-1 in cellular nuclear extracts obtained after LPS treatment. LPS-induced an 8.1- and 3.4-fold increase in NF-κB and AP-1 levels at 1 h in nuclear extracts, respectively, supporting activation of these transcription factors (Fig. 8, A and B). Nox2 silencing, but not Nox4 or Nox1 silencing, attenuated an LPS-mediated increase in nuclear translocation of these transcription factors by >60% (Fig. 8, A–C). Nox4 silencing has a minimal (<10%) effect on NF-κB activation alone (Fig. 8A). These data suggest that NF-κB and AP-1 activation in response to LPS in HPMECs is predominantly Nox2-dependent.
FIGURE 8.

Effect of Nox1, Nox2, and Nox4 siRNA on LPS-induced nuclear translocation NF-κB (p65) and AP-1 (c-jun) in HPMECs. A and B, NF-κB and AP-1 levels were quantified 60 min after LPS treatment in nuclear fractions obtained from control, LPS-treated, siNox1 + LPS-treated, siNox2 + LPS-treated, and siNox4 + LPS-treated HPMECs. Nuclear levels are expressed relative to the control. *, p < 0.001 (control versus LPS); **, p < 0.001 (LPS versus siNox2 + LPS); ***, p < 0.001 (LPS versus siNox4 + LPS); $, p < 0.001 (control versus LPS); $$, p < 0.001 (LPS versus siNox2 + LPS) (n = 3). C, histone expression in nuclear fractions was quantified by immunoblotting as a loading control for the experiments in A and B.
Effect of Nox2 Silencing on LPS-mediated Autocrine Angiogenesis in HPMECs
To determine whether Nox2 regulates LPS-mediated angiogenesis in HPMECs, we used both cell-based and CM experiments. LPS-induced tube and network formation was attenuated by >60% in Nox2-silenced cells (Fig. 9, A and B). Supplementing Nox2-silenced cells with recombinant Ang2 and VEGF-A restored LPS-mediated angiogenic tube and network formation (Fig. 9, A and B). Similarly, CM from LPS plus siNox2-treated cells showed a marked reduction in stimulating HPMEC angiogenic responses compared with CM from LPS-treated cells (Fig. 9, C and D). Supplementation of CM from Nox2-silenced cells with recombinant Ang2, recombinant VEGF-A, or both fully restored LPS-mediated angiogenic tube and network formation (Fig. 9, C and D). These data support the role of Nox2 in regulating LPS-induced, Ang2/VEGF-A-dependent angiogenesis in HPMECs.
FIGURE 9.

Effect of Nox2 inhibition on LPS-induced angiogenesis in HPMECs. A, fluorescent microscope images depicting angiogenic tube formation in control, LPS-treated, LPS + siNox2-treated, and LPS + siNox2 + rhAng2 + rhVEGF-A-treated cells. Images were captured at 5× magnification. B, graphical representation summarizing data from four different experiments in HPMECs (n = 4). *, p < 0.001 (control versus LPS tube and network formation); **, p < 0.001 (LPS versus LPS + siNox2 tube and network formation); ***, p < 0.001 (LPS + siNox2 versus LPS + siNox2 + rhAng2 + rhVEGF-A tube and network formation). C, HPMECs were treated with conditioned media from control (CM-control), LPS-treated (CM-LPS), and LPS + siNox2-treated (CM-LPS + siNox2) cells. All cells were treated with Ab-TLR4 before application of conditioned media. Recombinant VEGF-A and Ang2 were used to restore angiogenic responses in cells treated with conditioned media from LPS + siNox2-treated cells (CM-LPS + siNox2 + rhAng2 + rhVEGF-A). D, graphical representation summarizing data from three different experiments in HPMECs (n = 3). *, p < 0.001 (CM-Control versus CM-LPS tube and network formation); **, p < 0.001 (CM-LPS versus CM-LPS + siNox2 tube and network formation); ***, p < 0.001 (CM-LPS + siNox2 versus CM-LPS + siNox2 + rhAng2 tube and network formation); $, p < 0.001 (CM-LPS + siNox2 versus CM-LPS + siNox2 + rhVEGF-A tube and network formation); $$ p = 0.001 (CM-LPS + siNox2 versus CM-LPS + siNox2 + rhAng2 + rhVEGF-A tube and network formation).
