

Background: Adiponectin has been shown to limit liver fibrosis, but the molecular mechanisms remain unknown.
Results: In vitro and in vivo adiponectin increases TIMP-1 secretion, which binds to the CD63/β1-integrin complex to decrease FAK activity and stellate cell migration.
Conclusion: Adiponectin-promoted TIMP-1 plays an important role in limiting liver fibrosis.
Significance: Targeting adiponectin signaling could be a useful way to limit liver fibrosis.
Keywords: Adiponectin, Fibrosis, Liver, Tetraspanin, Tissue Inhibitor of Metalloproteinase (TIMP)
Abstract
Hepatic stellate cells (HSC) are central players in liver fibrosis that when activated, proliferate, migrate to sites of liver injury, and secrete extracellular matrix. Obesity, a known risk factor for liver fibrosis is associated with reduced levels of adiponectin, a protein that inhibits liver fibrosis in vivo and limits HSC proliferation and migration in vitro. Adiponectin-mediated activation of adenosine monophosphate-activated kinase (AMPK) inhibits HSC proliferation, but the mechanism by which it limits HSC migration to sites of injury is unknown. Here we sought to elucidate how adiponectin regulates HSC motility. Primary rat HSCs were isolated and treated with adiponectin in migration assays. The in vivo actions of adiponectin were examined by treating mice with carbon tetrachloride for 12 weeks and then injecting them with adiponectin. Cell and tissue samples were collected and analyzed for gene expression, signaling, and histology. Serum from patients with liver fibrosis was examined for adiponectin and tissue inhibitor of metalloproteinase-1 (TIMP-1) protein. Adiponectin administration into mice increased TIMP-1 gene and protein expression. In cultured HSCs, adiponectin promoted TIMP-1 expression and through binding of TIMP-1 to the CD63/β1-integrin complex reduced phosphorylation of focal adhesion kinase to limit HSC migration. In mice with liver fibrosis, adiponectin had similar effects and limited focal adhesion kinase phosphorylation. Finally, in patients with advanced fibrosis, there was a positive correlation between serum adiponectin and TIMP-1 levels. In sum, these data show that adiponectin stimulates TIMP-1 secretion by HSCs to retard their migration and contributes to the anti-fibrotic effects of adiponectin.
Introduction
Chronic liver injury results in hepatic fibrosis that can progress to cirrhosis, and complications including portal hypertension with variceal bleeding, liver failure, and hepatocellular carcinoma (1). At the molecular level, liver fibrosis results from the activation of hepatic stellate cells (HSCs)2 (2), which differentiate into highly proliferative and migratory myofibroblasts that accumulate in areas of hepatocyte apoptosis and necrosis. These cells secrete extracellular matrix (ECM) proteins including collagens and inhibit ECM degradation by the secretion of inhibitors, leading to the buildup of scar tissue within the liver (1, 3).
Obesity is associated with the development of non-alcoholic steatohepatitis (NASH), which is an established risk factor for the development of liver fibrosis (4). In part, this observation stems from the fact that adipose tissues secrete a number of adipokines that modulate hepatic inflammation, lipid metabolism, and fibrosis. Dominant among these is adiponectin, an abundant adipokine, which correlates negatively with body fat and fasting plasma insulin and that has been shown to protect against liver fibrosis development in vivo (4–6). Adiponectin null mice have more fibrosis than wild type mice after carbon tetrachloride (CCl4) treatment and adiponectin overexpression limits fibrosis. The application of adiponectin to HSCs in vitro reduces α-smooth muscle actin (αSMA) expression (a marker of HSC activation) and their proliferation and migration. Adiponectin responses via AMPK has been shown to be pivotal in modulating proliferation, but the mechanism by which this protein mediates HSC migration is unknown (5–7).
Tissue inhibitor of metalloproteinase-1 (TIMP-1) is secreted by HSCs during liver fibrosis, and serum and hepatic levels are increased in patients with liver fibrosis (8, 9). Conversely, reduced TIMP-1 levels are associated with the spontaneous resolution of liver fibrosis and increased matrix degradation (10). The conventional view has been that increased TIMP-1 levels inhibit matrix degradation to promote liver fibrosis. However, the role for TIMP-1 in liver fibrosis is not completely understood. For example, in TIMP-1 knock-out mice, CCl4 treatment is associated with enhanced fibrosis (11), suggesting a protective role. In contrast and in agreement with the consensus view, transgenic mice overexpressing TIMP-1 have enhanced liver fibrosis (12), and fibrosis resolution is reduced after the injury insult is removed (13).
However, apart from inhibiting MMP activity, TIMP-1 also possesses MMP-independent signaling. For example, in endothelial cells TIMP-1 can inhibit cell migration by inhibiting the activity of focal adhesion kinase (FAK) (14). Moreover, and of relevance to TIMP-1 functions during liver fibrosis, adiponectin as a dominant anti-fibrotic factor has been shown to up-regulate TIMP-1 in other cell types (15–17).
Given these conflicting observations, the purpose of the present study was to elucidate the role of adiponectin-induced TIMP-1 on HSC motility. Our results demonstrate that adiponectin increases TIMP-1 to initiate a MMP-independent signaling cascade through the CD63/β1-integrin protein complex and to inhibit FAK phosphorylation and HSC migration. Furthermore, we find in patients with liver fibrosis a positive association between adiponectin and TIMP-1 levels. Taken together, our findings reveal a novel functional role for TIMP-1 in the presence of adiponectin during liver fibrosis.
EXPERIMENTAL PROCEDURES
Reagents
Recombinant full-length murine adiponectin (fAd) was from Biovendor (Evropska, Czech Republic). Blocking anti-TIMP-1 and CD63 antibodies (azide-free) were from Santa Cruz (Santa Cruz Biotechnology, Santa Cruz, CA). Rat TIMP-1 recombinant protein was from R&D systems (Minneapolis, MN). Compound C, adenine 9-β-d-arabinofuranoside (AraA), and 5-aminoimidazole-4-carboxyamide ribonucleoside (AICAR) were from Calbiochem.
Animals
All studies were approved by the Western Sydney Local Health District Animal Ethics Committee. Mice were on the C57BL/6 background and housed under standard pathogen-free conditions with a 12-h light/dark cycle. To induce fibrosis, mice (6 each group) received 2 intraperitoneal injections per week for 12 weeks of carbon tetrachloride (300 μl/kg), as previously described (6), and then 3 days later were injected intraperitoneally with adiponectin (2 μg/g) or PBS, and the livers were harvested after 24 h.
