

Abstract
The small GTPase Rac1 has emerged as an important regulator of cell survival and apoptosis, but the mechanisms involved are not completely understood. In this report, constitutively active Rac1 is shown to stimulate the phosphorylation of the Bcl-2 family member Bad, thereby suppressing drug-induced caspase activation and apoptosis in human lymphoma cells. Rac1 activation leads to human Bad phosphorylation specifically at serine-75 (corresponding to murine serine-112) both in vivo and in vitro. Inhibition of constitutive and activated Rac1-induced Bad phosphorylation by a cell-permeable competitive peptide inhibitor representing this Bad phosphorylation site sensitizes lymphoma cells to drug-induced apoptosis. The data show further that endogenous protein kinase A is a primary catalyst of cellular Bad phosphorylation in response to Rac activation, while Akt is not involved. These findings define a mechanism by which active Rac1 promotes lymphoma cell survival and inhibits apoptosis in response to cancer chemotherapy drugs.
Rac GTPases, members of the Rho family of small GTP-binding proteins, play an important role in transducing signals generated on the surface of a cell to the nucleus. Upon stimulation by growth factors, Rac becomes activated and induces a variety of functional responses, including endogenous superoxide generation, reorganization of the actin cytoskeleton, gene transcription, cell cycle progression, and malignant transformation (6, 17, 35). It is also involved in controlling apoptosis (3, 7); expression of active Rac1 provides a survival signal to protect tumor cell lines or transformed fibroblasts from apoptosis (13, 24, 34, 36). Our recent data demonstrate that inactivation of Rac1 by caspase-mediated cleavage promotes apoptosis in human lymphoma cells, suggesting that native Rac1 activity interferes with the apoptotic process and needs to be diminished in order to maximize cell killing by chemotherapy drugs (46). However, the precise mechanism by which the Rac1 signaling pathway transduces a survival signal that inhibits apoptosis needs to be established. In most human tumors, Rac activity is upregulated either by overexpression of the protein, by mutations that render the protein resistant to normal regulatory systems (e.g., isoform Rac1b), or by alterations in associated regulator proteins (28, 37, 38). In addition, Rac is a downstream effector of the oncogenic Ras protein, which is known to participate in carcinogenesis in many human cancers (4, 15). Therefore, understanding the molecular basis of Rac-modulated cell survival and apoptosis might lead to strategies to improve anticancer therapy.
Induction of apoptosis by anticancer drugs involves disruption of mitochondria and the subsequent activation of the caspase protease cascade (25). This process is tightly regulated by Bcl-2 family proteins (41). Bad is a proapoptotic member of this family that has been implicated in coordinating the survival signals and the mitochondrial cell death machinery (5, 14, 44). Bad is normally phosphorylated and sequestered by binding to 14-3-3 in living cells. Apoptotic signals cause Bad dephosphorylation and release from 14-3-3. In the unphosphorylated state, Bad binds to Bcl-2 and Bcl-XL and inactivates their prosurvival function, resulting in mitochondrial dysfunction, cytochrome c release, caspase activation, and apoptotic death. The phosphorylation of endogenous Bad occurs under conditions in which growth factors are present to promote cell survival, suggesting that Bad phosphorylation is essential in order for survival factors to block apoptosis (10-12). Accordingly, transgenic mice carrying Bad mutants that lack its phosphorylation sites exhibit defects in growth factor-dependent survival (11). Thus, the reversible phosphorylation of Bad represents a critical sensor for survival signaling and a determinant for the outcome of apoptotic stimuli.
A number of different Ser/Thr protein kinases are known to phosphorylate Bad at different Ser residues in the protein. The most-studied phosphorylation sites occur at serine residues 112, 136, and 155 in the mouse protein (10, 11, 20, 27). These correspond to Ser-75, -99, and -118 in the human protein (16). Cyclic AMP (cAMP)-dependent protein kinase (PKA) reportedly phosphorylates Bad at murine Ser-112 and -155, while phosphatidylinositol 3 (PI 3)-kinase/Akt (PKB) is reported to phosphorylate murine Bad at Ser-136 (9, 33). The most recently identified Bad kinases are p-21-activated kinases (PAKs). While PAK1 and PAK2 were shown to phosphorylate murine Bad on both serines 112 and 136 (22, 39), PAK4 and PAK5 only phosphorylate murine Bad on Ser-112 (8, 19). While informative, one limitation of much of this work has been that it involves transfection and overexpression of Bad and the individual kinases, often to massive levels, which precludes the understanding of how the activity of Bad is regulated in the cellular context by specific signal transduction molecules that are activated by survival signals.
We hypothesized that the Rac pathway may suppress cell death and promote cell survival by causing Bad phosphorylation. In this report, we show that functional Rac GTPase is required for normal Bad phosphorylation in healthy, growing human lymphoma cells. Increased Rac1 signaling stimulates Bad phosphorylation at human Ser-75 in vitro and in vivo. When this phosphorylation is selectively inhibited, increased apoptosis occurs in response to chemotherapy drugs. Thus, Rac1 blocks drug-induced apoptosis at least in part by maintaining Bad in the phosphorylated state. Evidence is also provided to show that PKA phosphorylates Bad in response to activated Rac1, whereas Akt does not.
MATERIALS AND METHODS
Plasmids and transfections.
The pcDNA3.1(+)-hemagglutinin (HA)-Rac1 constructs, encoding HA-tagged human wild-type Rac1, constitutively active mutant Rac1V12, dominant-negative mutant Rac1N17, and caspase-3-resistant mutant Rac1D11E, were prepared as previously described (46). Transfection of Burkitt's lymphoma BL-41 cells was carried out by electroporation as described previously (26). Positive clones were selected with G418 (Mediatech, Herndon, Va.) and were tested for expression of Rac1 by anti-HA Western blotting. The stable cell lines were maintained at 37°C and 5% CO2 in RPMI 1640 medium containing 10% heat-inactivated fetal calf serum, 2 mM l-glutamine, 50 μM β-mercaptoethanol, and 1.5 mg of G418/ml.
Antibodies and reagents.
