

Background: Netrin-1 can either attract or repel neuronal growth cones.
Results: The ezrin/radixin/moesin proteins mediate interaction between protein kinase A (PKA) and deleted in colorectal cancer (DCC, a netrin receptor), which controls growth cone tropism.
Conclusion: Localized PKA signaling governs axon guidance behavior toward netrin.
Significance: This interaction provides insight into the signals governing axon pathfinding, neural development, and potentially other DCC-related functions.
Keywords: A Kinase Anchoring Protein (AKAP), Cell Signaling, Ezrin, Neurite Outgrowth, Protein Kinase A (PKA), Axon Guidance, Deleted in Colorectal Cancer (DCC), Netrin
Abstract
Netrin-1, acting through its principal receptor DCC (deleted in colorectal cancer), serves as an axon guidance cue during neural development and also contributes to vascular morphogenesis, epithelial migration, and the pathogenesis of some tumors. Several lines of evidence suggest that netrin-DCC signaling can regulate and be regulated by the cAMP-dependent protein kinase, PKA, although the molecular details of this relationship are poorly understood. Specificity in PKA signaling is often achieved through differential subcellular localization of the enzyme by interaction with protein kinase A anchoring proteins (AKAPs). Here, we show that AKAP function is required for DCC-mediated activation of PKA and phosphorylation of cytoskeletal regulatory proteins of the Mena/VASP (vasodilator-stimulated phosphoprotein) family. Moreover, we show that DCC and PKA physically interact and that this association is mediated by the ezrin-radixin-moesin (ERM) family of plasma membrane-actin cytoskeleton cross-linking proteins. Silencing of ERM protein expression inhibits DCC-PKA interaction, DCC-mediated PKA activation, and phosphorylation of Mena/VASP proteins as well as growth cone morphology and neurite outgrowth. Finally, although expression of wild-type radixin partially rescued growth cone morphology and tropism toward netrin in ERM-knockdown cells, expression of an AKAP-deficient mutant of radixin did not fully rescue growth cone morphology and switched netrin tropism from attraction to repulsion. These data support a model in which ERM-mediated anchoring of PKA activity to DCC is required for proper netrin/DCC-mediated signaling.
Introduction
Successful development of central and peripheral nervous systems requires axons to extend from their parent neurons and navigate through a complex extracellular environment to establish contact with their correct synaptic targets. The vanguard structure in this navigation is the growth cone, a specialized motile structure responsible for sampling, interpreting, and responding to extracellular cues in order to maintain its proper course (1–3). Over its course, the growth cone is beset on all sides by a complex mixture of soluble and insoluble factors that provide attractive and repulsive cues that guide the axon to and from appropriate environs (2–5). The major mechanism through which guidance cues exert their effects is through modulation of actin cytoskeletal dynamics (6–8). Specifically, attractant cues promote assembly and/or stabilization of F-actin within the portion of the growth cone closest to the source of attractant, whereas repulsive cues disassemble/destabilize proximal F-actin structures. Although the importance of actin cytoskeletal regulation during growth cone guidance is firmly established, the signal transduction pathways utilized by guidance cues to effect cytoskeletal regulation are still under intense investigation.
The netrin family of axon guidance cues have an evolutionarily conserved role in neural development (9, 10) and can variably regulate both chemoattraction and chemorepulsion of axons, axonal and dendritic branching, and whole-cell neuronal migration (9–11). Importantly, netrin functions extend well beyond neural development and axon guidance and have been directly implicated as regulators of vascular development, epithelial cell migration, and the pathogenesis of a growing variety of tumors (12–15). Two families of receptors are primarily responsible for mediating netrin signaling: the DCC2 (deleted in colorectal cancer) and UNC-5 proteins (9, 10, 12). The DCC family is required for both chemoattraction and chemorepulsion, whereas UNC-5 proteins act only in repulsion (9).
The cAMP-dependent protein kinase, PKA, has been shown to be both a regulator of and an effector for netrin/DCC signaling (2, 9, 10). However, the precise nature of the relationship between PKA and netrin/DCC signaling remains controversial. The ability of netrin to increase cAMP and activate PKA has been both inferred (16–18) and directly shown (19–21). However, other reports suggest that netrin neither increases cAMP nor activates PKA downstream of DCC (22, 23). The effect of PKA on actual axon behavior in response to netrin is similarly well established but controversial. For example, inhibition of PKA activity or modulation of the cAMP/cGMP ratio can convert the tropism of Xenopus spinal neurons and retinal axons from attraction to netrin into repulsion (16–18, 24). In contrast, in rat spinal commissural neurons, PKA does not appear to alter tropism but, rather, alters sensitivity to netrin gradients (22), possibly by increasing the amount of DCC at the cell surface (25). Finally, in rat dorsal root ganglion neurons, there appears to be a developmental switch wherein cAMP activates PKA to effect repulsion from netrin in adult neurons, whereas in embryonic neurons cAMP couples to a different effector-Epac (exchange protein activated by cAMP) to mediate attraction to netrin (21). Clearly, more work is needed to clarify the functional connections between PKA and netrin/DCC signaling.
Specificity in PKA signaling is achieved in large part through interaction with protein kinase A anchoring proteins (AKAPs), which localize PKA to various subcellular regions or structures and thereby couple a given stimulus to phosphorylation of a specific subset of local, relevant targets (26). We and others have shown that AKAP-mediated anchoring and localization of PKA to the leading edge of cells plays an important role in chemotaxis (27–31). In addition to localization, many AKAPs direct the assembly of multienzyme complexes that serve as preassembled circuits capable of integrating multiple, diverse input signals to control the phosphorylation of a given target(s) with exquisite specificity (26). Of note, many of the established effectors involved in netrin/DCC signaling are known to be regulated directly or indirectly by PKA (9, 10, 32, 33). The summed evidence connecting PKA functionally to netrin/DCC signaling along with the importance of anchoring in specifying PKA function prompted us to investigate whether this connection might involve AKAP-mediated anchoring of PKA to DCC.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
Rat monoclonal antibodies specific for ezrin, radixin, and moesin ((M11, R21, and M22; (34)) were obtained as hybridoma supernates from S. Tsukita (Osaka University) and used either undiluted or at a 1:5 dilution (determined empirically based on sample and application). Antibodies against DCC (AF5), the RIβ, RIIβ, RI, and RII subunits of PKA, AKAP79, Mena, and GFP were from BD Biosciences. Antibodies against ezrin, GST, and tubulin were from Sigma. Anti-ERM, -pThr567-Ezrin (which also recognizes the analogous modification on radixin and moesin), -VASP, -Ser(P)-157-VASP, and -phospho-PKA substrate were from Cell Signaling. Goat polyclonal anti-ERM as well as anti-PKA RIIα and polyclonal anti-DCC were from Santa Cruz Biotechnology. Anti-β1 integrin was a gift from Dr. M. Payet (University of Sherbrooke). Non-immune rabbit and mouse IgGs were from Jackson ImmunoResearch. Function-blocking anti-netrin1 antibodies were from R & D Systems or gifted from N. Lamarche-Vane and T. Kennedy (McGill University, Quebec CA). Horseradish peroxidase-conjugated secondary antibodies were from Calbiochem, whereas Alexa-fluor conjugated secondary antibodies and phalloidin were from Molecular Probes.
StHt31 was from Promega. The PKA inhibitor mixture (35) contained 200 μm Rp-cAMPs (Biolog), 1 μm mPKI (BIOSOURCE), 1 μm H89 (Calbiochem), and 1 μm KT5720 (Calbiochem). Purified, recombinant netrin-1 was from R&D Systems. Forskolin and most other ancillary chemicals were from Sigma. Culture media were from BD Biosciences and Invitrogen.
Cell Culture
The cell lines NG108-15 (rat neuroblastoma x mouse glioma hybrid), IMR-32 (human neuroblastoma), and HEK293 (human embryonic kidney epithelia) were from ATCC and routinely cultured as described by the supplier. Neuronal differentiation and neuritogenesis was induced by plating cells onto polylysine-coated surfaces in medium containing 2.5% fetal bovine serum (for NG108-15 cells) or serum-free medium containing 5 ng/ml netrin-1,5 ng/ml NGF, or 2 μm all-trans retinoic acid (for IMR-32 cells) and incubating for 16–24 h at 37 °C. Of note, we found that neuritogenesis in IMR-32 cells was far more efficient when cultured in reduced serum medium and plated on polylysine than when cultured in full serum or in reduced serum but on untreated surfaces.3 It should also be noted that although significant differentiation and neurite outgrowth in IMR-32 cells was observed within 24 h, the survival of these cells was severely compromised by prolonged culture in serum-free medium. Culture in reduced (2.5%) serum media supported longer survival but slower neuritogenesis.3
Primary hippocampal neurons were prepared from E18 rat tissue (Brain Bits, LLC) as follows. Hippocampi were allowed to settle to the bottom of their tube for ∼1 h at room temperature before replacing the supernatant medium with 250 of μl 0.25% trypsin per hippocampus and incubation for 15 min at 37 °C. Digestion was stopped with an equal volume of DMEM plus 10% FBS and gentle inversion to mix. After centrifugation at 80 × g, the supernatant was carefully aspirated and replaced with fresh DMEM, 10% FBS (0.5 ml per hippocampus), and the tissue was dissociated by triturating 20–30× through a barely fire-polished Pasteur pipette. After confirming cell viability by trypan blue exclusion, cells were seeded in DMEM, 10% FBS onto prepared coverslips or imaging dishes and incubated in a humidified 37 °C incubator with 5% CO2 for 2–4 h. Medium was replaced with Neurobasal medium containing 1× B27 supplements and incubated until reaching the desired morphological stage, typically late stage 2 to early stage 3 (12–24 h after plating).
Coverslips for immunofluorescence were cleaned by incubation in warm (∼75 °C) nitric acid overnight followed by extensive water washes, sonication for 30 min in absolute ethanol, and air drying in a sterile tissue culture dish. Cleaned coverslips and culture dishes for differentiated cultures were coated with polylysine (Sigma; either d or l; 30,000–70,000 Mr; 1 mg/ml in PBS) overnight at room temperature and washed with water before neuronal plating. In some cases polylysine-coated coverslips were subsequently coated with 5 μg/ml laminin (BD Transduction Laboratories) for an additional 24 h before washing and cell seeding.
Quantification of Filopodia, Neurites, and Neurite Length
The formation of filopodia in NG108-15 cells and the number and length of IMR-32 cell neurites were quantified from thresholded images by manual tracing using the NeuronJ plugin (36) for ImageJ (rsbweb.nih.gov).