Rat HSC Isolation and Cell Culture
HSCs from male Sprague-Dawley rat livers were isolated by in situ Pronase, collagenase perfusion, and then a single step Histodenz gradient step as previously reported (18). HSCs were maintained in Dulbecco's modified eagle medium containing 20% FBS. Human LX2 cells, a stellate cell line, were cultured in DMEN containing 20% FCS.
Migration Assay
Boyden chambers (BD Biosciences, 8-μm pore size) were coated with 50 μl of rat tail collagen I (2 mg/ml; BD Biosciences), and 5 × 104 activated HSCs were plated in 100 μl of serum-free medium containing either fAd (5 μg/ml), CD63, TIMP-1, or IgG antibodies (0.25 μg/ml). 400 μl of serum-free medium containing the chemoattractant MCP-1 (200 ng/ml; ProSpec) was supplied to the lower chamber. The cells were cultured for 8 h and then stained with hematoxylin and counted.
Enzyme-linked Immunosorbent Assay
ELISAs for rat TIMP-1 and human adiponectin and TIMP-1 were performed according to the manufacturer's instructions (R&D systems). For rats, activated HSCs (day 7) were serum-starved overnight and treated with fAd (5 μg/ml), and the supernatant was used for ELISA. Human serum TIMP-1 and adiponectin levels were measured from 28 patients with NASH (fibrosis stage 3 or 4) (19).
Cell Proliferation and Apoptosis Assays
Activated HSCs (day 7) were serum-starved overnight and treated with fAd (5 μg/ml), aminoimidazole-4-carboxyamide ribonucleoside (AICAR; 2 mm), Compound-C (6-[4-(2-piperidin-1-ylethoxy) phenyl]-3-pyridin-4-ylpyrazolo [1,5-a]pyrimidine) (20 μm), TIMP-1 or IgG antibodies (0.25 μg/ml) or rat TIMP-1 (500 ng/ml R&D systems) for 24 h. Cell proliferation or apoptosis was assessed using a bromodeoxyuridine (BrdU) kit (Roche Applied Science) or Caspase-3 Immunoassay/Activity kit (Calbiochem), respectively, according to the manufacturer's instructions.
RNA Isolation and Quantitative Real-time PCR
Total RNA from cells and livers was isolated with RNeasy (Qiagen). A260/280 ratios and RNA quantity were determined by a NanoDrop ND-1000 spectrophotometer. 1000 ng of total RNA was reverse-transcribed into cDNA using SuperScript III reverse transcriptase (Invitrogen). Quantification of mRNA expression was performed by quantitative real-time PCR using a Corbett 6000 rotor gene platform (Corbett). The primer pairs used for the amplification of genes of interest for mouse were SP-1 forward (5′-GCTATAGCAAACACCCCAGGT-3′) and reverse (5′-GATCAGGGCTGTTCTCTCCTT-3′) and TIMP-1 forward (5′-AAGGGCTACCAGAGCGATCA-3′) and reverse (5′-GGTATTGCCAGGTGCACAAAT-3′) and for human were TIMP1 forward (5′-ACTTCCACAGGTCCCACAAC-3′) and reverse (5′-CACTGTGCATTCCTCACAGC-3′), α-SMA forward (5′-CCGAGATCTCACCGACTACC-3′) and reverse (5′-TCCAGAGCGACATAGCACAG-3′), and GAPDH forward (5′-CGACCACTTTGTCAAGCTCA-3′ and reverse 5′-GGTTGAGCACAGGGTACTTTATT-3′).
Immunofluorescence
Mouse liver-frozen sections and cells were fixed with acetone or 1% paraformaldehyde in PBS, washed in PBS, blocked with Protein block (DAKO), incubated with antibodies against αSMA (Sigma), Ki67, FAK (Cell Signaling), CD63 (Serotec), or β1-integrin (Millipore) and the appropriate secondary. Apoptosis was performed with a TUNEL kit (Roche Applied Science), and slides were mounted with Prolong®Fold containing DAPI stain (Invitrogen). For staining of rat HSCs, a permeabilization step with 0.1% Triton X-100 for 10 min was included after paraformaldehyde fixation. Image acquisition was performed on a Leica DM LB confocal microscope (Leica, Germany) and analyzed with Image J.
Immunoprecipitation
For β1-integrin immunoprecipitation, subconfluent primary rat HSCs or LX-2 cells were lysed in buffer (50 mm HEPES, pH 7.4, 150 mm NaCl, 10% glycerol, 1% CHAPS (rat) or 1% Triton X-100 (LX-2), 1 mm EDTA) containing phosphatase inhibitors (50 mm NaF, 5 mm Na4P2O7, 2 mm Na3VO4) and a protease inhibitor mixture (Roche Applied Science), cleared by centrifugation, and quantified by the BCA protein assay (Thermo Scientific, Rockford, IL). 1 μg of recombinant rat or human TIMP-1 (R&D Systems) was incubated with 1 mg of total protein lysate for 2 h at 4 °C. Lysates were precleared for 2 h with Protein G-Sepharose 4B (Invitrogen) and then incubated with 1 μg of CD63 (Serotec) or β1-integrin antibody (Millipore) or isotype control prebound to Protein G-Sepharose 4B. The resin was then washed 4× with lysis buffer, and the bound complexes were eluted by reducing sample buffer (95 °C, 5 min). Proximity ligation assays were undertaken with rat HSCs and anti-CD63 (Serotec) and β1-integrin (Millipore) antibodies and subjected to the proximity ligation assays kit according to the manufacturer's instructions (Sigma).
Western Blot Analysis
Frozen liver tissue and activated HSCs treated with fAd (5 μg/ml), TIMP-1 (0.25 μg/ml), CD63 antibody (0.25 μg/ml), IgG antibody control (0.25 μg/ml), and/or rat TIMP-1 recombinant protein was lysed and blotted as previously described (21). Membranes were incubated with antibodies against AMPK, p-AMPK, FAK, pFAK Y397 (Cell Signaling), αSMA (Sigma), CD63 (Secrotec), TIMP-1, and β1-integrin (Millipore) overnight in TBS-Tween containing 5% BSA at 4 °C followed by HRP-conjugated secondary antibodies (Sigma) and visualization by chemiluminescence (SuperSignal® West Pico; Thermo Scientific). β-Actin was used as a loading control. Densitometry analysis was performed using NIH ImageJ software.
shRNA-mediated Knockdown of CD63
shRNAs were purchased from OpenBiosystems and lentiviruses (multiplicity of infection ≈ 10) was applied to activated HSCs cultured in DMEM containing FBS for 40 h. The efficiency of shRNA-mediated knockdown was evaluated by quantitative real-time PCR.
