

Background: A 25-base pair deletion in the MYBPC3 gene is the most prevalent hypertrophic cardiomyopathy-associated mutation among South Asian populations.
Results: Expression of mutant protein (cMyBP-CC10mut) in cultured adult rat cardiomyocytes led to improper localization and contractile dysfunction.
Conclusion: cMyBP-CC10mut expression is sufficient to cause contractile dysfunction.
Significance: The study sheds light on the etiology of this ethnic-specific hypertrophic cardiomyopathy mutation.
Keywords: Cardiomyopathy, Contractile Protein, Genetic Polymorphism, Heart Failure, Muscle Physiology
Abstract
Hypertrophic cardiomyopathy (HCM) results from mutations in genes encoding sarcomeric proteins, most often MYBPC3, which encodes cardiac myosin binding protein-C (cMyBP-C). A recently discovered HCM-associated 25-base pair deletion in MYBPC3 is inherited in millions worldwide. Although this mutation causes changes in the C10 domain of cMyBP-C (cMyBP-CC10mut), which binds to the light meromyosin (LMM) region of the myosin heavy chain, the underlying molecular mechanism causing HCM is unknown. In this study, adenoviral expression of cMyBP-CC10mut in cultured adult rat cardiomyocytes was used to investigate protein localization and evaluate contractile function and Ca2+ transients, compared with wild-type cMyBP-C expression (cMyBP-CWT) and controls. Forty-eight hours after infection, 44% of cMyBP-CWT and 36% of cMyBP-CC10mut protein levels were determined in total lysates, confirming equal expression. Immunofluorescence experiments showed little or no localization of cMyBP-CC10mut to the C-zone, whereas cMyBP-CWT mostly showed C-zone staining, suggesting that cMyBP-CC10mut could not properly integrate in the C-zone of the sarcomere. Subcellular fractionation confirmed that most cMyBP-CC10mut resided in the soluble fraction, with reduced presence in the myofilament fraction. Also, cMyBP-CC10mut displayed significantly reduced fractional shortening, sarcomere shortening, and relaxation velocities, apparently caused by defects in sarcomere function, because Ca2+ transients were unaffected. Co-sedimentation and protein cross-linking assays confirmed that C10mut causes the loss of C10 domain interaction with myosin LMM. Protein homology modeling studies showed significant structural perturbation in cMyBP-CC10mut, providing a potential structural basis for the alteration in its mode of interaction with myosin LMM. Therefore, expression of cMyBP-CC10mut protein is sufficient to cause contractile dysfunction in vitro.
Introduction
Hypertrophic cardiomyopathy (HCM)3 is the most commonly inherited cardiac disease. It is caused by mutations in genes encoding contractile proteins, leading to contractile dysfunction and subsequent hypertrophy. Affected individuals develop pump dysfunction and cardiac hypertrophy at a young age. Moreover, HCM is the most frequent cause of acute cardiac arrest in young people (1). In particular, mutations in the MYBPC3 gene are responsible for ∼40% of all HCM cases (2, 3). MYBPC3 encodes the thick filament protein cardiac myosin binding protein-C (cMyBP-C). HCM mutations in the MYBPC3 gene were first identified in 1995 (4, 5), and, to date, ∼200 mutations have been published (6). The precise functional and pathological consequences of MYBPC3 mutations are not fully characterized because mutant proteins are typically not found in cardiac tissue from affected patients (7, 8).
cMyBP-C is a critical regulator of cross-bridge cycling at its N terminus by altering the interaction between myosin, actin, and α-tropomyosin (α-TPM) (9, 10). The C-terminal amino acids of cMyBP-C are responsible for tight binding to the light meromyosin (LMM) region of myosin heavy chain (MyHC), as well as titin (11, 12), which provides sarcomeric stability and anchors cMyBP-C to the sarcomere (Fig. 1A). The most important regulatory role of cMyBP-C seems to be its effect on the cross-bridge cycling kinetics of sarcomere contraction (13, 14). It has been proposed that cMyBP-C acts as a structural constraint limiting cross-bridge formation and that phosphorylation of cMyBP-C accelerates cross-bridge kinetics, which is required for enhanced rates of relaxation and force development in diastole and systole, respectively (15, 16). cMyBP-C can be phosphorylated by PKA on at least four sites (9) and by glycogen synthase kinase3β (17), in addition to other kinases, such as PKC, PKD, ribosomal S6 kinase, and calcium/calmodulin-dependent kinase II (18).
FIGURE 1.

25-base pair deletion in MYBPC3. A, schematic representation of the 3′-end of MYBPC3 gene, showing the coding exons 30–34 (black), which contain the stop codon. The HCM-associated 25-base pair deletion in intron 32 leads to skipping of exon 33 and a frameshift. Because the original stop codon is missed by the frameshift, transcription of the entire exon 34 occurs, as well as part of the 3′-UTR. B, consequences of MYBPC3Δ25bp mutation on the protein level include changes in the C10 domain. C, in the mouse sequence, the C10 domain consists of amino acids 1177–1270. The MYBPC3Δ25bp mutation also causes a new sequence of amino acids starting at 1206 and is 15 amino acids shorter than the WT sequence. Only 2 of 50 amino acids are conserved between WT and C10 domain after amino acids 1206.
Among the many hundreds of individual mutations associated with HCM development, a highly prevalent 25-bp deletion mutation was recently discovered in the MYBPC3 gene (MYBPC3Δ25bp) (19–21). MYBPC3Δ25bp is estimated to occur in 55 million people worldwide (19). This mutation is pathogenic, because 90% of carriers over 60 years of age have developed HCM (19). Despite its high prevalence, the pathogenic mechanism is completely unknown. The 25-bp deletion leads to the skipping of exon 33 and a frameshift (Fig. 1B). The frameshift results in missing the stop codon at the beginning of exon 34, leading to the translation of exon 34, as well as part of the 3′-UTR. In the mouse sequence, this leads to replacement of the last 65 amino acids with a novel sequence of 50 amino acids (cMyBP-CC10mut) (Fig. 1C). Despite its pathogenicity and prevalence, the pathophysiological mechanism of this mutation is unknown. Most MYBPC3 mutations cause haploinsufficiency (8, 22), but cMyBP-CC10mut does not reduce total cMyBP-C levels (19). Therefore, it seems likely that the altered C10 domain is responsible for impairing the function of cMyBP-C by the so-called poison polypeptide effect. Although cMyBP-CC10mut has been associated with cardiomyopathies and an increased risk of heart failure, it is not known whether cMyBP-CC10mut expression alone is sufficient for the development of cardiomyopathy. Therefore, in this study, we aimed to determine in vitro whether cMyBP-CC10mut expression is, in fact, sufficient to impair protein stability and localization, thus causing contractile dysfunction in isolated rat cardiomyocytes. Our data did confirm the improper incorporation of cMyBP-CC10mut into the sarcomere by the loss of C10 domain interaction with the LMM region of MyHC. Under these conditions, cMyBP-CC10mut is less able to regulate cardiac contractility, thus providing evidence that HCM is likely caused by the cMyBP-CC10mut protein.
EXPERIMENTAL PROCEDURES
Isolating and Culturing of Adult Rat Cardiomyocytes
This study was conducted in accordance with the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health, using protocols approved by the Institutional Animal Care and Use Committee. Hearts were removed from heparinized and anesthetized adult male Sprague-Dawley rats (aged 3 months with average body weight of 300 g; Harlan Laboratories) and quickly mounted on a Radnotti perfusion apparatus. Cardiomyocytes were isolated as we described before (23). After isolation, Ca2+ concentration was increased stepwise to 1.3 mm. Isolated cardiomyocytes were plated on laminin-coated (10 μg/ml in PBS) 6-cm dishes for protein analysis, coverslips for cell shortening and Ca2+ transient measurements, or chamber slides for immunofluorescence studies. Isolation buffer was replaced by plating medium (Hanks' minimum essential medium; Invitrogen), 10% (v/v) fetal bovine serum, 10 mm 2,3-butanedione monoxime, and 100 units/ml penicillin/streptomycin). Cells (∼5 × 104 rod-shaped cells per dish) were plated on dishes and incubated at 95% O2, 2% CO2 for 1 h. Plating medium was replaced by culturing medium (Hanks' minimum essential medium, 10 mm 2,3-butanedione monoxime, 2 mm glucose, 0.1% (w/v) BSA, 1× insulin-transferrin-Selenium supplement (Sigma), and 100 units/ml penicillin/streptomycin preincubated at 37 °C, 95% O2, 2% CO2 for >1 h). Attached rod-shaped cells were counted with an eyepiece with a cell counting grid. Adenoviruses in multiplicity of infection were added after the cell count. Medium was changed daily, and cells were harvested 48 h after adenovirus infection.
Adenovirus Generation
Adenoviral vectors were generated in pShuttle-CMV utilizing the Stratagene AdEasy XL system (Stratagene, La Jolla, CA). Amplified adenoviruses (AV) were purified via ultracentrifugation through discontinuous cesium chloride gradients before dialysis into 10 mm Tris-HCl, 10 mm MgCl2 at pH 7.4 with 10% glycerol added as a cryopreservative. Titer was estimated by viral particle density at 260 nm (1 OD = 1012 particles/ml).
Adenoviral Infection of Smooth Muscle Cells
To create lysates containing 100% cMyc-cMyBP-C, A7r5 smooth muscle cells (ATCC, Manassas, VA) were used. These cells do not express cMyBP-C endogenously. A7r5 cells were plated on 60-mm2 dishes until 50% confluent and infected with cMyBP-CWT-AV (containing cMyc tag). Cells were cultured for 5 days.