Plasmids, RNAi Reagents, and Transfection
The plasmid expressing full-length rat DCC under control of the CMV promoter (pRK5-DCC) and its empty vector control (pRK5) were obtained from N. Lamarche-Vane (McGill University). Plasmids expressing shRNAs against human ezrin, radixin, and moesin (all in the pBS/U6 background) were obtained from Addgene. Vectors containing cDNAs for murine ezrin, moesin, and radixin (wild-type as well as the L421P and T564D mutants of radixin) were kind gifts from D. Barber (University of California San Francisco), S. Tsukita, G. Fenteany (University of Connecticut), and D. Altschuler (University of Pittsburgh). Of note, the sequences of all three murine ezrin/radixin/moesin (ERM) isoforms are naturally resistant to the human-specific shRNA plasmids described above and thus were cloned into pcDNA-based vectors and used in rescue experiments without further sequence modifications. Cells were transfected using FuGENE 6 (Roche Applied Science; 0.5–0.7 μg of DNA/35-mm dish and a lipid:DNA ratio of 4:1) and used 72–96 h after transfection of shRNA. The average extent of silencing was calculated using densitometric analysis of immunoblots, as the difference between the average intensity of the target in the two off-target control samples (e.g. ezrin in the sh-radixin and -moesin samples) and the intensity of the target in the target sample (e.g. ezrin in the sh-ezrin sample) divided by the average control intensity. For rescue experiments, 24 h after transfection with shRNA plasmids, cells were again transfected with a plasmid encoding mCherry along with empty pcDNA or vectors encoding murine ERM proteins. Efficiencies were typically >90% for initial shRNA plasmid transfection and ∼10–20% for secondary, rescue plasmid transfections. For re-plating, transfected cells were removed from their culture dish by gentle dissociation in PBS + 1 mm EDTA, diluted in their appropriate plating medium, collected by centrifugation, resuspended in plating medium, and seeded onto new polylysine-coated culture vessels for the indicated times. In our hands there were no discernable differences in the “kinetics” of neuritogenesis (assessed as the average time for cells to reach a given morphological stage) of freshly plated versus re-plated cells.
Western Blotting, Immunoprecipitations, and cAMP-agarose Pulldown Assays
Whole cell extracts for Western blotting were prepared by lysing cells in either 1× Laemmli sample buffer (4% SDS, 60 mm Tris, pH 6.8, 0.002% bromphenol blue) or ice-cold RIPA buffer (0.1% SDS, 0.5% Na deoxycholate, 1% Igepal CA-630, 50 mm Tris pH 7.8, 150 mm NaCl) containing protease and phosphatase inhibitors (5 μg/ml leupeptin, 10 mm 4-(2-aminoethyl)benzenesulfonyl fluoride, 5 μg/ml pepstatin, 10 μg/ml aprotinin, 1 mm sodium orthovanadate, and 50 mm NaF). Whole cell lysates were further processed by ice-bath sonication (Branson 2510) for 15 min followed by centrifugation at 16,873 × g for 15 min at 4 °C. Supernatants were collected, made to 1× Laemmli sample buffer (from a 2× or 5× stock) if needed, boiled for 5 min, and either stored at −20 °C or used immediately for SDS-PAGE. Samples were run on 10% SDS-PAGE gels for routine analysis and on gels containing 7.5% acrylamide, 0.25% methylene bisacrylamide for resolution of individual ERM family members.
Fractionation of cells into cytosolic, Triton-soluble, and Triton-insoluble fractions was done using a modification of a previously described method (38). Briefly, cells were detached with warm 1 mm EDTA in PBS, collected by centrifugation (500 × g for 5 min at room temperature), resuspended in ice-cold CSK buffer (10 mm PIPES, pH 6.8, 50 mm NaCl, 300 mm sucrose, 2 mm MgCl2, 1 mm MnCl2, and protease inhibitors; 1 ml CSK buffer/5 × 106 cells), and lysed by 20 strokes in a Dounce homogenizer on ice. Nuclei and intact cells were pelleted by centrifugation at 75 × g for 5 min at 4 °C. Cleared lysates were centrifuged at 30,000 × g for 30 min at 4 °C; the supernatant was designated the “cytosolic” fraction. The pellet was washed once with CSK buffer, resuspended in Triton lysis buffer (CSK buffer containing 1% Triton X-100, protease, and phosphatase inhibitors plus 0.25 μm calyculin A; 0.5 ml/5 × 106 cells), and incubated on ice for 15 min before centrifugation as described above. The supernatant was collected and designated the Triton-soluble fraction. The remaining pellet was resuspended by vigorous vortexing in RIPA buffer (supplemented with 0.25 μm calyculin A; 0.1 ml/5 × 106 cells), the lysate was clarified by centrifugation (which most often produced no visible pellet), and the supernatant was designated the Triton-insoluble fraction. Consistent with previous reports (39, 40), a significant portion of our target proteins (ezrin, PKA RII, and DCC) was found in the Triton X-100-insoluble fraction (data not shown). Therefore, this fraction was routinely used as the input for all co-immunoprecipitations and for cAMP-agarose pulldown assays. To ease in preparation of this fraction, the protocol described above was modified as follows. Cells were lysed directly in Triton lysis buffer, lysates were cleared of nuclei by low speed centrifugation, post-nuclear supernates were spun at 30,000 × g for 30 min at 4 °C, and the remaining pellets were dissolved in RIPA buffer and used for subsequent procedures. Protein concentration in all lysates (except for those lysed directly in Laemmli sample buffer) was determined using the bicinchonic acid assay (Pierce).
Immunoprecipitations were performed by preincubating target antibodies (1–5 μg/5 × 106 cells) with 25 μl of protein A/G-agarose beads (Santa Cruz Biotechnology) in RIPA buffer for 2 h at 4 °C. The beads were washed twice with RIPA buffer, added to lysates, and rocked end-over-end for 30–60 min. The pre-formation of high avidity, antibody-bound beads, and short incubation times were critical for successful isolation of the target protein-protein complexes. A similar approach was used for pulldown assays with cAMP-agarose (Sigma), in which 2–3 mg of soluble protein was incubated with 100 μl of the resin for 30 min. In some experiments, cAMP (25 μm final concentration) was added the lysates before the addition of the cAMP-agarose to act as a competitive inhibitor and control for nonspecific binding. For immunoprecipitations and pulldown assays, bead-bound proteins were eluted by boiling in Laemmli sample buffer, then stored at −20 °C or analyzed immediately by SDS-PAGE and immunoblotting.
PKA Assays
For assay of PKA activity in cell lysates, a previously-described method (41, 42) was used. Briefly, cell monolayers were rapidly washed twice with ice-cold PBS, covered in PKA assay buffer (25 mm β-glycerophosphate, pH 7.4, 1.25 mm EGTA, 10 mm MgCl2, 0.5 mm DTT), then snap-frozen on a pool of liquid nitrogen. Lysates were thawed on ice, scraped into tubes, sonicated, and clarified by centrifugation at 10,000 × g for 10 min at 4 °C. Supernatants were assayed for PKA activity by measuring transfer of radioactive phosphate from ATP into Kemptide (Sigma) in the presence or absence of purified PKI (Sigma). For assay of PKA activity in immunoprecipitates, the washed immunocomplexes were washed an additional time with 1× PKA assay buffer, then incubated with shaking for 30 min at 30 °C in PKA assay buffer containing 0.2 mg/ml of a purified, recombinant PKA substrate protein (GFP-R4S (43)) with or without 25 μm cAMP. Reactions were stopped by the addition of EDTA to a final concentration of 5 mm and centrifuged briefly, and supernatants were made 1× in Laemmli sample buffer before separation by SDS-PAGE and analysis by immunoblotting with phospho-PKA substrate antibody.
In Vitro RII-DCC Binding Assays
The C-terminal domain (amino acids 1122–1445) of rat DCC was subcloned from pRK5-DCC into pGEX6P3 using standard cloning techniques. pGEX2T-RdxL421P/T564D was made by replacing the BglII-HindIII fragment of pGEX2T-RdxT564D (G. Fenteany) with the same same fragment from pcDNA3-RdxL421P (D. Altschuler). GST and GST fusion proteins were expressed in DH5α Escherichia coli after induction with 0.4 mm isopropyl 1-thio-β-d-galactopyranoside for 12 h at 25 °C. Bacterial lysates were prepared using B-PER II (Pierce), and target proteins were collected by incubation of cleared lysates with glutathione-agarose beads (Sigma). Beads were washed extensively with lysis buffer and 1× PBS containing 0.25% Triton X-100 then either used as an affinity matrix for pulldown assays (GST and GST-DCC-cyto) or incubated overnight at room temperature with purified thrombin (GE Healthcare) to cleave the protein of interest (i.e. radixin (Rdx)) from the bead-bound GST. Thrombin was subsequently removed by incubation with p-aminobenzamidine-agarose (Sigma), and the remaining soluble Rdx proteins were used in pulldown assays. Recombinant PKA RIIα subunit, expressed from pET28b-RIIα (a gift from J. Scott (Vollum Institute)) and purified by cAMP-agarose chromatography, was mixed with RdxWT, RdxT564D, or RdxL421P/T564D and GST- or GST-DCC-CT-bound beads at a 5:1 protein weight ratio in PBS, 0.25% Triton X-100 for 30 min at room temperature with end-over-end mixing. A sample of each binding reaction was removed, mixed with an equal volume of 2× Laemmli sample buffer, and used as “input,” whereas the remainder of the reactions was washed extensively with PBS, 0.25% Triton X-100 before boiling in 1× Laemmli sample buffer. Input and bead-bound proteins were analyzed by SDS-PAGE and immunoblotting.
Netrin Microgradient Assays
IMR-32 cells were plated sparsely (1–2 × 103 cells per cm2) on polylysine-coated imaging dishes (ΔT4; Bioptechs) and differentiated in situ by incubation for 12–18 h in media containing 5 μm all-trans retinoic acid. Microscopic netrin-1 gradients were established using minor modifications to the method developed by Poo and co-workers (17, 44). Briefly, a stable gradient was created and maintained using a FemtoJet microinjection pump (Eppendorf) to apply constant pressure (150–200 hectopascals; ∼2–3 p.s.i.) to a glass micropipette, tipped with a polished 1-μm opening, and filled with netrin-1 (5 μg/ml in PBS). Using a Leica mechanical micromanipulator, the micropipette was positioned 100 μm from the center of a prospective growth cone at an angle of ∼45° relative to the axon shaft. Separate experiments using Texas Red-conjugated dextran as a gradient tracer confirmed that this configuration resulted in delivery of an effective concentration of ∼5 ng/ml netrin-1 to the growth cone. Phase contrast images were captured at 0 and 30 min after exposure to the gradient, and changes in growth cone morphology were assessed using a custom Fiji/ImageJ macro to generate and quantify protrusion-retraction analysis maps (code available upon request).
RESULTS
Expression of DCC Promotes PKA-dependent Filopodia Formation and AKAP-dependent Activation of PKA in NG108-15 Cells
To begin to determine whether netrin/DCC signaling could promote activation of PKA, we employed NG108-15 cells, a neuroblastoma x glioma hybrid neuronal cell line that expresses significant amounts of netrin-1 but no detectable DCC and has previously been used for functional DCC re-expression studies (45). Expression of DCC in these cells had significant morphological consequences, as transfected cells exhibited a striking increase in the number of filopodia at early times after re-plating onto polylysine (Fig. 1). This morphological effect was dependent on PKA activity, as the induction of filopodia in DCC-expressing cells was significantly inhibited by treatment with a PKA inhibitor mixture (PKAic). These data are consistent with earlier reports of DCC-dependent filopodia formation and activation of Rac and Cdc42 in NG108-15 cells (45) and DCC- and PKA-dependent induction of filopodia in netrin-stimulated NG108-15 cells and hippocampal neurons (35).