Patients and Data Collection
All 28 patients had biopsy-proven NASH with advanced fibrosis (F3: 61%) or cirrhosis (F4: 39%). All were Child-Pugh A with no signs of decompensation. Other liver diseases had been excluded by appropriate serology/history, and all patients were confirmed to drink less than 70 g of alcohol per week. Equal numbers of males and females (14/14) composed the cohort, and the mean age was 55 (±12) years. Patients were obese (mean body mass index 31 (±5.6) kg/m2) and markedly insulin-resistant (mean homeostasis model assessment of insulin resistance (HOMA-IR) 6.6). Mean values of important liver tests included: ALT 86 (±44) IU/liter, bilirubin 16 (±8.6) mmol/liter, albumin 44 (±3) g/liter, platelets 204 (±75) × 109/ml, international normalization ratio (INR) 1.0 (±0.27). All values are expressed as mean ± S.D. The study protocol was approved by the Human Ethics Committee of the Western Sydney Area Health Service, and written informed consent was obtained (19).
Data Analysis and Statistics
Quantitative data are expressed as the means ± S.E. All experiments were performed three times, and statistical analyses between control and treatment groups were evaluated using Student's t test. For multiple group analyses, statistical significance was determined using a one-way analysis of variance followed by a Tukey multiple group comparison test. For analyses using human samples, the strength of association between continuous variables was reported using Spearman rank correlations. For all analyses, significance was set at p ≤ 0.05.
RESULTS
Adiponectin Limits HSC Proliferation and Activation in Vivo but Increased TIMP-1
To validate adiponectin actions on HSCs in vivo, fibrosis was induced in wild type mice by 12 weeks of carbon tetrachloride treatment. Three days after the last treatment, the mice were treated with a single intraperitoneal injection of fAd (or vehicle) and harvested 24 h later. Over this short time frame we observed no change in sirius red staining for collagen between fAd or vehicle-injected groups (Fig. 1A). In contrast, a near 3-fold reduction in Ki67 staining that is colocalized with αSMA (Fig. 1B) and a 25% reduction in αSMA staining (Fig. 1C) were observed. Surprisingly, we did not observe any enhancement in HSC apoptosis through simultaneous staining for TUNEL and αSMA (Fig. 1D).
FIGURE 1.

Adiponectin reduces HSC proliferation and α-SMA expression in vivo. fAd injection promoted no change in Sirius red (collagen) staining (10× magnification) (A), reduced HSC proliferation as determined by Ki67 (green) and α-SMA (red) co-staining (bar, 50 μm) DAPI (blue) (B), decreased α-SMA (light gray) expression (10× magnification) (C), and no change in apoptosis as determined by TUNEL (TUN; green) and α-SMA (red) co-staining (Bar, 50 μm) (D). The arrows indicate a light green colocalized cell, and the inset represents a 2× magnified double-labeled cell. *, p value <0.05.
Wild type mice treated with adiponectin had 2.5-fold more TIMP-1 gene expression than mice treated with PBS (Fig. 2A). Analysis of TIMP-1 serum levels by ELISA revealed only a small non-significant increase in TIMP-1 levels in wild type mice treated with adiponectin (Fig. 2B). Western blot analysis of whole liver lysates revealed that fAd as expected promoted 2.0-fold more AMPK phosphorylation and surprisingly caused a significant reduction in the pFAK/FAK ratio compared with the placebo-treated mice. αSMA protein, a marker of activated fibrogenic HSCs, was reduced 3-fold in the livers of wild type mice injected with fAd compared with those injected with PBS (Fig. 2, C and D). These data demonstrate that in vivo adiponectin has limited effects on HSC cell viability, reduces HSCs proliferation and activation, promotes TIMP-1 secretion, and modulates AMPK and FAK phosphorylation.
FIGURE 2.

Adiponectin promotes TIMP-1 expression and inhibits FAK activity in vivo. fAd injection into mice increased hepatic TIMP-1 expression 3-fold (A), elevated serum TIMP-1 in WT (not significant) (B), and increased the hepatic p-AMPK/AMPK ratio and reduced the pFAK/FAK ratio and α-SMA protein levels as compared with placebo treated WT (C and D). E, the treatment of HSCs with fAd (0–15 μg/ml) promoted TIMP-1 gene expression. F, fAd (5 μg/ml) treatment of activated HSCs increased TIMP-1 protein secretion. G, after 6 h, the application of AMPK antagonists, compound C (C-C) (20 μm), or adenine 9-β-d-arabinofuranoside (AraA; 1 mm) with fAd (5 μg/ml) reduced TIMP-1 protein levels compared with fAd alone. p values <0.05, 0.001, and <0.0001 considered to be significant are represented with *, **, and ***, respectively. Plc, placebo.
Adiponectin Promotes TIMP-1 Expression in Activated HSCs
Although adiponectin promoted TIMP-1 expression in fibrotic livers and can promote TIMP-1 expression in cell lines and human monocytes, we evaluated TIMP-1 expression in rat-activated HSCs after fAd treatment (15–17). In dose-response experiments, adiponectin (0–15 μg/ml) robustly induced TIMP-1 gene expression 3–4-fold (Fig. 2E). On subsequent treatment with fAd (5 μg/ml), secreted TIMP-1 protein increased ∼4-fold to near 500 ng/ml compared with untreated cells (Fig. 2F).
Previous data have shown adiponectin to signal through AMPK in HSCs (7). To determine if adiponectin mediated TIMP-1 production through AMPK, we treated activated HSCs with fAd with or without the AMPK-specific antagonists CC or adenine 9-β-d-arabinofuranoside (AraA) and confirmed that signaling was mediated by AMPK (Fig. 2G).
Next we confirmed the anti-proliferative and pro-apoptotic activity of adiponectin on HSCs in vitro (5, 7). We found that fAd limited proliferation through AMPK, increased apoptosis, and reduced their activation (Fig. 3, A–D). Although TIMP-1 has been shown to inhibit apoptosis in HSCs (22), we next evaluated apoptosis and proliferation in the context of adiponectin induced TIMP-1. The joint application of fAd and TIMP-1 blocking antibodies to HSCs did not alter caspase 3 activity or proliferation. In the absence of fAd, the blockage of TIMP-1 increased apoptosis and did not affect proliferation. Rat recombinant TIMP-1 had no effect on proliferation (Fig. 3, E and F). These results indicate that adiponectin inhibits HSC activation in vitro and acts through AMPK to promote TIMP-1 secretion from HSCs, and TIMP-1 in the context of adiponectin does not influence HSC viability and proliferation.
FIGURE 3.