A mouse monoclonal anti-HA antibody was purchased from Covance Research Products (Berkeley, Calif.). Mouse monoclonal antibodies against human Bad (hBad), Bcl-2, catalytic subunit of protein kinase A (PKAC), Akt, and phospho-Akt-Ser 472/473 were purchased from BD Biosciences (Lexington, Ky.). Mouse monoclonal antibody specific for phospho-Bad-Ser-112 (7E11) and rabbit polyclonal anti-phospho-Bad-Ser-136, and -phospho-Bad-Ser-155 were obtained from Cell Signaling Technology (Beverly, Mass.). A monoclonal antibody against poly(ADP-ribose) polymerase (PARP) was from Oncogene Research Products (Boston, Mass.). The PKA peptide inhibitor (TTYADFIASGRTGRRNAIHD) and purified PKAC were from Promega (Madison, Wis.). Rp-cAMPS was from Alexis Biochemicals (San Diego, Calif.). A cell-permeable PKA inhibitor, myristoylated-GRTGRRNAI-NH2 (amide 14 to 22), and an Akt-specific inhibitor (SH-5) were obtained from Calbiochem. VP-16 (etoposide), GDP, and GTPγS were from Sigma. Bacterially expressed, purified recombinant (His)6-fusion proteins of full-length human Akt1/PKB, active recombinant human PAK2, and full-length murine Bad were from Upstate Biotechnology (Lake Placid, N.Y.). Human recombinant Rac1 proteins were expressed as (His)6-tagged fusions by the pET28a expression system and were loaded with GDP or GTPγS as described previously (45).
A peptide matching the 9 amino acids surrounding Ser-75 of hBad (amino acids 70 to 78) and a scrambled peptide from this sequence were custom synthesized by Biopeptide Corporation (San Diego, Calif.). The peptides were made cell permeable through coupling to the human immunodeficiency virus (HIV) Tat protein transduction domain (21). The purity of both peptides was greater than 95%, as determined by high-performance liquid chromatography. Peptide sequences were confirmed by mass spectrometry.
Apoptosis assay.
To induce apoptosis, cells were seeded at 5 × 105 cells/ml and were treated with VP-16 at 200 μg/ml. At indicated time points, cells were collected and washed sequentially with serum-containing medium, phosphate-buffered saline, and annexin V buffer (10 mM HEPES [pH 7.4], 140 mM NaCl, and 2.5 mM CaCl2). These samples were then incubated at room temperature for 15 min with annexin V-fluorescein isothiocyanate (FITC) (Pharmingen) and propidium iodide (PI) at final concentrations of 2.5 and 5.0 μg/ml, respectively. After being washed once with annexin V buffer, the cells were resuspended in 500 μl of annexin V buffer and were analyzed (10,000 cells per sample) by flow cytometry. The percentage of apoptotic cells was determined by assessing the cells that were bound to annexin V-FITC but were negative for PI staining.
In vitro kinase assays.
The Bad kinase assays were carried out in the presence of 1 μg of recombinant murine His-Bad protein in a kinase buffer (20 mM Tris-HCl [pH 7.5], 300 μM ATP, 2 mM EGTA, 50 mM MgCl2, 1 mM dithiothreitol) containing protease inhibitors (1:50 dilution of the Protease Inhibitor Cocktail Set III; Calbiochem) and phosphatase inhibitors (25 mM NaF, 1 mM Na3VO4, 20 mM glycerophosphate). The reaction was initiated by the addition of cell extracts or a purified kinase. The mixture was incubated at 30°C for 10 min and was terminated by the addition of sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer. Phosphorylated Bad was detected by Western blotting using antibodies specific to murine phospho-Bad at Ser-112, -136, or -155 and to human phospho-Bad at Ser-75, -99, and -118, respectively. To test the effect of Rac GTPase activity on the kinase activities, recombinant glutathione S-transferase-tagged human Rac1 that was preloaded with GDP or GTPγS was added to the reaction mixture (45).
The kinase activity of cAMP-dependent protein kinase (PKA) was determined by a nonradioactive PepTag assay (Cat.V5340; Promega), which utilizes a brightly colored, fluorescent peptide substrate that is highly specific for PKA. Phosphorylation by PKA alters the net charge of the substrate from +1 to −1, thereby allowing the phosphorylated and nonphosphorylated versions of the substrate to be separated by electrophoresis on an agarose gel. The phosphorylated species migrates toward the positive electrode, while the nonphosphorylated substrate migrates toward the negative electrode. To start the reaction, Bl-41 whole lysates (0.2 to 2 μg in 10 μl) were incubated with PKA reaction mixture (25 μl) at room temperature for 30 min. The reactions were terminated by placing the tubes in a 95°C heating block for 10 min. After adding 80% glycerol (1 μl), the samples were loaded onto an agarose gel (0.8% agarose in 50 mM Tris-HCl, pH 8.0) and were separated in the same buffer at 100 V for 15 min. The bands were visualized under UV light. For quantitation of the PepTag results, the phosphorylated bands were cut out and heated at 95°C until the gel slice melted. The hot agarose solution (125 μl) was transferred to a tube containing 75 μl of Gel Solubilization Solution (Promega) and 50 μl of glacial acetic acid, and the absorbance was measured at 570 nm on a plate reader (Dynatech).
Akt kinase activity was measured by Stressgen's nonradioactive PKB (Akt) kinase assay kit (Victoria, British Columbia, Canada), which is based on a solid-phase enzyme-linked immunosorbent assay that utilizes an Akt-specific peptide as a substrate and a polyclonal antibody that recognizes the phosphorylated form of the substrate. Briefly, purified Akt or whole-cell extract was incubated at 30°C for 40 min in the precoated Akt substrate plate. The reaction was terminated by the addition of a phosphospecific substrate antibody, followed by incubation with an anti-rabbit immunoglobulin G horseradish peroxidase-conjugated antibody. Antibody binding was quantified colorimetrically (405 nm) by using 2,2′-Azino-bis 3-methylbenzthiazoline-6-sulfonic acid (TMB) and a 96-well microplate reader. The assay was validated by using purified Akt and adjusting the amount of cell extract to fall in the linear range of the standard curve.
Western blot analysis.
Equal amounts of cell lysates (20 μg per lane) were resolved by electrophoresis using a 4 to 12% NuPAGE Bis-Tris gel (Invitrogen) and were transferred to nitrocellulose membranes (Millipore) for immunoblotting. When necessary, the membrane was stripped by Restore Western Blot Stripping buffer (Pierce) and reprobed with appropriate antibodies. Immunocomplexes were visualized by enhanced chemiluminescence or SuperSignal reagent (Pierce).