Real Time PCR Analysis
Cultured adult rat ventricular cardiomyocytes were used to extract total mRNA by homogenization, using TRIzol and a Mini-Beadbeater (Biospec). Then chloroform precipitation and purification of total mRNA were performed using an RNA binding column (Bio-Rad 732-6830). Next, cDNA was synthesized using 1 μg of purified total mRNA and an iScript cDNA synthesis kit (Bio-Rad 170-8891). Quantitative PCR was performed with TaqMan primers against MYBPC3 (IDT Mm.PT.53a.2930640) and with a TaqMan GAPDH primer sequence (Applied Biosystems, Mm00486742m1) as a reference gene. Quantitative PCR and normalized gene expression were calculated as we described previously (22).
Relative Quantification of AV-derived cMyBP-C
Increasing amounts of A7r5 cells + cMyc-cMyBP-C lysate were loaded on a gel, together with lysates from rat cardiomyocytes infected with cMyBP-CWT and cMyBP-CC10mut AV. Proteins were transferred to hybond polyvinylidene difluoride membranes (GE Healthcare Life Sciences). Aspecific binding was blocked by incubating the membranes in SEA Block buffer (Thermo Scientific, Waltham, MA) for 1 h. Membrane was incubated simultaneously with mouse monoclonal antibody against cMyBP-C (E7; Santa Cruz Biotechnology Inc., Dallas, TX) and rabbit polyclonal antibody against cMyc (Roche). Cy3-conjugated anti-mouse IgG and Cy5-conjugated anti-rabbit IgG were used as secondary antibodies. Cy3 and Cy5 fluorescent signals were obtained on a Typhoon 9200 scanner (GE Healthcare Life Sciences). From the increasing amounts of A7r5 + cMyc-cMyBP-C lysates, a standard curve was made for both cMyBP-C (Cy3) and cMyc (Cy5) signals. When within the linear range, the linear regression line will give the following equations.
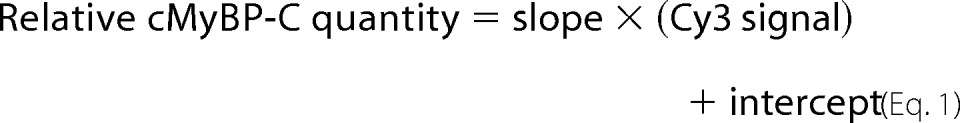 |
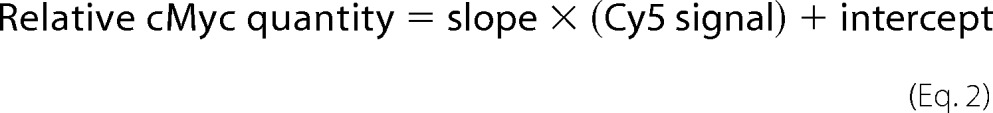 |
The percentage of AV-derived cMyBP-C (cMyc) in total cMyBP-C (cMyBP-C) in the cardiomyocytes infected with cMyBP-CWT and cMyBP-CC10mut is calculated as (relative quantity cMyc/Relative quantity cMyBP-C) × 100%.
Immunoblotting
Dishes were washed twice with PBS, and cells were harvested in SDS loading buffer. Immunoblotting was performed against cMyBP-C and its phosphorylation level at Ser-273, Ser-282, and Ser-302 sites, cMyc-tag, GAPDH, and α-TPM, as we described previously (24).
Unloaded Shortening and Ca2+ Transient Measurements
Forty-eight hours after AV infection with a high AV dose, which led to ∼50% mutant protein expression, cells were slowly adapted to Tyrode's solution (135 mm NaCl, 4 mm KCl, 1 mm CaCl2, 1 mm MgCl2, 10 mm glucose, and 10 mm HEPES, pH 7.4). After extensive washing to remove all 2,3-butanedione monoxime, cells were loaded with a Ca2+-sensitive fluorescent dye, Indo-1AM (Invitrogen), for 15 min. Excess dye was removed by washing with Tyrode's solution. Experiments were carried out at room temperature. Cells were electrically paced with pulses of 20V, 4 ms duration at 0.5 Hz. Sarcomere length was measured simultaneously with Ca2+ transients, recorded, and analyzed with IonOptix software as we previously described (24).
Subcellular Fractionation of Cultured Cardiomyocytes
Forty-eight hours after AV infection with a high AV dose, which led to ∼50% mutant protein expression, cultured cardiomyocytes were harvested in F60 + 1% (v/v) Triton X-100 (100 μl per 35-mm2 dish). A 25-μl aliquot was taken (total fraction), and the remainder was centrifuged (14,000 × g, 5 min, 4 °C). The supernatant was collected (soluble fraction), and the pellet (myofilament fraction) was homogenized in loading buffer.
Immunofluorescence Analysis
Immunofluorescence experiments on cultured adult cardiomyocytes were performed as we described previously (24). Primary antibodies used were mouse monoclonal anti-cMyc (Sigma) and rabbit polyclonal anti-α-actinin, both at a dilution of 1:200, and incubated overnight at 4 °C. Secondary antibody incubations were done with anti-rabbit IgG Alexa 488 and anti-mouse IgG Alexa 568 at a dilution of 1:100 for 1 h at room temperature. Immunofluorescence images were obtained on a Zeiss Axiovert 100 microscope (Carl Zeiss Microimaging, Thornwood, NY).
Recombinant Protein Expression
To generate recombinant protein encoding for human myosin LMM, the gene corresponding to this region was cloned into pET 28a+ with a His-tag. DNA encoding for mouse cardiac C8-C10WT (UniProt accession number O70468; residues 967–1270) and C8-C10mut (residues 967–1256) was subcloned into the pETite vector from the ExpressoTM SUMO T7 cloning and expression system from Lucigen® according to the manufacturer's protocol. Plasmids were confirmed with DNA sequencing. The C8-C10 plasmids were transformed into Escherichia coli HI-Control BL21 (DE3) (Lucigen, Middleton, WI) in accordance with the manufacturer's protocols. Routinely, cells were grown at 37 °C in Luria-Bertani medium containing 30 μg/ml kanamycin and shaken (250 rpm) until optical cell densities (A600) reached 0.8–1.0. Cells were induced with 0.5 mm isopropyl β-d-thiogalactopyranoside and grown at 30 °C. Cells were harvested 20 h postinduction by centrifugation at 4 °C for 30 min at 3700 × g. Cell pellets containing recombinant C8-C10WT or C8-C10mut were resuspended in buffer on ice containing 8 m urea, 500 mm NaCl, 10 mm imidazole, 20 mm Tris-HCl, and 1 mm phenylmethanesulfonyl fluoride at pH 8.0. Suspensions were lysed by sonication (FB-705) at 35 mA on ice for 15 s on and 45 s off for a total on time of 3 min. The lysate was centrifuged at 16,000 × g for 30 min at 4 °C. Soluble supernatant was loaded onto a 5-ml HisTrap HP Ni-Sepharose resin (GE Healthcare Life Sciences). Proteins were eluted using the same buffer with the addition of 500 mm imidazole. Eluted proteins were dialyzed against buffer at room temperature containing 6 m urea, 150 mm NaCl, 25 mm Tris, and 1 mm PMSF at pH 8.0. Protein concentrations were determined at A280 using a NanoDrop spectrometer (Thermo Scientific, Waltham, MA). Molecular extinction coefficients were individually calculated for C8-C10WT and C8-C10mut using the online ExPASy ProtParam tool (25), which is based on the method as described by Gill and von Hippel (26). The molecular extinction coefficient for C8-C10WT was 47900 m−1 cm−1, whereas that for C8-C10mut was 46,410 m−1 cm−1. Based on these values, both C8-C10WT and C8-C10mut recombinant protein concentrations were calculated and used in the co-sedimentation assay. Protein samples were stored at −20 °C. Both wild-type and C10 mutants were confirmed by Western blot analysis using C10 domain-specific polyclonal antibody and C10mut-specific custom monoclonal antibody (clone ID 8560-1-4M-4K14-1_120915; AbMART, Arlington, MA).
Co-sedimentation Assay
To determine whether C10mut alters C10 domain interaction with the myosin LMM region, a co-sedimentation assay was performed with either C8-C10WT or C8–10mut and myosin LMM 2.7–4 (amino acids 1554–1935) recombinant proteins as described previously by Flashman et al. (12). Purified recombinant proteins of C8-C10WT or C8–10mut and myosin LMM recombinant proteins were prespun in a Beckman TLA 100 rotor at 60,000 rpm for 20 min at 4 °C to remove particulates and precipitated proteins and then dialyzed with co-sedimentation buffer (20 mm imidazole, pH 7.4, 180 mm KCl, 1 mm MgCl2, 1 mm EGTA, and 1 mm DTT). Standard curves were generated to convert the intensity ratios of known amounts of recombinant C8-C10 proteins (4–40 pmol) and myosin LMM (40 pmol) in each pellet to molar ratios (mol of C8-C10/mol of myosin LMM; C8-C10/myosin LMM mol/mol ratio). To determine the binding affinity, myosin LMM (5 μm) was then mixed with various concentrations of recombinant C8-C10 proteins (1–30 μm) in Eppendorf tubes in co-sedimentation buffer, followed by incubation at 37 °C for 30 min and centrifugation for 30 min at 100,000 rpm at 4 °C in a TLA100 rotor in an Optima TLX Beckman ultracentrifuge (Beckman Coulter, Fullerton, CA). The supernatant was then removed, and the pellet was washed in the co-sedimentation buffer. The pellet was then redissolved in 20 μl of 2× SDS-PAGE loading buffer (Bio-Rad) and analyzed using SDS-PAGE. The gel was stained in Coomassie Brilliant Blue and imaged on a Chemidoc XRS (Bio-Rad) for protein bands. The image files (TIFF format) were then used to quantify the band intensities using ImageJ (National Institutes of Health, Bethesda, MD). Following the assay, bound C8-C10 was determined by comparing the band intensity ratios from each pellet fraction to the corresponding molar ratios provided in the standard curve. Saturation binding curves were generated by plotting the bound C8-C10 molar ratios versus the total C8-C10 concentrations added using the one-site binding (hyperbola) feature with a nonlinear regression curve fit in GraphPad Prism version 6.00 for Mac OS X (GraphPad Software, San Diego, CA). The maximum molar binding ratio (Bmax) and dissociation constant (Kd) for each assay were then determined as previously described by Shaffer et al. (27).