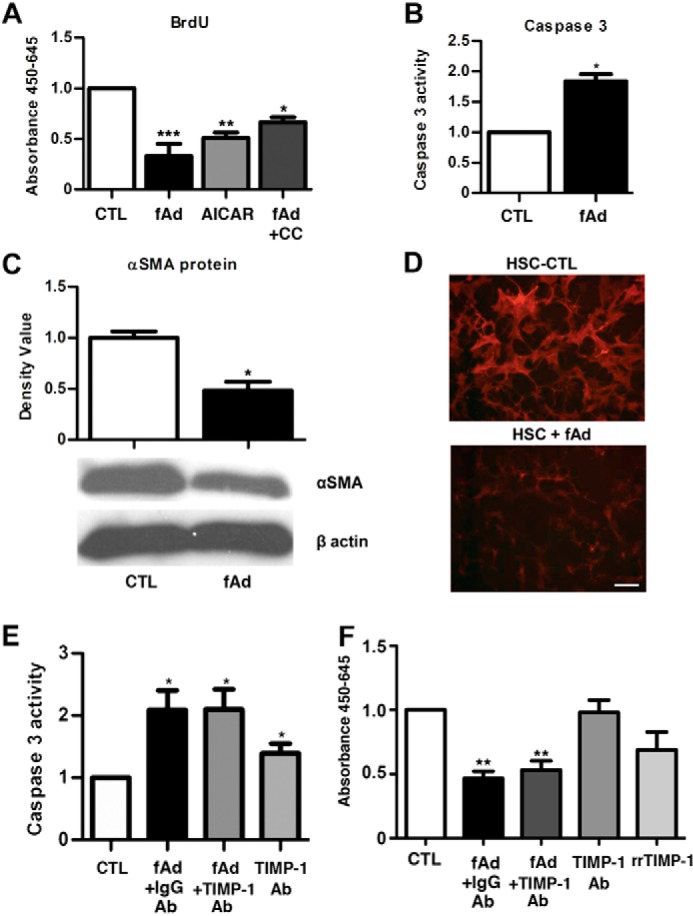
Adiponectin reverses the fibrotic phenotype of activated HSCs. A, treatment of HSCs with fAd (5 μg/ml) or AMPK agonist aminoimidazole-4-carboxyamide ribonucleoside (AICAR; 2 mm) for 24 h resulted in a 3- and 2-fold respective reduction in BrdU incorporation. Application of the antagonist compound C (CC; 20 μm) with fAd for 24 h reversed proliferation. B, fAd treatment of HSCs increased caspase 3 activity 2-fold. C, Western blot analysis showed that αSMA protein was down-regulated 2-fold in fAd treated compared with control HSCs. D, immunofluorescence staining showed α-SMA (red) was greater in untreated than fAd treated HSCs. E, fAd treatment increased caspase 3 activity 2-fold, and IgG control and TIMP-1 blocking antibodies had no effect. In untreated HSCs TIMP-1 antibodies increased caspase 3 activity 1.4-fold. F, fAd reduced proliferation and IgG control, and TIMP-1 blocking antibodies had no effect. In untreated HSCs, TIMP-1 antibodies and rrTIMP-1 had no significant effect. p values <0.05, 0.001 and <0.0001 considered to be significant are represented with *, **, and ***, respectively (bar, 100 μm).
TIMP-1 through the CD63/β1-Integrin Complex Impairs HSC Motility
It has previously been reported that TIMP-1 regulates FAK activity through MMP-independent signaling by binding to the tetraspanin CD63/β1-integrin complex (23). We investigated whether this complex was present in HSCs. Western blot analysis confirmed that activated rat HSCs express β1-integrin, FAK, and CD63 (Fig. 4A). Moreover, confocal analyses of single-activated rat HSCs illustrated that FAK and CD63 and β1-integrin and CD63 abundantly colocalize in the cytoplasm (Fig. 4B). We then undertook proximity ligation assays. Proximity ligation assays utilizes oligonucleotide-ligated antibodies that when brought in close proximity, due to the binding of epitopes on proteins engaged in complex formation, initiate a rolling circle amplification that can be detected with Cy3-labeled probes, appearing as fluorescent spots (24). Employing specific CD63 and β1-integrin antibodies, there was robust association compared with control (Fig. 4, C and D). Finally, to confirm this association, respective CD63 and β1-integrin immunoprecipitations were performed with rat-activated HSCs and human LX2 (a human HSC cell line) lysates. Western blotting of isolated CD63 immunocomplexes from rat HSCs revealed that β1-integrin and rat recombinant TIMP-1 associated with CD63 (Fig. 4E). Through quantitative PCR we confirmed that LX2 cells reacted similarly to fAd and after treatment had decreased αSMA (2-fold) and increased TIMP-1 expression (2.7-fold) (Fig. 4, F–G). Western blotting of isolated β1-integrin immunocomplexes from LX2 cells revealed that CD63, FAK, and human recombinant TIMP-1 were associated with β1-integrin (Fig. 4H). Limited binding was observed in IgG controls. These data demonstrate that CD63, β1-integrin, FAK, and TIMP-1 form a complex in HSCs.
FIGURE 4.

β1-integrin, CD63, and FAK form a complex in HSCs. A, Western blot shows that rat HSCs express β1-integrin, CD63, and FAK. B, immunofluorescence and confocal analysis shows that FAK (green) and CD63 (red) and that β1-integrin (green) and CD63 colocalize (yellow), respectively (bar, 20 μm). C and D, proximity ligand assay (PLA) detects the complex of CD63 and β1-integrin (red) in rat HSCs. p value = 0.001 (**) (bar, 20 μm). E, CD63 immunoprecipitations (IP) of primary rat cells pulled down CD63, β1-integrin, and rat recombinant TIMP-1. fAd treatment of LX2 cells reduced the gene expression of αSMA 1.7-fold (F) and increased TIMP-1 expression by 2.7-fold (G). H, immunoprecipitation of β1-integrin from human LX2 cells pulled down CD63, FAK, and human recombinant TIMP-1.
It has been previously shown that CD63 can modulate HSC motility (25), yet the mechanistic role of TIMP-1 and CD63 in HSC migration has not been clarified. Therefore, to test the role of TIMP-1 binding to CD63 on HSC motility we performed migration assays using a modified Boyden chamber assay. Adiponectin treatment resulted in an ∼80% reduction in migration. Co-treatment with CD63 blocking antibodies in the absence of fAd did not impair HSC motility. However, on fAd treatment, anti-CD63 significantly repressed the inhibitory activity of fAd on migration by 3-fold compared with fAd treatment alone (Fig. 5A). In separate experiments co-treatment with fAd and a TIMP-1 blocking antibody significantly increased HSC migration 2-fold compared with fAd and fAd+ IgG antibody-treated cells (Fig. 5B). To further clarify the roles of TIMP-1 and CD63 in HSC signaling, we treated activated HSCs with fAd and either TIMP-1 or CD63 blocking antibodies and evaluated FAK phosphorylation. Untreated HSCs expressed prominent pFAK, whereas treatment with fAd reduced the pFAK/FAK protein ratio 4-fold. Co-treatment with blocking antibodies against TIMP-1 or CD63 reversed fAd inhibition of pFAK/FAK significantly by 2-fold (Fig. 5, C and D).