RESULTS
Rac1-dependent signaling pathways stimulate phosphorylation of Bad at Ser-75.
The main goal of these studies was to determine whether Rac1 inhibits cell death, at least in part, by modulating the phosphorylation and activity of Bad. Our working hypothesis was that Rac1 is required for cell survival and that elimination of Rac1 activity through caspase cleavage is required for maximal apoptosis to occur. Because Bad is antiapoptotic in its phosphorylated state, we focused on characterizing the phosphorylation state of Bad in healthy, growing cells. Because the cellular studies employ human cells while the cell-free studies use purified recombinant murine Bad, the Ser residue numbers differ between experiments; i.e., Ser-112, -136, and -155 for murine Bad and Ser-75, -99, and -118 for hBad. However, the sequences in both species are exactly homologous and cross-react with the respective phosphorylation site-specific antibodies.
We first determined whether cells containing higher or lower levels of Rac activity phosphorylate the different phosphorylation sites in Bad to different degrees. Stable transfectants of several Rac1 mutants were generated in human Burkitt's lymphoma BL-41 cells. The genes transfected and expressed were the dominant-negative mutant Rac1N17, the constitutively active mutant Rac1V12, and the caspase-3-resistant mutant Rac1D11 (46). Empty vector was used for control transfections. Western blot analysis of the HA-tagged mutant proteins suggested that roughly equal amounts of Rac1N17, Rac1V12, and Rac1D11 protein were expressed in the respective cell lines (Fig. 1A). To examine the phosphorylation of Bad, extracts from the transfected cells were subjected to immunoblot analysis using antibodies specific for the known phosphorylation sites in Bad. In parental BL-41 cells growing under normal conditions, Bad phosphorylation was only detected on Ser-75. Significantly more Ser-75-specific phosphorylation was observed in cells expressing constitutively active Rac1V12 and caspase-resistant Rac1D11, while the opposite was true in the cells expressing the dominant-negative Rac1N17 mutant. In contrast, neither Ser-99 nor Ser-118 phosphorylation was detected in control BL-41 cells or in cells expressing the Rac1 mutants. As will be shown below, the difference in phosphorylation of the different sites on Bad did not result from a reduced sensitivity of the antibodies for p-99 and p-118 (see Fig. 4), and all of the cell lines expressed comparable levels of Bad protein (Fig. 1B). These results suggested that Rac1 activity selectively upregulates hBad Ser-75 phosphorylation in vivo. Because Ser-75 was the only residue found to be phosphorylated constitutively in healthy, growing cells, it is the focus of the remainder of the studies on the regulation of Bad phosphorylation by Rac GTPases.
FIG. 1.

Phosphorylation of hBad at Ser-75 is modulated by Rac1. (A) Rac1 protein expression in human BL-41 Burkitt's lymphoma cells stably expressing vector alone, HA-tagged Rac1N17 (dominant-negative mutant), HA-Rac1V12 (constitutively active mutant), or HA-Rac1D11 (caspase-3-resistant mutant). Cell extracts were separated on Bis-Tris SDS-PAGE (4 to 12% acrylamide), transferred to a nitrocellulose membrane, and immunoblotted with antibodies against HA. (B) Levels of phosphorylated hBad in transfected cells lines. The immunoblots were probed with antibodies that specifically recognize residues phosphorylated at Ser-75 (p-75-hBad), Ser-99 (p-99-hBad), and Ser-118 (p-118-hBad). Equal loading was confirmed by reprobing the membrane with antibodies to Bad or α-actin. (C) In vitro phosphorylation of Bad by cell extracts. Purified recombinant His-tagged murine Bad (mBad; 1 μg) was incubated in standard kinase buffer with various amounts (20 to 2,000 ng) of extracts from healthy BL-41 cells as the source of kinase(s). The reactions were stopped by addition of SDS sample buffer and were analyzed for Bad phosphorylation by Western blot immunoassay using site-specific anti-phospho-Bad antibodies (see Materials and Methods). (D) Effect of Rac1 GTPase activity on phosphorylation of Bad. Recombinant glutathione S-transferase-tagged Rac1 was preloaded with GDP or GTPγS and then added (final concentration, 2.0 μM) to a reaction mixture containing 1 μg of His-tagged murine Bad and lysis buffer (upper panel) or 50 ng of BL-41 extract (lower panel). The phosphorylation reactions were carried out under the conditions described for panel B. The results are representative of three independent experiments.
FIG. 4.
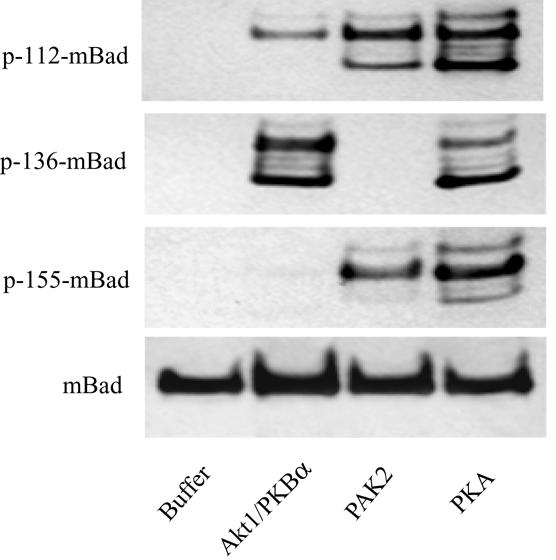
In vitro phosphorylation of Bad by purified protein kinases. Purified recombinant His-Bad protein (1 μg) was incubated in kinase reaction buffer (10 min at 30°C) containing the following different purified, recombinant protein kinases (1 μg): AKT1/PKBα, PAK2, and the PKAC (see Materials and Methods). Phosphorylation of murine Bad (mBad) on different serine residues was determined by immunoblot analysis with site-specific anti-phospho-Bad antibodies.