DISCUSSION
Recent studies demonstrate the importance of pulmonary endothelial Ang2 expression to lung inflammation and vascular injury in BPD and other diseases (29, 30). However, the mechanisms by which Ang2 is regulated in response to systemic sepsis remain unclear. In this study, we report a novel role for Nox-dependent signaling in the regulation of proinflammatory Ang2 expression in pulmonary endothelial cells. We demonstrate that Nox2 regulates LPS-mediated Ang2 and VEGF-A expression as well as Ang2- and VEGF-A-dependent autocrine angiogenesis in HPMECs. We propose that Nox2 regulates LPS-mediated IKKβ and p38 phosphorylation, resulting in the nuclear translocation of NF-κB and AP-1, transcriptional induction of VEGF-A and Ang2, and altered angiogenesis in HPMECs (Fig. 10). Validating the importance of this mechanism in animal models of BPD will further support the biological relevance of our findings.
FIGURE 10.
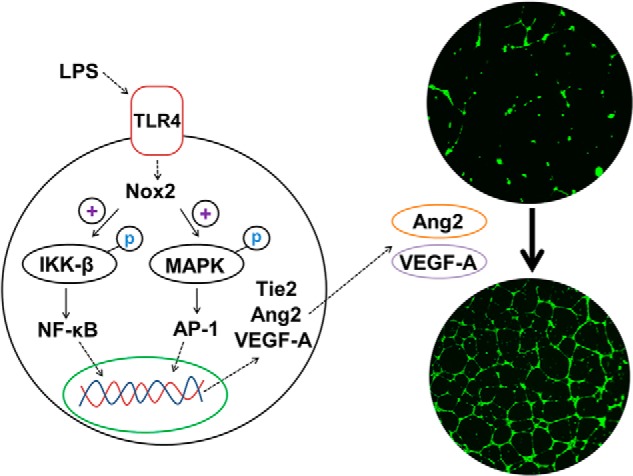
Proposed mechanism for Nox2-dependent regulation of LPS mediated Angiopoietin signaling and angiogenesis in HPMECs.
The temporal increase in Ang2 and VEGF-A expression observed with LPS in HPMECs is consistent with rodent and human studies showing increased expression of Ang2 or VEGF-A in the whole lung or systemic circulation after LPS administration or systemic sepsis, respectively (12, 31–33). Our data indicate that pulmonary microvascular endothelial cells respond to bacterial ligands with robust Ang2, Tie2, and VEGF-A expression. Previous works on the relationships between Tie2 expression and sepsis have noted disparate results. Our data are consistent with van der Heijden et al. (34) and Yang et al. (35), who showed that Tie2 expression in the systemic circulation was elevated in sepsis, but are in contrast with results reported by Mofarrahi et al. (12) and David et al. (36), who showed decreased lung Tie2 gene expression after LPS administration. The differences in these results could be explained by the fact that, although our data were obtained in HPMECs, the above mentioned studies examined Tie2 expression in the whole lung or systemic circulation. Our data on mouse lung endothelial cells showing that Tie2 and Ang2 expression is induced after systemic LPS support our in vitro results. Similar to our findings, in a model of tumor necrosis factor α-induced microvascular injury, Willam et al. (37) noted an up-regulation of Tie2 in human umbilical vein endothelial cells and coronary microvascular endothelial cells. The presence of AP-1 binding sites in the promoter region of the human Tie2 gene further supports our data (28). Increased expression of Ang2, VEGF-A, and Tie2 in parallel with NF-κB and AP-1 activation is consistent with the establishment of a pro-inflammatory endothelial phenotype in HPMECs after LPS.