FIGURE 5.

Adiponectin-stimulated TIMP-1 inhibits HSC migration and FAK activity. In migration assays rat HSCs were treated with combinations of fAd (5 μg/ml), IgG, CD63, and TIMP-1 antibodies (0.25 μg/ml). A, fAd inhibited invasion 4-fold. The blocking CD63 antibody had no effect alone but limited the action of fAd on migration. Control IgG had no effect. B, co-treatment with fAd and the TIMP-1 Ab reversed the inhibitory effect of fAd on migration. C and D, Western blot revealed the pFAK/FAK ratio to be significantly down-regulated after fAd treatment. Co-treatment with CD63 or TIMP-1 blocking antibodies partially reversed the inhibitory effect of fAd on pFAK/FAK. E, CD63 gene expression was reduced by 55% in HSCs transduced with CD63 shRNA. Scrambled shRNA (scram) did not affect CD63 expression. F–G, CD63 shRNA lentivirus-transduced cells treated with fAd (5 μg/ml) showed a 50% reduction in pFAK/FAK ratio in scram shRNA treated cells and a 30% reduction with CD63 shRNA. pFAK/FAK levels increased 1.4-fold in CD63 shRNA-transduced cells compared with scram shRNA. p values <005, 0.001 and <0.0001 considered to be significant are represented with *, **, and ***, respectively.
To further substantiate the role of CD63, we transduced activated HSCs with lentivirus expressing shRNA against rat CD63 and observed a 55% knock-down in CD63 transcript (Fig. 5E). Evaluation of the action of fAd on transduced HSCs with lentivirus confirmed that CD63 knock-down significantly increased the pFAK/FAK ratio compared with scrambled shRNA (Fig. 5, F–G). These data show that adiponectin-stimulated TIMP-1 binds CD63 to modulate FAK activity and HSC cellular motility in vitro.
TIMP-1 Protein Parallels Adiponectin Levels in Patients with NASH
To support our observations of a causal association between adiponectin and TIMP-1 levels, we measured their serum levels by ELISA from patients with NASH and advanced liver fibrosis (Table 1). When patients with low (<5 μg/ml) and high (>10 μg/ml) serum adiponectin were analyzed, TIMP-1 levels were found to be higher (373 ± 15 ng/ml) in those with high adiponectin versus those with low adiponectin (255 ± 14 ng/ml, p < 0.001). Finally, using Spearman rank analysis we observed that TIMP-1 levels strongly and positively correlated with serum adiponectin (R = 0.71, p < 0.001)(Fig. 6, A and B).
TABLE 1.
ELISA data showing the low (<5 μg/ml) and high (>10 μg/ml) serum adiponectin and TIMP-1 levels for individual patients
| Adiponectin concentration | Adiponectin | TIMP-1 |
|---|---|---|
| μg/ml | ng/ml | |
| Low, <5 μg/ml | 2.03 | 224.81 |
| 2.69 | 298.15 | |
| 3.05 | 290.4 | |
| 3.3 | 209.3 | |
| 3.81 | 212.28 | |
| 3.97 | 234.35 | |
| 4.21 | 304.12 | |
| 4.55 | 341.68 | |
| 4.6 | 249.26 | |
| 4.63 | 247.47 | |
| 4.73 | 333.34 | |
| 4.8 | 217.65 | |
| 4.81 | 153.84 | |
| 5.03 | 259.39 | |
| Mean | 4.02 | 255.43 |
| S.E. | 0.25 | 14.10 |
| High, >10 μg | ||
| 10.04 | 353.01 | |
| 10.49 | 334.53 | |
| 10.52 | 271.32 | |
| 11.73 | 389.99 | |
| 11.95 | 346.46 | |
| 12.75 | 453.2 | |
| 13.58 | 546.82 | |
| 13.67 | 311.27 | |
| 14.08 | 386.41 | |
| 14.47 | 307.69 | |
| 15.88 | 344.67 | |
| 22.31 | 384.02 | |
| 25.32 | 407.88 | |
| 28.61 | 381.64 | |
| Mean | 15.39 | 372.78 |
| S.E. | 1.55 | 18.21 |
FIGURE 6.

Positive correlation between serum TIMP-1 and adiponectin levels. A, ELISA revealed increased serum TIMP-1 in patients with high versus low serum adiponectin (>10 μg/ml serum adiponectin; n = 14; TIMP-1: 372 ± 15 ng/ml, versus <5 μg/ml serum adiponectin; n = 14; TIMP-1: 255 ± 14 ng/ml, p value <0.05). B, linear regression revealed a positive correlation (Spearman correlation co-efficient 0.71) between serum TIMP-1 and adiponectin levels. C, model representing the action of adiponectin on HSCs leading to an increase in AMPK phosphorylation, the subsequent release and binding of TIMP-1 to CD63, and dephosphorylation of FAK. The net effect was to decrease HSC proliferation and migration.
DISCUSSION
A poorly answered question in hepatic biology is the role of TIMP-1 in liver fibrosis. In other biological systems, TIMP-1 limits cell migration through MMP-independent signaling suggesting that in the context of liver fibrosis, TIMP-1 function may be more complex than previously considered. Moreover, it is well known that adiponectin can limit HSC migration; however, the mechanism for this effect is unknown. Here we have answered important questions pertaining to the roles of TIMP-1 and adiponectin in fibrosis. We show 1) that despite well described anti-fibrotic effects, adiponectin can promote TIMP-1 expression in vitro, in vivo, and in patients, 2) that this effect is mediated by adiponectin-induced AMPK signaling, and 3) that adiponectin-stimulated TIMP-1 impairs HSC motility through binding to the CD63/β1-integrin complex to inhibit FAK phosphorylation. These data in sum suggest that in the presence of adiponectin, TIMP-1 levels are elevated and may protect against liver fibrosis by limiting HSC migration to sites of injury (Fig. 6C).
In examining the role of adiponectin on HSC function, we unexpectedly found that TIMP-1 expression was robustly up-regulated in the liver by adiponectin. Serum levels of TIMP-1 were not significantly elevated, reflecting perhaps that the effects are principally paracrine and autocrine in nature. Histology of the livers after 24 h of adiponectin treatment revealed unchanged sirius red staining and reduced αSMA, suggesting that adiponectin reduces HSC activation in vivo. On inspection by staining for TUNEL and Ki67, we observed that adiponectin reduced proliferation but had limited effects on cell viability. It is possible that apoptotic HSCs may have been removed through phagocytosis; however, as adiponectin promotes a strong anti-inflammatory reaction to inhibit macrophage function (26), the most likely explanation is that in vivo, adiponectin predominantly reduces HSC proliferation.
