

Background: Little is known about the interaction between GLP-1 and the heptahelical core domain of GLP1R.
Results: GLP-1 Asp9 and Gly4 interact with the evolutionarily conserved residues in extracellular loop 3.
Conclusion: Ligand binding pocket formed by evolutionarily conserved residues in the GLP1R core domain.
Significance: This study highlights the mechanism underlying high affinity interaction between GLP-1 and the binding pocket of the receptor.
Keywords: Diabetes, Evolution, G Protein-coupled Receptor (GPCR), Molecular Modeling, Signaling, GLP-1, GLP1R, Ligand Selectivity
Abstract
Glucagon-like peptide-1 (GLP-1) plays a pivotal role in glucose homeostasis through its receptor GLP1R. Due to its multiple beneficial effects, GLP-1 has gained great attention for treatment of type 2 diabetes and obesity. However, little is known about the molecular mechanism underlying the interaction of GLP-1 with the heptahelical core domain of GLP1R conferring high affinity ligand binding and ligand-induced receptor activation. Here, using chimeric and point-mutated GLP1R, we determined that the evolutionarily conserved amino acid residue Arg380 flanked by hydrophobic Leu379 and Phe381 in extracellular loop 3 (ECL3) may have an interaction with Asp9 and Gly4 of the GLP-1 peptide. The molecular modeling study showed that Ile196 at transmembrane helix 2, Met233 at ECL1, and Asn302 at ECL2 of GLP1R have contacts with His1 and Thr7 of GLP-1. This study may shed light on the mechanism underlying high affinity interaction between the ligand and the binding pocket that is formed by these conserved residues in the GLP1R core domain.
Introduction
Glucagon-like peptide-1 (GLP-1)4 is generated by tissue-specific posttranslational processing of preproglucagon in intestinal L-cells and plays a key role in glucose homeostasis (1). Activation of the GLP-1 receptor (GLP1R) in pancreatic β-cells by GLP-1 potentiates glucose-dependent insulin secretion (2, 3). In addition to its insulinotropic effects, GLP-1 promotes growth, survival, and differentiation of β-cells (4, 5). Furthermore, GLP-1 slows down gastric emptying and promotes satiety. Thus, sustained activation of GLP1R results in weight loss (6, 7). Because of these multiple beneficial effects that regulate blood glucose concentration and body weight, GLP-1 is a promising therapeutic agent for the treatment of type 2 diabetes mellitus and obesity. However, circulating GLP-1 is rapidly degraded by dipeptidyl peptide-IV and cannot be administered orally due to its peptidergic chemical nature (8, 9). Thus, there is a great need to develop orally active small molecules that can act on GLP1R (10, 11). The delineation of high affinity ligand-receptor binding and receptor activation will contribute to the development of such molecules.
GLP1R is a member of the class B G protein-coupled receptor (GPCR) family, which includes the glucagon receptor (GCGR) subfamily consisting of five members GCGR, GLP1R, GLP2R, glucose-dependent insulinotropic polypeptide receptor (GIPR), and glucagon-related peptide receptor (GCRPR) (12–15). In addition, growth hormone-releasing hormone receptor (GHRHR), secretin receptor (SCTR), vasoactive intestinal peptide receptor 1 (VPAC1 receptor (VPAC1R)), VPAC2R, and pituitary adenylate cyclase-activating polypeptide receptor (PAC1 receptor, PAC1R) share amino acid sequence similarity with members of the GCGR subfamily (13, 16, 17). Class B GPCRs have a relatively long (∼120 amino acids) N-terminal extracellular domain (ECD) with an α-helix at the N terminus and two antiparallel β sheets stabilized by three disulfide bonds and a salt bridge (18–21). The peptide ligands for this receptor family also share a common structure consisting of a random coiled N terminus followed by an α-helix (20, 22, 23).
According to the two-domain model for class B GPCR activation, the second half of the α-helix of the peptide binds to the N-terminal ECD of the receptor (24–26). This induces a secondary interaction between the N-terminal moiety of the peptide and the receptor core domain consisting of transmembrane helices (TMHs) and extracellular loops (ECLs). This secondary interaction confers receptor activation and G protein coupling (27–29). The interactions between the N-terminal ECD of GLP1R and the second half of the α-helical part of GLP-1 and exendin-4 have been elucidated by x-ray crystallography (20, 22). However, a crystal structure for the ligand-bound receptor core domain is not yet available. This structure would provide useful information for understanding the mechanism of ligand-induced receptor activation.
Alanine scanning analysis revealed that His1, Gly4, Thr7, and Asp9 in the N-terminal portion of GLP-1 are important for receptor binding and activation (30, 31). Recently, by using chimeric GLP1R/GIPR together with chimeric GLP-1/GIP peptides, we identified interactions of His1 and Thr7 of GLP-1 with Ile196/Lys 197 at TMH2, Met233 at ECL1, and Asn302 at ECL2 of GLP1R (32). These results demonstrated the evolutionary pressure to conserve critical residues for ligand binding and activation of GLP1R (33). However, this study did not fully account for the ligand binding pocket of GLP1R, which may require additional residues, probably located at ECL3, for interaction with Gly4 and Asp9 of GLP-1.
In the present study we constructed chimeric GLP1Rs in which the GLP1R ECL3 was replaced with the VPAC1R ECL3, which has a markedly different amino acid sequence. This chimeric receptor responded poorly to GLP-1, showing the importance of ECL3 for ligand-induced receptor activation. Additional experiments in which single amino acid mutations were introduced into GLP1R ECL3 revealed that the evolutionarily conserved basic residue Arg380 and the flanking hydrophobic residues Leu379 and Phe381 were likely to mediate interactions with Gly4 and Asp9 of GLP-1. Based on this observation and our previous result (33), we propose that the ligand binding pocket of GLP1R is formed by evolutionarily conserved residues in TMH2, ECL1, ECL2, and ECL3.
EXPERIMENTAL PROCEDURES
Peptides
Wild-type GLP-1, glucagon, GCRP, GIP, GLP-2, and modified peptides were synthesized by AnyGen (Gwangju, Korea). The amino acid sequences of these peptides are shown in Table 1 and Fig. 7A.
TABLE 1.
Amino acid sequences of modified GLP-1 peptides
Gly4 and Asp9 of GLP-1 were replaced with charged amino acids. Modified positions are shown in bold.
| Peptides | |||
|---|---|---|---|
| GLP-1 | 1HAEGTFTSDV | 11SSYLEGQAAK | 21EFIAWLVKGR |
| [Asp4]GLP-1 | 1HAEDTFTSDV | 11SSYLEGQAAK | 21EFIAWLVKGR |
| [Glu4]GLP-1 | 1HAEETFTSDV | 11SSYLEGQAAK | 21EFIAWLVKGR |
| [His4]GLP-1 | 1HAEHTFTSDV | 11SSYLEGQAAK | 21EFIAWLVKGR |
| [Lys4]GLP-1 | 1HAEKTFTSDV | 11SSYLEGQAAK | 21EFIAWLVKGR |
| [Arg4]GLP-1 | 1HAERTFTSDV | 11SSYLEGQAAK | 21EFIAWLVKGR |
| [Glu9]GLP-1 | 1HAEGTFTSEV | 11SSYLEGQAAK | 21EFIAWLVKGR |
| [Lys9]GLP-1 | 1HAEGTFTSKV | 11SSYLEGQAAK | 21EFIAWLVKGR |
| [Arg9]GLP-1 | 1HAEGTFTSRV | 11SSYLEGQAAK | 21EFIAWLVKGR |
FIGURE 7.

The potencies of wild-type and [Arg9]peptide toward GLP1R-related receptors and their mutant receptors. A, amino acid sequence alignments of GLP-1 and its related peptides and ligand-interacting domains in the receptors. Residues involved in the GLP-1-GLP1R interaction are connected by lines. The residues colored in blue represent sequences that are conserved across paralogs. Residues in red are sequences that are conserved within orthologs of vertebrates, including mouse, anole, chicken, Xenopus tropicalis, medaka, fugu, Tetraodon, stickleback, and zebrafish. The residues in black are variable sequences. Amino acids with similar biochemical properties, such as T/S, K/R, I/L/V, E/D, and F/Y, are considered to be conserved. B, the potencies of glucagon and [Arg9]glucagon toward GCGR and the GCGR[R378D] mutant. C, the potencies of GCRP and [Arg9]GCRP toward GCRPR and the GCRPR[R379D] mutant. D, ligand selectivity of GIPR and GIPR[R362D] mutant for GIP and [Arg9]GIP. E, the potencies of GLP-2 and [Arg9]GLP-2 toward GLP2R and the GLP2R[K414D] mutant. The data on the sigmoid curves represent the means ± S.E. of at least three independent experiments.