The mechanisms underlying the regulation of proinflammatory Ang2 expression in the endothelium by reactive oxygen species (ROS) remain unknown. In HPMECs, LPS-induced superoxide formation and Ang2/VEGF-A expression were inhibited by Nox2 silencing. The presence of Nox2 subunits in HPMECs as well as coimmunoprecipitation of p47phox with gp91phox after LPS treatment support activation of the Nox2 complex. Studies with chemical inhibitors that inhibit Nox (VAS2870) or quench superoxide (PEG-superoxide dismutase) suggest that superoxide formation contributes to LPS-mediated induction of angiogenic markers. In conjunction with our Nox2 silencing data, these results support a role for Nox2-induced ROS in LPS-mediated Ang2 and VEGF-A expression. A direct role for Nox in inflammatory Ang2 expression has not been shown before. In a hemangioma model, Bhandarkar et al. (38) demonstrated that lentiviral knockdown of Nox4 impaired Ang2 expression in polyoma-transformed brain endothelial cells. Although their data support our observation that Nox isoforms can regulate Ang2 expression, their tumor model is likely representative of Ets-driven Ang2 expression, whereas our inflammatory model is more reflective of AP-1/NF-κB-driven Ang2 up-regulation. Although other investigators have shown that VEGF signaling in the endothelium activates Nox, we demonstrate that LPS-induced VEGF expression in lung endothelial cells is Nox-2-dependent (39). Oh et al. (40) have reported previously that, in microglial cells, Nox indirectly regulates VEGF expression through hypoxia-inducible factor 1 (HIF-1) up-regulation. We did not examine HIF-1α specifically in HPMECs because our model did not involve hypoxic conditions and because we were probing proinflammatory VEGF-A expression. Depending on the nature of the stimulus, endothelial VEGF can be up-regulated by transcription factors belonging to the Ets family, NF-κB, and HIF-1α, among others (26). Although Ang1/Tie2-dependent angiogenesis has been reported to involve generation of ROS, the role of Nox isoforms in LPS-mediated Tie2 expression has not been evaluated before (41). Our data suggest that, similar to proinflammatory VEGF-A and Ang2 induction in endothelial cells, Tie2 expression in response to LPS is also modulated by Nox2 in HPMECs.
To examine the mechanisms underlying Nox-mediated regulation of Ang2 and VEGF expression in HPMECs, we examined the IKKβ/NF-κB and MAPK/AP-1 pathways activated in canonical TLR signaling (24). In HPMECs, phosphorylation of IKKβ and the MAP kinase p38 induced by LPS was attenuated by inhibiting Nox2. Although LPS stimulated JNK phosphorylation, our data regarding Nox-silencing and JNK phosphorylation were not selective and not significant. Prior work from Loukili et al. (42) and Menden et al. (13) has demonstrated that Nox isoforms can regulate proinflammatory IKKβ phosphorylation. Although the involvement of ROS in LPS-mediated MAP kinase activation has been reported in other cell types, the specific isoform of Nox that regulates p38 and JNK phosphorylation in lung endothelial cells remains unknown (43–45). Our data support a role for Nox2 in mediating LPS-induced p38 phosphorylation in HPMECs. Peng et al. (43) showed that LPS-mediated TNF-α expression in cardiomyocytes was dependent on gp91phox and p38 kinase activation. Similarly, Wu et al. (46) and Patel et al. (45) showed Nox2 and Nox4 mediated LPS-dependent MAPK activation in aortic smooth muscle cells and skeletal muscle microvascular endothelial cells, respectively. Our data regarding HPMECs are consistent with the studies mentioned above and support the regulation of proinflammatory MAPK activation by Nox isoforms. The mechanisms underlying the regulation of IKKβ and p38 phosphorylation by Nox2 in HPMECs were not examined in this study because this was not the focus of our experiments. Whether these effects are mediated through TGF-β-activated kinase 1 (TAK1) or represent direct effects on IKKβ and p38 remains to be elucidated. The involvement of ROS in the activation of transcription factors NF-κB and AP-1 has been shown before (47). We examined the nuclear translocation of NF-κB and AP-1 (as a marker of activation) in HPMECs because Ang2, VEGF-A, and Tie2 can be induced transcriptionally by NF-κB and AP-1 (25, 27, 28). We noted that Nox2 silencing, but not Nox4 or Nox1 silencing, inhibited LPS-mediated superoxide formation and NF-κB/AP-1 nuclear translocation. These data support a role for Nox2-dependent ROS in proinflammatory NF-κB and AP-1 activation in HPMECs.