FIGURE 1.

Expression of DCC promotes filopodia formation and promotes activation of PKA and AKAP-dependent phosphorylation of Mena/VASP in NG108-15 neuroblastoma x glioma cells. A, NG108-15 cells were co-transfected with a plasmid encoding EGFP and either an empty plasmid (pRK5) or a plasmid encoding full-length rat DCC (pRK5-DCC) then replated onto polylysine-coated coverslips for 1 h in the absence or presence of a PKA inhibitor mixture (PKAic; see “Experimental Procedures”). Cells were fixed and stained with phalloidin and GFP-positive cells were visualized. B, NG108-15 cells were transfected as in A then replated in the absence or presence of the DCC-blocking AF5 antibody or PKA inhibitors (PKAic) onto polylysine-coated coverslips for 2 h. Cells were fixed and stained with phalloidin, and the number of filopodia per cell (n > 20 cells per condition) was measured. The bars represent the means and S.E from three different transfections. C, NG108-15 cells were transfected with empty pRK5 or with pRK5-DCC. Twenty-four hours later cells were replated onto polylysine/laminin-coated coverslips for 24 h in the absence (−) or presence of a function-blocking DCC antibody (anti-DCC; AF5). Whole cell extracts were analyzed by SDS-PAGE and immunoblotting using a phospho-PKA substrate antibody. D, NG108-15 cells were transfected with pRK5 (Ctrl) or pRK5-DCC and replated as above. For one sample, DCC-expressing cells plated in the absence of AF5 antibody were treated with PKA inhibitors (PKAic) for 30 min. PKA activity was assessed in an in vitro kinase assay. Data represent the means ± S.D. for three separate transfections. E and F, NG108-15 cells were transfected and cultured as in A. Where indicated, cells were replated in the presence of a DCC function-blocking antibody (AF5) or were treated for 30 min with PKA inhibitors (PKAic) or 50 μm concentrations of a cell-permeable peptide inhibitor of PKA anchoring (StHt31). Whole cell extracts were analyzed by immunoblotting with antibodies against total VASP or against the phosphorylated form of Ser-157 (pS157). The slower-migrating species in the total VASP blot corresponds to the Ser(P)-157 form. Note also that the Ser(P)-157 antibody cross-reacts with Mena at ∼95 kDa (41).
The PKA dependence of DCC-induced filopodia formation suggested that PKA was indeed activated by the expression of DCC. To test this we first examined PKA activity in control- and DCC-expressing cells by immunoblotting cell extracts with a phospho-PKA substrate antibody, raised against peptides comprising the phosphorylated form of the PKA consensus site. Expression of DCC increased PKA activity, as assessed by increased signal intensity and complexity in phospho-PKA substrate antibody reactivity in extracts from DCC-expressing cells (Fig. 1C). Inclusion of a DCC function-blocking antibody (AF5) blocked PKA activation, underscoring the receptor specificity of the effect. DCC-dependent activation of PKA was also evidenced by direct measurement of PKA activity using a specific in vitro kinase assay (Fig. 1D). To determine if this DCC-mediated activation of PKA could affect specific substrates that might contribute to the morphological responses observed, we examined the phosphorylation state of Mena/VASP (mammalian enabled/vasodilator-stimulated phosphoprotein) proteins. Mena/VASP family members are important cytoskeletal regulatory proteins that have been implicated in netrin/DCC-dependent filopodia formation (35) and are bona fide PKA substrates (32, 35, 46). PKA preferentially phosphorylates VASP at Ser-157 (and the analogous position in Mena), a modification that can be assessed in immunoblots by electrophoretic mobility shift and by the use of phospho-site-specific antibodies (e.g. (41)). Using both approaches, expression of DCC promoted phosphorylation of both VASP and Mena at their consensus PKA sites (Fig. 1, C and D). This modification was indeed mediated by PKA, as DCC-induced phosphorylation was completely inhibited by treatment of DCC-expressing cells with the PKAic. Interestingly, VASP phosphorylation was also almost completely blocked by treatment with StHt31 (Fig. 1F), a cell-permeable inhibitor of PKA-AKAP interaction, demonstrating that both PKA activity and AKAP-mediated PKA anchoring are required for DCC-driven VASP phosphorylation.
DCC Immunoprecipitates Contain PKA Activity
The requirement for AKAP function for DCC-dependent activation of PKA led us to investigate whether PKA might be physically associated with the receptor itself. To begin to test this hypothesis, we tested for the presence of PKA activity in DCC immunoprecipitates from lysates of embryonic rat brain. The addition of cAMP to DCC-associated proteins led to an increase in the phosphorylation of an optimized PKA substrate (Fig. 2, A and B). This phosphorylation was comparable to the level seen upon the addition of cAMP to immunoprecipitates of a known PKA anchoring protein, AKAP79 (Fig. 2A). Inclusion of the specific peptide inhibitor PKI in the reactions eliminated the cAMP-induced phosphorylation, confirming the DCC-associated kinase activity as PKA (Fig. 2B).
FIGURE 2.
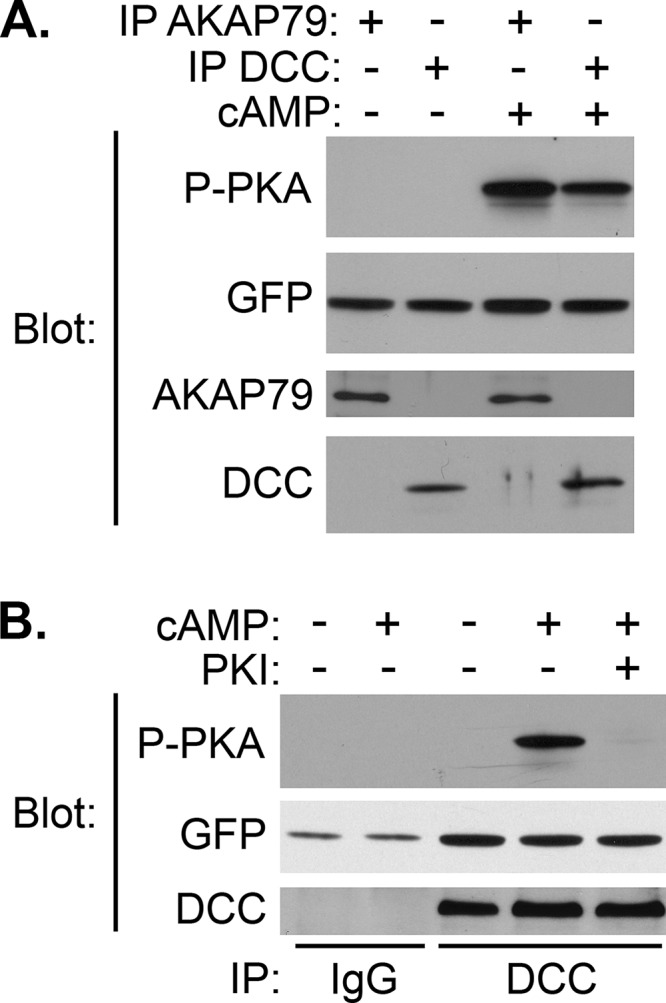
DCC immunoprecipitates contain PKA activity. A and B, E18 rat brains were homogenized in modified RIPA buffer and immunoprecipitated (IP) with antibodies against AKAP79 or DCC or with non-immune IgG as indicated. Immunoprecipitates were incubated with PKA substrate (GFP-R4S) in the absence or presence of cAMP and PKI peptide as indicated. Reaction products were separated by SDS-PAGE and analyzed by immunoblotting with the indicated antibodies to assess immunoprecipitation, substrate levels, and substrate phosphorylation.
DCC Co-immunoprecipitates with but Does Not Directly Bind to PKA
The requirement of AKAP function for DCC-driven phosphorylation of VASP (Fig. 1D) and the presence of PKA activity in DCC immunoprecipitates (Fig. 2) suggested that DCC couples, directly or indirectly, to PKA, a hypothesis we tested through co-immunoprecipitation experiments. When lysates made from E18 rat hippocampal tissue were immunoprecipitated with either control IgG or antibody against the PKA RII subunit, DCC was specifically detected in the RII immunoprecipitate (Fig. 3A). To ensure the specificity of this interaction, we performed the converse co-immunoprecipitation using an anti-DCC antibody in lysates from control- and DCC-expressing NG108-15 cells. PKA RII subunit was found specifically in DCC-containing immunoprecipitates and not in control IgG immunoprecipitates or in DCC immunoprecipitates from control transfected cells lacking DCC (Fig. 3B). The PKA-DCC interaction was partially blocked by treating cells with a function-blocking DCC antibody and was essentially completely disrupted by treatment with StHt31 (Fig. 3B). Additional co-immunoprecipitations demonstrated that DCC could associate with both type I and type II PKA R subunits (Fig. 3C). Inspection of the primary sequence of DCC did not reveal any regions with appreciable homology to the amphipathic helical R subunit binding region present in all known AKAPs (26). The only region in DCC that even approximated this sequence was its proposed transmembrane domain, which would be expected to be completely unavailable for binding to a cytosolic protein. Indeed, pulldown experiments using purified, recombinant PKA RIIα subunit and the cytoplasmic domain of DCC fused to GST did not show evidence of a direct interaction between these proteins (data not shown). Together, these data demonstrate that DCC interacts with PKA in vivo and suggest that this interaction is indirect and likely involves one or more intermediate proteins that serve as AKAPs.
FIGURE 3.