We next examined the ability of Rac1 activity to modulate phosphorylation of recombinant murine Bad protein by cell extracts isolated from the lymphoma cells. Purified murine Bad becomes highly phosphorylated in a dose-dependent manner when incubated with the endogenous cellular kinases (Fig. 1C). Saturation of Ser-112 phosphorylation was achieved with as little as 200 ng of whole lysate protein, while no Ser-136 or Ser-155 phosphorylation was detected under these conditions. Phosphorylation of Bad at Ser-136 and Ser-155 required roughly 10-fold more lysate than did Ser-112 phosphorylation. These results indicate that human lymphoma cells exhibit an intrinsic capability to phosphorylate Bad and that the endogenous kinase activity is relatively specific for Ser-112.
The effect of Rac GTPase activity on endogenous Bad phosphorylation was examined by using purified Rac1 protein that had either been activated by preloading with GTPγS or inhibited by preloading with GDP. Addition of Rac1-GTPγS to the cell lysates enhanced phosphorylation of murine Bad at Ser-112 by approximately fivefold, whereas Rac1-GDP had no detectable effect on Bad phosphorylation (Fig. 1D). In contrast, neither Rac1 form stimulated Bad phosphorylation on Ser-75 in the absence of endogenous kinases (Fig. 1D, upper panel). Together these results demonstrate that the active form of Rac1 can stimulate site-specific kinases to phosphorylate Bad both in vivo and in vitro.
Active Rac1 inhibits drug-induced caspase activation and apoptosis.
In previous studies (46) we showed that Rac1 needs to be cleaved and inactivated by caspases in order for maximal apoptosis to occur in response to chemotherapy drugs. The finding that active Rac1 triggers Bad Ser-75 phosphorylation suggested that a Rac1-dependent signaling pathway involving Bad phosphorylation might be responsible for suppressing drug-induced apoptosis. To investigate this possibility, we took advantage of a transfected BL-41 cell line that expresses a Rac1 mutant (Rac1D11E) that is resistant to caspase-3 cleavage and apoptosis (46). As shown in Fig. 2A, cells transfected with Rac1D11E and treated with the chemotherapy drug VP-16 (etoposide) show 30 and 55% less apoptosis at 3 and 6 h, respectively, than control cells, as determined by annexin V staining. Quantitative data from several separate experiments are shown in Fig. 2B and demonstrate that both Rac1V12 and Rac1D11E prevented the cells from undergoing apoptosis induced by VP-16. Note that Rac1V12 is also relatively resistant to caspase-3 cleavage and hence remains active in cells treated with VP-16 (46). Similar prosurvival effects of activated Rac1 were observed previously in JLP-119 lymphoma cells (46). Activation of caspase-3 followed a commensurate pattern, as demonstrated by the cleavage of procaspase-3 (Fig. 2C) and PARP (Fig. 2D), which is a known caspase-3 substrate. By 6 h, a significant amount caspase-3 activation had occurred in both control cells and cells expressing Rac1N17, and PARP cleavage was almost complete. In contrast, in cells expressing Rac1V12 and Rac1D11E, caspase-3 activation and PARP cleavage were significantly delayed.
FIG. 2.

Rac1 activity modulates apoptosis induced by VP-16. (A) Apoptosis was assessed in control BL-41 cells and in BL-41 cells transfected with the caspase-3-resistant Rac1 mutant Rac1D11E by fluorescence-activated cell sorting analysis using FITC-annexin V and PI (see Materials and Methods and reference 46 for details). Cells were treated with 200 μg of VP-16/ml for 3 or 6 h and then were harvested. The percentage of each cell population is shown. Cells in the lower right fluorescence-activated cell sorting quadrant represent apoptotic cells. (B) Quantification of VP-16-induced apoptosis (as described for panel A) in BL-41 cells expressing endogenous Rac1 only, dominant-negative Rac1N17, constitutively active Rac1V12, or the caspase-3-resistant mutant Rac1D11E, as indicated. The results represent the means ± standard deviations from three independent experiments. (C and D) Measurement of procaspase-3 and PARP cleavage in control and Rac1-transfected cell lines expressing the various mutant proteins after treatment with VP-16 as indicated by Western blot immunoassay using anti-caspase-3 (C) and anti-PARP (D) antibodies. Caspase activity is indicated by the appearance of the p17/p12 caspase-3 cleavage fragments (D) and the 85-kDa PARP cleavage product. *, nonspecific band. (E) The level of endogenous Bad phosphorylation in apoptotic cells was assessed by subjecting whole-cell lysates to Western blot immunoassay using antibodies specific for p-Ser-75. All data are representative of at least three independent experiments.
The status of Bad phosphorylation during apoptosis was assessed by immunoblot analysis using phosphorylation site-specific antibodies (Fig. 2E). In control cells, Bad phosphorylation on Ser-75 decreased over time after VP-16 treatment. In cells expressing Rac1V12, however, there was a significant delay in the decrease in Bad Ser-75 phosphorylation. A stronger delay was observed in the cells expressing Rac1D11E, which is the most caspase-resistant mutant. These data show that increased Rac1 function correlates with increased Bad phosphorylation and leads to protection from apoptotic cell death.
Inhibition of Bad phosphorylation on Ser-75 sensitizes lymphoma cells to drug-induced apoptosis.
If Rac1 enhances lymphoma cell survival by stimulating Bad phosphorylation, then selective inhibition of Bad phosphorylation should enhance the apoptotic response to drug treatment. To establish this linkage, we designed a cell-permeable peptide comprised of the Ser-75 phosphorylation site (amino acids 70 to 78) of hBad coupled to the protein transduction domain (RKKRRQRRR) (PTD) of the HIV-1 transactivator protein TAT (Fig. 3A). This PTD has been used previously to enhance delivery of exogenous proteins or peptides into living cells (21). A second peptide containing the same PTD but a scrambled Bad phosphorylation sequence was prepared and used for control experiments. Initially, the effects of the peptides on Bad phosphorylation were tested in vitro by using purified PKA and recombinant purified murine Bad as a substrate. As shown in Fig. 3B, addition of the hBad-Ser75 peptide to this assay resulted in a dose-dependent inhibition of Bad phosphorylation at Ser-112. At 100 μM peptide, Bad phosphorylation was inhibited by ∼90%, with a concomitant appearance of the phosphorylated peptide product. In contrast, the control peptide with the scrambled sequence had relatively little effect on PKA-induced Bad phosphorylation. We next determined whether the hBad-Ser75 peptide would inhibit Bad phosphorylation and promote apoptosis in vivo. When control cells or cells expressing Rac1D11 were treated with 500 μM hBad-Ser75 peptide, a significant reduction in Bad phosphorylation on serine-75 was observed (Fig. 3C). Furthermore, treatment with the hBad-Ser75 peptide increased VP-16-induced apoptosis in control and Rac1D11 cells by two- and fourfold, respectively (Fig. 3D), thereby restoring the level of apoptosis to that observed in control VP-16-treated cells. In sharp contrast, Rac1N17-expressing cells, which are relatively weak in their ability to phosphorylate Bad (Fig. 1B) and are already hypersensitive to VP-16-induced apoptosis, are not sensitive to the hBad-Ser75 peptide. Taken together these results indicate that Rac1-regulated Bad phosphorylation on Ser-75 is an important mediator of the survival signaling pathway in lymphoma cells. Cells expressing Rac1D11 are, nonetheless, still relatively resistant to apoptosis even when Bad phosphorylation is diminished (Fig. 3D), suggesting that phosphorylation of Bad alone may not be sufficient for lymphoma cell survival. It may be that additional survival signaling molecules are required to fully confer Rac1-mediated cell survival.