Plasmids
The cDNA encoding human GLP1R was originally purchased from Benebiosis (Seoul, Korea) and subcloned into the mammalian expression vector pcDNA3 (Invitrogen). Human VPAC1R, GLP2R, and GCGR in pcDNA3 were obtained from BRNscience Inc. (Seoul, Korea). Human GIPR was kindly provided by Dr. Bernard Thorens (Institute of Pharmacology and Toxicology, Switzerland). Chicken GCRPR was cloned from a White Leghorn hen brain cDNA library (15). The CRE-luc vector, which contains four copies of CRE (TGACGTCA), was obtained from Stratagene (La Jolla, CA).
Construction of Chimeras and Mutants
To swap domains between GLP1R and VPAC1R, individual cDNA fragments of interest were amplified by PCR with Pfu polymerase (ELPIS Biotech., Daejeon, Korea) and two specific primers. One primer corresponded to the 5′ or 3′ end of the receptor cDNAs, and the other primer corresponded to the region of overlap between the two receptors. One fragment was obtained from GLP1R, and the other was from VPAC1R. Both fragments were subjected to a second round of PCR to generate chimeric cDNAs. All of the chimeric constructs were cloned into the pcDNA3 expression vector at the HindIII and XhoI sites. The single and double mutants were constructed by PCR-based site-directed mutagenesis and cloned into pcDNA3 at the HindIII and XhoI sites. The DNA sequences of the chimeras and mutants were verified by automatic sequencing.
Cell Transfection and Luciferase Assays
HEK293T cells were maintained in Dulbecco's modified Eagle's media (DMEM) in the presence of 10% fetal bovine serum. For luciferase assays, cells (2.5 × 104) were plated in 48-well plates 1 day before transfection and transfected with Effectene reagent (Qiagen, Chatsworth, CA) according to the manufacturer's instructions. Approximately 48 h after transfection, cells were treated with the respective ligands for 6 h. Cells were harvested 6 h after ligand treatment. Luciferase activities were determined in cell extracts with a luciferase assay system according to the standard methods for the Synergy 2 Multi-Mode Microplate Reader (BioTek, Winooski, VT).
Binding Assay
GLP-1 and [Arg9]GLP-1 were radioiodinated by the chloramine-T method and purified by chromatography on a Sephadex G-25 column (Sigma) in 0.01 m acetic acid and 0.1% bovine serum albumin (BSA). Cells (1.2 × 105) were transfected with wild-type or mutant receptors (300 ng of DNA/well in 12-well plates) with Effectene (Qiagen). Forty-eight hours later, cells were washed and incubated for 1 h at 37 °C with binding buffer (serum-free DMEM with 0.1% BSA, pH 7.4) containing 100,000 cpm 125I-Labeled ligand (equivalent to ∼30 nm) in the presence of various concentrations of cold ligand. Relative expression levels of receptors were determined using 500,000 cpm 125I-labeled ligand (a concentration for submaximal binding toward the wild-type receptor) in the presence of 10 μm cold ligand. Cells were washed twice with ice-cold Dulbecco's PBS. The radioactivity of the cell lysate resolved in 1% SDS and 0.2 m NaOH was determined on a Wallac 1489 Wizard 3 γ-counter (PerkinElmer Life Sciences).
Data Analysis
Data analysis was performed by nonlinear regression with a sigmoid dose-response curve. Agonist concentrations that induced half-maximal stimulation (EC50) were calculated with GraphPad PRISM4 software (GraphPad Software Inc., San Diego, CA). All data are presented as the means ± S.E. of at least three independent experiments. Group means were compared by Student's t test or one-way analysis of variance followed by the Bonferroni's multiple comparison test. p < 0.05 was considered to be statistically significant.
Molecular Modeling
A homology model for GLP1R and GLP-1 interaction was built with the homology modeling program MODELLER 9v11 (34) and was based on the crystal structures of two class B GPCRs (Protein Data Bank codes 4k5y, and 4l6r) and the ligand-bound ECD of human GLP1R and GIPR (Protein Data Bank codes 3iol and 2qkh). The sequence of GLP1R was manually aligned to the GPCRs of the crystal structures based on TMHs predicted by TMHMM Server v.2.0 (35) and evolutionarily conserved residues. During homology modeling, a disulfide bond was forced between Cys226 and Cys296 of GLP1R. The distance restraints between the following residue pairs were introduced: His1 of GLP-1 ∼ Asn302 of GLP1R; Thr7 of GLP-1 ∼ Ile196, Lys197, Met233, and Met302 of GLP1R; Gly4 and Asp9 of GLP-1 ∼ Arg380 of GLP1R. An α-helical secondary structure was enforced for each TMH. The variable target function method with the very thorough option and molecular dynamics optimization with the thorough option were applied during model building. All structural analyses and figure preparations were performed with ICM Version 3.7–3b (Molsoft, San Diego, CA) and Ligplot+ Version 1.4.3 (36). The electrostatic potential of the protein surface was calculated with the REBEL method, which solves the Poisson equation with the boundary element method (37). Structures for the mutants were generated by exchanging residues with other amino acids and minimizing energy locally.
RESULTS
The Second Half of GLP1R ECL3 Interacts with GLP-1
We previously exchanged TMH2, ECL1, and ECL2 of GLP1R and GIPR to create chimeric receptors. The peptide ligands for these receptors showed significantly altered potencies and affinities for the receptor chimera (32), demonstrating that residues in TMH2, ECL1, and ECL2 contribute to interactions between GLP-1 and GLP1R. However, exchanging ECL3 of GLP1R with that of GIPR did not affect the potency of GLP-1. This is likely due to a relatively high degree (47%) of sequence similarity between ECL3 of GLP1R and that of GIPR. Indeed, the ECL3 regions of GLP1R and its paralogous receptors GIPR, GLP2R, GCGR, and GCRPR exhibit considerable similarities in amino acid length and sequence (32). In our previous study with GLP1R/GIPR chimeric receptors, we were unable to identify the residues in GLP1R ECL3 that interact with the peptide ligand. In this study we employed VPAC1R, which is phylogenetically closer to the GLP1R subfamily than other class B GPCR subfamilies, such as parathyroid hormone receptor, corticotropin-releasing hormone receptor, and calcitonin receptor subfamilies (13). Similarly, the amino acid sequence and secondary structure of the VIP peptide family are very similar to those of the GLP-1 peptide family (13).
To determine if ECL3 contributes to the interaction between GLP1R and GLP-1, the sequence from the start of ECL3 to the C terminus of GLP1R was replaced with that of VPAC1R to create the G/V[E3-C] receptor (Fig. 1). Cells expressing the G/V[E3-C] receptor were treated with increasing concentrations of GLP-1. This replacement greatly reduced the potency of GLP-1. In contrast, replacement with the GIPR ECL3 to C-terminal regions (GL/I6) (32) did not alter the potency of GLP-1 (Fig. 1). This result suggests that the sequence comprising ECL3 to TMH7 of GLP1R is important for GLP-1 interaction.
FIGURE 1.
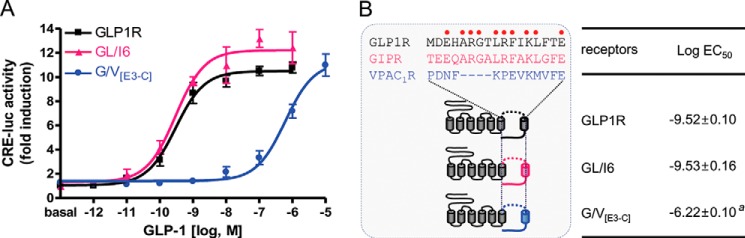
GLP-1 potencies toward GLP1R chimeric receptors. A, the GLP-1 potency of the wild-type and chimeric GLP1Rs with the sequence from ECL3 to C terminus of GIPR (GL/I6) or of VPAC1R (G/V[E3-C]). B, schematic diagram for chimeric receptors and log EC50 values of GLP-1. Amino acid sequences of ECL3 of GLP1R, GIPR, and VPAC1R were aligned above the receptor structures. The conserved residues between GLP1R and GIPR are indicated as red dots. The data on the sigmoidal curves and log EC50 values represent the means ± S.E. of at least three independent experiments with triplicates. a, versus wild-type GLP1R (p < 0.05).