Cross-linking Studies
To verify co-sedimentation results, cross-linking studies were performed to determine whether C10mut alters C10 interaction with myosin LMM region as described previously by Dihazi and Sinz (28). 6 μm of recombinant proteins encoding C8–10WT and C8-C10mut of cMyBP-C and myosin LMM regions were used in a 400-μl reaction with 60 μm (10-fold) of the cross-linking reagent bis[sulfosuccinimidyl] suberate (Thermo Fisher Scientific, Inc., Rockford, IL) and 20 mm HEPES buffer, pH 7.7. The reaction was incubated at room temperature for 60 min and terminated by adding 5 μl of 50 mm Tris, pH 7.5. A control reaction without cross-linking reagent was prepared in the same manner without bis[sulfosuccinimidyl] suberate. The reaction samples were then run on SDS-PAGE, and the results were analyzed.
Homology Modeling
Tertiary structure prediction for C10WT and C10mut was accomplished using SWISS-MODEL Workspace (29, 30). The primary amino acid sequences of C10WT and C10mut were used as targets, and multiple template structures were identified. The top scoring homology models were selected for C10WT and C10mut. Model quality was assessed using QMEAN4 (31) scores, and each homology model was further subjected to PROCHECK analysis using the PDBsum server (32).
Statistical Analysis
Statistical analysis was performed using GraphPad Prism (version 6). Statistical difference between groups was first determined using one-way analysis of variance, followed by Tukey's post hoc test. A p value <0.05 was considered to be statistically significant.
RESULTS
Adenoviral-induced Expression of cMyBP-CC10mut in Adult Rat Cardiomyocytes
To express cMyBP-CC10mut in adult rat cardiomyocytes, AV infection was used in the present study. Cultured cardiomyocytes were infected with different levels of multiplicity of infection (MOI) of AV encoding cMyc-tagged cMyBP-CWT or cMyBP-CC10mut complementary DNA. To determine whether cMyBP-C transcript levels were increased in a MOI-dependent manner after 48 h of AV infection, real time PCR was performed to determine total transcript levels of cMyBP-C, followed by normalization with the expression level of housekeeping gene GAPDH. As expected, a significant increase in total cMyBP-C transcripts was achieved after 48 h of AV transduction (Fig. 2A). Next, profiles of total protein lysates were determined by SDS-PAGE, followed by SYPRO staining. Results showed that no gross changes were observed in overall protein expression after AV transduction (Fig. 2B). However, Western blot analyses with antibody against cMyc-tag revealed that an AV dose-dependent increase in expression of cMyBP-CWT and cMyBP-CC10mut exogenous proteins was seen (Fig. 2C). Quantitative data revealed that similar expression levels of exogenous cMyBP-CWT or cMyBP-CC10mut could be achieved 48 h postinfection (Fig. 2D). Viral expression of cMyBP-CWT seemed to lead to replacement of endogenous cMyBP-C, rather than addition, because total cMyBP-C levels were unchanged after AV infection, whereas in cMyBP-CC10mut, at last partial addition was observed because total cMyBP-C levels increased at high MOI (Fig. 2E).
FIGURE 2.
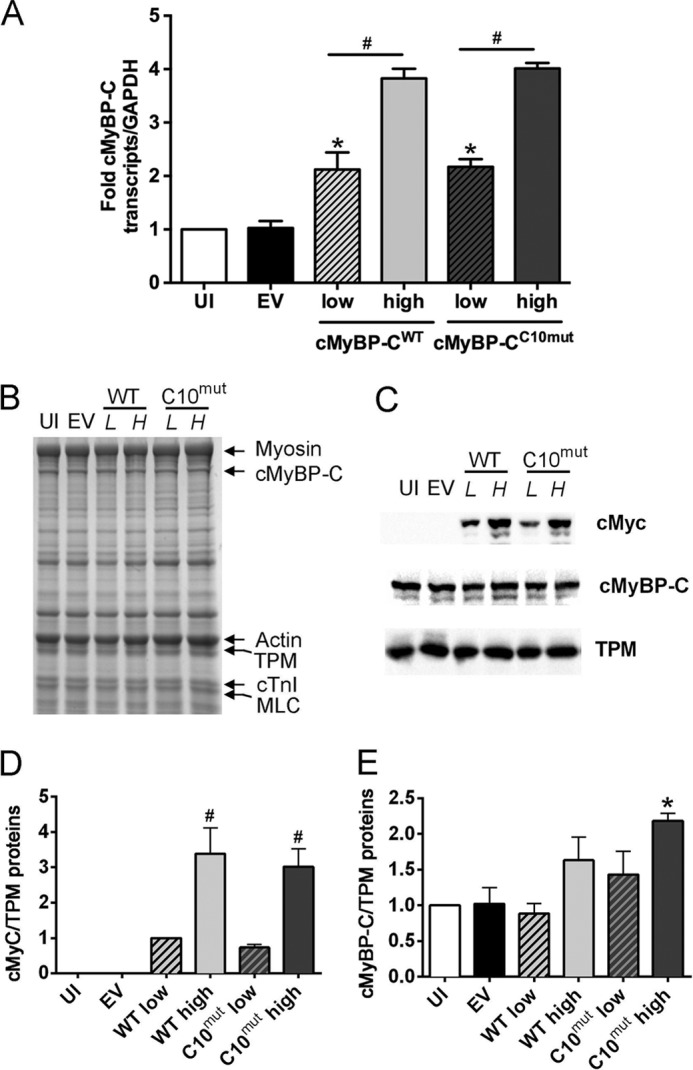
Adenoviral expression of cMyBP-CC10mut in cultured isolated rat cardiomyocytes. Isolated rat cardiomyocytes were cultured without infection (uninfected, UI), with adenovirus and without protein coding sequence (empty vector, EV), with cMyBP-CWT, or with cMyBP-CC10mut. Adenovirus-derived cMyBP-C (WT and C10mut) are N-terminally cMyc-tagged to distinguish AV-derived cMyBP-C from endogenous cMyBP-C. Low (L) and high (H) MOI of cMyBP-CWT and cMyBP-CC10mut were used to achieve dose-dependent protein expression. Cells were harvested 48 h postinfection. A, transcript levels of cMyBP-C relative to GAPDH expression and UI controls show a significant increase in cMyBP-C transcripts in a MOI-dependent manner. B, representative Coomassie-stained gel of total lysates of cultured cardiomyocytes show no apparent differences in overall protein levels. C, immunoblotting for cMyc (virus-derived cMyBP-C), cMyBP-C (endogenous + virus-derived cMyBP-C), and α-TPM as a loading control. D, quantification of cMyc levels, showing that similar levels of cMyBP-CWT and cMyBP-CC10mut were expressed 48 h postinfection. E, quantification of cMyBP-C levels, normalized with the levels of α-TPM, showed no significant increases in total cMyBP-C levels except cMyBP-CC10mut high MOI expression. The data in A, C, and D were averages ± S.E. from three independent experiments. *, p < 0.01 versus empty vector/uninfected; #, p < 0.01 versus respective low MOI virus titers (n = 4–6).
Quantification of cMyBP-CC10mut Expression
The mass difference between cMyBP-CC10mut and endogenous cMyBP-C was too small to resolve on SDS-PAGE (cMyBP-CWT: 140,632 Da; cMyBP-CC10mut: 138,466 Da). Therefore, quantification of the amount of expression of the AV-derived cMyBP-CC10mut was performed using a standard series (Fig. 3). The cMyc-tagged cMyBP-CWT was expressed in rat A7r5 smooth muscle cells, which had no endogenous cMyBP-C expression. Increasing amounts of these lysates were applied to a gel together with AV-infected cardiomyocytes. Subsequently, the amount of cMyBP-C (endogenous + AV-derived protein) and cMyc (only AV-derived protein) was measured with dual color fluorescent immunoblotting (Fig. 3A). Slopes of the cMyBP-C and cMyc standard series were used to calculate the percentage of cMyc-tagged cMyBP-C in total cMyBP-C (Fig. 3B). After 48 h of infection, AV-virus-derived cMyBP-CWT made up 44 ± 13% of total cMyBP-C, and cMyBP-CC10mut accounted for 36 ± 10% of total cMyBP-C (Fig. 3C).
FIGURE 3.