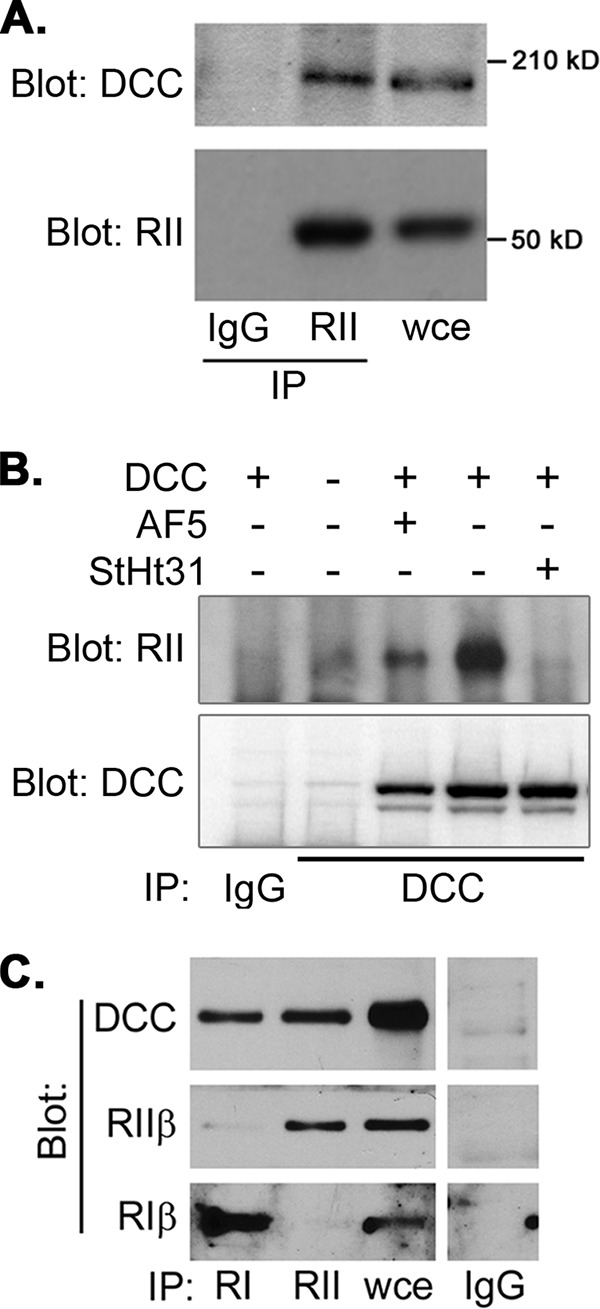
DCC co-immunoprecipitates with PKA RI and RII subunits. A, lysates were prepared from E18 rat hippocampi and immunoprecipitated (IP) with control IgG or antibody against RII subunits of PKA as indicated. Immunoprecipitates and whole cell extract (wce) were analyzed by SDS-PAGE and immunoblotting with the indicated antibodies. B, NG108-15 cells transfected with empty vector or pRK5-DCC were replated on polylysine-coated coverslips in the presence or absence of anti-DCC antibody (AF5) or StHt31. Lysates were prepared, immunoprecipitated with control IgG or anti-DCC antibody, and analyzed by immunoblotting with the indicated antibodies. C, lysates from E18 rat hippocampi were immunoprecipitated with antibodies against PKA RI or RII subunits or with non-immune IgG, and immunoprecipitates were analyzed by immunoblotting with the indicated antibodies.
DCC and PKA Interact with the ERM Proteins Ezrin, Radixin, and Moesin
The ERM family of proteins coordinates the physical coupling of the cortical actin cytoskeleton with plasma membrane and also regulates the transduction of signals emanating from transmembrane receptors (47–49). Not surprisingly then, the ERM proteins have also been shown to have important and diverse function in neurons, especially in growth cone dynamics (e.g. Refs. 50–53; for review, see Ref. 54). Moreover, all three ERM proteins have been shown to serve as AKAPs (55–59), with ezrin capable of functioning as a dual-specificity AKAP (60). Importantly, at least one ERM protein, ezrin, has been shown to interact with DCC in both colon cancer cells (39) and neuronal cells (61). In the latter report ezrin was demonstrated to exist in, and be activated by, a netrin-regulated complex with DCC in growth cone filopodia and was further demonstrated to be required for netrin-mediated cortical axon outgrowth (61).
Despite these previous reports a ternary complex containing all three proteins, ezrin, DCC, and PKA, has never been demonstrated. We, therefore, confirmed that endogenous ezrin could indeed interact with endogenous DCC in netrin-stimulated (but not unstimulated) IMR-32 cells (Fig. 4A) and exogenously expressed DCC in NG108-15 cells (Fig. 4B) in which DCC is activated by endogenously expressed netrin-1. These data are consistent with published observations showing the necessity of activation of DCC for its interaction with ERM proteins (61). We then investigated whether ezrin, DCC, and PKA RII could be found together in a higher order complex. Immunoprecipitation of each protein resulted in the co-immunoprecipitation of the other two (Fig. 4B). Control immunoprecipitations using non-immune IgG confirmed the specificity of the pulldown assays (Fig. 4, A and B). These data show that DCC, ezrin, and PKA form a complex in vivo and suggest that ezrin may indeed serve as a DCC-associated AKAP.
FIGURE 4.

Interactions between PKA, ERM proteins, and DCC. PKA and DCC interact with all three ERM proteins. A and B, lysates prepared from IMR-32 cells in the absence or presence of 500 ng/ml netrin (A) or NG108-15 cells transfected with pRK5-DCC (B) were immunoprecipitated (IP) with control IgG or antibodies against ezrin (Ezr), DCC, or PKA RII subunits as indicated. Immunoprecipitates were analyzed by SDS-PAGE and immunoblotting with the indicated antibodies. C, left panel, whole cell lysate from differentiated, stage II-III hippocampal neurons (Hp) was separated by low percentage/high cross-linking SDS-PAGE and analyzed by immunoblotting with a pan-ERM antibody. The positions of ezrin (Ezr), radixin (Rxn), and moesin (Msn) are indicated. Right panel, whole cell lysates from differentiated NG108-15 cells (NG), IMR-32 cells (IMR), and hippocampal neurons (Hp) in triplicate were separated on a single, low percentage/high cross-linking SDS-PAGE gel and transferred to nitrocellulose. The membrane was cut into thirds, and each was analyzed by immunoblotting with antibodies specific for ezrin, radixin, and moesin. D, Triton-insoluble lysate from IMR-32 cells was incubated with cAMP-agarose in the absence or presence of 20 μm cAMP. After extensive washing, bead-bound proteins were eluted in sample buffer and analyzed alongside unfractionated whole cell extract (wce) by SDS-PAGE and immunoblotting as described in A. E, Triton-insoluble lysates from E18 rat brain were immunoprecipitated with non-immune IgG or anti-DCC antibody, and immunoprecipitates were analyzed by immunoblotting with antibodies against ezrin (Ezr), radixin (Rxn), and moesin (Msn) as indicated.
The fact that all three ERM proteins have important roles in growth cone dynamics, serve as AKAPs, and have high sequence homology in the region known to mediate interaction with DCC (39, 47, 61) suggests that all three ERM proteins may contribute to the coupling of PKA to DCC in neuronal cells. To investigate this possibility, we first determined the expression of all three ERM proteins across several experimental systems. Using pan-ERM as well as isoform-specific antibodies, we found that all three ERM proteins are expressed in early-stage primary hippocampal neurons (Fig. 4C, left), as reported previously (53), as well as in the NG108-15 and IMR-32 cell lines (Fig. 4C, right). Also, consistent with their conserved and previously established role as AKAPs, all three ERM proteins were pulled out of IMR-32 cells lysates by cAMP-agarose (Fig. 4D); this interaction was blocked by the addition of high concentrations of exogenous cAMP (Fig. 4D) or Ht31 (1 μm; data not shown) to the pulldown, confirming that the presence of ERM proteins in the pulldown is due to interaction with cAMP-binding PKA R subunits. Finally, all three ERM proteins were also detectable in DCC immunoprecipitates from embryonic rat brain (Fig. 4E). Together, these data demonstrate the expression of ezrin, radixin, and moesin in our experimental systems and further show that all three proteins can bind both PKA and DCC and thus likely contribute to the coupling of those two proteins.
ERM Proteins Are Required for Physical and Functional Coupling of PKA to DCC
To demonstrate that ERM proteins mediate the interaction between DCC and PKA, we endeavored to silence the expression of all three ERM proteins and assess the effects on DCC-PKA interaction. IMR-32 human neuroblastoma cells were used for subsequent experiments for the following reasons: they express all of the proteins of interest (Fig. 4C), they have been used previously to great effect for analyses of DCC function (40, 62), they can be differentiated easily and form axonal processes with well formed growth cones (63–65), and finally, they allow ERM-silencing and rescue experiments to be performed with unaltered murine homologs, which are naturally resistant to commercially available human-specific RNAi reagents. Transfection of IMR-32 cells with shRNA plasmids targeting individual ERM proteins confirmed the specificity and efficacy of the reagents; for each plasmid, there was at least a 75% knockdown in the expression of the target ERM protein with no significant effect on the other two (Fig. 5A). In addition, analysis by immunoblotting with a pan-ERM antibody showed that expression of all ERM proteins was nearly eliminated by co-transfection of the three plasmids (Fig. 5B). Importantly, this triple knockdown also eliminated the co-immunoprecipitation between DCC and PKA R subunits (Fig. 5B), confirming that ERM proteins are required for linking PKA to DCC.
FIGURE 5.
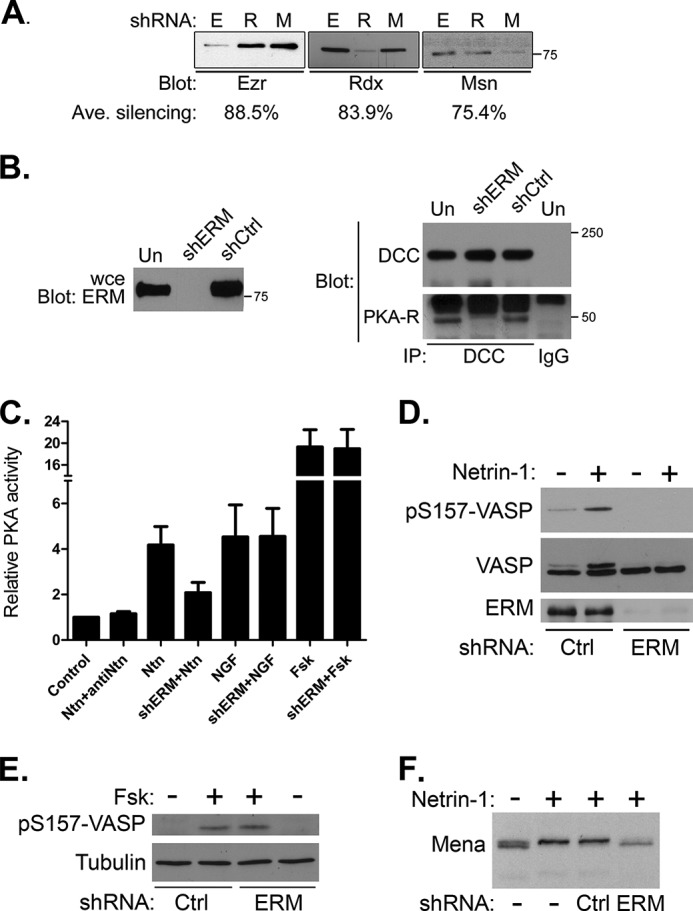
Silencing ERM protein expression ablates DCC-PKA interaction and specifically blocks netrin-induced PKA activity and phosphorylation of downstream cytoskeletal targets. A, IMR-32 cells were transfected for 72 h with plasmids expressing shRNA against ezrin (E), radixin (R), or moesin (M), then harvested and analyzed by SDS-PAGE and immunoblotting. The average percent silencing was determined from three separate transfections and is indicated below the panels. B, Triton-insoluble protein from netrin-stimulated, untransfected IMR-32 cells (Un), cells transfected with empty vector (shCtrl) or co-transfected with shRNA plasmids targeting ezrin, radixin, and moesin (shERM) were immunoprecipitated (IP) with antibodies against DCC or with non-immune IgG as indicated. Immunoprecipitated proteins as well as whole cell extracts (wce) were analyzed by SDS-PAGE and immunoblotting with antibodies against DCC or PKA-RI. C, IMR-32 cells were transfected with three shRNA-expressing plasmids targeting ezrin, radixin, and moesin (shERM) or with empty plasmid, then replated onto polylysine-coated dishes and cultured in serum-free media containing 2 μm retinoic acid for 12 h. Differentiated cells were then stimulated for 20 min with 1 μg/ml netrin-1 with or without the prior addition of a function-blocking netrin antibody (antiNtn) to the cells. Control- and shERM-transfected cells were also treated with 20 ng/ml NGF or 20 μm forskolin (Fsk) as indicated. PKA activity was measured using an in vitro kinase assay as described under “Experimental Procedures.” Data are presented as relative kinase activity, with the activity in untreated, control-transfected cells (Control) assigned a value of 1 and represent the means ± S.D. from three independent transfections. D–F, control- and shERM-transfected IMR-32 cells were cultured and stimulated as in C, and whole cell extracts were analyzed by SDS-PAGE and immunoblotting with a pan-ERM antibody (ERM) or antibodies against total or phospho-Ser-157 VASP (VASP; pS157-VASP), tubulin, or Mena as indicated.