Angiogenesis involves endothelial cell migration, proliferation, and sprouting of new vessels (48). We used an in vitro assay to determine the relevance of Nox2-mediated Ang2 and VEGF-A expression in LPS-induced angiogenesis (23). In HPMECs, LPS-induced Ang2 and VEGF-A expression stimulated angiogenic tube and network formation in an autocrine manner. Nox2 silencing or conditioned media from Nox2-silenced cells attenuated LPS-mediated angiogenic responses, demonstrating the importance of Nox2 in regulating proinflammatory, Ang2-dependent angiogenesis. Although Nox4, Nox1, and Nox2 have been reported to mediate the angiogenic response to hypoxia and growth factors such as VEGF and fibroblast growth factor, the Nox isoform involved in endotoxin- and Ang2-mediated signaling has not been characterized before (49, 50). Interestingly, supplementation with Ang2 or VEGF-A restored angiogenic responses in Nox2-silenced cells in equal measure, showing that Ang2 alone is sufficient to mediate angiogenesis during inflammation. Our results are consistent with prior work showing that Ang2-dependent endothelial cell migration and sprouting of new vessels requires the presence of inflammatory cytokines or VEGF (7, 11). Examination of the implications of LPS-mediated pulmonary endothelial Ang2, VEGF-A, and Tie2 expression to vascular remodeling in BPD will have to be pursued in animal models.
In summary, we demonstrate that Nox2 regulates Ang2 and VEGF-A expression in pulmonary endothelial cells through the IKKβ/NF-κB and MAPK/AP-1 pathways. We also show that Ang2 and VEGF-A mediate LPS-induced angiogenic responses in an autocrine fashion. To the best of our knowledge, this is one of the initial reports to demonstrate regulation of Ang2 expression and proinflammatory angiogenesis by Nox-dependent signaling. Increased Ang2 expression in the systemic circulation or in the lung has been associated with mortality in humans with sepsis, severity of acute lung injury, and with development of BPD in premature infants (30, 31, 51). Although we showed that mouse lung endothelial cells express increased Ang2 and Tie2 after systemic LPS, the rest of our data were obtained in primary cells in vitro, and, therefore, verification of the results of this study in animal models expressing angiogenic factors in a tissue-restricted manner will enable us to better understand the significance of these findings. This assumes translational significance because inhibiting Ang2 using antibodies or modulating Nox2 activity are emerging as promising strategies to decrease lung injury in bacterial sepsis (51, 52).
Acknowledgments
We thank Dr. Neil Hogg, Ph.D. (Departments of Redox Biology and Biophysics, Medical College of Wisconsin) for general guidance.
This work was supported, in whole or in part, by National Institutes of Health Grants 1R03HD062693-01A1 and 8KL2TR000056 (to V. S.) and HL102745 (to S. C. and R. R.).
- BPD
- bronchopulmonary dysplasia
- Nox
- NADPH oxidase
- HPMEC
- human pulmonary microvascular endothelial cell
- ECM
- endothelial cell medium
- VAS2870
- 3-benzyl-7-(2-benzoxazolyl) thio-1,2,3,-triazolo (4,5-d) pyrimidine
- CM
- conditioned medium
- ROS
- reactive oxygen species
- IKKβ
- inhibitor of κ-B kinase β.
REFERENCES
- 1. Thébaud B., Abman S. H. (2007) Bronchopulmonary dysplasia: where have all the vessels gone? Roles of angiogenic growth factors in chronic lung disease. Am. J. Respir. Crit. Care Med. 175, 978–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abman S. H. (2001) Bronchopulmonary dysplasia: “a vascular hypothesis.” Am. J. Respir. Crit. Care Med. 164, 1755–1756 [DOI] [PubMed] [Google Scholar]
- 3. Jobe A. J. (1999) The new BPD: an arrest of lung development. Pediatr. Res. 46, 641–643 [DOI] [PubMed] [Google Scholar]
- 4. Cordero L., Ayers L. W., Davis K. (1997) Neonatal airway colonization with Gram-negative bacilli: association with severity of bronchopulmonary dysplasia. Pediatr. Infect. Dis. J. 16, 18–23 [DOI] [PubMed] [Google Scholar]