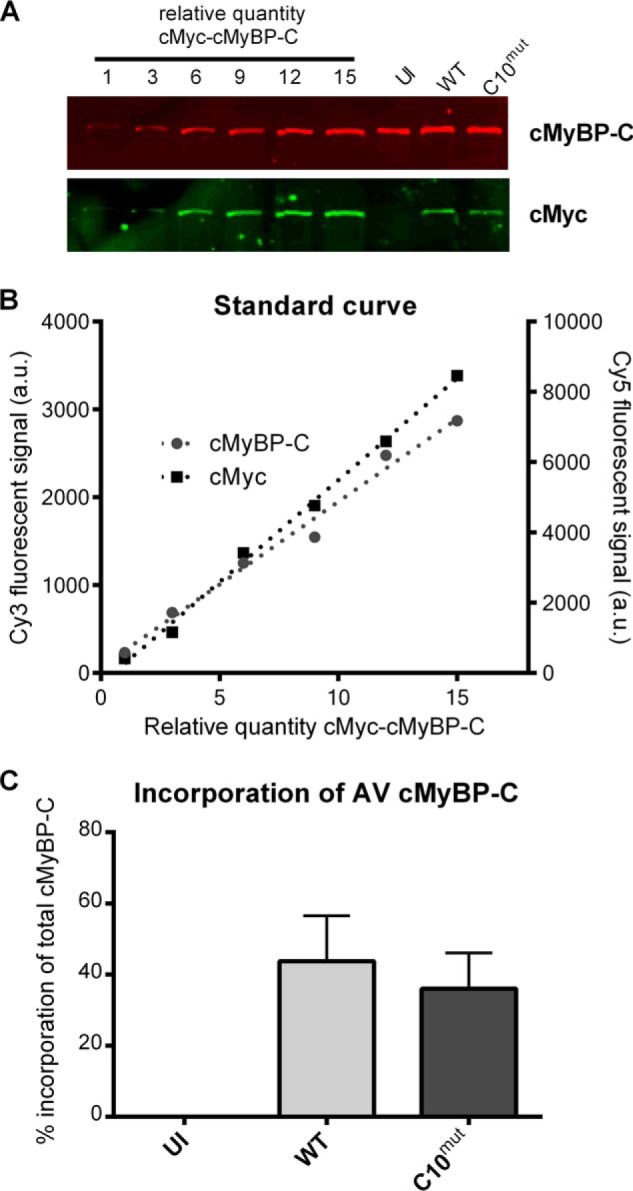
Relative quantification of AV-derived cMyBP-C. A, A7r5 smooth muscle cells with no endogenous cMyBP-C were infected with cMyBP-CWT-AV. A7R5 cell lysates were loaded in different amounts on gel, as well as cultured cardiomyocytes infected with cMyBP-CWT or cMyBP-CC10mut. After transfer, membranes were simultaneously incubated with cMyc (rabbit polyclonal) and cMyBP-C (mouse monoclonal) antibodies. Cy3-labeled anti-mouse IgG and Cy5-labeled anti-rabbit IgG secondary antibodies were used to measure total cMyBP-C (endogenous + virus-derived) and cMyc (virus-derived). B, Cy3 and Cy5 samples from A7r5 samples loaded in different amounts were used to make a standard curve. These signals were used to calculate percentages of virus-derived (cMyc) signal to total cMyBP-C (cMyBP-C). C, standard curve was used to calculate percentage of virus-derived cMyBP-C to total cMyBP-C present in total lysates 48 h postinfection without virus (uninfected, UI), with cMyBP-CWT-AV or with cMyBP-CC10mut-AV. The data are presented as means ± S.E. of three independent experiments.
Mislocalization of cMyBP-CC10mut in the Sarcomere
To assess whether AV-derived exogenous cMyBP-CWT and cMyBP-CC10mut proteins properly localized in the C-zone of the sarcomere, immunofluorescence staining was performed using rabbit polyclonal antibodies against N′-region of cMyBP-C and monoclonal antibody against cMyc, followed by confocal microscopy. Co-localization was determined by line scans using ImageJ software (National Institute of Health). Results show that expression of AV-derived cMyBP-CWT protein properly localized in the C-zone by the appearance of two distinct bands (doublets) between Z-lines, as visualized by α-actinin staining (Fig. 4, A and C), representing a characteristic localization pattern of cMyBP-C. Staining with cMyBP-C antibody, which recognizes both endogenous cMyBP-C and AV-derived cMyBP-CWT exogenous, showed exact co-localization with cMyc staining in the cMyBP-CWT-infected cells (Fig. 4, B and D), confirming that cMyc-tagged exogenous cMyBP-CWT protein properly localized in the C-zone of the sarcomere. In contrast, cMyBP-CC10mut localization was characterized by the absence of doublet patterns between Z-lines with either cMyBP-C or cMyc antibody. Furthermore, cMyc staining of cMyBP-CC10mut showed co-localization with α-actinin at the Z-line (Fig. 4, A, indicated with arrows, and C), which is an unusual localization pattern. Co-staining of cMyBP-CC10mut protein with cMyBP-C and cMyc antibodies showed only limited overlap in the C-zone (Fig. 4, B and D), confirming minimal localization of cMyBP-CC10mut in the C-zone of the sarcomere and more mislocalization to the Z-line in the sarcomere. Because cMyBP-CC10mut is mislocalized more in the Z-line, these data indicate that cMyBP-CC10mut targeting may contribute to the contractile dysfunction.
FIGURE 4.

Determination of localization of AV-derived cMyBP-C with immunofluorescence. Cultured rat cardiomyocytes infected for 48 h with cMyBP-CWT, cMyBP-CC10mut, or uninfected (UI). A, cells were stained for cMyc (green) and α-actinin (red) so that co-localization of signal would give a yellow signal in the merged images. In cells infected with cMyBP-CWT, the classical staining pattern of two cMyBP-C bands between Z-lines (stained with α-actinin) was seen, indicating proper localization of AV-virus-derived cMyBP-CWT. In uninfected cells, no nonspecific cMyc signal was detected. In cells infected with AV-expressing cMyBP-CC10mut, co-localization of cMyc and α-actinin signal was observed (arrows), with only minor signal between Z-lines. B, cells were stained for cMyc (green) and cMyBP-C (red). Note that the cMyBP-C antibody will bind to endogenous cMyBP-C, as well as AV-derived cMyBP-CWT and cMyBP-CC10mut. In UI and cMyBP-CWT, cMyBP-C signal showed doublet staining, with no staining in the Z-lines. cMyBP-C signal in cMyBP-CC10mut-infected cells showed the two cMyBP-C bands, with additional staining in the Z-line. Merging of the cMyc signal and the cMyBP-C signal mainly showed co-localization in the Z-line (arrows), with almost no overlaps in the C-zones. C, fluorescent line scans of figures in A show that cMyBP-CWT (green) forms a doublet between two Z-lines (red), as is expected for cMyBP-C. In contrast, cMyBP-CC10mut (green) only shows overlap within the Z-line marker α-actinin (red). D, fluorescent line scans of figures in B show that cMyBP-CWT (green, stained with cMyc) overlaps completely with total cMyBP-C (red, stains both endogenous and AV-derived cMyBP-C). In contrast, cMyBP-CC10mut (green) shows only minor overlap with endogenous cMyBP-C (red).
Subcellular Localization of cMyBP-CC10mut: Subcellular Fractionation
To quantify the amount of cMyBP-CC10mut that localizes to the myofilament fraction, subcellular fractionation of the cultured cardiomyocytes was performed. Cardiomyocyte lysates (total fraction) were separated into detergent-soluble fraction (mainly cytosolic fraction) and myofilament fraction (Fig. 5A). Fractionation was shown to be successful with GAPDH (cytosolic marker) and α-TPM (myofilament marker) exclusively present in the expected fractions (Fig. 5B). In uninfected cardiomyocytes and cardiomyocytes infected with EV, no cMyBP-C was seen in the soluble fraction (Fig. 5C). In contrast, cMyBP-C was observed in the soluble fraction in the cMyBP-CWT-infected cells and especially cMyBP-CC10mut-infected cells (Fig. 5C). The higher level of soluble cMyBP-C in the cMyBP-CC10mut AV cells compared with cMyBP-CWT AV cells resulted from reduced incorporation of cMyBP-CC10mut into the sarcomere (Fig. 5, C and D), as illustrated by the reduced cMyc signal.
FIGURE 5.