Subsequent examination of signaling in adiponectin-treated mice illustrated that the adipokine surprisingly reduces FAK phosphorylation that is downstream of AMPK phosphorylation. It is well known that adiponectin can regulate AMPK activity to affect cell proliferation and metabolism, but adiponectin regulation of FAK is completely novel. FAK is an important kinase that is associated with cellular adhesion and movement, and inhibition studies have shown FAK to be necessary in HSC migration (27). Additionally, previous studies have shown TIMP-1 can inhibit cell migration by inhibiting FAK activity (14). We, therefore, reasoned that perhaps adiponectin-induced TIMP-1 and FAK were related in the context of HSC function.
Many studies have shown that TIMP-1 is pro-fibrogenic, yet we find that adiponectin, a well known and potent inhibitor of liver fibrosis, increased TIMP-1 levels. To address this, we examined the function of TIMP-1 on HSC apoptosis and proliferation in the context of adiponectin and found that TIMP-1 had a limited role in modulating HSC viability and turnover in vitro. This highly controversial observation in terms of current views on the role of TIMP-1 in fibrosis led us to speculate on the potential biological relationship between adiponectin, TIMP-1, and FAK during liver fibrosis. Previous studies have shown that TIMP-1 can have MMP-independent signaling through CD63 (Tspan 30), a member of the tetraspanins (23). These molecules are hydrophobic proteins containing four transmembrane α-helices, two extracellular loops, and a short cytoplasmic tail. Expressed by a variety of cells, CD63 is present in late endosomes, lysosomes, secretory vesicles, and on the cell surface (28). Biochemical studies have illustrated that tetraspanin proteins can interact with proteins including major histocompatibility complex, growth factor receptors, immunoglobulins, and of relevance to CD63, integrins (29–32). Integrins are expressed by HSCs and regulate HSC proliferation and apoptosis (33). Moreover, previous studies have identified that CD63 and β1-integrin are expressed on HSCs, and the targeting of these proteins in function-blocking experiments can inhibit HSC migration (25). Here through exhaustive analyses, we found by visual, proximity ligation assay and immunoprecipitation studies that CD63 and β1-integrin interact in HSCs. Furthermore, it is known that β1-integrin interacts with FAK, a major regulator of cellular motility (34), and we show that this complex is also present in HSCs. Moreover, we confirmed that TIMP-1 could bind to CD63 and β1-integrin immunoprecipitates and have, therefore, established the presence of the CD63/β1-integrin complex in HSCs.
At the mechanistic level, to demonstrate that the CD63/β1-integrin complex can affect HSC function, we performed migration assays in conjunction with specific function blocking antibodies and shRNA. Furthermore, as CD63 interacts with β1-integrin, which in turn interacts with FAK, we examined FAK phosphorylation to demonstrate a motility signaling change precipitated by the addition of adiponectin. Consistently, we observed with blocking antibodies to CD63 or TIMP-1 or with CD63 shRNA that FAK phosphorylation was increased compared with adiponectin treatment alone, suggesting that reduced HSC migration in the presence of adiponectin is modulated by FAK. Collectively, these data illustrate a novel pathway of adiponectin-TIMP-1-induced inhibition of HSC migration.
The prevailing view is that elevated TIMP-1 levels are causally associated with promoting liver fibrosis, and numerous studies suggest an association of TIMP-1 with the extent of fibrosis (9, 35). In other reports, TIMP-1 levels have been included in non-invasive algorithms to classify the extent of fibrosis (36, 37). In support of such a role, animal models have shown that TIMP-1 overexpression promotes liver fibrosis (12) and retards fibrosis resolution (13), and the administration of TIMP-1 blocking antibodies attenuates fibrosis (38). In addition, during the process of fibrosis resolution, reduced TIMP-1 levels result in increased MMP activity and matrix degradation (39, 40). Here we show that in the presence of adiponectin, TIMP-1 levels are unexpectedly elevated but that this in fact may assist in limiting fibrosis by reducing HSC invasion and migration through the extracellular matrix.
Our findings may have relevance for all forms of advanced liver fibrosis as several studies now indicate that irrespective of etiology, adiponectin (19, 41–43) and TIMP-1 levels are elevated (8, 9, 20). If indeed adiponectin is causally associated with increasing TIMP-1 in these patients, then we would expect that those with low serum adiponectin and advanced fibrosis would have lower levels of TIMP-1 protein. Indeed, in extending the strength and veracity of this link from our in vitro and in vivo observations in mice, this is what we clearly observed in a subset of patients with advanced fibrosis. It is, therefore, possible that as adiponectin levels increase during the later stages of liver fibrosis HSC proliferation, and migration may be impaired by adiponectin-mediated elevations in TIMP-1.
In conclusion, our study demonstrates that adiponectin can promote the expression of TIMP-1 and has revealed a new mechanism through which adiponectin can impair liver fibrosis (Fig. 6C). Given these findings, it is possible that the utilization of specific small molecule adiponectin receptor agonists or substances to increase serum adiponectin levels could be a possible strategy to effectively treat liver fibrosis.
This project was supported by the Robert W. Storr Bequest to the Sydney Medical Foundation, University of Sydney, National Health and Medical Research Council of Australia (NHMRC) Program Grant 1053206, NHMRC Project Grant 632630 (to L. H. and J. G.), and Cancer Council NSW Project Grant 1069733 (to L. H.).
- HSC
- hepatic stellate cell
- NASH
- non-alcoholic steatohepatitis
- αSMA
- α-smooth muscle actin
- AMPK
- AMP-activated kinase
- TIMP-1
- tissue inhibitor of metalloproteinase-1
- FAK
- focal adhesion kinase
- pFAK
- phosphorylated FAK
- fAd
- full-length murine adiponectin.