FIG. 3.

A competitive inhibitor of hBad Ser-75 phosphorylation sensitizes lymphoma cells to VP-16-induced apoptosis. (A) Sequence of cell-permeable hBad-Ser-75 peptide and the control (scrambled) peptide. The hBad-Ser-75 peptide contains the serine-75 phosphorylation site (amino acids 70 to 78) of hBad. The PTD (underlined) derived from the HIV-1 Tat protein promotes cell entry. (B) Effects of hBad Ser-75 peptide on Bad phosphorylation in vitro. Recombinant murine Bad (mBad; 0.5 μg) was incubated in standard PKA assay buffer with active PKAC (20 ng) and increasing amounts (1 to 100 μM) of peptides in a final volume of 70 μl for 10 min at 30°C. The reactions were stopped by addition of SDS sample buffer and were analyzed for Bad phosphorylation by Western blotting with anti-pSer-112-mBad antibodies. (C) Effects of peptides on Bad phosphorylation in vivo. Cells were treated with 500 μM hBad-Ser75 or scrambled peptide for 2 h and were analyzed for the status of Bad phosphorylation on Ser-75. (D) Effect of hBad Ser-75 peptide on the sensitivity of lymphoma cells to VP-16-induced apoptosis. Cells were pretreated with 500 μM peptide for 1 h and then were incubated with VP-16 (200 μg/ml) for an additional 2.5 h. Apoptosis was assessed by annexin V-fluorescence-activated cell sorting analysis. The results represent the means ± standard deviations from three independent experiments. MW, molecular size.
Site-specific phosphorylation of Bad by different protein kinases.
Having demonstrated a role for Rac1 in regulating Bad phosphorylation, we then attempted to identify the effector proteins through which Rac1 exerts its activity. Several different protein kinases are known to have the capacity to phosphorylate Bad, with various levels of specificity for murine Ser residues 112, 136, and 155 (8, 9, 19, 20, 22). These include Akt, cAMP-dependent protein kinase (PKA), and members of the PAKs. We compared the phosphorylation site specificity of these kinases by using antibodies specific for phosphorylated murine Ser-112, Ser-136, and Ser-155. As shown in Fig. 4, purified, constitutively active Akt1/PKBα phosphorylated recombinant murine Bad in vitro on both Ser-112 and Ser-136 with a strong preference for the latter residue, while no Ser-155 phosphorylation was observed. This result is consistent with previous reports showing that Ser-136 is the major site of phosphorylation of Bad by Akt (10, 12). In contrast, purified, constitutively active PAK2, which was previously shown to phosphorylate Bad at Ser-112 and Ser-136 (22), preferentially induced phosphorylation of Bad on Ser-112 and Ser-155 without phosphorylation of Ser-136. PKAC, which was reported previously to phosphorylate Bad on either Ser-112 (20) or Ser-155 (27), phosphorylated all three serine residues to similar degrees. Thus, Akt, PKA, and PAK2 exhibit differential specificities for Bad residues Ser-112, Ser-136, and Ser-155.
Inhibition of PKA activity blocks Rac1-stimulated phosphorylation of Bad.
To examine whether PKA might be responsible for the constitutive phosphorylation of Bad at Ser-112 in growing, healthy lymphoma cells, two different PKA inhibitors were employed: a PKA-specific inhibitory peptide and the cAMP antagonist Rp-cAMPS. As shown in Fig. 5A, when extracts from healthy BL-41 cells were assayed for in vitro kinase activity, phosphorylation of Ser-112 was seen in control extracts but not in extracts containing either of the PKA inhibitors. To confirm the presence of PKA activity in the BL-41 cell extracts, we employed a fluorescent peptide substrate assay that is specific for PKA activity (PepTag A1). The utility of the assay was validated by using PKAC. As illustrated in Fig. 5B, nonphosphorylated substrate migrates toward the negative electrode while the phosphorylated peptide migrates toward the positive electrode. Addition of increasing amounts of PKA catalytic subunit resulted in a systematic shift in mobility, reflecting an increase in the phosphorylated substrate. Whole-cell lysates from normal lymphoma cells showed a basal PKA activity that was stimulated by addition of exogenous cAMP. This activity was blocked completely by both PKA-specific inhibitory peptide and Rp-cAMPS (Fig. 5C).
FIG. 5.

Inhibition of cellular PKA activity blocks Bad phosphorylation. (A) BL-41 cell extracts (200 ng of total protein) were assayed for Bad kinase activity in the absence or presence of a PKA peptide inhibitor (20 μM) or Rp-cAMPS (100 μM). (B) PepTag assay for determining PKA activity. PepTag A1 peptide (2 μg), a PKA-specific substrate, was incubated in standard PKA assay buffer with various amounts (1 to 24 ng) of active PKAC in a final volume of 25 μl for 30 min at room temperature. The reactions were stopped and then subjected to agarose gel electrophoresis. The phosphorylated peptide substrate migrates toward the positive electrode (+), while the nonphosphorylated peptide migrates toward the negative electrode (−). (C) Detection of PKA-specific kinase activity in cell lysates. Extracts (2 μg) from healthy BL-41 cells were subjected to the PepTag PKA assay as described for panel B in the absence (lane 3) or presence (lanes 4 to 6) of cAMP. The PKA peptide inhibitor (20 μM) or Rp-cAMPS (100 μM) was added where indicated (lanes 5 and 6). Lanes 1 and 2 show the negative (buffer only) and positive controls (with 10 ng purified PKAC). The data represent one of two independent experiments.