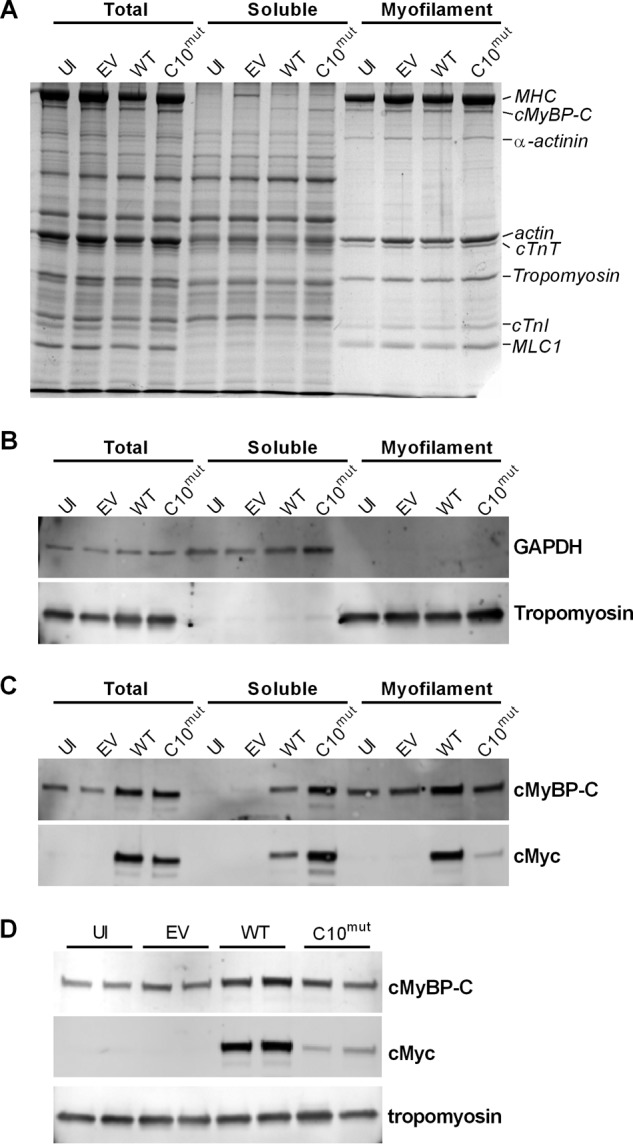
Determination of localization of AV-derived cMyBP-C with subcellular fractionation. Cultured cardiomyocytes were homogenized in buffer containing 1% Triton X-100 and centrifuged. Supernatant was named soluble, whereas the pellet was named myofilament fraction. A, Coomassie-stained SDS-polyacrylamide gel of total, soluble, and myofilament fractions. B, high fractionation efficiency was achieved as shown by using markers for cytosol (GAPDH) and myofilament (α-TPM). C, dual color immunoblots incubated with antibodies against cMyBP-C (endogenous + virus-derived cMyBP-C) and cMyc (virus-derived cMyBP-C). No cMyBP-C was detected in soluble fraction of uninfected (UI) or empty vector (EV) fractions, whereas it was detected in cMyBP-CWT and, especially, in cMyBP-CC10mut. cMyc signal shows the lower incorporation of cMyBP-CC10mut into the sarcomere. D, immunoblots of myofilament fractions of UI, EV, cMyBP-CWT, and cMyBP-CC10mut cells showed that cMyBP-CC10mut incorporated into the sarcomere at a lower level than cMyBP-CWT.
Effect of cMyBP-CC10mut Expression on Unloaded Shortening and Ca2+ transients of Isolated Cardiomyocytes
For functional analysis of the effect of cMyBP-CC10mut, unloaded shortening experiments were performed in cultured cardiomyocytes. Two days after AV infection, cells were loaded with the Ca2+-sensitive fluorophore indo1-AM, and both cell shortening and Ca2+ transients were measured in cells stimulated at 0.5 Hz (Fig. 6A). cMyBP-CC10mut expression significantly reduced contractility (Fig. 6, B and C), velocity of shortening (Fig. 6D), and relaxation (Fig. 6E). These changes in contractility were not explained by changes in calcium handling (Fig. 7A), because neither calcium transient amplitude (Fig. 7B) nor calcium transient decay (Fig. 7C) was changed. Similar results for sarcomere shortening and Ca2+ transients were obtained when cells were paced at 1 Hz (data not shown). Next, to determine whether expression of cMyBP-CC10mut alters total phosphorylation levels of cMyBP-C at Ser-273, Ser-282, and Ser-302, Western blot analyses were performed using phospho-specific antibodies. The results showed that the total cMyBP-C phosphorylation level at Ser-273, Ser-282, and Ser-302 was not significantly altered by the expression of cMyBP-CC10mut compared with control groups (Fig. 7D). Taken together, these data suggest that expression of cMyBP-CC10mut in adult rat cardiomyocytes in vitro is sufficient to cause contractile dysfunction without affecting Ca2+ transients or phosphorylation status.
FIGURE 6.

Contractile function after cMyBP-CC10mut expression. Isolated rat cardiomyocytes were uninfected (UI) or infected with either cMyBP-CWT or cMyBP-CC10mut and cultured for 48 h. Unloaded shortening was measured in these cells. A, example of isolated rat cardiomyocyte in which sarcomere length and Ca2+ transients were measured simultaneously. B and C, fractional shortening was reduced in cMyBP-CC10mut-infected cells, compared with uninfected or cMyBP-CWT-expressing cells. D and E, the kinetics of contraction was also disturbed after cMyBP-CC10mut expression, with slower shortening velocity (D) and relaxation velocity (E) compared with uninfected cells and cMyBP-CWT. The data are means ± S.E. of four independent experiments. #, p < 0.05 in cMyBP-CC10mut versus uninfected; *, p < 0.05 in cMyBP-CC10mut versus cMyBP-CWT.
FIGURE 7.

Calcium transients and cMyBP-C phosphorylation levels in cultured cardiomyocytes. Concomitant with unloaded shortening, Ca2+ transients were measured. Cells were loaded with the Ca2+-sensitive fluorescent dye Indo-1. Ca2+ release into the cytosol was determined by measuring the ratio between Ca2+-bound Indo signal (at 405 nm) and Ca2+-unbound Indo signal (at 480 nm). A, representative traces of Ca2+ transients of uninfected (UI), cMyBP-CWT- and cMyBP-CC10mut-infected cells. B, no changes were seen in amplitude of the Ca2+ transient. C, Ca2+ reuptake kinetics (displayed as tau) was also unaffected by cMyBP-CC10mut expression. The data are means ± S.E. of four independent experiments. D, representative Western blot analyses of cMyBP-C phosphorylation at Ser-273 (Ser-273p), Ser-282 (Ser-282p), and Ser-302 (Ser-302p) sites using phospho-specific antibodies. cMyBP-C levels, as measured by Ponceau S staining, and α-TPM levels, as assessed by Western blot, were used as loading controls. 10 μg of total cultured cardiomyocyte lysate was used to perform the Western blot analyses. Quantitative data showed no significant difference among all the groups (data not shown).
cMyBP-CC10mut Failed to Interact with LMM Region of MyHC
Previous studies determined that the C10 domain of cMyBP-C interacts with the myosin LMM region (12), which is necessary for cMyBP-C stability in the sarcomere. In the present study, we hypothesized that cMyBP-CC10mut has lost its ability to interact with the myosin LMM region. To test this hypothesis, three recombinant proteins, including C8-C10WT, C8-C10mut, and myosin LMM, were generated and purified by the Ni-NTA-agarose system (Fig. 8A). Both C8-C10WT and C8-C10mut were confirmed by Western blot analyses using C10WT- and C10mut-specific antibodies (Fig. 8B). Then a co-sedimentation assay was performed using either recombinant proteins of C8-C10WT or C8-C10mut with myosin LMM recombinant proteins. Results showed that C8-C10WT, but not C8-C10mut, interacts with myosin LMM protein (Fig. 8, C–E). Furthermore, a dose-dependent interaction was determined using C8-C10WT recombinant proteins with a dissociation constant (Kd) of 9.8 ± 1.8 μm and a molar binding ratio (Bmax) of 1.14 ± 0.08 (Fig. 8, F and G). In agreement with previous studies (12), our results showed that C8-C10WT strongly interacts with the myosin LMM region at 1:1 (mol/mol) equimolar concentration. In contrast, the presence of C10mut caused the loss of C8-C10mut interaction with myosin LMM (Fig. 8G); therefore, no binding affinities could be determined, suggesting a possible mechanism for the development of HCM disease through impaired sarcomere anchoring. To validate these results, a cross-linking assay was carried out with bis[sulfosuccinimidyl] suberate cross-linking agent, and the products were resolved on a 12% SDS-PAGE (Fig. 8H). As expected, C8-C10WT protein interacted with myosin LMM and showed an upper band of ∼75 kDa in a position, which is absent in the cMyBP-CC10mut reaction, confirming that cMyBP-CC10mut has completely lost C10 domain interaction with myosin LMM.
FIGURE 8.

cMyBP-CC10mut has lost C10 domain interaction with myosin LMM. A, SDS-PAGE analysis followed by Coomassie Blue staining demonstrated the purity of His-tagged recombinant C8-C10WT, C8-C10mut, and myosin LMM proteins. B, various concentrations of C8-C10 recombinant proteins were used to show purity using anti-C10WT rabbit antibodies and anti-C10mut monoclonal antibodies. Ponceau staining was used to verify equal loading on the nitrocellulose membrane. C, prior to the co-sedimentation assay, recombinant proteins were individually tested for their sedimentation capacity. Net protein content before centrifugation (total (T)) and after centrifugation in the supernatants (S) and pellets (P) was determined by SDS-PAGE and Coomassie Blue staining of the gels. D, co-sedimentation of either C8-C10WT or C8-C10mut with LMM proteins. All protein concentrations were 6 μm in the assay at 250,000 × g for 10 min at 4 °C. Co-sedimentation between C8-C10WT and LMM indicates interaction, as illustrated by the presence of C8-C10WT in the pellet, whereas C8-C10mut showed little binding. E, quantitative data summarized the amount of C8-C10WT and C8-C10mut proteins in the supernatants (S) and pellets (P) after centrifugation (n = 3). *, p < 0.001. F, a representative standard curve showing a range of known molar concentrations (4–40 pmol) of C8-C10WT to myosin LMM (40 pmol) on the x axis that were blotted against the SDS-PAGE band intensity on the y axis. G, co-sedimentation assay was performed with varying concentration (1–30 μm) of either C8-C10WT or C8-C10mut to 5 μm of myosin LMM to determine the Kd and Bmax values. The Kd and Bmax values for C8-C10WT (circles) were of 9.8 ± 1.8 and 1.14 ± 0.08 mol/mol (n = 5, means±S.E.). Because C8-C10mut (squares) did not bind to myosin LMM, no significant Kd and Bmax values could be determined (n = 5, means ± S.E.). H, cross-linking analysis of either C8-C10WT or C8-C10mut with LMM showed cross-linking of C8-C10WT with LMM (arrow), indicating interaction, whereas C8-C10mut did not cross-link to LMM. BS3, bis[sulfosuccinimidyl] suberate.