The GLP-1-interacting motifs in ECL3-TMH7 were further narrowed down by generating additional GLP1R/VPAC1R chimeric receptors. Chimeric receptors included the VPAC1R sequence from the second half (b) of ECL3 to the C terminus (G/V[E3b-C]), from TMH7 to the C terminus of VPAC1R (G/V[T7-C]), the entire VPAC1R ECL3 (G/V[E3ab]), or the second half of VPAC1R ECL3 (G/V[E3b]) (Fig. 2). The potency of GLP-1 for each of the chimeric receptors was measured. G/V[T7-C], which retained the second half of GLP1R ECL3, responded to GLP-1 in a similar manner to that of the wild-type GLP1R (Fig. 2). However, chimeric receptors G/V[E3b-C], G/V-E[E3ab], G/V[E3-C], and G/V[E3b], in which the second half of GLP1R ECL3 was absent, responded very poorly to GLP-1 (Fig. 2). These results suggest that the second half of ECL3 mediates the interaction of GLP1R with GLP-1.
FIGURE 2.
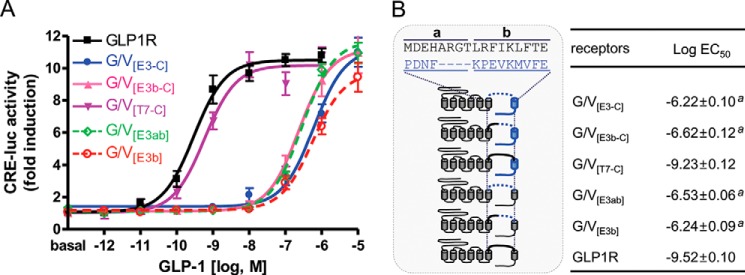
GLP-1-interacting motif in ECL3 and TMH7 of GLP1R. A, sigmoidal curves showing GLP-1 potency toward the wild-type or chimeric GLP1Rs, which have different parts of ECL3 and the C terminus of VPAC1R. B, schematic diagram for chimeric receptors and log EC50 values of GLP-1. The VPAC1R ECL sequence was divided into a and b domains. The data on the sigmoidal curves and log EC50 values are presented as the means ± S.E. of at least three independent experiments. a, versus wild-type GLP1R (p < 0.05).
Leu379, Arg380, and Phe381 in ECL3 of GLP1R Are Involved in GLP-1 Interaction
The amino acid residues Leu379, Arg380, Phe381, and Leu384 in the second half of GLP1R ECL3 are found in the corresponding positions of GIPR but not in VPAC1R, which has Lys, Pro, Glu, and Met in these positions, respectively (Fig. 1B). To investigate whether these amino acid residues were responsible for specific interactions with GLP-1, the residues were substituted for those in G/V[E3b], which had a marginal response to GLP-1. Single residue-substituted mutants, such as K379L, P380R, and E381F in G/V[E3b], increased the GLP-1 potency compared with that of G/V[E3b]. The Arg380 substitution ([P380R]G/V[E3b]) showed the greatest increase in GLP-1 potency out of all of the single residue mutations (Fig. 3). However, the M384L mutant did not increase the potency of GLP-1. Compared with [P380R]G/V[E3b], the additional substitutions, K379L/P380R (KP-LR) and K379L/P380R/E381F (KPE-LRF), further increased the GLP-1 potency by 10- and 100-fold, respectively (Fig. 3). K379L/P380R/E381F exhibited ligand-responsive behavior similar to that of wild-type GLP1R. However, additional mutations of Met to Leu384 did not significantly augment the potency of GLP-1 (Fig. 3). These results suggest that the specific structure formed by basic Arg380 flanked by the two hydrophobic bulky residues, Leu379 and Phe381, is likely critical for direct contact with the N-terminal moiety of the GLP-1 peptide.
FIGURE 3.
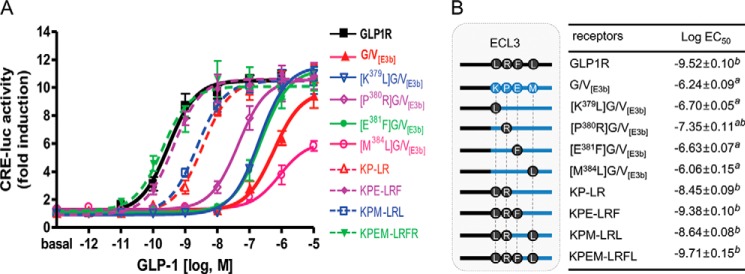
Identification of GLP-1-interacting residues in the second half of GLP1R ECL3. A, the potency of GLP-1 toward chimeric GLP1R, which has the second half of VPAC1R ECL3 (G/V[E3b]), or mutant G/V[E3b], into which residues from GLP1R ECL3 were introduced. B, schematic diagram for ECL3 mutations and log EC50 values of GLP-1. The diagram shows that amino acid residues Leu379, Arg380, Phe381, and/or Leu384 were reintroduced into G/V[E3b]. The data on the sigmoidal curves and EC50 values represent the means ± S.E. of at least three independent experiments. a, versus wild-type GLP1R (p < 0.05); b, versus G/V[E3b] (p < 0.05).
Asp9 of GLP-1 May Interact with Arg380 of GLP1R
Substitutions at each of the residues in GLP-1 with Ala indicates that N-terminal residues His1, Gly4, Thr7, and Asp9 are crucial for either maintaining the secondary structure of the peptide or for interaction with the receptor (30, 31). Our previous observation suggests that His1 and Thr7 of GLP-1 may contact Asn302 in ECL2 and Ile196 in the upper half of THM2 of GLP1R (32). Interestingly, molecular modeling studies based on this biochemical observation suggested that Gly4 and Asp9 in GLP-1 interacted with residues in ECL3 of GLP1R. To address this possibility, we generated mutant GLP1Rs in which the basic Arg380 was changed to acidic Asp (R380D) or neutral Gly (R380G), and hydrophobic Leu379 and Phe381 were modified to either basic (L379R and F381R) or acidic (L379E and F381E) residues (Fig. 4). All of the mutants except for F381R exhibited decreased responsiveness and affinity to GLP-1 (Table 2). In particular, the GLP-1 potency of the R380D mutant receptor was reduced by >1000-fold compared with that of wild-type GLP1R (Fig. 4). In contrast, the basic Arg substitution in F381R GLP1R maintained a high affinity for GLP-1. To identify the amino acid residues in GLP-1 that interact with Arg380 of GLP1R, we generated modified GLP-1 peptides in which Gly4 was replaced with acidic (Asp and Glu) or basic (His, Lys, and Arg) residues and Asp9 was changed to a basic Lys or Arg residue or to an acidic Glu (Table 1). The potencies of the modified peptides were determined for wild-type and R380D mutant GLP1R (Table 2). All of the modified GLP-1 peptides except for [Glu9]GLP-1 exhibited substantially decreased potencies for wild-type GLP1R (Table 2 and Fig. 5A). These data indicate the importance of Gly4 and Asp9 in GLP-1 for receptor binding and activation. It is of interest to note that [Arg4]-, [Lys9]-, and [Arg9]GLP-1 had increased potencies for R380D GLP1R compared with wild-type GLP-1. Among all of the peptides, the potency of [Arg9]GLP-1 on R380D GLP1R was the greatest (Table 2 and Fig. 5B). We have confirmed these results using a pGlosensorTM-22F system that directly examines cAMP production in response to ligand stimulation (14). The results (data not shown) are similar in tendency to that of Fig. 5 such that the mutant peptides showed decreased potency and efficacy toward wild-type receptors while exhibiting higher potency or efficacy toward R380D GLP1R than wild-type peptide. These changes represent reciprocal mutations of charged residues in the ligand and receptor, and the result suggests a possible interaction of Asp9 of GLP-1 with Arg380 of GLP1R. To corroborate this, we performed a ligand-receptor binding assay. Iodinated [Arg9]GLP-1 exhibited significantly lower binding affinity for wild-type GLP1R but relatively high affinity for R380D, which had low affinity for wild-type GLP-1 (Fig. 5, C and D). We also examined the potency and affinity of the mutant GLP-1 peptides for the mutant receptors, L379R, L379E, F381R, and F381E (Table 2). However, none of the mutant peptides exhibited potencies and affinities higher than that of wild-type GLP-1 toward the mutant receptors. These results indicated that Leu379 and Phe381 do not directly interact with the ligand but may be important for maintaining a receptor conformation that favors ligand binding. To determine the expression levels of wild-type and mutant receptors, we performed a binding assay using submaximal concentration of radiolabeled ligands (Table 2). Additionally, Western blot and confocal microscope for GFP-conjugated wild-type and mutant receptors reveals that all receptors seem to be stably expressed in the cells (data not shown).