REFERENCES
- 1. Lotersztajn S., Julien B., Teixeira-Clerc F., Grenard P., Mallat A. (2005) Hepatic fibrosis: molecular mechanisms and drug targets. Annu. Rev. Pharmacol. Toxicol. 45, 605–628 [DOI] [PubMed] [Google Scholar]
- 2. Lee U. E., Friedman S. L. (2011) Mechanisms of hepatic fibrogenesis. Best Pract. Res. Clin. Gastroenterol. 25, 195–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Safadi R., Friedman S. L. (2002) Hepatic fibrosis: role of hepatic stellate cell activation. MedGenMed 4, 27. [PubMed] [Google Scholar]
- 4. Marra F., Aleffi S., Bertolani C., Petrai I., Vizzutti F. (2005) Adipokines and liver fibrosis. Eur. Rev. Med. Pharmacol. Sci. 9, 279–284 [PubMed] [Google Scholar]
- 5. Ding X., Saxena N. K., Lin S., Xu A., Xu A., Srinivasan S., Anania F. A. (2005) The roles of leptin and adiponectin: a novel paradigm in adipocytokine regulation of liver fibrosis and stellate cell biology. Am. J. Pathol. 166, 1655–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kamada Y., Tamura S., Kiso S., Matsumoto H., Saji Y., Yoshida Y., Fukui K., Maeda N., Nishizawa H., Nagaretani H., Okamoto Y., Kihara S., Miyagawa J., Shinomura Y., Funahashi T., Matsuzawa Y. (2003) Enhanced carbon tetrachloride-induced liver fibrosis in mice lacking adiponectin. Gastroenterology 125, 1796–1807 [DOI] [PubMed] [Google Scholar]
- 7. Adachi M., Brenner D. A. (2008) High molecular weight adiponectin inhibits proliferation of hepatic stellate cells via activation of adenosine monophosphate-activated protein kinase. Hepatology 47, 677–685 [DOI] [PubMed] [Google Scholar]
- 8. Murawaki Y., Ikuta Y., Idobe Y., Kitamura Y., Kawasaki H. (1997) Tissue inhibitor of metalloproteinase-1 in the liver of patients with chronic liver disease. J. Hepatol. 26, 1213–1219 [DOI] [PubMed] [Google Scholar]
- 9. Benyon R. C., Iredale J. P., Goddard S., Winwood P. J., Arthur M. J. (1996) Expression of tissue inhibitor of metalloproteinases 1 and 2 is increased in fibrotic human liver. Gastroenterology 110, 821–831 [DOI] [PubMed] [Google Scholar]
- 10. Iredale J. P., Benyon R. C., Pickering J., McCullen M., Northrop M., Pawley S., Hovell C., Arthur M. J. (1998) Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J. Clin. Invest. 102, 538–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang H., Lafdil F., Wang L., Yin S., Feng D., Gao B. (2011) Tissue inhibitor of metalloproteinase 1 (TIMP-1) deficiency exacerbates carbon tetrachloride-induced liver injury and fibrosis in mice: involvement of hepatocyte STAT3 in TIMP-1 production. Cell Biosci. 1, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yoshiji H., Kuriyama S., Miyamoto Y., Thorgeirsson U. P., Gomez D. E., Kawata M., Yoshii J., Ikenaka Y., Noguchi R., Tsujinoue H., Nakatani T., Thorgeirsson S. S., Fukui H. (2000) Tissue inhibitor of metalloproteinases-1 promotes liver fibrosis development in a transgenic mouse model. Hepatology 32, 1248–1254 [DOI] [PubMed] [Google Scholar]
- 13. Yoshiji H., Kuriyama S., Yoshii J., Ikenaka Y., Noguchi R., Nakatani T., Tsujinoue H., Yanase K., Namisaki T., Imazu H., Fukui H. (2002) Tissue inhibitor of metalloproteinases-1 attenuates spontaneous liver fibrosis resolution in the transgenic mouse. Hepatology 36, 850–860 [DOI] [PubMed] [Google Scholar]
- 14. Akahane T., Akahane M., Shah A., Connor C. M., Thorgeirsson U. P. (2004) TIMP-1 inhibits microvascular endothelial cell migration by MMP-dependent and MMP-independent mechanisms. Exp. Cell Res. 301, 158–167 [DOI] [PubMed] [Google Scholar]
- 15. Kumada M., Kihara S., Ouchi N., Kobayashi H., Okamoto Y., Ohashi K., Maeda K., Nagaretani H., Kishida K., Maeda N., Nagasawa A., Funahashi T., Matsuzawa Y. (2004) Adiponectin specifically increased tissue inhibitor of metalloproteinase-1 through interleukin-10 expression in human macrophages. Circulation 109, 2046–2049 [DOI] [PubMed] [Google Scholar]
- 16. Stearns M. E., Fudge K., Garcia F., Wang M. (1997) IL-10 inhibition of human prostate PC-3 ML cell metastases in SCID mice: IL-10 stimulation of TIMP-1 and inhibition of MMP-2/MMP-9 expression. Invasion Metastasis 17, 62–74 [PubMed] [Google Scholar]
- 17. Stearns M. E., Wang M., Hu Y., Garcia F. U. (2003) Interleukin-10 activation of the interleukin-10E1 pathway and tissue inhibitor of metalloproteinase-1 expression is enhanced by proteasome inhibitors in primary prostate tumor lines. Mol. Cancer Res. 1, 631–642 [PubMed] [Google Scholar]
- 18. Wang J., Leclercq I., Brymora J. M., Xu N., Ramezani-Moghadam M., London R. M., Brigstock D., George J. (2009) Kupffer cells mediate leptin-induced liver fibrosis. Gastroenterology 137, 713–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. van der Poorten D., Samer C. F., Ramezani-Moghadam M., Coulter S., Kacevska M., Schrijnders D., Wu L. E., McLeod D., Bugianesi E., Komuta M., Roskams T., Liddle C., Hebbard L., George J. (2013) Hepatic fat loss in advanced nonalcoholic steatohepatitis: are alterations in serum adiponectin the cause? Hepatology 57, 2180–2188 [DOI] [PubMed] [Google Scholar]
- 20. Boeker K. H., Haberkorn C. I., Michels D., Flemming P., Manns M. P., Lichtinghagen R. (2002) Diagnostic potential of circulating TIMP-1 and MMP-2 as markers of liver fibrosis in patients with chronic hepatitis C. Clin. Chim. Acta 316, 71–81 [DOI] [PubMed] [Google Scholar]
- 21. Hebbard L. W., Garlatti M., Young L. J., Cardiff R. D., Oshima R. G., Ranscht B. (2008) T-cadherin supports angiogenesis and adiponectin association with the vasculature in a mouse mammary tumor model. Cancer Res. 68, 1407–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Murphy F. R., Issa R., Zhou X., Ratnarajah S., Nagase H., Arthur M. J., Benyon C., Iredale J. P. (2002) Inhibition of apoptosis of activated hepatic stellate cells by tissue inhibitor of metalloproteinase-1 is mediated via effects on matrix metalloproteinase inhibition: implications for reversibility of liver fibrosis. J. Biol. Chem. 277, 11069–11076 [DOI] [PubMed] [Google Scholar]