C10WT and C10mut Tertiary Structure Analyses Using Homology Modeling
No high resolution structures are available for cMyBP-C or the C10 domain. Therefore, protein homology modeling using SWISS-MODEL Workspace was used to predict the tertiary structures of C10WT and C10mut. Template sequences (Protein Data Bank codes 1U2H and 2J8H) were identified for C10WT and C10mut, respectively. Each template selected for modeling had a minimum of 28% sequence identity to and 58% sequence coverage for the C10 domain. In the C10mut model, the C-terminal portion was built in as a loop because this region had lower sequence coverage with the selected templates as compared with C10WT. Valid models of C10WT and C10mut were determined by QMEAN4 and PROCHECK analyses, and top scoring models were selected as representative structures. Both C10WT and C10mut models had >90% of residues in the most favorable regions as indicated by PROCHECK analyses. Based on the secondary structure content visible in each model, C10WT largely consisted of β-sheet secondary structure (Fig. 9A). In direct contrast, C10mut exhibited a marked reduction in β-sheet composition (Fig. 9B). Additionally, the C10WT model exhibited a well folded global structure, whereas C10mut was less structurally defined, particularly in the C-terminal region.
FIGURE 9.
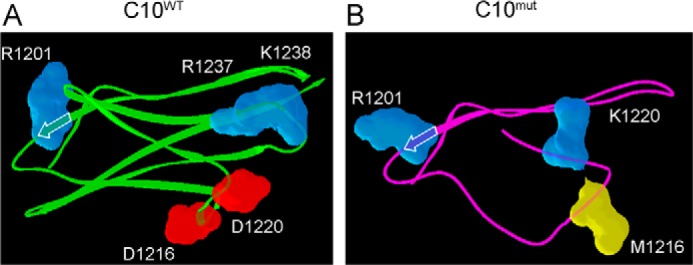
Homology models of C10WT and C10mut domains. The top scoring homology models were selected as representative structures for the C10 domains. β-Sheets are indicated by arrows. LMM-binding residues in C10WT (A) are labeled. The corresponding amino acids in C10mut (B) are labeled.
DISCUSSION
The outcomes of this study showed that the expression of cMyBP-CC10mut is sufficient to cause contractile impairment in isolated rat cardiomyocytes in vitro. This reduced contractility was caused by impaired myofilament function, because no differences were observed in amplitude and kinetics of Ca2+ transients. Improper localization of cMyBP-CC10mut occurs, as illustrated by 1) reduced incorporation in the myofilament fraction, 2) predominant Z-disc localization versus wild-type localization to the C-zone, and 3) loss of cMyBP-CC10mut interaction with the myosin LMM region. These data suggest that cMyBP-CC10mut localizes improperly in the sarcomere and may bind to other sarcomeric proteins, thus acting as a “poison polypeptide” to reduce contractility.
cMyBP-CC10mut Fails to Incorporate into the Sarcomere
The 25-bp deletion in intron 32 of MYBPC3 leads to skipping of exon 33 and a frameshift. The consequences of this mutation on the protein level are changes in most residues of the C-terminal domain, i.e. the C10 domain. For correct incorporation into the C-zone of the sarcomere, domains C7–C10 are important, with domain C10 being vital for incorporation (33). This localization is mediated through interactions with the LMM region of MyHC (11, 34) and titin (35, 36). The interaction between titin C-zone immunoglobulin domains and MyBP-C occurs within domains C8-C10 (35). The LMM of MyHC makes up the rod region of the thick filament, and interaction with cMyBP-C occurs through the C10 domain of cMyBP-C (34, 37, 38). Specifically, a set of five charged amino acids were identified to be responsible for LMM binding (34). On LMM, the region stretching from amino acids 1554 to 1581 was found to be the binding domain of C10 (12). Based on the predicted LMM-binding region and the structural insights derived from homology modeling studies for the C10 domain, the tertiary structure altered by cMyBP-CC10mut could be expected to reduce affinity for LMM. Of the five identified amino acids necessary for LMM binding (34), only Arg-1201 remains unchanged in cMyBP-CC10mut. Asp-1216, Asp-1220, Arg-1237, and Lys-1238 are all changed. The negatively charged Asp-1216 and Asp-1220 are changed to uncharged methionine and the positively charged lysine, respectively, whereas the positively charged Arg-1237 and Lys-1238 are changed to the uncharged phenylalanine and serine. In our homology models, the LMM-binding surface was diminished, and the global fold was destabilized in C10mut as compared with C10WT. Therefore, we proposed that cMyBP-CC10mut alters the incorporation of cMyBP-C into the sarcomere. Transgenic expression of a cMyc-tagged cMyBP-C in mouse hearts does not result in any aberrant phenotype and does not affect the overall stoichiometry of cMyBP-C or other sarcomeric proteins (39). Similarly, although cMyBP-CWT transcripts were increased in a MOI-dependent manner, total cMyBP-CWT protein levels in the cardiomyocytes were unchanged in vitro, suggesting that cMyBP-C stoichiometry was maintained by the overexpression of cMyBP-CWT transcripts using AV. In contrast, cMyBP-CC10mut protein levels were significantly increased in the cardiomyocytes, indicating that cMyBP-CC10mut protein incorporation and regulatory pathways were altered by cMyBP-CC10mut. To further support these findings, cMyBP-CC10mut levels in the myofilament fraction were found to be much lower than those of cMyBP-CWT, and levels of cMyBP-CC10mut were higher in the cytoplasm. Moreover, cMyBP-CC10mut only had minor C-zone staining and appeared more at the Z-disc (Fig. 4, A and C), indicating a lack of C-terminal interaction with myosin LMM, leading, in turn, to the differential localization of cMyBP-CC10mut in the sarcomere. However, it is unknown whether this Z-line localization has direct functional implications. Interestingly, cross-linking and co-sedimentation experiments confirm that cMyBP-CC10mut failed to interact with the LMM region in vitro. Taken together, then, cMyBP-CC10mut mutation causes sufficient structural changes in the C10 domain to reduce its affinity for LMM and prevent proper localization of the protein. Partial incorporation of cMyBP-CC10mut full-length protein through its N-terminal region in the sarcomere fraction may result from its interaction with other proteins.
cMyBP-CC10mut Is Sufficient to Cause Contractile Dysfunction in Vitro
Approximately 40% of identified HCM mutations are in MYBPC3 (2, 3), of which 70% are either truncation or frameshift mutations, making it the most common cause of HCM. To date, ∼200 individual mutations in MYBPC3 have been identified, with the prevalence of individual mutations being low (6). Notable exceptions to this limited distribution of individual mutations can be found, including, for example, the 2373insG founder mutation, which is responsible for almost 25% of all HCM mutations in The Netherlands (40), or the recently described c.927–2A>G founder mutation that occurs in almost 60% of Icelandic HCM patients (41). However, the most prevalent HCM-associated mutation is the 25-bp deletion mutation, which is estimated to occur in 55 million people worldwide (19). This deletion mutation was first identified in 2003 (21) and occurs almost exclusively in South Asian populations (19, 42). Previous studies have only shown an association between the MYBPC3 25-bp deletion and cardiomyopathies. In the present study, we show that expression of cMyBP-CC10mut in isolated cardiomyocytes from healthy rats is enough to cause contractile dysfunction. The contractile dysfunction could not be explained by changes in Ca2+ transients; therefore, such dysfunction likely results from direct impairment of sarcomeric function. Even though the C10 domain mutation prevents proper anchoring in the C-zone of the mutant protein, the N terminus is still functional, indicating possible interaction of cMyBP-CC10mut with actin and myosin leading to improper contraction. To support this notion, the phosphorylation level of cMyBP-C at Ser-273, Ser-282, and Ser-302 sites, which are located in the N-terminal region (M-domain) of cMyBP-C, was preserved in the cMyBP-CC10mut-expressing cardiomyocytes. These data suggest that a normal level of cMyBP-CC10mut phosphorylation at these sites is sufficient to regulate its dynamic interaction with actin and myosin S2 region, even though cMyBP-CC10mut is not present in the C-zone. Thus, it is possible that the N-terminal of cMyBP-CC10mut transiently interacts with myosin and actin in and outside the C-zone, preventing normal regulation by cMyBP-C. This could lead to contractile dysfunction because incubation of an N-terminal cMyBP-C fragment in permeabilized human myocardium caused contractile dysfunction by preventing endogenous cMyBP-C from regulating cross-bridge cycling (10). Furthermore, severe cardiac dysfunction, hypertrophy, and ultimately heart failure developed in a transgenic mouse model expressing the same N-terminal fragment (43). Part of the contractile dysfunction might be caused by changes in Ca2+ sensitivity of force development. N-terminal fragments have been shown to cause alterations in Ca2+ sensitivity, with some decreasing it (10), whereas other fragments have been shown to cause an increase (44, 45). Such a disruptive effect of cMyBP-CC10mut expression on contractile function might be explained by interference with the still functional N terminus of cMyBP-C. Our studies clearly determined that 40% expression of cMyBP-CC10mut is sufficient to result in contractile dysfunction.
cMyBP-CC10mut Disease Mechanism
Reports in the literature suggest that cMyBP-C HCM-associated mutant proteins cause disease through poison polypeptide effects, haploinsufficiency, or ubiquitin-proteasome system impairment. Most of the truncation mutations in the C-terminal portion of cMyBP-C lead to haploinsufficiency (6), as the truncated proteins were never found in the heart (7, 8). This leads to a reduced cMyBP-C protein level, because the healthy allele can only partly compensate. If the carriers are homozygous, then C-terminally truncated mutations result in a cMyBP-C null phenotype, leading to HCM at early ages (46). In patients with the MYBPC3Δ25bp mutation, it was reported that no mutant protein could be detected in heart tissue (19). However, this was based on protein analysis in two tissue samples and on the assumption that the mutant protein would have a different migration speed on SDS-PAGE. Because the difference in molecular mass between cMyBP-C (140,762 Da) and cMyBP-CC10mut (computed molecular mass, 139,405 Da) is less than 1%, the two proteins are likely to run at the same height. In fact, in our experiments, this did happen (Fig. 2B). Therefore, experiments with mutant-specific antibodies are needed to truly determine whether and how much mutant protein is present in human myocardium.