FIGURE 4.
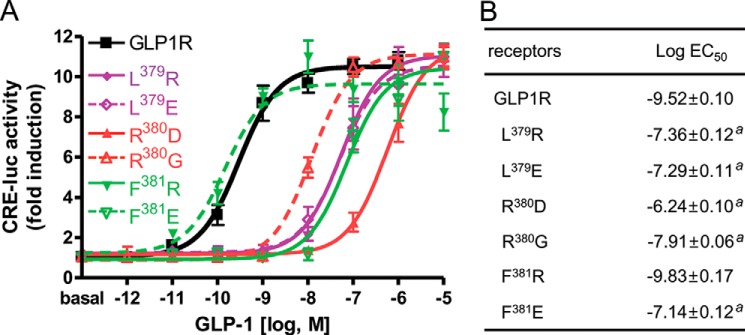
Mutation of GLP-1-interacting residues in the second half of GLP1R ECL3. A, sigmoidal curves of GLP-1-induced receptor activation. Amino acid residues Leu379, Arg380, Phe381, or Leu384 in ECL3 were mutated to either basic or acidic residues. B, the log EC50 values of mutant receptors. The data on the sigmoidal curves and EC50 values are presented as the means ± S.E. of at least three independent experiments. a, versus wild-type GLP1R (p < 0.05).
TABLE 2.
Ligand affinities and potencies of wild-type GLP1R and GLP1R mutants
Expression of receptors is shown as % maximal level of wild-type GLP1R. Binding affinity is presented as log IC50. The potencies of the modified GLP-1 peptides are shown as log EC50. The Emax values of each mutant in response to modified peptides were not significantly different from the Emax value of wild-type GLP1R to GLP-1.
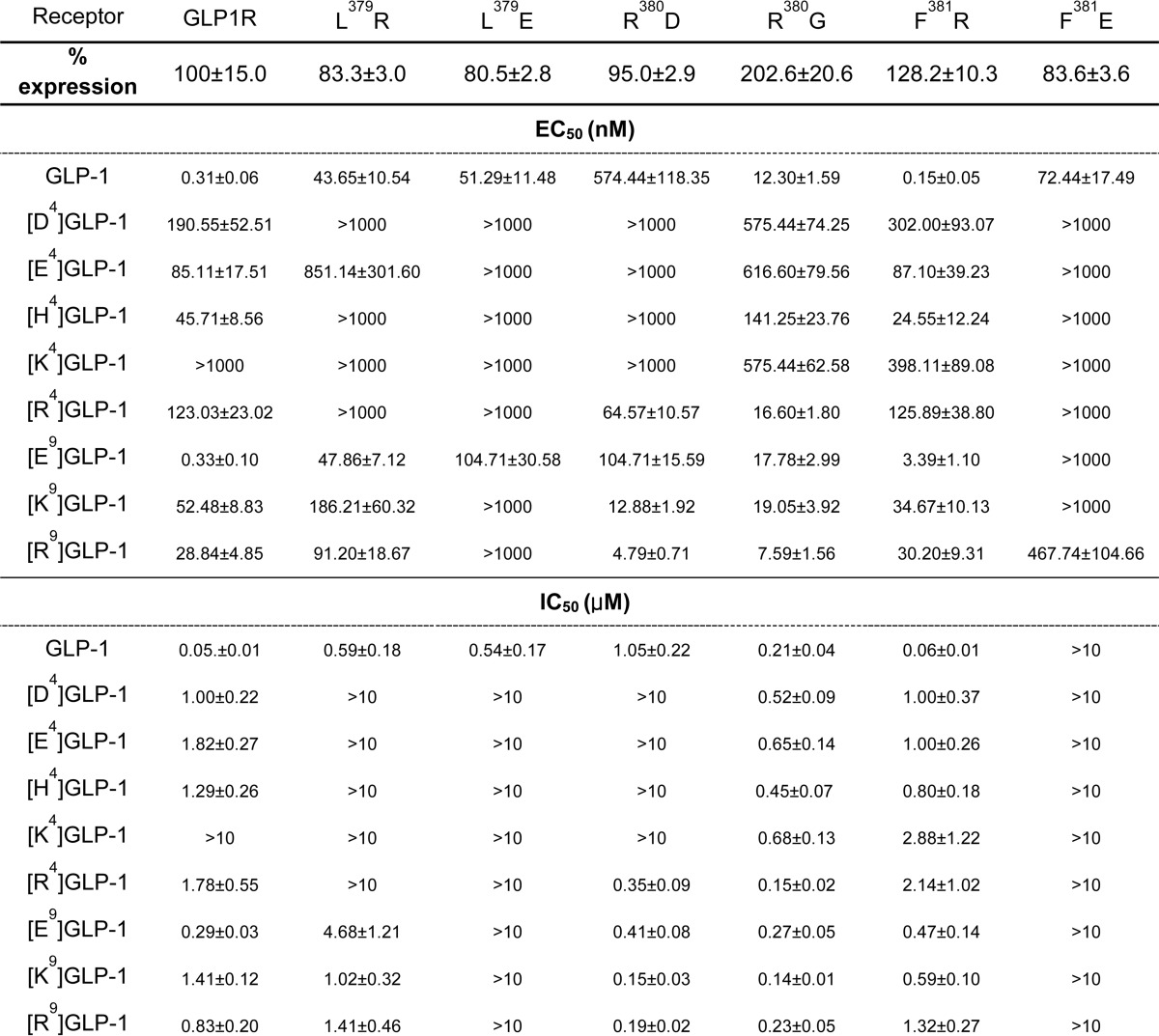
FIGURE 5.

Effect of GLP-1 complementary mutation on the R380D GLP1R mutant. A, ligand potency of mutant GLP-1s in which Gky4 and Asp9 were replaced with basic residues, toward wild-type GLP1R. B, ligand potency of mutant GLP-1 peptides toward the R380D mutant. C, binding analysis of wild-type and [Arg9]GLP-1 peptides to GLP1R. D, ligand binding affinity of GLP-1 complementary mutation to mutant R380D. The results are presented as the means ± S.E. of at least three independent experiments.
Molecular Modeling Shows Direct Interaction between Asp9 of GLP-1 and Arg380 of GLP1R
The GLP-1-GLP1R interaction was examined on a three-dimensional atomic scale using a homology model. The crystal structures of the ligand-bound ECD of human GLP1R and human GIPR served as structural templates of the ECD modeling of GLP1R. The core domain structure was built based on the crystal structures of two class B GPCRs, human GCGR (38) and corticotropin-releasing factor 1 receptor (39). Although these two crystal structures represent inactive structures with antagonists, a lesson from the class A GPCR structures is that there is no significant structural difference in the ligand binding sites between agonist-bound and antagonist-bound structures, whereas there are large conformational changes in the G protein binding regions (40). Sequence alignment between GLP1R and the GPCRs of the crystal structures was done manually with predicted TMHs and evolutionarily conserved residues. Seven distance restraints between GLP-1 and GLP1R were introduced during homology modeling as listed under “Experimental Procedures,” and extensive optimization was performed.