We next investigated whether ERM proteins are required for PKA activity and PKA-mediated phosphorylation of cytoskeletal regulators during netrin/DCC signaling. Netrin signaling through DCC in IMR-32 cells promoted activation of PKA, although unlike NG108-15 cells, this activation required the addition of recombinant netrin-1 to the culture medium (see Fig. 5, C, D, and F, and “Experimental Procedures”). This requirement for exogenous netrin is in line with prior observations (40) and is likely due a lower amount of endogenous netrin produced by these cells compared with NG108-15 cells overexpressing exogenous DCC (45). Importantly, the netrin- and DCC-dependent activation of PKA was significantly reduced (∼50%, p < 0.002) by silencing ERM expression (Fig. 5C), and this effect was specific for the netrin/DCC pathway, as ERM silencing had no effect on PKA activation in response to nerve growth factor or allosteric activation of adenylyl cyclase by forskolin (Fig. 5C). Furthermore, although ERM knockdown had no effect on VASP phosphorylation in response to forskolin (Fig. 5E), it completely inhibited phosphorylation of VASP and Mena (ascertained by phospho-specific antibody reactivity or electrophoretic mobility shift) in response to netrin-1 (Fig. 5, D and F). Thus, we conclude that ERM proteins are required for netrin/DCC signaling through PKA.
Silencing of ERM Protein Expression Disrupts Cell and Growth Cone Morphology and Neuritogenesis
During the course of these ERM-silencing experiments, we observed a significant and consistent effect on the morphology of shERM-transfected cells even while growing in non-differentiating, serum-containing growth medium. Specifically, shERM-transfected cells exhibited few lamellae or lamellipodia but had numerous microspikes and retraction fibers (Fig. 6A); these structures were qualitatively distinct from classical filopodia (e.g. the structures observed and quantified in Fig. 1), as they did not emanate from lamellipodia. This was not seen to the same extent or with the same consistency in experiments where any single ERM was knocked down alone (data not shown). A similar, striking morphological effect was seen in IMR-32 cells that were induced to undergo neuronal differentiation in response to either netrin-1 or all-trans retinoic acid, both of which have been used previously to promote neurite outgrowth in these cells (40, 65). Although neuritogenesis occurred more efficiently (i.e. neuritic processes formed faster and grew longer) in response to all-trans retinoic acid than to netrin, the general effects of ERM protein knockdown were quite similar (Fig. 6A). Specifically, although there was no effect on the number of neurites that formed, those neurites that did form in the absence of ERMs were significantly shorter than in control cultures and were typically devoid of well developed growth cones (Fig. 6B). These data are consistent with previous reports demonstrating dramatic effects of silencing radixin and/or moesin, but not necessarily ezrin, on axonal process formation and growth cone morphology and dynamics (51, 53).
FIGURE 6.

Silencing ERM protein expression alters cell morphology, neuritogenesis, and growth cone morphology; rescue by wild-type but not AKAP-deficient radixin. A, IMR-32 cells were transfected with empty plasmid (Ctrl) or with shRNA plasmids targeting ezrin, radixin, and moesin (shERM) for 72 h. Cells were then re-plated onto uncoated glass coverslips in regular growth medium containing 10% serum (Glass/10% FBS) or onto poly-l-lysine-coated coverslips (PLL) in serum-free medium containing 5 μm all-trans retinoic acid (atRA) or 500 ng/ml netrin-1 (Ntn). Cells were fixed 12 h after re-plating and stained with Alexa 488-phallodin and DAPI to visualize F-actin and nuclei, respectively, then imaged by epifluorescence microscopy (scale bar = 20 μm; note that the panels in the last column have been magnified 2× to better visualize the growth cones). In some instances the images are purposely overexposed to better visualize the fine details in growth cones and other structures. B, the number of neurites per nucleus, neurite length, and the percent of neurites exhibiting well defined growth cones (% GC+ neurites) were determined from three separate transfections and after differentiation in the presence of netrin-1. Data are presented as the averages ± S.E. (n ≥ 40) and were analyzed by a two-tailed, unpaired t test (*, p = 0.022; **, p < 0.001). C, radixin, but not ezrin or moesin, rescues growth cone morphology in ERM-silenced IMR-32 cells. IMR-32 cells were transfected with empty silencing plasmid (shCtrl) or with shRNA plasmids targeting ezrin, radixin, and moesin (shERM) for 24 h, then co-transfected again with a plasmid expressing mCherry (red) along with an empty expression vector (vector) or plasmids expressing wild-type ezrin (Ezr), moesin (Msn), or radixin (Rdx) or an AKAP-deficient point mutant of radixin (RdxL421P) for 36 h. Cells were then re-plated onto poly-l-lysine-coated coverslips in serum-free medium containing 500 ng/ml netrin-1 for 12 h before fixing and staining with Alexa 488-phallodin (shown in inverted grayscale) to visualize F-actin and general growth cone morphology. D, quantification of growth cone area (in μm2) in cells transfected as indicated. n ≥ 12 growth cones total from three separate transfections (*, p = 0.0132 versus shCtrl and p < 0.0005 versus all other conditions). E, phospho-mimetic T564D Rdx, but not WT Rdx, anchors PKA RIIα to the DCC cytoplasmic domain in vitro. Purified, recombinant RIIα, and either wild-type (WT) or T564D (564D) radixin (Rdx) were combined with purified GST fused to either full-length DCC cytoplasmic domain (WT) or to the cytoplasmic domain lacking the P3 region (ΔP3). Complexes were collected on glutathione beads and analyzed along with a portion of the input mixtures by SDS-PAGE and immunoblotting with the indicated antibodies. F, T564D Rdx, but not L421P/T564D Rdx, anchors PKA RIIα to the DCC cytoplasmic domain in vitro. Purified, recombinant RIIα, and either T564D or L421P/T564 Rdx were combined with purified GST-DCC cytoplasmic domain. Complexes were collected on glutathione beads and analyzed along with a portion of the input mixtures by SDS-PAGE and immunoblotting with the indicated antibodies.
Ezrin, radixin, and moesin are multifunctional proteins with complex physical and regulatory interactions (47, 54) beyond their roles as AKAPs, and therefore, silencing the expression of all three proteins promotes loss of all ERM functions in aggregate, not just ERM-mediated PKA anchoring. Therefore, to dissect out the discrete role of ERM-mediated PKA anchoring in events downstream of netrin/DCC signaling, we attempted rescue experiments in which AKAP-deficient mutants of all three ERM proteins were reintroduced into ERM-silenced cells. These experiments, which require a six-plasmid transfection at high efficiency, were not surprisingly unfruitful. We, therefore, determined whether any single ERM protein had the ability to rescue the morphological phenotype observed in shERM cells and found that expression of radixin, but not ezrin or moesin, was best able to restore growth cone morphology in ERM-silenced IMR-32 cells (Fig. 6, C and D).
To determine whether the AKAP domain of radixin contributed to the altered growth cone dynamics observed in shERM knockdown cells, we restored expression with a point mutant L421P that disrupts the AKAP binding domain (66). Loss of AKAP function severely compromised the ability of radixin to restore growth cone morphology (Fig. 6, C and D). To confirm that the L421P mutation only disrupted the ability of radixin to bind to PKA and not DCC, in vitro binding assays were performed using purified proteins. In the course of optimizing the conditions for these in vitro experiments, we found that radixin needed to be in its open and active conformation, elicited in vivo by phosphorylation of Thr at position 564, in order for it to bind to the cytoplasmic tail of DCC, consistent with previous observations (61). Mutation of the threonine in this position to aspartic acid (T564D) is known to mimic phosphorylation in all ERM proteins (47). In agreement with this, recombinant T564D-radixin exhibited markedly enhanced binding specifically to the DCC cytoplasmic domain as compared with wild-type radixin and also supported the coupling of PKA to DCC (Fig. 6E). Importantly, introduction of the L421P mutation into T564D-radixin did not impact the ability of radixin to bind to the DCC cytoplasmic tail in vitro but ablated its ability to anchor PKA RII subunits to DCC (Fig. 6F). These results confirm that the AKAP function of radixin is required for linking PKA to DCC in vitro and suggest that ERM-mediated PKA anchoring contributes to normal growth cone morphology.
Loss of Radixin-mediated PKA Anchoring Converts Netrin-mediated Growth Cone Attraction to Repulsion
Given the effects of loss of radixin-mediated AKAP function on growth cone morphology, we next investigated whether this function might also impact growth cone dynamics. To do this, we assessed growth cone turning behavior in response directional gradients of netrin-1 in shERM-IMR-32 cells expressing either wild-type or AKAP-deficient (L421P) radixin. It should be noted here that although these triple-knockdown cells formed neuritic extensions and growth cones, the speed and extent of overall neurite elaboration and growth cone “turning” in these cells was greatly reduced compared with wild-type cells. Thus, to assess the role of ERM-mediated anchoring of PKA to DCC on directional responses to netrin-1, growth cones from shERM/RdxWT and shERM/RdxL421P cells were imaged before and after gradient stimulation, and directional growth cone responses were assessed by calculating protrusion and retraction indexes of growth cones toward and away from the gradient (Fig. 7). In the absence of netrin-1, the profile of protrusion and retraction of shERM/RdxWT and shERM/RdxL421P cells were similar in that neither showed any directional bias (Fig. 7, A and C). In response to netrin gradients, shERM/RdxWT cells displayed increased protrusion toward netrin and increased retraction on the contralateral side of the growth cone, as expected for a response to an attractive guidance cue. The response of the RdxL421P-expressing cells, however, was significantly different and somewhat unexpected. Rather than failing to show an attractive response, growth cones in shERM/RdxL421P cells were instead consistently repelled by netrin gradients, displaying increased protrusion away from the gradient and increased retraction proximal to the gradient (Fig. 7C). Taken together, these observations demonstrate that ERM proteins are required for coupling PKA to DCC and that ERM-mediated anchoring of PKA plays an important role in directional responses toward netrin-1.