- 5. Kallapur S. G., Jobe A. H. (2006) Contribution of inflammation to lung injury and development. Arch. Dis. Child. 91, F132–F135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Augustin H. G., Koh G. Y., Thurston G., Alitalo K. (2009) Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat. Rev. Mol. Cell Biol. 10, 165–177 [DOI] [PubMed] [Google Scholar]
- 7. Fiedler U., Augustin H. G. (2006) Angiopoietins: a link between angiogenesis and inflammation. Trends Immunol. 27, 552–558 [DOI] [PubMed] [Google Scholar]
- 8. Hayes A. J., Huang W. Q., Mallah J., Yang D., Lippman M. E., Li L. Y. (1999) Angiopoietin-1 and its receptor Tie-2 participate in the regulation of capillary-like tubule formation and survival of endothelial cells. Microvasc. Res. 58, 224–237 [DOI] [PubMed] [Google Scholar]
- 9. Hato T., Kimura Y., Morisada T., Koh G. Y., Miyata K., Tabata M., Kadomatsu T., Endo M., Urano T., Arai F., Araki K., Suda T., Kobayashi K., Oike Y. (2009) Angiopoietins contribute to lung development by regulating pulmonary vascular network formation. Biochem. Biophys. Res. Commun. 381, 218–223 [DOI] [PubMed] [Google Scholar]
- 10. Ziegler T., Horstkotte J., Schwab C., Pfetsch V., Weinmann K., Dietzel S., Rohwedder I., Hinkel R., Gross L., Lee S., Hu J., Soehnlein O., Franz W. M., Sperandio M., Pohl U., Thomas M., Weber C., Augustin H. G., Fässler R., Deutsch U., Kupatt C. (2013) Angiopoietin 2 mediates microvascular and hemodynamic alterations in sepsis. J. Clin. Invest. 10.1172/JCI66549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lobov I. B., Brooks P. C., Lang R. A. (2002) Angiopoietin-2 displays VEGF-dependent modulation of capillary structure and endothelial cell survival in vivo. Proc. Natl. Acad. Sci. U.S.A. 99, 11205–11210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mofarrahi M., Nouh T., Qureshi S., Guillot L., Mayaki D., Hussain S. N. (2008) Regulation of angiopoietin expression by bacterial lipopolysaccharide. Am. J. Physiol. 294, L955–L963 [DOI] [PubMed] [Google Scholar]
- 13. Menden H., Tate E., Hogg N., Sampath V. (2013) LPS mediated endothelial activation in pulmonary endothelial cells: role of Nox2-dependent IKK-β phosphorylation. Am. J. Physiol. Lung Cell Mol. Physiol. 304, L445–L455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bedard K., Krause K. H. (2007) The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 87, 245–313 [DOI] [PubMed] [Google Scholar]
- 15. Park H. S., Chun J. N., Jung H. Y., Choi C., Bae Y. S. (2006) Role of NADPH oxidase 4 in lipopolysaccharide-induced proinflammatory responses by human aortic endothelial cells. Cardiovasc. Res. 72, 447–455 [DOI] [PubMed] [Google Scholar]
- 16. Takac I., Schröder K., Brandes R. P. (2012) The Nox family of NADPH oxidases: friend or foe of the vascular system? Curr. Hypertens. Rep. 14, 70–78 [DOI] [PubMed] [Google Scholar]
- 17. Miyoshi T., Yamashita K., Arai T., Yamamoto K., Mizugishi K., Uchiyama T. (2010) The role of endothelial interleukin-8/NADPH oxidase 1 axis in sepsis. Immunology 131, 331–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pfaffl M. W. (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Griendling K. K., Minieri C. A., Ollerenshaw J. D., Alexander R. W. (1994) Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 74, 1141–1148 [DOI] [PubMed] [Google Scholar]
- 20. Teng R.-J., Eis A., Bakhutashvili I., Arul N., Konduri G. G. (2009) Increased superoxide production contributes to the impaired angiogenesis of fetal pulmonary arteries with in utero pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 297, L184–L195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cifuentes-Pagano E., Meijles D. N., Pagano P. J. (2014) The quest for selective Nox inhibitors and therapeutics: challenges, triumphs and pitfalls. Antioxid. Redox Signal. 20, 2741–2754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Veronese F. M., Caliceti P., Schiavon O., Sergi M. (2002) Polyethylene glycol-superoxide dismutase, a conjugate in search of exploitation. Adv. Drug Deliv. Rev. 54, 587–606 [DOI] [PubMed] [Google Scholar]