The final model provided interesting explanations for the effects of the GLP1R mutations. Initially, GLP1R Arg380 was located near Asp9 and Gly4 of GLP-1 (Fig. 6A). The interaction between the side chains of GLP1R Arg380 and GLP-1 Asp9 is likely due to an electrostatic attraction. Electrostatic surface charge analyses of wild-type and R380D GLP1R revealed the importance of the ionic charge at position 380 (Fig. 6, B and C). Arg380 is located at the entrance of the binding pocket formed by the ECLs and TMHs and conveys a positively charged electrostatic surface, permitting negatively charged Asp9 or Glu9 in GLP-1 to access this binding pocket for binding. The positive surface charge of the binding pocket may repel Lys9 or Arg9 residues of the mutant GLP-1 peptides. In contrast, mutation of Arg380 in GLP1R to Asp abolishes the positive surface charge of the area and creates a negative surface charge, repelling the acidic Asp9 or Glu9 in GLP-1 from the binding pocket. However, this acidic surface charge may permit [Arg9]GLP-1 or [Lys9]GLP-1 to interact with the mutant receptor (Fig. 5, B and D). Gly4 of GLP-1 is located close to Arg380 of GLP1R so that an introduction of a side chain by mutation of the residue would affect the binding of the ligand to the receptor. For instance, [Arg4]GLP-1 has a very low potency and affinity to the wild-type receptor, but it has a relatively high potency and affinity toward the R380G mutant receptor (Table 2). Thus, it can be postulated that a repulsion between GLP-1 Arg4 and GLP1R Arg380 results in the low affinity of [Arg4]GLP-1 toward the wild-type receptor, whereas the Arg4 mutation in the GLP-1 peptide may provide relatively high affinity to the R380G mutant receptor.
FIGURE 6.

Molecular model showing the interaction between GLP-1 and the core of GLP1R. A homology model of the GLP1R and GLP-1 interaction was built based on the crystal structures of class B GPCR cores and the ECD of human GLP1R and GIPR in complex with GLP-1 and GIP, respectively. The model is consistent with the experimental results in this study. A, the enlarged view shows the residues that are important for interactions between GLP-1 (yellow) and GLP1R (cyan). The carbon, nitrogen, and oxygen atoms of the residues are colored gray, blue, and red, respectively. The orientation of each TMH (TM1 to TM7) is indicated. The electrostatic potentials of GLP1R wild-type (B) and R380D mutant (C) are shown with the binding pockets formed by ECLs and TMHs at the center. The positive and negative electrostatic values are colored blue and red, respectively. The atoms of Asp9 and Arg9 of GLP-1 are shown in a ball-and-chain diagram in B and C, respectively. Residue 380 is located at the entrance of the binding pocket and interacts with Asp9 of GLP-1. The atoms that are located within 6 Å from the side chains of Leu379 (in violet) are shown in a ball-and-chain model to show the extensive interaction network of the residue (D). Leu379 has contacts with many neighboring residues in TMH6, ECL3, and TMH7 such that mutation of Leu379 would result in a structural change that affects the binding of GLP-1. In contrast, the side chain of Phe381 protrudes away from the binding pocket so that mutation of the residue would have little structural change (E).
The hydrophobic residue Leu379 may not directly interact with the peptide ligand but may contribute to receptor conformation. For example, the side chain of Leu379 was within 6 Å of Phe367, Met371, Asp372, and Glu373 of TMH6, His374, Gly375, Gly377, Thr378, Arg380, and Phe381 of ECL3, and Ile382, Leu384, Phe385, and Leu388 of TMH7 (Fig. 6D). Thus, mutation of Leu379 to a bulky Arg may substantially alter receptor conformation and attenuate ligand binding (Fig. 4). Our model, however, suggests that the side chain of Phe381 was orientated away from the binding pocket and interacted with only several residues of ECL3, enduring a substantial change to Arg381 (Fig. 6E). The combined cationic nature of Arg at 380 and 381 is well tolerated but demonstrates preference of Asp9 relative to Glu9 of GLP-1. However, mutation to negatively charged residues at Phe381 would affect the binding of the ligand significantly because the electrostatic attraction between the side chains of the receptor may disorient the side chain of Arg380.
Role of Arg380-corresponding Basic Residues in Related Receptors
The basic Arg380 of GLP1R is well conserved in the equivalent positions of the related paralogous receptors, GCGR (Arg378), GCRPR (Arg379), GLP2R (Lys414), and GIPR (Arg362). Similarly, Gly at position 4 and acidic residue Asp or Glu at position 9 are common for GLP-1 and its related peptides, glucagon, GCRP, GLP-2, and GIP (Fig. 7A). This observation suggests that the GLP-1 peptide and receptor families may share a common mechanism of interaction between acidic Asp/Glu9 and/or Gly4 of the peptide and basic Arg380 in ECL3 of the corresponding receptor. To address this possibility, we generated mutant GCGR, GCRPR, GLP2R, and GIPR, in which the basic Arg (or Lys) in ECL3 was replaced with Asp. Wild-type and mutant receptors were treated with the corresponding peptide ligands or mutant peptides, in which the acidic residue at position 9 was changed to Arg. All of the [Arg9]peptide showed decreased potencies for the corresponding wild-type receptors, GCGR, GCRPR, GIPR, and GLP2R (Fig. 7, B–E). Mutation of Arg378 to Asp in GCGR greatly reduced the potency of wild-type glucagon. Interestingly, the mutant [Arg9]glucagon exhibited higher potency for this mutant receptor compared with wild-type glucagon (Fig. 7B). Likewise, mutation of Arg379 to Asp in GCRPR decreased the potency of wild-type GCRP but increased the potency of [Arg9]GCRP by 10-fold compared with that of wild-type GCRP (Fig. 7C). However, mutation of Lys414 to Asp in GLP2R and Arg362 to Asp in GIPR partially reduced responsiveness to the wild-type peptide ligands. Furthermore, Arg9-exchanged peptide ligands exhibited weaker activities on the mutant receptors compared with that of the wild-type peptide ligands. However, [Arg9]GIP has a higher potency at R362D (EC50 = −9.10) than it does at the wild-type receptor (EC50 = −8.45), although the differences in potency between [Arg9]GIP toward R362D and wild-type GIPR are not statistically significant. Thus, a possible interaction between Asp9 of GIP and Arg362 of GIPR cannot be excluded (Fig. 7, D and E).
DISCUSSION
The interaction between the N terminus of GLP-1 and the core domain of GLP1R is important for ligand-induced receptor activation (41, 42). Thus, many approaches, such as alanine scanning, photoaffinity labeling, and molecular docking, have been attempted to identify specific amino acids that are required for this interaction (28, 43–47). Mutation mapping studies suggest that charged amino acids (Lys197, Asp198, Lys202, Asp215, Arg227, and Lys288) and conserved residues (Met204, Tyr205, and Trp306) are likely to be important for GLP-1-induced receptor activation (24, 28, 43, 44, 48). However, these studies do not explain how individual residues in the peptide interact with residues in the receptor. Furthermore, most of these residues are highly conserved within the class B GPCRs, including VPACR, CRFR, CALCR, and PTHR. This suggests that these residues are more important for maintaining the basic receptor architecture than for conferring selective ligand interaction (45, 47, 49). Experiments with photolabile probes for GLP-1 provide only partial information regarding the spatial approximation of the residues in the peptide and receptor that interact with each other. Phe6 of GLP-1 is in close proximity to Tyr145 of GLP1R according to that study (46). Recent results from another study that used the same method showed discrepancies compared with crystallography experiments. For example, although GLP-1 Ala18 was proposed to be located close to Glu133 of the ECD of GLP1R by photoaffinity labeling (45), the ligand-bound crystal structure of the GLP1R ECD shows a hydrophobic interaction between GLP-1 Ala18 and Leu32 of the ECD (20, 21). Molecular docking studies with biochemical analyses have suggested possible ligand binding pockets in the GLP1R core domain (47, 50, 51). However, these modeling approaches failed to provide a common consensus for the ligand binding pocket. Instead, these approaches differed in their identification of residues that contact the ligand. Thus, stricter biochemical analyses with an appropriate strategy may be helpful for constructing a more reliable ligand-bound receptor model.
Although GLP-1 and its related peptides share a high degree of sequence identity and structural similarity, they generally exhibit specific binding to their own receptors with some cross-reactivity toward paralogous receptors (32, 52). This observation suggests that there are distinct amino acid residues among paralogs of the peptide and receptor that mediate selective interactions with their own partners. Thus, our study is based on the concept of coevolution of the peptide ligand and receptor family. Evolutionary pressure preserves the amino acid residues that are essential for the basic protein structure and for primary interactions between ligand and receptor family members. Specific changes in the amino acid sequence permit selective interaction between a ligand-receptor pair (53–57). In the former case, residues are conserved across paralogous members. However, in latter case, residues are conserved within orthologous members and differ from those of paralogs (33). This hypothesis is supported by recent crystal structures for the ligand-bound ECD of GLP1R and GIPR (18, 20, 21) and by molecular docking models for the ligand-bound ECD of GCGR and GLP2R (58, 59). The hydrophobic face of the peptides is formed by the conserved residues, Phe22, Ile/Val23, Trp25, and Leu26, and points toward the hydrophobic binding cavity of the ECD. The ECD hydrophobic binding cavity is formed by conserved hydrophobic residues, which are localized primarily in the N-terminal α-helix, at the end of the β2 strand, and in the loop between the β3 and β4 strands. These evolutionarily conserved residues may contribute to primary interactions across these peptide and receptor family members (27, 52). In contrast, the residues located at positions 16–20 vary across paralogous peptides but are conserved among orthologs of each paralog (33). These conserved residues may account for the selectivity and strong interaction between each peptide-receptor pair (18, 21).