FIGURE 7.

Loss of ERM-mediated PKA anchoring converts netrin-induced growth cone attraction to repulsion. A, IMR-32 cells serially transfected with shERM plasmids and rescue plasmids expressing either wild-type (WT) or AKAP-deficient (L421P) radixin were plated onto poly-l-lysine-coated coverslips in serum-free medium containing 5 μm all-trans retinoic acid for 12 h. Phase contrast images were acquired before and 30 min after exposure to a gradient of either BSA (Control) or netrin-1 (5 μg/ml in the pipette; position indicated by the arrowheads) and processed using a custom ImageJ macro to generate protrusion-retraction analysis maps (PRAM). B and C, directional growth cone responses were quantified by calculating the protrusion or retraction index toward (Proximal) or away from (Distal) to the gradient source. P, protrusion; R, retraction; O, overlap. Summed data are plotted in C as the mean ± S.D. for at least five cells from two separate experiments.
DISCUSSION
As the principal neuronal receptor for netrin, DCC is required for both chemoattractive and chemorepulsive responses. In both cases, netrin/DCC signaling must ultimately interface with machinery that regulates cytoskeletal dynamics in order to drive the turning responses of neural growth cones. It is also well established that the response of neurons to netrin-1 is both qualitatively and quantitatively regulated by cAMP and PKA signaling, although the exact manner and mechanism through which this regulation manifests is unclear. The current data show that the ezrin/radixin/moesin proteins play an important role in this signaling paradigm by physically and functionally coupling PKA to DCC, an interaction that appears to help determine the tropism of growth cone guidance responses. Our results are in excellent complement with observations from Lamarche-Vane and co-workers (61), demonstrating that ERM proteins are phosphorylated downstream of netrin-1 in a manner dependent on DCC, Src-family kinases, and Rho GTPase activity and that ERM protein function is required for axon outgrowth in primary cortical neurons .
Our observations that netrin/DCC signaling activates PKA are consistent with some earlier reports in which levels of cAMP or PKA activity increase in response to netrin (19, 20) but contradict others in which no such responses could be measured (22, 23). In these latter studies, only modest increases in cAMP/PKA activity were seen even in response to 10–20 μm forskolin. Often, the magnitude of effect of physiologic activators of cAMP/PKA signaling is significantly lower than seen with even these modest concentrations of forskolin (e.g. Fig. 5C). Thus, the conditions used to assess PKA activity in earlier studies may not have been sensitive enough to see a physiologically relevant rise in cAMP/PKA. Of course, differences in experimental systems may also play a role and may be potentially informative as to the molecular bases of cell- and tissue-specific responses. Nonetheless, the current data still do not provide a mechanism through which netrin/DCC signaling activates PKA. It is possible that the DCC-mediated activation of PKA observed in our studies is a result of cross-talk between DCC and another, cAMP/PKA-activating pathway. Here, an intriguing but controversial possibility would be the A2b adenosine receptor (A2BAR). A2BAR directly couples to the Gs heterotrimeric G-protein and thus activates adenylyl cyclases (67) and has also been shown to interact, albeit perhaps indirectly, with both ezrin and PKA (68). However, although compelling evidence demonstrates that A2BAR is dispensable for netrin-dependent axon guidance in vivo (69), other compelling yet contradictory evidence implicates A2BAR as a co-receptor with DCC for netrin (19) and as an important modulator of growth cone guidance toward and away from netrin (16). Clearly, additional rigorous experimentation and discussion are needed to address these and other controversies in the field.
Regardless of the mechanism of PKA activation, it is particularly interesting to note that the current data showing that specific loss of ERM-mediated anchoring of PKA switches growth cone tropism from attraction to repulsion (Fig. 7) and are in excellent agreement with previous reports that established the paradigm wherein inhibition or loss of cAMP/PKA signaling converts growth cone attraction to netrin into repulsion (16, 17, 20, 21). This paradigm is also not without contention, however, as other investigators have found that PKA signaling tunes the sensitivity to netrin but does not alter tropism (22). However, there is considerable variation among the experimental systems used throughout these reports, and it is likely, one might even say reassuring, that neurons and neuronal cell lines of different origins might exhibit different responses to a given guidance cue. For netrin-mediated responses, this is likely a product of the balance of expression of various types of netrin receptors. Although DCC mediates growth cone attraction to netrin-1, repulsion from netrin is mediated through UNC5 proteins, either alone or in concert with DCC, and there is considerable evidence to suggest that the relative levels of DCC and UNC5 proteins expressed either in total or on the cell surface controls both the sensitivity and the trajectory of growth cones in netrin gradients (9, 10, 22). It should be noted here that UNC5H is expressed in IMR-32 cells (Ref. 62 and data not shown), the cells used in the current study to show that uncoupling PKA from ERM proteins and DCC switches netrin tropism from attraction to repulsion. Although recently solved crystal structures of netrin-receptor complexes (70, 71) provide intriguing possibilities, the exact molecular mechanism through which this dramatic switch in cellular response takes place and the manner in which PKA and ERM proteins might contribute to the switch remains unclear.
Their role as AKAPs notwithstanding, the ERM proteins have important roles in organizing the cell cortex and related functions in a wide variety of neuronal and non-neuronal cell types (47, 54). Not surprisingly then, the ERM proteins have a large number of binding partners, including many cell-surface receptors and elements of the cell cortex and actin cytoskeleton (47). Their ability to function as AKAPs would, therefore, provide the possibility of juxtaposition of PKA near any or all of these partners. Thus, it is possible that the current results obtained with the AKAP-deficient RdxL421P mutant are not due solely to loss of PKA anchoring to DCC but due to the aggregate loss of ERM-mediated anchoring to several targets. Resolving this would require the identification of mutations in ERMs or DCC that specifically disrupt only DCC-ERM interaction while leaving all other interactions intact. Given the number and diversity of interacting partners for both ERMs and DCC, the identification of such mutations would likely require extraordinary effort. However, given that PKA is indeed physically and functionally coupled to DCC, that ERM proteins are required for this coupling, and that expression of the AKAP-deficient RdxL421P exerts an effect on directional growth cone responses to netrin gradients that mimics previously reported effects of inhibition of PKA, we contend that PKA anchoring to DCC itself likely plays a major role in the results reported herein.
This contention would suggest that ERM-anchored PKA activity helps regulate signaling events in close molecular proximity to the DCC receptor protein itself that govern the balance between attractive and repulsive responses to netrin gradients. Indeed, one of the most important contributions of AKAP-mediated anchoring to PKA-dependent signaling is through the molecular juxtapositioning of PKA and specific pools of substrates (26). In this regard, it is intriguing to note that a considerable number of proteins implicated in netrin/DCC signaling are known to be regulated, directly or indirectly, by PKA (9, 10, 32, 72). These include but are not limited to the non-receptor tyrosine kinases FAK, Src, and Fyn (42, 73–78), the adaptor protein Nck (79, 80), the Rho family GTPases Rac and Cdc42 (27, 45, 81–83) and their effector PAK (p21-activated kinase) (42, 84), and as shown here, the Mena/VASP proteins (35, 41, 46). Also of particular interest is the well established cross-talk between cAMP/PKA and Ca2+ signaling in cell migration (33) and the observed intricate interplay between localized cAMP (and perhaps PKA) and Ca2+ signaling in growth cone turning responses (18, 85), although additional work is needed to determine the relative contributions of PKA versus other cAMP effectors (principally, the Epacs (exchange proteins activated by cAMP) (21, 85)) in this context. The well established ability of PKA to exert multifaceted control over exocytosis machinery in neuronal and non-neuronal cells (37, 86) is also noteworthy in light of the demonstrated ability of PKA to mobilize DCC from intracellular stores in a exocytosis-dependent manner (25). It is interesting here to consider the ability of ERM proteins themselves to control the abundance, distribution, and innate function of a variety of membrane-bound receptors (47, 48). Finally, it is also important to consider that although the ERM proteins share considerable homology, they are thought to exert non-redundant functions in those cells expressing more than one family member (47–49). As shown here and elsewhere (50–54), growth cone morphology and dynamics appeared to be more directly and/or efficiently regulated by radixin than either moesin or ezrin, supporting these proteins' distinct functions. Therefore, although all three ERM proteins have the capacity to couple PKA to DCC, each family member may impart its own unique contributions to this interaction, e.g. through distinct localization or subpopulations of binding partners. Thus, an important future effort will be to identify which factors and/or signaling pathways associated with DCC are modified and regulated by ERM-anchored PKA and to determine how modification of these targets controls growth cone tropism in netrin gradients.
Thus, although important questions still remain to be answered, our current results help demonstrate that localized cAMP/PKA signaling, coordinated by ERM proteins, plays an important role in the DCC-mediated neuronal response to netrin and suggest that receptor-proximal signaling events contribute to the regulation of growth cone attraction and repulsion during axon guidance.
Acknowledgments
We thank Nathalie Lamarche-Vane and Tim Kennedy (McGill University) for reagents, excellent discussions, and advice, Sachiko Tsukita (Osaka University) for anti-ezrin, -radixin, and -moesin hybridoma supernatants and cDNAs, Daniel Altschuler (University of Pittsburgh), Diane Barber (University of California San Francisco), and Gabriel Fenteany (University of Connecticut) for ERM plasmids, Marcel Payet (University of Sherbrooke) for β1 integrin antibody, and John Scott (Howard Hughes Medical Institute and University of Washington) for PKA R subunit plasmids.
This work was supported, in whole or in part, by National Institutes of Health Grants R01GM074204 and R01GM097495 (to A. K. H.) and P20RR016435 (to R. Parsons and A. K. H., COBRE (Centers of Biomedical Research Excellence) Project 1).
A. K. Howe, unpublished observations.
- DCC
- deleted in colorectal cancer
- PKA
- cAMP-dependent protein kinase
- AKAP
- A-kinase anchoring protein
- ERM
- ezrin-radixin-moesin
- Mena
- mammalian enabled
- VASP
- vasodilator-stimulated phosphoprotein
- PKAic
- PKA inhibitor cocktail
- RI
- PKA regulatory subunit type I
- RII
- PKA regulatory subunit type II
- StHt31
- stearated Ht31 peptide
- RIPA buffer
- radioimmune precipitation assay buffer
- Rdx
- radixin
- A2BAR
- RA2b adenosine receptor
- VASP
- vasodilator-stimulated phosphoprotein.