This PKA-specific assay was then employed to determine the ability of Rac1 to modulate PKA activity in BL-41 cells. To this end, we measured constitutive PKA activity levels in whole-cell extracts from cells transfected either with constitutively active or with dominant-negative Rac1 (Rac1V12 or Rac1D11 and Rac1N17, respectively). As shown in Fig. 6A, PKA activity was increased by approximately twofold in cells expressing Rac1V12 or Rac1D11 and decreased by ∼60% in Rac1N17-expressing cells compared to corresponding levels in control cells. These data suggested that Rac1 GTPase can activate PKA (either directly or indirectly). In support of this conclusion, addition of active Rac1-GTPγS protein to BL-41 cell extracts stimulated basal PKA activity by approximately twofold. In contrast, addition of inactive Rac1-GDP had no effect on basal PKA activity (Fig. 6B).
FIG. 6.

Rac1 stimulates PKA activity. (A) Equal amounts of extracts (2 μg) from healthy control BL-41 cells or from cell lines stably transfected with Rac1N17, Rac1V12, or Rac1D11 were subjected to the PepTag assay as described in the legend to Fig. 5. (B) Kinase activity was quantified by spectrophotometric analysis (absorbance at 570 nm) of the phosphorylated bands and is expressed relative to control (Vector alone) activity. (B) Purified Rac1 protein (2 μg) was preloaded with either GDP or GTPγS to inhibit or activate Rac1 activity, respectively. The Rac1 protein was then added to healthy BL-41 extracts and tested for an effect on PKA activity, which is expressed relative to the activity of active PKAC (10 ng). (C) Stable Rac1D11-expressing cells were treated for 60 min with an increasing concentration of a specific myristoylated PKA inhibitor. Whole-cell extracts were subjected to Western blotting to assess hBad Ser-75 phosphorylation. (D) Stable cell lines were pretreated with 10 μM PKA inhibitor prior to adding VP-16 (200 μg/ml). Apoptosis was allowed to proceed for 3 h. The percentage of apoptotic cells was assessed by fluorescence-activated cell sorting as described in the legend to Fig. 2. The data are representative of three independent assays.
To determine whether PKA activity is required for Rac1-mediated Bad phosphorylation in vivo, Rac1D11-expressing cells were treated with a cell-permeable peptide inhibitor of PKA. As shown in Fig. 6C, the PKA inhibitor decreased the phosphorylation of endogenous Bad in a dose-dependent manner. At 50 μM, approximately 70% of Bad phosphorylation was inhibited in Rac1D11 cells (based on densitometry analysis of the bands). These results implicate PKA as a principal Bad kinase that acts in response to Rac1 activation in lymphoma cells. Accordingly, pretreatment with 50 μM PKA inhibitor resulted in significantly more apoptosis in control and Rac1D11 cells (Fig. 6D), respectively, reinforcing the conclusion that Rac1-stimulated Bad phosphorylation plays a role in mediating lymphoma cell survival.
Akt does not appear to be involved in Bad phosphorylation following Rac1 activation. Akt is a Ser/Thr kinase that responds to PI 3-kinase activation and is thought to suppress apoptosis following a variety of stimuli, in part through phosphorylation of Bad (9, 10, 12, 23). Rac1 was shown previously to mediate PI 3-kinase activation of Akt and to promote cell survival in COS-7 cells (31) or human natural killer cells (23). In hematopoietic cells, it is the Rac2 isoform that appears to be responsible for Akt activation and survival (43). To determine whether Akt might be responsible for the Bad phosphorylation observed in our experimental system, we first assessed the levels of active (phosphorylated) Akt in BL-41 lymphoma cells containing differing levels of Rac1 activity. No alteration in endogenous Akt activity was observed in cells expressing Rac1V12, Rac1D11, or Rac1N17, as determined by Western blots using anti-phospho-Akt (Ser-473) antibodies that specifically detect the active form of the kinase (Fig. 7A). Similarly, an enzymatic assay for Akt-specific activity showed no difference between cells expressing the different forms of Rac1, while an Akt-specific inhibitor (SH-5) almost completely inhibited in vitro Akt activation (Fig. 7B). This result suggested that Rac-1-induced phosphorylation of hBad at Ser-75 is Akt independent in human lymphoma cells. In support of this conclusion, immunodepletion of Akt from lysates of Rac1V12 cells had no effect on Bad kinase activity (Fig. 7C). Moreover, addition of a selective cell-permeable Akt inhibitor did not affect endogenous Bad phosphorylation on Ser-75, even though Akt autophosphorylation and activation were almost completely blocked (Fig. 7D). Accordingly, treatment with the Akt inhibitor did not enhance the sensitivity of the cells to VP-16 induced apoptosis (Fig. 7E). Collectively, these results indicate that Akt is not the principal kinase responsible for the phosphorylation of Bad in response to Rac1 activation.
FIG. 7.

Akt activity is not responsive to Rac1 activity. (A) The activation state of Akt in stable lymphoma cells expressing Rac1 mutants was assessed by measuring the level of Akt phosphorylation by using an antibody against phospho-Akt (Ser-473). Equal loading was assessed by reprobing the membrane with an anti-Akt antibody. (B) Akt-specific kinase activities were measured by a colorimetric Akt kinase assay as described in Materials and Methods. The kinase reactions were carried out with equal amounts of cell extracts from the indicated cell lines. The positive control contains purified recombinant Akt. Inhibition of Akt activity in cell extracts was accomplished with the Akt-specific inhibitor SH-5. (C) Phosphorylation of Bad by whole (left lane) or Akt-depleted (right lane) cell extracts was assessed as described in the legend to Fig. 1. (D) Stable Rac1D11-expressing cell lines were treated with increasing amounts of Akt inhibitor (SH-5) for 1 h. Whole-cell lysates were analyzed by Western blotting with the indicated phosphospecific antibodies to detect Akt phosphorylation and activation and Bad phosphorylation. (E) Effect of the Akt inhibitor on the sensitivity of the lymphoma cells to VP-16-induced apoptosis. Cells were pretreated with 10 μM Akt inhibitor (SH-5) for 1 h and then were incubated with VP-16 (200 g/ml) for an additional 3 h. Apoptosis was assessed by the annexin V-fluorescence-activated cell sorting assay. The results represent the means ± standard deviations from three independent experiments.