In the N-terminal portion of GLP-1, His1, Gly4, Thr7, and Asp9 are important for receptor binding and activation (30, 31). His1 and Thr7 are proposed to interact with evolutionarily conserved residues in GLP1R, which are Ile196/Lys197 in TMH2, Met233 in ECL1, and Asn302 in ECL2 (32). The current study suggests that Gly4 and Asp9 are likely to have close contacts with Arg380 flanked by hydrophobic Leu379 and Phe381 in ECL3. According to the model, Arg380 provides the basic surface of the binding pocket, which allows the acidic Asp9 of GLP-1 to enter the pocket. In contrast, mutation of Arg380 to Asp acidifies the binding pocket, which interferes with binding of GLP-1 Asp9 but permits interaction with [Arg9]GLP-1. Leu379 and Phe381 may affect either the receptor conformation or surface charge of the binding pocket to promote ligand binding.
Interestingly, His1, Gly4, Thr7, and Asp9 of GLP-1 are highly conserved across the related peptides, glucagon, GCRP, and GLP-2, although GIP has Tyr1 and Ile7. Thus, similarities in the ligand binding pocket structures of GLP1R, GCGR, GCRPR, and GLP2R may accommodate the conserved residues in the N-terminal moieties of the peptides. Indeed, mutation of Arg378 to Asp in GCGR greatly reduced glucagon potency. This mutation effect was partially compensated by glucagon containing an Arg9 residue. Likewise, mutation of Arg379 to Asp in GCRPR greatly reduced the potency of wild-type GCRP. However, this mutation was tolerant to GCRP that contained an Arg9 residue. Furthermore, GLP-1 and GCRP can cross-interact with their evolutionary related receptors, although the affinity or potency of cross-interaction is lower than that of the natural cognate interaction (14). These observations may support the concept that GLP1R, GCGR, and GCRPR have similar ligand binding pockets formed by evolutionarily conserved residues in the core domain. However, these receptors may also have distinct structures formed by ortholog-specific residues in the core domain that interact with ortholog-specific residues in the corresponding peptide. For example, in addition to the highly common residues Gly4, Phe6, The7, and Asp/Glu9, glucagon-specific residues, Ser2, Gln3, Tyr10, and Lys12, are also important for receptor binding and activation (60, 61). Indeed, a recent glucagon-docked GCGR modeling based on the antagonist-bound GCGR crystal structure revealed putative interactions of these glucagon-specific residues with GCGR (38). However, this model did not show direct interaction between Asp9 of glucagon and Arg379 of GCRPR. This is likely due to the distance restraints between glucagon and GCGR being based on photo-cross-linking studies between GLP-1 and GLP1R (49).
GLP2R and GIPR are expected to have binding pocket structures that are distinct from that of GLP1R. GLP2R has different residues, Val230 and Leu267, at the corresponding positions of GLP1R Ile196 and Met233. In addition, GLP2R has Ala at the equivalent position of Leu379 of GLP1R. According to our modeling data, the hydrophobic bulky side chain of Leu379 contacts TMH6 and TMH7, contributing to the receptor conformation that favors ligand binding. Thus, replacing Leu379 in GLP2R with an Ala, which has a shorter side chain, may affect the structure of the binding pocket. Alanine scanning of GLP-2 showed that the residues at positions 2, 5, 6, and 17 are important for receptor activation (62), indicating that receptor-interacting residues in GLP-2 differ from those of GLP-1. Because GIP contains Tyr1 and Ile7 and GIPR possesses Ser, Thr, and Val at the corresponding positions of Ile196, Met233, and Asn302 in GLP1R, the binding pocket structure of GIPR will be markedly different from that of GLP1R. Thus, mutations at Lys414 in GLP2R and Arg362 in GIPR did not critically affect ligand binding.
In summary, our study suggests that residues Gly4 and Asp9 in the GLP-1 N terminus and Asp380 in ECL3 of GLP1R confer ligand-induced receptor activation. Furthermore, this interaction appears to be conserved in glucagon/GCGR and GCRP/GCRPR pairs. Together with our previous study demonstrating interactions between His1 and Thr7 in GLP-1 and Ile196, Met233, and Asn302 in GLP1R (32, 32), this study sheds light on the mechanism underlying the high affinity interaction between the GLP-1 peptide family and the binding pocket in the receptor that is formed by evolutionarily conserved residues. Identifying the structure of the ligand-binding pocket is important for in silico virtual screening of small molecules that activate GLP1R, which could be further developed by medicinal chemistry in treatments for diabetes and obesity.
This work was supported by grants from Basic Science Research Program (2013R1A1A2010481) of the National Research Foundation of Korea and Korea-South Africa Collaboration Program (2012K1A3A1A09033014) of the National Research Foundation of South Africa.
- GLP-1
- glucagon-like peptide-1
- GLP1R
- GLP-1 receptor
- GLP2R
- GLP-2, receptor
- ECD
- extracellular domain
- ECL
- extracellular loop
- GCGR
- glucagon receptor
- GCRPR
- glucagon-related peptide (GCRP) receptor
- GIP
- glucose-dependent insulinotropic polypeptide
- GIPR
- GIP receptor
- GPCR
- G protein-coupled receptor
- TMH
- transmembrane helix
- VPAC1R
- VPAC1 receptor
- PAC1R
- PAC1 receptor.