- 23. Medhora M., Dhanasekaran A., Pratt P. F., Jr., Cook C. R., Dunn L. K., Gruenloh S. K., Jacobs E. R. (2008) Role of JNK in network formation of human lung microvascular endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 294, L676–L685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Akira S., Uematsu S., Takeuchi O. (2006) Pathogen recognition and innate immunity. Cell 124, 783–801 [DOI] [PubMed] [Google Scholar]
- 25. Hegen A., Koidl S., Weindel K., Marmé D., Augustin H. G., Fiedler U. (2004) Expression of angiopoietin-2 in endothelial cells is controlled by positive and negative regulatory promoter elements. Arterioscler. Thromb. Vasc. Biol. 24, 1803–1809 [DOI] [PubMed] [Google Scholar]
- 26. Pagès G., Pouysségur J. (2005) Transcriptional regulation of the Vascular Endothelial Growth Factor gene: a concert of activating factors. Cardiovasc. Res. 65, 564–573 [DOI] [PubMed] [Google Scholar]
- 27. Jośko J., Mazurek M. (2004) Transcription factors having impact on vascular endothelial growth factor (VEGF) gene expression in angiogenesis. Med. Sci. Monit. 10, RA89–RA98 [PubMed] [Google Scholar]
- 28. Hewett P. W., Daft E. L., Murray J. C. (1998) Cloning and partial characterization of the human tie-2 receptor tyrosine kinase gene promoter. Biochem. Biophys. Res. Commun. 252, 546–551 [DOI] [PubMed] [Google Scholar]
- 29. Bhandari V., Choo-Wing R., Lee C. G., Zhu Z., Nedrelow J. H., Chupp G. L., Zhang X., Matthay M. A., Ware L. B., Homer R. J., Lee P. J., Geick A., de Fougerolles A. R., Elias J. A. (2006) Hyperoxia causes angiopoietin 2-mediated acute lung injury and necrotic cell death. Nat. Med. 12, 1286–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Thomas W., Seidenspinner S., Kramer B. W., Wirbelauer J., Kawczyńska-Leda N., Szymankiewicz M., Speer C. P. (2011) Airway angiopoietin-2 in ventilated very preterm infants: association with prenatal factors and neonatal outcome. Pediatr. Pulmonol. 46, 777–784 [DOI] [PubMed] [Google Scholar]
- 31. Kümpers P., van Meurs M., David S., Molema G., Bijzet J., Lukasz A., Biertz F., Haller H., Zijlstra J. G. (2009) Time course of angiopoietin-2 release during experimental human endotoxemia and sepsis. Crit. Care 13, R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Medford A. R., Douglas S. K., Godinho S. I., Uppington K. M., Armstrong L., Gillespie K. M., van Zyl B., Tetley T. D., Ibrahim N. B., Millar A. B. (2009) Vascular Endothelial Growth Factor (VEGF) isoform expression and activity in human and murine lung injury. Respir. Res. 10, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Karmpaliotis D., Kosmidou I., Ingenito E. P., Hong K., Malhotra A., Sunday M. E., Haley K. J. (2002) Angiogenic growth factors in the pathophysiology of a murine model of acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 283, L585–L595 [DOI] [PubMed] [Google Scholar]
- 34. van der Heijden M., van Nieuw Amerongen G. P., van Hinsbergh V. W., Groeneveld A. B. (2010) The interaction of soluble Tie2 with angiopoietins and pulmonary vascular permeability in septic and nonseptic critically ill patients. Shock 33, 263–268 [DOI] [PubMed] [Google Scholar]
- 35. Yang J., Huang J., Zhang Y. Z., Chen L. (2010) Tie2 mRNA in peripheral blood: a new marker to assess damage of endothelial cells in a rat model of sepsis. Chin. J. Traumatol. 13, 308–312 [PubMed] [Google Scholar]