DISCUSSION
In this report, we provide evidence that the Rac1 small GTPase is a key upstream regulator of Bad phosphorylation, thereby providing a molecular mechanism for how Rac1 inhibits apoptosis in response to cancer chemotherapy drugs. We also show that the only Bad phosphorylation site relevant for survival of these human lymphoma cells occurs on Ser-75 in the hBad protein. Finally, we show that Rac1-induced Bad Ser-75 phosphorylation is catalyzed by PKA but not Akt. These conclusions are supported by the following data: (i) of the three major Ser residues known to be phosphorylated in hBad, only Ser-75 is found to be phosphorylated in healthy, growing Burkitt's lymphoma cells; (ii) constitutively active Rac1 mutants (Rac1V12 or Rac1D11E) stimulate the phosphorylation of Bad specifically on Ser-75 and inhibit apoptosis in response to VP-16; (iii) a Rac1 dominant-negative mutant (Rac1N17) fails to stimulate phosphorylation of Bad, and cells expressing this mutant are more susceptible to apoptosis than cells expressing wild-type or constitutively active Rac1; (iv) inhibition of Bad phosphorylation by a cell-permeable competitive peptide inhibitor representing the Bad Ser-75 phosphorylation site sensitizes lymphoma cells to drug-induced apoptosis; (v) the selective inhibition of hBad Ser-75 phosphorylation by a selective PKA inhibitor diminishes the ability of Rac1 to prevent apoptosis in response to chemotherapy drugs, while the selective inhibition of Akt has no such effect; (vi) when kinase activity in cell extracts is studied, the selective inhibition of PKA diminishes the phosphorylation of Bad—inhibition or selective immunodepletion of Akt in cell extracts has no such effect; (vii) active (GTP-bound) Rac1 stimulates PKA to phosphorylate Bad while inactive (GDP-bound) Rac1 lacks this activity. These findings will be discussed in greater detail below.
Numerous previous studies have demonstrated that the reversible phosphorylation of Bad plays a pivotal role in controlling apoptosis (5, 11, 14). This is thought to be mediated through the reversible binding of Bad to antiapoptotic Bcl-2 family proteins. In particular, it has been shown that the phosphorylation of murine Bad on Ser-112 or Ser-136 prevents its association with Bcl-2 or Bcl-XL, leaving these proteins free to exert their antiapoptotic function (42, 44). Consistent with these findings, Bcl-2 was enriched in the mitochondria of cells expressing Rac1V12, in which a higher level of phosphorylated Bad existed (data not shown). While Bad is phosphorylated in a variety of cell contexts, the upstream regulatory mechanisms that control Bad phosphorylation are not fully understood. A number of cell survival pathways that lead to Bad phosphorylation have been described. The best characterized of these is the PI 3-kinase/Akt pathway that has been shown to induce the phosphorylation of Bad on Ser-136 in response to stimulation of survival factors such as platelet-derived growth factor and interleukin-3 (9, 12, 33). In addition to the PI 3-kinase pathway, recent evidence has implicated the Ras/Raf cascade in the control of Bad phosphorylation (18). The results presented here demonstrate that Rac1 activation leads to the phosphorylation of Bad at Ser-75 in human lymphoma cells. However, while there is a connection between Rac signaling and the PI 3-kinase/Akt pathway (29), Rac appears to be activated in our system in a PI 3-kinase-independent manner, as has been observed with other cell types (1, 32). The relative contributions and interactions that the different survival pathways make in controlling Bad activity in different cell settings remains to be more fully elucidated.
Rac1-stimulated phosphorylation of Bad contributes to its antiapoptotic function; there is a significant inhibition of apoptosis in BL-41 cells upon the expression of activated Rac1 mutants, and the increased cell survival correlates with the ability of these mutants to induce the phosphorylation of Bad on Ser-75. Inhibition of Bad phosphorylation in response to activated Rac1 counters the antiapoptotic activity of the protein. In parallel, the caspase-3 activation triggered by VP-16 is prevented or delayed in these cells, as determined by procaspase-3 and PARP cleavage. Our previous data showed that Rac1 is a caspase substrate that becomes cleaved during drug-induced apoptosis (46). Hence, Rac1 prevents its own degradation and inactivation by stimulating Bad phosphorylation and inhibiting caspase activation. Interestingly, a caspase-resistant mutant, Rac1D11E, appears to be constitutively active in terms of stimulating Bad phosphorylation. Expression of Rac1D11E leads to an even stronger protective effect than that of the constitutively active Rac1V12, which is less resistant to caspase-mediated cleavage (46). Because many of the growth factors that activate Rac proteins could simultaneously induce the phosphorylation of Bad, the Rac1-stimulated phosphorylation of Bad at Ser-75 may represent a general mechanism by which growth factor receptors deliver a survival signal that leads to the inhibition of apoptosis. However, these results do not rule out the possibility that Rac1 promotes cell survival by other mechanisms in addition to that mediated by the phosphorylation of Bad. Indeed, prevention of Rac1-stimulated Bad phosphorylation only partially protected cells from drug-induced apoptosis. In this respect, Rac1 and Rac2 have been shown by others to promote cell survival through the activation of NF-κB (24) or generation of reactive oxygen species via NADPH oxidase (2, 13), which suggests that Rac GTPases regulate cell survival by at least one other mechanism.
Through an effort to identify the kinases that are capable of phosphorylating Bad at Ser-75, we show by a number of criteria that PKA is likely a principal kinase responsible for the site-specific phosphorylation of Bad in response to Rac1 activation in human lymphoma cells. Among an expanding family of Bad kinases, PKA has been shown to phosphorylate murine Bad at Ser-112 (8, 19, 20, 22, 39). However, the published evidence on the site specificity of PKA is inconsistent. One study showed that PKA primarily phosphorylates murine Bad at Ser-112 (20), and a separate report suggested that Ser-155 was phosphorylated preferentially by PKA in vitro and was the only residue in Bad that became phosphorylated when HEK-293 cells were exposed to cAMP-elevating agents (27). In the present study, we show that the three residues are almost equally susceptible to phosphorylation by PKAC but that only the human Ser-75 residue is phosphorylated in vivo. Specific inhibition of the kinase activity of PKA in the cell lysates blocked Bad phosphorylation. Intriguingly, Rac1 appears able to modulate the activity of PKA, although the underlying mechanism is unknown. This finding is important because it suggests a novel mechanism for PKA activation by factors other than cAMP.