Conversely, with missense mutations and mutations with partial modifications like cMyBP-CC10mut, the mechanism of HCM disease development is entirely different. The full-length protein is expressed and either completely or partially incorporated into the sarcomere, which would then cause poison polypeptide effects that result in contractile dysfunction. Based on the results obtained from the present in vitro study, cMyBP-CC10mut could act as such a poison polypeptide. Although cMyBP-CC10mut is expressed and localized improperly, it affects contraction without changing the levels of cMyBP-C in the myofilament. This effect could be mediated by its still intact N terminus, which, if not anchored correctly into the sarcomere, would cause contractile dysfunction (10, 43).
An alternative, albeit not mutually exclusive, pathomechanism for cMyBP-CC10mut could be through impairment of the ubiquitin-proteasome system (UPS). cMyBP-C mutants have been described to be targeted by the UPS (47, 48). The large supply of mutant protein can overwhelm the UPS, eventually leading to impairment of the system (47, 49). Indeed HCM patients with mutations have lower proteasome activity than patients in whom no mutation was identified (50). The impairment of the UPS may have a number of consequences that could contribute to the disease process. It could lead to an accumulation of malfunctioning proteins, including cMyBP-C. It could also lead to the formation of aggregates of unfolded proteins. Because the UPS system decreases in the heart with age (51), UPS impairment could also be an explanation for the marked age-dependent increase in disease penetrance in MYBPC3Δ25bp mutation carriers (19).
In conclusion, we have, for the first time, shown that expression of the HCM-associated cMyBP-CC10mut is sufficient to cause contractile dysfunction in cultured adult rat cardiomyocytes in vitro. The mutated C10 domain of the protein prevents proper localization and anchoring of the protein in the sarcomere. Further studies are in progress using both genetically engineered mouse models and human induced pluripotent stem cell-derived cardiomyocytes to define the physiology of the disease mechanism. Determining the pathogenic mechanism, which will help in developing therapeutic approaches, is the first priority to prevent the development of heart failure in millions of carriers worldwide.
This work was supported, in whole or in part, by National Institutes of Health Grants R01HL105826 and K02HL114749 (to S. S.). This work was also supported by American Heart Association Midwest Affiliate Research Programs Grant-in-Aid 14GRNT20490025 and Postdoctoral Fellowships 13POST17220009 (to D. W. D. K.) and 13POST14720024 (to S. G.).
- HCM
- hypertrophic cardiomyopathy
- AV
- adenoviruses
- LMM
- light meromyosin
- MyHC
- myosin heavy chain
- α-TPM
- α-tropomyosin
- MOI
- multiplicity of infection
- UPS
- ubiquitin-proteasome system.
REFERENCES
- 1. Maron B. J., Doerer J. J., Haas T. S., Tierney D. M., Mueller F. O. (2009) Sudden deaths in young competitive athletes: analysis of 1866 deaths in the United States, 1980–2006. Circulation 119, 1085–1092 [DOI] [PubMed] [Google Scholar]
- 2. Alcalai R., Planer D., Culhaoglu A., Osman A., Pollak A., Lotan C. (2007) Acute coronary syndrome vs nonspecific troponin elevation: clinical predictors and survival analysis. Arch. Intern. Med. 167, 276–281 [DOI] [PubMed] [Google Scholar]
- 3. Richard P., Charron P., Carrier L., Ledeuil C., Cheav T., Pichereau C., Benaiche A., Isnard R., Dubourg O., Burban M., Gueffet J. P., Millaire A., Desnos M., Schwartz K., Hainque B., Komajda M. (2003) Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 107, 2227–2232 [DOI] [PubMed] [Google Scholar]
- 4. Bonne G., Carrier L., Bercovici J., Cruaud C., Richard P., Hainque B., Gautel M., Labeit S., James M., Beckmann J., Weissenbach J., Vosberg H. P., Fiszman M., Komajda M., Schwartz K. (1995) Cardiac myosin binding protein-C gene splice acceptor site mutation is associated with familial hypertrophic cardiomyopathy. Nat. Genet. 11, 438–440 [DOI] [PubMed] [Google Scholar]
- 5. Watkins H., Conner D., Thierfelder L., Jarcho J. A., MacRae C., McKenna W. J., Maron B. J., Seidman J. G., Seidman C. E. (1995) Mutations in the cardiac myosin binding protein-C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nat. Genet. 11, 434–437 [DOI] [PubMed] [Google Scholar]
- 6. Harris S. P., Lyons R. G., Bezold K. L. (2011) In the thick of it: HCM-causing mutations in myosin binding proteins of the thick filament. Circ. Res. 108, 751–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rottbauer W., Gautel M., Zehelein J., Labeit S., Franz W. M., Fischer C., Vollrath B., Mall G., Dietz R., Kübler W., Katus H. A. (1997) Novel splice donor site mutation in the cardiac myosin binding protein-C gene in familial hypertrophic cardiomyopathy. Characterization of cardiac transcript and protein. J. Clin. Invest. 100, 475–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van Dijk S. J., Dooijes D., dos Remedios C., Michels M., Lamers J. M., Winegrad S., Schlossarek S., Carrier L., ten Cate F. J., Stienen G. J., van der Velden J. (2009) Cardiac myosin binding protein-C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation 119, 1473–1483 [DOI] [PubMed] [Google Scholar]
- 9. Barefield D., Sadayappan S. (2010) Phosphorylation and function of cardiac myosin binding protein-C in health and disease. J. Mol. Cell Cardiol. 48, 866–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Witayavanitkul N., Ait Mou Y., Kuster D. W., Khairallah R. J., Sarkey J., Govindan S., Chen X., Ge Y., Rajan S., Wieczorek D. F., Irving T., Westfall M. V., de Tombe P. P., Sadayappan S. (2014) Myocardial infarction-induced N-terminal fragment of cardiac myosin-binding protein C (cMyBP-C) impairs myofilament function in human myocardium. J. Biol. Chem. 289, 8818–8827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Starr R., Offer G. (1978) The interaction of C-protein with heavy meromyosin and subfragment-2. Biochem. J. 171, 813–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Flashman E., Watkins H., Redwood C. (2007) Localization of the binding site of the C-terminal domain of cardiac myosin binding protein-C on the myosin rod. Biochem. J. 401, 97–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stelzer J. E., Dunning S. B., Moss R. L. (2006) Ablation of cardiac myosin binding protein-C accelerates stretch activation in murine skinned myocardium. Circ. Res. 98, 1212–1218 [DOI] [PubMed] [Google Scholar]
- 14. Stelzer J. E., Patel J. R., Walker J. W., Moss R. L. (2007) Differential roles of cardiac myosin-binding protein C and cardiac troponin I in the myofibrillar force responses to protein kinase A phosphorylation. Circ. Res. 101, 503–511 [DOI] [PubMed] [Google Scholar]
- 15. Tong C. W., Stelzer J. E., Greaser M. L., Powers P. A., Moss R. L. (2008) Acceleration of crossbridge kinetics by protein kinase A phosphorylation of cardiac myosin binding protein-C modulates cardiac function. Circ. Res. 103, 974–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Korte F. S., McDonald K. S., Harris S. P., Moss R. L. (2003) Loaded shortening, power output, and rate of force redevelopment are increased with knockout of cardiac myosin binding protein-C. Circ. Res. 93, 752–758 [DOI] [PubMed] [Google Scholar]
- 17. Kuster D. W., Sequeira V., Najafi A., Boontje N. M., Wijnker P. J., Witjas-Paalberends E. R., Marston S. B., Dos Remedios C. G., Carrier L., Demmers J. A., Redwood C., Sadayappan S., van der Velden J. (2013) GSK3β phosphorylates newly identified site in the proline-alanine-rich region of cardiac myosin-binding protein C and alters cross-bridge cycling kinetics in human: short communication. Circ. Res. 112, 633–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bardswell S. C., Cuello F., Kentish J. C., Avkiran M. (2012) cMyBP-C as a promiscuous substrate: phosphorylation by non-PKA kinases and its potential significance. J. Muscle Res. Cell Motil. 33, 53–60 [DOI] [PubMed] [Google Scholar]