- 36. David S., Ghosh C. C., Kümpers P., Shushakova N., Van Slyke P., Khankin E. V., Karumanchi S. A., Dumont D., Parikh S. M. (2011) Effects of a synthetic PEG-ylated Tie-2 agonist peptide on endotoxemic lung injury and mortality. Am. J. Physiol. Lung Cell Mol. Physiol. 300, L851–L862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Willam C., Koehne P., Jürgensen J. S., Gräfe M., Wagner K. D., Bachmann S., Frei U., Eckardt K. U. (2000) Tie2 receptor expression is stimulated by hypoxia and proinflammatory cytokines in human endothelial cells. Circ. Res. 87, 370–377 [DOI] [PubMed] [Google Scholar]
- 38. Bhandarkar S. S., Jaconi M., Fried L. E., Bonner M. Y., Lefkove B., Govindarajan B., Perry B. N., Parhar R., Mackelfresh J., Sohn A., Stouffs M., Knaus U., Yancopoulos G., Reiss Y., Benest A. V., Augustin H. G., Arbiser J. L. (2009) Fulvene-5 potently inhibits NADPH oxidase 4 and blocks the growth of endothelial tumors in mice. J. Clin. Invest. 119, 2359–2365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Blanchetot C., Boonstra J. (2008) The ROS-NOX connection in cancer and angiogenesis. Crit. Rev. Eukaryot. Gene Expr. 18, 35–45 [DOI] [PubMed] [Google Scholar]
- 40. Oh Y. T., Lee J. Y., Yoon H., Lee E. H., Baik H. H., Kim S. S., Ha J., Yoon K.-S., Choe W., Kang I. (2008) Lipopolysaccharide induces hypoxia-inducible factor-1 α mRNA expression and activation via NADPH oxidase and Sp1-dependent pathway in BV2 murine microglial cells. Neurosci. Lett. 431, 155–160 [DOI] [PubMed] [Google Scholar]
- 41. Chen J.-X., Zeng H., Lawrence M. L., Blackwell T. S., Meyrick B. (2006) Angiopoietin-1-induced angiogenesis is modulated by endothelial NADPH oxidase. Am. J. Physiol. Heart Circ. Physiol. 291, H1563–H1572 [DOI] [PubMed] [Google Scholar]
- 42. Loukili N., Rosenblatt-Velin N., Rolli J., Levrand S., Feihl F., Waeber B., Pacher P., Liaudet L. (2010) Oxidants positively or negatively regulate nuclear factor κB in a context-dependent manner. J. Biol. Chem. 285, 15746–15752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Peng T., Lu X., Feng Q. (2005) NADH oxidase signaling induces cyclooxygenase-2 expression during lipopolysaccharide stimulation in cardiomyocytes. FASEB J. 19, 293–295 [DOI] [PubMed] [Google Scholar]
- 44. Fakler C. R., Wu B., McMicken H. W., Geske R. S., Welty S. E. (2000) Molecular mechanisms of lipopolysaccharide induced ICAM-1 expression in A549 cells. Inflamm. Res. 49, 63–72 [DOI] [PubMed] [Google Scholar]
- 45. Patel D. N., Bailey S. R., Gresham J. K., Schuchman D. B., Shelhamer J. H., Goldstein B. J., Foxwell B. M., Stemerman M. B., Maranchie J. K., Valente A. J., Mummidi S., Chandrasekar B. (2006) TLR4-NOX4-AP-1 signaling mediates lipopolysaccharide-induced CXCR6 expression in human aortic smooth muscle cells. Biochem. Biophys. Res. Commun. 347, 1113–1120 [DOI] [PubMed] [Google Scholar]
- 46. Wu F., Tyml K., Wilson J. X. (2008) iNOS expression requires NADPH oxidase-dependent redox signaling in microvascular endothelial cells. J. Cell Physiol. 217, 207–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Brigelius-Flohé R., Flohé L. (2011) Basic principles and emerging concepts in the redox control of transcription factors. Antioxid. Redox Signal. 15, 2335–2381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Carmeliet P. (2000) Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 6, 389–395 [DOI] [PubMed] [Google Scholar]
- 49. Ushio-Fukai M. (2006) Redox signaling in angiogenesis: role of NADPH oxidase. Cardiovasc. Res. 71, 226–235 [DOI] [PubMed] [Google Scholar]
- 50. Ushio-Fukai M., Urao N. (2009) Novel role of NADPH oxidase in angiogenesis and stem/progenitor cell function. Antioxid. Redox Signal. 11, 2517–2533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. van Meurs M., Kümpers P., Ligtenberg J. J., Meertens J. H., Molema G., Zijlstra J. G. (2009) Bench-to-bedside review: angiopoietin signaling in critical illness: a future target? Crit. Care 13, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Carnesecchi S., Pache J. C., Barazzone-Argiroffo C. (2012) NOX enzymes: potential target for the treatment of acute lung injury. Cell. Mol. Life Sci. 69, 2373–2385 [DOI] [PMC free article] [PubMed] [Google Scholar]