REFERENCES
- 1. Mojsov S., Heinrich G., Wilson I. B., Ravazzola M., Orci L., Habener J. F. (1986) Preproglucagon gene expression in pancreas and intestine diversifies at the level of post-translational processing. J. Biol. Chem. 261, 11880–11889 [PubMed] [Google Scholar]
- 2. Fehmann H. C., Göke R., Göke B. (1995) Cell and molecular biology of the incretin hormones glucagon-like peptide-I and glucose-dependent insulin releasing polypeptide. Endocr. Rev. 16, 390–410 [DOI] [PubMed] [Google Scholar]
- 3. Moon M. J., Kim H. Y., Park S., Kim D. K., Cho E. B., Hwang J. I., Seong J. Y. (2011) Insulin contributes to fine-tuning of the pancreatic β-cell response to glucagon-like peptide-1. Mol. Cells 32, 389–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Drucker D. J. (2003) Glucagon-like peptides: regulators of cell proliferation, differentiation, and apoptosis. Mol. Endocrinol. 17, 161–171 [DOI] [PubMed] [Google Scholar]
- 5. List J. F., Habener J. F. (2004) Glucagon-like peptide 1 agonists and the development and growth of pancreatic β-cells. Am. J. Physiol. Endocrinol. Metab. 286, E875–E881 [DOI] [PubMed] [Google Scholar]
- 6. Nauck M. A., Heimesaat M. M., Behle K., Holst J. J., Nauck M. S., Ritzel R., Hüfner M., Schmiegel W. H. (2002) Effects of glucagon-like peptide 1 on counterregulatory hormone responses, cognitive functions, and insulin secretion during hyperinsulinemic, stepped hypoglycemic clamp experiments in healthy volunteers. J. Clin. Endocrinol. Metab. 87, 1239–1246 [DOI] [PubMed] [Google Scholar]
- 7. Zander M., Madsbad S., Madsen J. L., Holst J. J. (2002) Effect of 6-week course of glucagon-like peptide 1 on glycaemic control, insulin sensitivity, and β-cell function in type 2 diabetes: a parallel-group study. Lancet 359, 824–830 [DOI] [PubMed] [Google Scholar]
- 8. Mentlein R., Gallwitz B., Schmidt W. E. (1993) Dipeptidyl-peptidase IV hydrolyses gastric inhibitory polypeptide, glucagon-like peptide-1(7–36)amide, peptide histidine methionine and is responsible for their degradation in human serum. Eur. J. Biochem. 214, 829–835 [DOI] [PubMed] [Google Scholar]
- 9. Kieffer T. J., McIntosh C. H., Pederson R. A. (1995) Degradation of glucose-dependent insulinotropic polypeptide and truncated glucagon-like peptide 1 in vitro and in vivo by dipeptidyl peptidase IV. Endocrinology 136, 3585–3596 [DOI] [PubMed] [Google Scholar]
- 10. Knudsen L. B., Kiel D., Teng M., Behrens C., Bhumralkar D., Kodra J. T., Holst J. J., Jeppesen C. B., Johnson M. D., de Jong J. C., Jorgensen A. S., Kercher T., Kostrowicki J., Madsen P., Olesen P. H., Petersen J. S., Poulsen F., Sidelmann U. G., Sturis J., Truesdale L., May J., Lau J. (2007) Small-molecule agonists for the glucagon-like peptide 1 receptor. Proc. Natl. Acad. Sci. U.S.A. 104, 937–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sloop K. W., Willard F. S., Brenner M. B., Ficorilli J., Valasek K., Showalter A. D., Farb T. B., Cao J. X., Cox A. L., Michael M. D., Gutierrez Sanfeliciano S. M., Tebbe M. J., Coghlan M. J. (2010) Novel small molecule glucagon-like peptide-1 receptor agonist stimulates insulin secretion in rodents and from human islets. Diabetes 59, 3099–3107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang Y., Meng F., Zhong Y., Huang G., Li J. (2012) Discovery of a novel glucagon-like peptide (GCGL) and its receptor (GCGLR) in chickens: evidence for the existence of GCGL and GCGLR genes in nonmammalian vertebrates. Endocrinology 153, 5247–5260 [DOI] [PubMed] [Google Scholar]
- 13. Hwang J. I., Moon M. J., Park S., Kim D. K., Cho E. B., Ha N., Son G. H., Kim K., Vaudry H., Seong J. Y. (2013) Expansion of secretin-like G protein-coupled receptors and their peptide ligands via local duplications before and after two rounds of whole-genome duplication. Mol. Biol. Evol. 30, 1119–1130 [DOI] [PubMed] [Google Scholar]
- 14. Park C. R., Moon M. J., Park S., Kim D. K., Cho E. B., Millar R. P., Hwang J. I., Seong J. Y. (2013) A novel glucagon-related peptide (GCRP) and its receptor GCRPR account for coevolution of their family members in vertebrates. PLoS ONE 8, e65420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hwang J. I., Yun S., Moon M. J., Park C. R., Seong J. Y. (2014) Molecular evolution of GPCRs: GLP1/GLP1 receptors. J. Mol. Endocrinol. 52, T15–T27 [DOI] [PubMed] [Google Scholar]
- 16. Harmar A. J. (2001) Family-B G-protein-coupled receptors. Genome Biol. 2, REVIEWS3013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Oh D. Y., Kim K., Kwon H. B., Seong J. Y. (2006) Cellular and molecular biology of orphan G protein-coupled receptors. Int. Rev. Cytol. 252, 163–218 [DOI] [PubMed] [Google Scholar]
- 18. Parthier C., Kleinschmidt M., Neumann P., Rudolph R., Manhart S., Schlenzig D., Fanghänel J., Rahfeld J. U., Demuth H. U., Stubbs M. T. (2007) Crystal structure of the incretin-bound extracellular domain of a G protein-coupled receptor. Proc. Natl. Acad. Sci. U.S.A. 104, 13942–13947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tan Y. V., Couvineau A., Murail S., Ceraudo E., Neumann J. M., Lacapère J. J., Laburthe M. (2006) Peptide agonist docking in the N-terminal ectodomain of a class II G protein-coupled receptor, the VPAC1 receptor. Photoaffinity, NMR, and molecular modeling. J. Biol. Chem. 281, 12792–12798 [DOI] [PubMed] [Google Scholar]
- 20. Runge S., Thøgersen H., Madsen K., Lau J., Rudolph R. (2008) Crystal structure of the ligand-bound glucagon-like peptide-1 receptor extracellular domain. J. Biol. Chem. 283, 11340–11347 [DOI] [PubMed] [Google Scholar]
- 21. Underwood C. R., Garibay P., Knudsen L. B., Hastrup S., Peters G. H., Rudolph R., Reedtz-Runge S. (2010) Crystal structure of glucagon-like peptide-1 in complex with the extracellular domain of the glucagon-like peptide-1 receptor. J. Biol. Chem. 285, 723–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Neidigh J. W., Fesinmeyer R. M., Prickett K. S., Andersen N. H. (2001) Exendin-4 and glucagon-like-peptide-1: NMR structural comparisons in the solution and micelle-associated states. Biochemistry 40, 13188–13200 [DOI] [PubMed] [Google Scholar]
- 23. Alaña I., Malthouse J. P., O'Harte F. P., Hewage C. M. (2007) The bioactive conformation of glucose-dependent insulinotropic polypeptide by NMR and CD spectroscopy. Proteins 68, 92–99 [DOI] [PubMed] [Google Scholar]
- 24. Al-Sabah S., Donnelly D. (2003) A model for receptor-peptide binding at the glucagon-like peptide-1 (GLP-1) receptor through the analysis of truncated ligands and receptors. Br. J. Pharmacol. 140, 339–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. López de Maturana R., Willshaw A., Kuntzsch A., Rudolph R., Donnelly D. (2003) The isolated N-terminal domain of the glucagon-like peptide-1 (GLP-1) receptor binds exendin peptides with much higher affinity than GLP-1. J. Biol. Chem. 278, 10195–10200 [DOI] [PubMed] [Google Scholar]
- 26. Dong M., Li Z., Zang M., Pinon D. I., Lybrand T. P., Miller L. J. (2003) Spatial approximation between two residues in the mid-region of secretin and the amino terminus of its receptor. Incorporation of seven sets of such constraints into a three-dimensional model of the agonist-bound secretin receptor. J. Biol. Chem. 278, 48300–48312 [DOI] [PubMed] [Google Scholar]
- 27. Runge S., Wulff B. S., Madsen K., Bräuner-Osborne H., Knudsen L. B. (2003a) Different domains of the glucagon and glucagon-like peptide-1 receptors provide the critical determinants of ligand selectivity. Br. J. Pharmacol. 138, 787–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. López de Maturana R., Treece-Birch J., Abidi F., Findlay J. B., Donnelly D. (2004) Met-204 and Tyr-205 are together important for binding GLP-1 receptor agonists but not their N-terminally truncated analogues. Protein Pept Lett. 11, 15–22 [DOI] [PubMed] [Google Scholar]
- 29. Castro M., Nikolaev V. O., Palm D., Lohse M. J., Vilardaga J. P. (2005) Turn-on switch in parathyroid hormone receptor by a two-step parathyroid hormone binding mechanism. Proc. Natl. Acad. Sci. U.S.A. 102, 16084–16089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Adelhorst K., Hedegaard B. B., Knudsen L. B., Kirk O. (1994) Structure-activity studies of glucagon-like peptide-1. J. Biol. Chem. 269, 6275–6278 [PubMed] [Google Scholar]
- 31. Gallwitz B., Witt M., Paetzold G., Morys-Wortmann C., Zimmermann B., Eckart K., Fölsch U. R., Schmidt W. E. (1994) Structure/activity characterization of glucagon-like peptide-1. Eur. J. Biochem. 225, 1151–1156 [DOI] [PubMed] [Google Scholar]