- 23. Jung K. K., Liu X. W., Chirco R., Fridman R., Kim H. R. (2006) Identification of CD63 as a tissue inhibitor of metalloproteinase-1 interacting cell surface protein. EMBO J. 25, 3934–3942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Söderberg O., Gullberg M., Jarvius M., Ridderstråle K., Leuchowius K. J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L. G., Landegren U. (2006) Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995–1000 [DOI] [PubMed] [Google Scholar]
- 25. Mazzocca A., Carloni V., Sciammetta S., Cordella C., Pantaleo P., Caldini A., Gentilini P., Pinzani M. (2002) Expression of transmembrane 4 superfamily (TM4SF) proteins and their role in hepatic stellate cell motility and wound healing migration. J. Hepatol. 37, 322–330 [DOI] [PubMed] [Google Scholar]
- 26. Mandal P., Park P.-H., McMullen M. R., Pratt B. T., Nagy L. E. (2010) The anti-inflammatory effects of adiponectin are mediated via a heme oxygenase-1-dependent pathway in rat Kupffer cells. Hepatology 51, 1420–1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reif S., Lang A., Lindquist J. N., Yata Y., Gabele E., Scanga A., Brenner D. A., Rippe R. A. (2003) The role of focal adhesion kinase-phosphatidylinositol 3-kinase-Akt signaling in hepatic stellate cell proliferation and type I collagen expression. J. Biol. Chem. 278, 8083–8090 [DOI] [PubMed] [Google Scholar]
- 28. Pols M. S., Klumperman J. (2009) Trafficking and function of the tetraspanin CD63. Exp. Cell Res. 315, 1584–1592 [DOI] [PubMed] [Google Scholar]
- 29. Chirco R., Liu X. W., Jung K. K., Kim H. R. (2006) Novel functions of TIMPs in cell signaling. Cancer Metastasis Rev. 25, 99–113 [DOI] [PubMed] [Google Scholar]
- 30. Mannion B. A., Berditchevski F., Kraeft S. K., Chen L. B., Hemler M. E. (1996) Transmembrane-4 superfamily proteins CD81 (TAPA-1), CD82, CD63, and CD53 specifically associated with integrin α4β1 (CD49d/CD29). J. Immunol. 157, 2039–2047 [PubMed] [Google Scholar]
- 31. Berditchevski F., Bazzoni G., Hemler M. E. (1995) Specific association of CD63 with the VLA-3 and VLA-6 integrins. J. Biol. Chem. 270, 17784–17790 [DOI] [PubMed] [Google Scholar]
- 32. Berditchevski F., Odintsova E. (1999) Characterization of integrin-tetraspanin adhesion complexes: role of tetraspanins in integrin signaling. J. Cell Biol. 146, 477–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhou X., Murphy F. R., Gehdu N., Zhang J., Iredale J. P., Benyon R. C. (2004) Engagement of αvβ3 integrin regulates proliferation and apoptosis of hepatic stellate cells. J. Biol. Chem. 279, 23996–24006 [DOI] [PubMed] [Google Scholar]
- 34. Zachary I., Rozengurt E. (1992) Focal adhesion kinase (p125FAK): a point of convergence in the action of neuropeptides, integrins, and oncogenes. Cell 71, 891–894 [DOI] [PubMed] [Google Scholar]
- 35. Herbst H., Wege T., Milani S., Pellegrini G., Orzechowski H. D., Bechstein W. O., Neuhaus P., Gressner A. M., Schuppan D. (1997) Tissue inhibitor of metalloproteinase-1 and -2 RNA expression in rat and human liver fibrosis. Am. J. Pathol. 150, 1647–1659 [PMC free article] [PubMed] [Google Scholar]
- 36. Younossi Z. M., Page S., Rafiq N., Birerdinc A., Stepanova M., Hossain N., Afendy A., Younoszai Z., Goodman Z., Baranova A. (2011) A biomarker panel for non-alcoholic steatohepatitis (NASH) and NASH-related fibrosis. Obes. Surg. 21, 431–439 [DOI] [PubMed] [Google Scholar]
- 37. Rosenberg W. M., Voelker M., Thiel R., Becka M., Burt A., Schuppan D., Hubscher S., Roskams T., Pinzani M., Arthur M. J., and European Liver Fibrosis Group (2004) Serum markers detect the presence of liver fibrosis: a cohort study. Gastroenterology 127, 1704–1713 [DOI] [PubMed] [Google Scholar]
- 38. Parsons C. J., Bradford B. U., Pan C. Q., Cheung E., Schauer M., Knorr A., Krebs B., Kraft S., Zahn S., Brocks B., Feirt N., Mei B., Cho M. S., Ramamoorthi R., Roldan G., Ng P., Lum P., Hirth-Dietrich C., Tomkinson A., Brenner D. A. (2004) Antifibrotic effects of a tissue inhibitor of metalloproteinase-1 antibody on established liver fibrosis in rats. Hepatology 40, 1106–1115 [DOI] [PubMed] [Google Scholar]
- 39. Iredale J. P., Benyon R. C., Arthur M. J., Ferris W. F., Alcolado R., Winwood P. J., Clark N., Murphy G. (1996) Tissue inhibitor of metalloproteinase-1 messenger RNA expression is enhanced relative to interstitial collagenase messenger RNA in experimental liver injury and fibrosis. Hepatology 24, 176–184 [DOI] [PubMed] [Google Scholar]
- 40. Zhou X., Hovell C. J., Pawley S., Hutchings M. I., Arthur M. J., Iredale J. P., Benyon R. C. (2004) Expression of matrix metalloproteinase-2 and -14 persists during early resolution of experimental liver fibrosis and might contribute to fibrolysis. Liver Int. 24, 492–501 [DOI] [PubMed] [Google Scholar]
- 41. Tietge U. J., Böker K. H., Manns M. P., Bahr M. J. (2004) Elevated circulating adiponectin levels in liver cirrhosis are associated with reduced liver function and altered hepatic hemodynamics. Am. J. Physiol. Endocrinol. Metab. 287, E82–E89 [DOI] [PubMed] [Google Scholar]
- 42. Tacke F., Wüstefeld T., Horn R., Luedde T., Srinivas Rao A., Manns M. P., Trautwein C., Brabant G. (2005) High adiponectin in chronic liver disease and cholestasis suggests biliary route of adiponectin excretion in vivo. J. Hepatol. 42, 666–673 [DOI] [PubMed] [Google Scholar]
- 43. Salman T. A., Allam N., Azab G. I., Shaarawy A. A., Hassouna M. M., El-Haddad O. M. (2010) Study of adiponectin in chronic liver disease and cholestasis. Hepatol. Int. 4, 767–774 [DOI] [PMC free article] [PubMed] [Google Scholar]