REFERENCES
- 1. Mueller B. K. (1999) Growth cone guidance: first steps towards a deeper understanding. Annu. Rev. Neurosci. 22, 351–388 [DOI] [PubMed] [Google Scholar]
- 2. Huber A. B., Kolodkin A. L., Ginty D. D., Cloutier J. F. (2003) Signaling at the growth cone: ligand-receptor complexes and the control of axon growth and guidance. Annu. Rev. Neurosci. 26, 509–563 [DOI] [PubMed] [Google Scholar]
- 3. Lowery L. A., Van Vactor D. (2009) The trip of the tip: understanding the growth cone machinery. Nat. Rev. Mol. Cell Biol. 10, 332–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Goodman C. S. (1996) Mechanisms and molecules that control growth cone guidance. Annu. Rev. Neurosci. 19, 341–377 [DOI] [PubMed] [Google Scholar]
- 5. Guan K. L., Rao Y. (2003) Signalling mechanisms mediating neuronal responses to guidance cues. Nat. Rev. Neurosci. 4, 941–956 [DOI] [PubMed] [Google Scholar]
- 6. Gallo G., Letourneau P. C. (2004) Regulation of growth cone actin filaments by guidance cues. J. Neurobiol. 58, 92–102 [DOI] [PubMed] [Google Scholar]
- 7. Dent E. W., Gupton S. L., Gertler F. B. (2011) The growth cone cytoskeleton in axon outgrowth and guidance. Cold Spring Harb. Perspect. Biol. 3, a001800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gomez T. M., Letourneau P. C. (2014) Actin dynamics in growth cone motility and navigation. J. Neurochem. 129, 221–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barallobre M. J., Pascual M., Del Río J. A., Soriano E. (2005) The Netrin family of guidance factors: emphasis on Netrin-1 signalling. Brain Res. Brain Res. Rev. 49, 22–47 [DOI] [PubMed] [Google Scholar]
- 10. Round J., Stein E. (2007) Netrin signaling leading to directed growth cone steering. Curr. Opin. Neurobiol. 17, 15–21 [DOI] [PubMed] [Google Scholar]
- 11. Kennedy T. E., Tessier-Lavigne M. (1995) Guidance and induction of branch formation in developing axons by target-derived diffusible factors. Curr. Opin. Neurobiol. 5, 83–90 [DOI] [PubMed] [Google Scholar]
- 12. Arakawa H. (2004) Netrin-1 and its receptors in tumorigenesis. Nat. Rev. Cancer 4, 978–987 [DOI] [PubMed] [Google Scholar]
- 13. Mehlen P., Furne C. (2005) Netrin-1: when a neuronal guidance cue turns out to be a regulator of tumorigenesis. Cell. Mol. Life Sci. 62, 2599–2616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nikolopoulos S. N., Giancotti F. G. (2005) Netrin-integrin signaling in epithelial morphogenesis, axon guidance, and vascular patterning. Cell Cycle 4, e131–e135 [PubMed] [Google Scholar]
- 15. Chédotal A. (2007) Chemotropic axon guidance molecules in tumorigenesis. Prog. Exp. Tumor Res. 39, 78–90 [DOI] [PubMed] [Google Scholar]
- 16. Shewan D., Dwivedy A., Anderson R., Holt C. E. (2002) Age-related changes underlie switch in netrin-1 responsiveness as growth cones advance along visual pathway. Nat. Neurosci. 5, 955–962 [DOI] [PubMed] [Google Scholar]
- 17. Ming G. L., Song H. J., Berninger B., Holt C. E., Tessier-Lavigne M., Poo M. M. (1997) cAMP-dependent growth cone guidance by netrin-1. Neuron 19, 1225–1235 [DOI] [PubMed] [Google Scholar]
- 18. Nishiyama M., Hoshino A., Tsai L., Henley J. R., Goshima Y., Tessier-Lavigne M., Poo M. M., Hong K. (2003) Cyclic AMP/GMP-dependent modulation of Ca2+ channels sets the polarity of nerve growth-cone turning. Nature 423, 990–995 [DOI] [PubMed] [Google Scholar]
- 19. Corset V., Nguyen-Ba-Charvet K. T., Forcet C., Moyse E., Chédotal A., Mehlen P. (2000) Netrin-1-mediated axon outgrowth and cAMP production requires interaction with adenosine A2b receptor. Nature 407, 747–750 [DOI] [PubMed] [Google Scholar]
- 20. Höpker V. H., Shewan D., Tessier-Lavigne M., Poo M., Holt C. (1999) Growth-cone attraction to netrin-1 is converted to repulsion by laminin-1. Nature 401, 69–73 [DOI] [PubMed] [Google Scholar]
- 21. Murray A. J., Tucker S. J., Shewan D. A. (2009) cAMP-dependent axon guidance is distinctly regulated by Epac and protein kinase A. J. Neurosci. 29, 15434–15444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moore S. W., Kennedy T. E. (2006) Protein kinase A regulates the sensitivity of spinal commissural axon turning to netrin-1 but does not switch between chemoattraction and chemorepulsion. J. Neurosci. 26, 2419–2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Manitt C., Thompson K. M., Kennedy T. E. (2004) Developmental shift in expression of netrin receptors in the rat spinal cord: predominance of UNC-5 homologues in adulthood. J. Neurosci. Res. 77, 690–700 [DOI] [PubMed] [Google Scholar]
- 24. Song H., Ming G., He Z., Lehmann M., McKerracher L., Tessier-Lavigne M., Poo M. (1998) Conversion of neuronal growth cone responses from repulsion to attraction by cyclic nucleotides. Science 281, 1515–1518 [DOI] [PubMed] [Google Scholar]
- 25. Bouchard J. F., Moore S. W., Tritsch N. X., Roux P. P., Shekarabi M., Barker P. A., Kennedy T. E. (2004) Protein kinase A activation promotes plasma membrane insertion of DCC from an intracellular pool: a novel mechanism regulating commissural axon extension. J. Neurosci. 24, 3040–3050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Michel J. J., Scott J. D. (2002) AKAP mediated signal transduction. Annu. Rev. Pharmacol. Toxicol. 42, 235–257 [DOI] [PubMed] [Google Scholar]
- 27. Howe A. K., Baldor L. C., Hogan B. P. (2005) Spatial regulation of the cAMP-dependent protein kinase during chemotactic cell migration. Proc. Natl. Acad. Sci. U.S.A. 102, 14320–14325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lim C. J., Han J., Yousefi N., Ma Y., Amieux P. S., McKnight G. S., Taylor S. S., Ginsberg M. H. (2007) α4 integrins are type I cAMP-dependent protein kinase-anchoring proteins. Nat. Cell Biol. 9, 415–421 [DOI] [PubMed] [Google Scholar]
- 29. Lim C. J., Kain K. H., Tkachenko E., Goldfinger L. E., Gutierrez E., Allen M. D., Groisman A., Zhang J., Ginsberg M. H. (2008) Integrin-mediated protein kinase A activation at the leading edge of migrating cells. Mol. Biol. Cell 19, 4930–4941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Paulucci-Holthauzen A. A., Vergara L. A., Bellot L. J., Canton D., Scott J. D., O'Connor K. L. (2009) Spatial distribution of protein kinase A activity during cell migration is mediated by A-kinase anchoring protein AKAP Lbc. J. Biol. Chem. 284, 5956–5967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McKenzie A. J., Campbell S. L., Howe A. K. (2011) Protein kinase A activity and anchoring are required for ovarian cancer cell migration and invasion. PloS ONE 6, e26552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Howe A. K. (2004) Regulation of actin-based cell migration by cAMP/PKA. Biochim. Biophys. Acta 1692, 159–174 [DOI] [PubMed] [Google Scholar]
- 33. Howe A. K. (2011) Cross-talk between calcium and protein kinase A in the regulation of cell migration. Current opinion in cell biology 23, 554–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hirao M., Sato N., Kondo T., Yonemura S., Monden M., Sasaki T., Takai Y., Tsukita S. (1996) Regulation mechanism of ERM (ezrin/radixin/moesin) protein/plasma membrane association: possible involvement of phosphatidylinositol turnover and Rho-dependent signaling pathway. J. Cell Biol. 135, 37–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lebrand C., Dent E. W., Strasser G. A., Lanier L. M., Krause M., Svitkina T. M., Borisy G. G., Gertler F. B. (2004) Critical role of Ena/VASP proteins for filopodia formation in neurons and in function downstream of netrin-1. Neuron 42, 37–49 [DOI] [PubMed] [Google Scholar]
- 36. Meijering E., Jacob M., Sarria J. C., Steiner P., Hirling H., Unser M. (2004) Design and validation of a tool for neurite tracing and analysis in fluorescence microscopy images. Cytometry A 58, 167–176 [DOI] [PubMed] [Google Scholar]
- 37. Evans G. J., Morgan A. (2003) Regulation of the exocytotic machinery by cAMP-dependent protein kinase: implications for presynaptic plasticity. Biochem. Soc. Trans. 31, 824–827 [DOI] [PubMed] [Google Scholar]
- 38. Rivard R. L., Birger M., Gaston K. J., Howe A. K. (2009) AKAP-independent localization of type-II protein kinase A to dynamic actin microspikes. Cell Motil. Cytoskeleton 66, 693–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Martín M., Simon-Assmann P., Kedinger M., Martin M., Mangeat P., Real F. X., Fabre M. (2006) DCC regulates cell adhesion in human colon cancer derived HT-29 cells and associates with ezrin. Eur. J. Cell Biol. 85, 769–783 [DOI] [PubMed] [Google Scholar]
- 40. Petrie R. J., Zhao B., Bedford F., Lamarche-Vane N. (2009) Compartmentalized DCC signalling is distinct from DCC localized to lipid rafts. Biol. Cell 101, 77–90 [DOI] [PubMed] [Google Scholar]
- 41. Howe A. K., Hogan B. P., Juliano R. L. (2002) Regulation of vasodilator-stimulated phosphoprotein phosphorylation and interaction with Abl by protein kinase A and cell adhesion. J. Biol. Chem. 277, 38121–38126 [DOI] [PubMed] [Google Scholar]
- 42. Howe A. K., Juliano R. L. (2000) Regulation of anchorage-dependent signal transduction by protein kinase A and p21-activated kinase. Nat. Cell Biol. 2, 593–600 [DOI] [PubMed] [Google Scholar]
- 43. Yang F., Liu Y., Bixby S. D., Friedman J. D., Shokat K. M. (1999) Highly efficient green fluorescent protein-based kinase substrates. Anal. Biochem. 266, 167–173 [DOI] [PubMed] [Google Scholar]
- 44. Lohof A. M., Quillan M., Dan Y., Poo M. M. (1992) Asymmetric modulation of cytosolic cAMP activity induces growth cone turning. J. Neurosci. 12, 1253–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shekarabi M., Kennedy T. E. (2002) The netrin-1 receptor DCC promotes filopodia formation and cell spreading by activating Cdc42 and Rac1. Mol. Cell Neurosci. 19, 1–17 [DOI] [PubMed] [Google Scholar]
- 46. Krause M., Dent E. W., Bear J. E., Loureiro J. J., Gertler F. B. (2003) Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Annu. Rev. Cell Dev. Biol. 19, 541–564 [DOI] [PubMed] [Google Scholar]