- 19. Dhandapany P. S., Sadayappan S., Xue Y., Powell G. T., Rani D. S., Nallari P., Rai T. S., Khullar M., Soares P., Bahl A., Tharkan J. M., Vaideeswar P., Rathinavel A., Narasimhan C., Ayapati D. R., Ayub Q., Mehdi S. Q., Oppenheimer S., Richards M. B., Price A. L., Patterson N., Reich D., Singh L., Tyler-Smith C., Thangaraj K. (2009) A common MYBPC3 (cardiac myosin binding protein-C) variant associated with cardiomyopathies in South Asia. Nat. Genet. 41, 187–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kuster D. W., Sadayappan S. (2014) MYBPC3's alternate ending: consequences and therapeutic implications of a highly prevalent 25 bp deletion mutation. Pflugers Arch. 466, 207–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Waldmüller S., Sakthivel S., Saadi A. V., Selignow C., Rakesh P. G., Golubenko M., Joseph P. K., Padmakumar R., Richard P., Schwartz K., Tharakan J. M., Rajamanickam C., Vosberg H. P. (2003) Novel deletions in MYH7 and MYBPC3 identified in Indian families with familial hypertrophic cardiomyopathy. J. Mol. Cell Cardiol. 35, 623–636 [DOI] [PubMed] [Google Scholar]
- 22. Barefield D., Kumar M., de Tombe P. P., Sadayappan S. (2014) Contractile dysfunction in a mouse model expressing a heterozygous MYBPC3 mutation associated with hypertrophic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 306, H807–H815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sarkey J. P., Chu M., McShane M., Bovo E., Ait Mou Y., Zima A. V., de Tombe P. P., Kartje G. L., Martin J. L. (2011) Nogo-A knockdown inhibits hypoxia/reoxygenation-induced activation of mitochondrial-dependent apoptosis in cardiomyocytes. J. Mol. Cell Cardiol. 50, 1044–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Govindan S., Sarkey J., Ji X., Sundaresan N. R., Gupta M. P., de Tombe P. P., Sadayappan S. (2012) Pathogenic properties of the N-terminal region of cardiac myosin binding protein-C in vitro. J. Muscle Res. Cell Motil. 33, 17–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wilkins M. R., Gasteiger E., Bairoch A., Sanchez J. C., Williams K. L., Appel R. D., Hochstrasser D. F. (1999) Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 112, 531–552 [DOI] [PubMed] [Google Scholar]
- 26. Gill S. C., von Hippel P. H. (1989) Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182, 319–326 [DOI] [PubMed] [Google Scholar]
- 27. Shaffer J. F., Kensler R. W., Harris S. P. (2009) The myosin-binding protein C motif binds to F-actin in a phosphorylation-sensitive manner. J. Biol. Chem. 284, 12318–12327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dihazi G. H., Sinz A. (2003) Mapping low-resolution three-dimensional protein structures using chemical cross-linking and Fourier transform ion-cyclotron resonance mass spectrometry. Rapid Commun. Mass Spectrom. 17, 2005–2014 [DOI] [PubMed] [Google Scholar]
- 29. Arnold K., Bordoli L., Kopp J., Schwede T. (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 30. Biasini M., Bienert S., Waterhouse A., Arnold K., Studer G., Schmidt T., Kiefer F., Cassarino T. G., Bertoni M., Bordoli L., Schwede T. (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, W252–W258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Benkert P., Tosatto S. C., Schomburg D. (2008) QMEAN: A comprehensive scoring function for model quality assessment. Proteins 71, 261–277 [DOI] [PubMed] [Google Scholar]
- 32. de Beer T. A., Berka K., Thornton J. M., Laskowski R. A. (2014) PDBsum additions. Nucleic Acids Res. 42, D292–D296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gilbert R., Kelly M. G., Mikawa T., Fischman D. A. (1996) The carboxyl terminus of myosin binding protein-C (MyBP-C, C-protein) specifies incorporation into the A-band of striated muscle. J. Cell Sci. 109, 101–111 [DOI] [PubMed] [Google Scholar]
- 34. Miyamoto C. A., Fischman D. A., Reinach F. C. (1999) The interface between MyBP-C and myosin: site-directed mutagenesis of the CX myosin-binding domain of MyBP-C. J. Muscle Res. Cell Motil. 20, 703–715 [DOI] [PubMed] [Google Scholar]
- 35. Freiburg A., Gautel M. (1996) A molecular map of the interactions between titin and myosin binding protein-C: implications for sarcomeric assembly in familial hypertrophic cardiomyopathy. Eur. J. Biochem. 235, 317–323 [DOI] [PubMed] [Google Scholar]
- 36. Fürst D. O., Vinkemeier U., Weber K. (1992) Mammalian skeletal muscle C-protein: purification from bovine muscle, binding to titin and the characterization of a full-length human cDNA. J. Cell Sci. 102, 769–778 [DOI] [PubMed] [Google Scholar]
- 37. Okagaki T., Weber F. E., Fischman D. A., Vaughan K. T., Mikawa T., Reinach F. C. (1993) The major myosin binding domain of skeletal muscle MyBP-C (C protein) resides in the COOH-terminal, immunoglobulin C2 motif. J. Cell Biol. 123, 619–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Welikson R. E., Fischman D. A. (2002) The C-terminal IgI domains of myosin-binding proteins C and H (MyBP-C and MyBP-H) are both necessary and sufficient for the intracellular crosslinking of sarcomeric myosin in transfected non-muscle cells. J. Cell Sci. 115, 3517–3526 [DOI] [PubMed] [Google Scholar]
- 39. Sadayappan S., Gulick J., Osinska H., Barefield D., Cuello F., Avkiran M., Lasko V. M., Lorenz J. N., Maillet M., Martin J. L., Brown J. H., Bers D. M., Molkentin J. D., James J., Robbins J. (2011) A critical function for Ser-282 in cardiac myosin binding protein-C phosphorylation and cardiac function. Circ. Res. 109, 141–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Alders M., Jongbloed R., Deelen W., van den Wijngaard A., Doevendans P., Ten Cate F., Regitz-Zagrosek V., Vosberg H. P., van Langen I., Wilde A., Dooijes D., Mannens M. (2003) The 2373insG mutation in the MYBPC3 gene is a founder mutation, which accounts for nearly one-fourth of the HCM cases in the Netherlands. Eur. Heart J. 24, 1848–1853 [DOI] [PubMed] [Google Scholar]
- 41. Adalsteinsdottir B., Teekakirikul P., Maron B. J., Burke M. A., Gudbjartsson D. F., Holm H., Stefansson K., DePalma S. R., Mazaika E., McDonough B., Danielsen R., Seidman J. G., Seidman C. E., Gunnarsson G. T. (2014) Nationwide study on hypertrophic cardiomyopathy in iceland: evidence of a MYBPC3 founder mutation. Circulation 130, 1158–1167 [DOI] [PubMed] [Google Scholar]
- 42. Simonson T. S., Zhang Y., Huff C. D., Xing J., Watkins W. S., Witherspoon D. J., Woodward S. R., Jorde L. B. (2010) Limited distribution of a cardiomyopathy-associated variant in India. Ann. Hum. Genet. 74, 184–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Razzaque M. A., Gupta M., Osinska H., Gulick J., Blaxall B. C., Robbins J. (2013) An endogenously produced fragment of cardiac myosin-binding protein C is pathogenic and can lead to heart failure. Circ. Res. 113, 553–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Herron T. J., Rostkova E., Kunst G., Chaturvedi R., Gautel M., Kentish J. C. (2006) Activation of myocardial contraction by the N-terminal domains of myosin binding protein-C. Circ. Res. 98, 1290–1298 [DOI] [PubMed] [Google Scholar]
- 45. Harris S. P., Rostkova E., Gautel M., Moss R. L. (2004) Binding of myosin binding protein-C to myosin subfragment S2 affects contractility independent of a tether mechanism. Circ. Res. 95, 930–936 [DOI] [PubMed] [Google Scholar]
- 46. Zahka K., Kalidas K., Simpson M. A., Cross H., Keller B. B., Galambos C., Gurtz K., Patton M. A., Crosby A. H. (2008) Homozygous mutation of MYBPC3 associated with severe infantile hypertrophic cardiomyopathy at high frequency among the Amish. Heart 94, 1326–1330 [DOI] [PubMed] [Google Scholar]
- 47. Bahrudin U., Morisaki H., Morisaki T., Ninomiya H., Higaki K., Nanba E., Igawa O., Takashima S., Mizuta E., Miake J., Yamamoto Y., Shirayoshi Y., Kitakaze M., Carrier L., Hisatome I. (2008) Ubiquitin-proteasome system impairment caused by a missense cardiac myosin-binding protein C mutation and associated with cardiac dysfunction in hypertrophic cardiomyopathy. J. Mol. Biol. 384, 896–907 [DOI] [PubMed] [Google Scholar]
- 48. Sarikas A., Carrier L., Schenke C., Doll D., Flavigny J., Lindenberg K. S., Eschenhagen T., Zolk O. (2005) Impairment of the ubiquitin-proteasome system by truncated cardiac myosin binding protein C mutants. Cardiovasc. Res. 66, 33–44 [DOI] [PubMed] [Google Scholar]
- 49. Schlossarek S., Englmann D. R., Sultan K. R., Sauer M., Eschenhagen T., Carrier L. (2012) Defective proteolytic systems in Mybpc3-targeted mice with cardiac hypertrophy. Basic Res. Cardiol. 107, 235. [DOI] [PubMed] [Google Scholar]
- 50. Predmore J. M., Wang P., Davis F., Bartolone S., Westfall M. V., Dyke D. B., Pagani F., Powell S. R., Day S. M. (2010) Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies. Circulation 121, 997–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bulteau A. L., Szweda L. I., Friguet B. (2002) Age-dependent declines in proteasome activity in the heart. Arch. Biochem. Biophys. 397, 298–304 [DOI] [PubMed] [Google Scholar]