- 32. Moon M. J., Kim H. Y., Park S., Kim D. K., Cho E. B., Park C. R., You D. J., Hwang J. I., Kim K., Choe H., Seong J. Y. (2012) Evolutionarily conserved residues at glucagon-like peptide-1 (GLP-1) receptor core confer ligand-induced receptor activation. J. Biol. Chem. 287, 3873–3884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Moon M. J., Park S., Kim D. K., Cho E. B., Hwang J. I., Vaudry H., Seong J. Y. (2012) Structural and molecular conservation of glucagon-like Peptide-1 and its receptor confers selective ligand-receptor interaction. Front. Endocrinol. (Lausanne) 3, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sali A., Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 35. Krogh A., Larsson B., von Heijne G., Sonnhammer E. L. (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580 [DOI] [PubMed] [Google Scholar]
- 36. Laskowski R. A., Swindells M. B. (2011) LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model 51, 2778–2786 [DOI] [PubMed] [Google Scholar]
- 37. Totrov M., Abagyan R. (2001) Rapid boundary element solvation electrostatics calculations in folding simulations: successful folding of a 23-residue peptide. Biopolymers 60, 124–133 [DOI] [PubMed] [Google Scholar]
- 38. Siu F. Y., He M., de Graaf C., Han G. W., Yang D., Zhang Z., Zhou C., Xu Q., Wacker D., Joseph J. S., Liu W., Lau J., Cherezov V., Katritch V., Wang M. W., Stevens R. C. (2013) Structure of the human glucagon class B G-protein-coupled receptor. Nature 499, 444–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hollenstein K., Kean J., Bortolato A., Cheng R. K., Doré A. S., Jazayeri A., Cooke R. M., Weir M., Marshall F. H. (2013) Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature 499, 438–443 [DOI] [PubMed] [Google Scholar]
- 40. Katritch V., Cherezov V., Stevens R. C. (2012) Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol. Sci. 33, 17–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Thorens B., Porret A., Bühler L., Deng S. P., Morel P., Widmann C. (1993) Cloning and functional expression of the human islet GLP-1 receptor. Demonstration that exendin-4 is an agonist and exendin-(9–39) an antagonist of the receptor. Diabetes 42, 1678–1682 [DOI] [PubMed] [Google Scholar]
- 42. Montrose-Rafizadeh C., Yang H., Rodgers B. D., Beday A., Pritchette L. A., Eng J. (1997) High potency antagonists of the pancreatic glucagon-like peptide-1 receptor. J. Biol. Chem. 272, 21201–21206 [DOI] [PubMed] [Google Scholar]
- 43. Xiao Q., Jeng W., Wheeler M. B. (2000) Characterization of glucagon-like peptide-1 receptor-binding determinants. J. Mol. Endocrinol. 25, 321–335 [DOI] [PubMed] [Google Scholar]
- 44. López de Maturana R., Donnelly D. (2002) The glucagon-like peptide-1 receptor binding site for the N terminus of GLP-1 requires polarity at Asp198 rather than negative charge. FEBS Lett. 530, 244–248 [DOI] [PubMed] [Google Scholar]
- 45. Chen Q., Pinon D. I., Miller L. J., Dong M. (2009) Molecular basis of glucagon-like peptide 1 docking to its intact receptor studied with carboxyl-terminal photolabile probes. J. Biol. Chem. 284, 34135–34144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen Q., Pinon D. I., Miller L. J., Dong M. (2010) Spatial approximations between residues 6 and 12 in the amino-terminal region of glucagon-like peptide 1 and its receptor: a region critical for biological activity. J. Biol. Chem. 285, 24508–24518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lin F., Wang R. (2009) Molecular modeling of the three-dimensional structure of GLP-1R and its interactions with several agonists. J. Mol. Model 15, 53–65 [DOI] [PubMed] [Google Scholar]
- 48. Koole C., Wootten D., Simms J., Miller L. J., Christopoulos A., Sexton P. M. (2012) Second extracellular loop of human glucagon-like peptide-1 receptor (GLP-1R) has a critical role in GLP-1 peptide binding and receptor activation. J. Biol. Chem. 287, 3642–3658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Miller L. J., Chen Q., Lam P. C., Pinon D. I., Sexton P. M., Abagyan R., Dong M. (2011) Refinement of glucagon-like peptide 1 docking to its intact receptor using mid-region photolabile probes and molecular modeling. J. Biol. Chem. 286, 15895–15907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Coopman K., Wallis R., Robb G., Brown A. J., Wilkinson G. F., Timms D., Willars G. B. (2011) Residues within the transmembrane domain of the glucagon-like peptide-1 receptor involved in ligand binding and receptor activation: modelling the ligand-bound receptor. Mol. Endocrinol. 25, 1804–1818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kirkpatrick A., Heo J., Abrol R., Goddard W. A., 3rd. (2012) Predicted structure of agonist-bound glucagon-like peptide 1 receptor, a class B G protein-coupled receptor. Proc. Natl. Acad. Sci. U.S.A. 109, 19988–19993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Moon M. J., Kim H. Y., Kim S. G., Park J., Choi D. S., Hwang J. I., Seong J. Y. (2010) Tyr1 and Ile7 of glucose-dependent insulinotropic polypeptide (GIP) confer differential ligand selectivity toward GIP and glucagon-like peptide-1 receptors. Mol. Cells 30, 149–154 [DOI] [PubMed] [Google Scholar]
- 53. Acharjee S., Do-Rego J. L., Oh D. Y., Oh da Y., Ahn R. S., Choe H., Vaudry H., Kim K., Seong J. Y., Kwon H. B. (2004) Identification of amino acid residues that direct differential ligand selectivity of mammalian and nonmammalian V1a type receptors for arginine vasopressin and vasotocin. Insights into molecular coevolution of V1a type receptors and their ligands. J. Biol. Chem. 279, 54445–54453 [DOI] [PubMed] [Google Scholar]
- 54. Wang C., Yun O., Maiti K., Oh D. Y., Oh da Y., Kim K. K., Chae C. H., Lee C. J., Seong J. Y., Kwon H. B. (2004) Position of Pro and Ser near Glu7.32 in the extracellular loop 3 of mammalian and nonmammalian gonadotropin-releasing hormone (GnRH) receptors is a critical determinant for differential ligand selectivity for mammalian GnRH and chicken GnRH-II. Mol. Endocrinol. 18, 105–116 [DOI] [PubMed] [Google Scholar]
- 55. Li J. H., Choe H., Wang A. F., Maiti K., Wang C., Salam A., Chun S. Y., Lee W. K., Kim K., Kwon H. B., Seong J. Y. (2005) Extracellular loop 3 (EL3) and EL3-proximal transmembrane helix 7 of the mammalian type I and type II gonadotropin-releasing hormone (GnRH) receptors determine differential ligand selectivity to GnRH-I and GnRH-II. Mol. Pharmacol. 67, 1099–1110 [DOI] [PubMed] [Google Scholar]
- 56. Cho H. J., Acharjee S., Moon M. J., Oh D. Y., Vaudry H., Kwon H. B., Seong J. Y. (2007) Molecular evolution of neuropeptide receptors with regard to maintaining high affinity to their authentic ligands. Gen. Comp. Endocrinol. 153, 98–107 [DOI] [PubMed] [Google Scholar]
- 57. Lee Y. R., Tsunekawa K., Moon M. J., Um H. N., Hwang J. I., Osugi T., Otaki N., Sunakawa Y., Kim K., Vaudry H., Kwon H. B., Seong J. Y., Tsutsui K. (2009) Molecular evolution of multiple forms of kisspeptins and GPR54 receptors in vertebrates. Endocrinology 150, 2837–2846 [DOI] [PubMed] [Google Scholar]
- 58. Venneti K. C., Hewage C. M. (2011) Conformational and molecular interaction studies of glucagon-like peptide-2 with its N-terminal extracellular receptor domain. FEBS Lett. 585, 346–352 [DOI] [PubMed] [Google Scholar]
- 59. Koth C. M., Murray J. M., Mukund S., Madjidi A., Minn A., Clarke H. J., Wong T., Chiang V., Luis E., Estevez A., Rondon J., Zhang Y., Hötzel I., Allan B. B. (2012) Molecular basis for negative regulation of the glucagon receptor. Proc. Natl. Acad. Sci. U.S.A. 109, 14393–14398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Perret J., Van Craenenbroeck M., Langer I., Vertongen P., Gregoire F., Robberecht P., Waelbroeck M. (2002) Mutational analysis of the glucagon receptor: similarities with the vasoactive intestinal peptide (VIP)/pituitary adenylate cyclase-activating peptide (PACAP)/secretin receptors for recognition of the ligand's third residue. Biochem. J. 362, 389–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Runge S., Gram C., Brauner-Osborne H., Madsen K., Knudsen L. B., Wulff B. S. (2003) Three distinct epitopes on the extracellular face of the glucagon receptor determine specificity for the glucagon amino terminus. J. Biol. Chem. 278, 28005–28010 [DOI] [PubMed] [Google Scholar]
- 62. DaCambra M. P., Yusta B., Sumner-Smith M., Crivici A., Drucker D. J., Brubaker P. L. (2000) Structural determinants for activity of glucagon-like peptide-2. Biochemistry 39, 8888–8894 [DOI] [PubMed] [Google Scholar]