- 47. Fehon R. G., McClatchey A. I., Bretscher A. (2010) Organizing the cell cortex: the role of ERM proteins. Nat. Rev. Mol. Cell Biol. 11, 276–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McClatchey A. I., Fehon R. G. (2009) Merlin and the ERM proteins–regulators of receptor distribution and signaling at the cell cortex. Trends Cell Biol. 19, 198–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Neisch A. L., Fehon R. G. (2011) Ezrin, Radixin and Moesin: key regulators of membrane-cortex interactions and signaling. Curr. Opin. Cell Biol. 23, 377–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Birgbauer E., Dinsmore J. H., Winckler B., Lander A. D., Solomon F. (1991) Association of ezrin isoforms with the neuronal cytoskeleton. J. Neurosci. Res. 30, 232–241 [DOI] [PubMed] [Google Scholar]
- 51. Castelo L., Jay D. G. (1999) Radixin is involved in lamellipodial stability during nerve growth cone motility. Mol. Biol. Cell 10, 1511–1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gonzalez-Agosti C., Solomon F. (1996) Response of radixin to perturbations of growth cone morphology and motility in chick sympathetic neurons in vitro. Cell Motil. Cytoskeleton 34, 122–136 [DOI] [PubMed] [Google Scholar]
- 53. Paglini G., Kunda P., Quiroga S., Kosik K., Cáceres A. (1998) Suppression of radixin and moesin alters growth cone morphology, motility, and process formation in primary cultured neurons. J. Cell Biol. 143, 443–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ramesh V. (2004) Merlin and the ERM proteins in Schwann cells, neurons, and growth cones. Nat. Rev. Neurosci. 5, 462–470 [DOI] [PubMed] [Google Scholar]
- 55. Dransfield D. T., Bradford A. J., Smith J., Martin M., Roy C., Mangeat P. H., Goldenring J. R. (1997) Ezrin is a cyclic AMP-dependent protein kinase anchoring protein. EMBO J. 16, 35–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Grönholm M., Vossebein L., Carlson C. R., Kuja-Panula J., Teesalu T., Alfthan K., Vaheri A., Rauvala H., Herberg F. W., Taskén K., Carpén O. (2003) Merlin links to the cAMP neuronal signaling pathway by anchoring the RIβ subunit of protein kinase A. J. Biol. Chem. 278, 41167–41172 [DOI] [PubMed] [Google Scholar]
- 57. Ruppelt A., Mosenden R., Grönholm M., Aandahl E. M., Tobin D., Carlson C. R., Abrahamsen H., Herberg F. W., Carpén O., Taskén K. (2007) Inhibition of T cell activation by cyclic adenosine 5′-monophosphate requires lipid raft targeting of protein kinase A type I by the A-kinase anchoring protein ezrin. J. Immunol. 179, 5159–5168 [DOI] [PubMed] [Google Scholar]
- 58. Semenova I., Ikeda K., Ivanov P., Rodionov V. (2009) The protein kinase A-anchoring protein moesin is bound to pigment granules in melanophores. Traffic 10, 153–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sun F., Hug M. J., Bradbury N. A., Frizzell R. A. (2000) Protein kinase A associates with cystic fibrosis transmembrane conductance regulator via an interaction with ezrin. J. Biol. Chem. 275, 14360–14366 [DOI] [PubMed] [Google Scholar]
- 60. Jarnaess E., Ruppelt A., Stokka A. J., Lygren B., Scott J. D., Taskén K. (2008) Dual specificity A-kinase anchoring proteins (AKAPs) contain an additional binding region that enhances targeting of protein kinase A type I. J. Biol. Chem. 283, 33708–33718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Antoine-Bertrand J., Ghogha A., Luangrath V., Bedford F. K., Lamarche-Vane N. (2011) The activation of ezrin-radixin-moesin proteins is regulated by netrin-1 through Src kinase and RhoA/Rho kinase activities and mediates netrin-1-induced axon outgrowth. Mol. Biol. Cell 22, 3734–3746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Delloye-Bourgeois C., Fitamant J., Paradisi A., Cappellen D., Douc-Rasy S., Raquin M. A., Stupack D., Nakagawara A., Rousseau R., Combaret V., Puisieux A., Valteau-Couanet D., Bénard J., Bernet A., Mehlen P. (2009) Netrin-1 acts as a survival factor for aggressive neuroblastoma. J. Exp. Med. 206, 833–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gupta M., Notter M. F., Felten S., Gash D. M. (1985) Differentiation characteristics of human neuroblastoma cells in the presence of growth modulators and antimitotic drugs. Brain Res. 351, 21–29 [DOI] [PubMed] [Google Scholar]
- 64. Rossino P., Defilippi P., Silengo L., Tarone G. (1991) Up-regulation of the integrin α1/β1 in human neuroblastoma cells differentiated by retinoic acid: correlation with increased neurite outgrowth response to laminin. Cell Regul. 2, 1021–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Poongodi G. L., Suresh N., Gopinath S. C., Chang T., Inoue S., Inoue Y. (2002) Dynamic change of neural cell adhesion molecule polysialylation on human neuroblastoma (IMR-32) and rat pheochromocytoma (PC-12) cells during growth and differentiation. J. Biol. Chem. 277, 28200–28211 [DOI] [PubMed] [Google Scholar]
- 66. Hochbaum D., Barila G., Ribeiro-Neto F., Altschuler D. L. (2011) Radixin assembles cAMP effectors Epac and PKA into a functional cAMP compartment: role in cAMP-dependent cell proliferation. J. Biol. Chem. 286, 859–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ralevic V., Burnstock G. (1998) Receptors for purines and pyrimidines. Pharmacol. Rev. 50, 413–492 [PubMed] [Google Scholar]
- 68. Sitaraman S. V., Wang L., Wong M., Bruewer M., Hobert M., Yun C. H., Merlin D., Madara J. L. (2002) The adenosine 2b receptor is recruited to the plasma membrane and associates with E3KARP and Ezrin upon agonist stimulation. J. Biol. Chem. 277, 33188–33195 [DOI] [PubMed] [Google Scholar]
- 69. Stein E., Zou Y., Poo M., Tessier-Lavigne M. (2001) Binding of DCC by netrin-1 to mediate axon guidance independent of adenosine A2B receptor activation. Science 291, 1976–1982 [DOI] [PubMed] [Google Scholar]
- 70. Xu K., Wu Z., Renier N., Antipenko A., Tzvetkova-Robev D., Xu Y., Minchenko M., Nardi-Dei V., Rajashankar K. R., Himanen J., Tessier-Lavigne M., Nikolov D. B. (2014) Neural migration. Structures of netrin-1 bound to two receptors provide insight into its axon guidance mechanism. Science 344, 1275–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Finci L. I., Krüger N., Sun X., Zhang J., Chegkazi M., Wu Y., Schenk G., Mertens H. D., Svergun D. I., Zhang Y., Wang J. H., Meijers R. (2014) The crystal structure of netrin-1 in complex with DCC reveals the bifunctionality of netrin-1 as a guidance cue. Neuron 83, 839–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Shabb J. B. (2001) Physiological substrates of cAMP-dependent protein kinase. Chem. Rev. 101, 2381–2411 [DOI] [PubMed] [Google Scholar]
- 73. Li W., Lee J., Vikis H. G., Lee S. H., Liu G., Aurandt J., Shen T. L., Fearon E. R., Guan J. L., Han M., Rao Y., Hong K., Guan K. L. (2004) Activation of FAK and Src are receptor-proximal events required for netrin signaling. Nat. Neurosci. 7, 1213–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Liu G., Beggs H., Jürgensen C., Park H. T., Tang H., Gorski J., Jones K. R., Reichardt L. F., Wu J., Rao Y. (2004) Netrin requires focal adhesion kinase and Src family kinases for axon outgrowth and attraction. Nat. Neurosci. 7, 1222–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ren X. R., Ming G. L., Xie Y., Hong Y., Sun D. M., Zhao Z. Q., Feng Z., Wang Q., Shim S., Chen Z. F., Song H. J., Mei L., Xiong W. C. (2004) Focal adhesion kinase in netrin-1 signaling. Nat. Neurosci. 7, 1204–1212 [DOI] [PubMed] [Google Scholar]
- 76. Abrahamsen H., Vang T., Taskén K. (2003) Protein kinase A intersects SRC signaling in membrane microdomains. J. Biol. Chem. 278, 17170–17177 [DOI] [PubMed] [Google Scholar]
- 77. Meriane M., Tcherkezian J., Webber C. A., Danek E. I., Triki I., McFarlane S., Bloch-Gallego E., Lamarche-Vane N. (2004) Phosphorylation of DCC by Fyn mediates Netrin-1 signaling in growth cone guidance. J. Cell Biol. 167, 687–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yeo M. G., Oh H. J., Cho H. S., Chun J. S., Marcantonio E. E., Song W. K. (2011) Phosphorylation of Ser 21 in Fyn regulates its kinase activity, focal adhesion targeting, and is required for cell migration. J. Cell Physiol. 226, 236–247 [DOI] [PubMed] [Google Scholar]
- 79. Park D., Rhee S. G. (1992) Phosphorylation of Nck in response to a variety of receptors, phorbol myristate acetate, and cyclic AMP. Mol. Cell. Biol. 12, 5816–5823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Li X., Meriane M., Triki I., Shekarabi M., Kennedy T. E., Larose L., Lamarche-Vane N. (2002) The adaptor protein Nck-1 couples the netrin-1 receptor DCC (deleted in colorectal cancer) to the activation of the small GTPase Rac1 through an atypical mechanism. J. Biol. Chem. 277, 37788–37797 [DOI] [PubMed] [Google Scholar]
- 81. Li X., Saint-Cyr-Proulx E., Aktories K., Lamarche-Vane N. (2002) Rac1 and Cdc42 but not RhoA or Rho kinase activities are required for neurite outgrowth induced by the Netrin-1 receptor DCC (deleted in colorectal cancer) in N1E-115 neuroblastoma cells. J. Biol. Chem. 277, 15207–15214 [DOI] [PubMed] [Google Scholar]
- 82. Feoktistov I., Goldstein A. E., Biaggioni I. (2000) Cyclic AMP and protein kinase A stimulate Cdc42: role of A(2) adenosine receptors in human mast cells. Mol. Pharmacol. 58, 903–910 [DOI] [PubMed] [Google Scholar]
- 83. O'Connor K. L., Mercurio A. M. (2001) Protein kinase A regulates Rac and is required for the growth factor-stimulated migration of carcinoma cells. J. Biol. Chem. 276, 47895–47900 [DOI] [PubMed] [Google Scholar]
- 84. Shekarabi M., Moore S. W., Tritsch N. X., Morris S. J., Bouchard J. F., Kennedy T. E. (2005) Deleted in colorectal cancer binding netrin-1 mediates cell substrate adhesion and recruits Cdc42, Rac1, Pak1, and N-WASP into an intracellular signaling complex that promotes growth cone expansion. J. Neurosci. 25, 3132–3141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Nicol X., Hong K. P., Spitzer N. C. (2011) Spatial and temporal second messenger codes for growth cone turning. Proc. Natl. Acad. Sci. U.S.A. 108, 13776–13781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Seino S., Shibasaki T. (2005) PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiol. Rev. 85, 1303–1342 [DOI] [PubMed] [Google Scholar]