Members of the PAK family are immediate Rac GTPase effector proteins, and they have also been shown to limit apoptosis through phosphorylation of Bad on Ser-112 and Ser-136 (8, 19, 22, 39). However, no one has yet shown which PAK isozymes phosphorylate critical Bad serine residues in response to Rac1 activation. In preliminary studies, we also found that immunodepletion of PAK2 or PAK4, but not PAK1, diminishes Bad phosphorylation in vitro (data not shown). Moreover, constitutively active PAK2 catalyzed phosphorylation of recombinant murine Bad at Ser-112 and Ser-155 in vitro while Ser-136 was untouched (see Fig. 4). These preliminary results confirm those of the previous reports showing that some, but not all, of the PAK isozymes are Bad kinases. Further experiments will be required to determine whether the PAKs phosphorylate Bad in response to Rac1 activation in a manner that inhibits apoptosis.
The finding that activated forms of Rac1 induce phosphorylation and inactivation of Bad may be of clinical importance, as Rac1 and its family members are frequently activated in human cancers (28, 37, 38). Their effect on the function of Bad may contribute to the oncogenic properties of upregulated Rac. Therefore, it will be of interest to determine whether Bad phosphorylation at Ser-75 is elevated in human cancers showing functional activation of the Rac signaling pathway. Recent studies suggested that the relative level of expression and phosphorylation of Bad might play an important role in the outcome of cancer chemotherapy (30, 40). Our work adds to this hypothesis by suggesting that interference with the function of Rac proteins and their signaling components might provide a useful anticancer strategy.
REFERENCES
- 1.Akasaki, T., H. Koga, and H. Sumimoto. 1999. Phosphoinositide 3-kinase-dependent and -independent activation of the small GTPase Rac2 in human neutrophils. J. Biol. Chem. 274:18055-18059. [DOI] [PubMed] [Google Scholar]
- 2.Archer, H., and D. Bar-Sagi. 2002. Ras and Rac as activators of reactive oxygen species (ROS). Methods Mol. Biol. 189:67-73. [DOI] [PubMed] [Google Scholar]
- 3.Aznar, S., and J. C. Lacal. 2001. Rho signals to cell growth and apoptosis. Cancer Lett. 165:1-10. [DOI] [PubMed] [Google Scholar]
- 4.Bar-Sagi, D., and A. Hall. 2000. Ras and Rho GTPases: a family reunion. Cell 103:227-238. [DOI] [PubMed] [Google Scholar]
- 5.Bergmann, A. 2002. Survival signaling goes BAD. Dev. Cell. 3:607-608. [DOI] [PubMed] [Google Scholar]
- 6.Bokoch, G. M. 2000. Regulation of cell function by Rho family GTPases. Immunol. Res. 21:139-148. [DOI] [PubMed] [Google Scholar]
- 7.Coleman, M. L., and M. F. Olson. 2002. Rho GTPase signalling pathways in the morphological changes associated with apoptosis. Cell Death. Differ. 9:493-504. [DOI] [PubMed] [Google Scholar]
- 8.Cotteret, S., Z. M. Jaffer, A. Beeser, and J. Chernoff. 2003. p21-activated kinase 5 (Pak5) localizes to mitochondria and inhibits apoptosis by phosphorylating BAD. Mol. Cell. Biol. 23:5526-5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Datta, S. R., A. Brunet, and M. E. Greenberg. 1999. Cellular survival: a play in three Akts. Genes Dev. 13:2905-2927. [DOI] [PubMed] [Google Scholar]
- 10.Datta, S. R., H. Dudek, X. Tao, S. Masters, H. Fu, Y. Gotoh, and M. E. Greenberg. 1997. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91:231-241. [DOI] [PubMed] [Google Scholar]
- 11.Datta, S. R., A. M. Ranger, M. Z. Lin, J. F. Sturgill, Y. C. Ma, C. W. Cowan, P. Dikkes, S. J. Korsmeyer, and M. E. Greenberg. 2002. Survival factor-mediated BAD phosphorylation raises the mitochondrial threshold for apoptosis. Dev. Cell. 3:631-643. [DOI] [PubMed] [Google Scholar]
- 12.del Peso, L., M. Gonzalez-Garcia, C. Page, R. Herrera, and G. Nunez. 1997. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science 278:687-689. [DOI] [PubMed] [Google Scholar]
- 13.Deshpande, S. S., P. Angkeow, J. Huang, M. Ozaki, and K. Irani. 2000. Rac1 inhibits TNF-alpha-induced endothelial cell apoptosis: dual regulation by reactive oxygen species. FASEB J. 14:1705-1714. [DOI] [PubMed] [Google Scholar]
- 14.Downward, J. 1999. How BAD phosphorylation is good for survival. Nat. Cell Biol. 1:E33-E35. [DOI] [PubMed] [Google Scholar]
- 15.Downward, J. 2003. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer. 3:11-22. [DOI] [PubMed] [Google Scholar]
- 16.Eisenmann, K. M., M. W. VanBrocklin, N. A. Staffend, S. M. Kitchen, and H. M. Koo. 2003. Mitogen-activated protein kinase pathway-dependent tumor-specific survival signaling in melanoma cells through inactivation of the proapoptotic protein Bad. Cancer Res. 63:8330-8337. [PubMed] [Google Scholar]
- 17.Etienne-Manneville, S., and A. Hall. 2002. Rho GTPases in cell biology. Nature 420:629-635. [DOI] [PubMed] [Google Scholar]
- 18.Fang, X., S. Yu, A. Eder, M. Mao, R. C. Bast, Jr., D. Boyd, and G. B. Mills. 1999. Regulation of BAD phosphorylation at serine 112 by the Ras-mitogen-activated protein kinase pathway. Oncogene 18:6635-6640. [DOI] [PubMed] [Google Scholar]
- 19.Gnesutta, N., J. Qu, and A. Minden. 2001. The serine/threonine kinase PAK4 prevents caspase activation and protects cells from apoptosis. J. Biol. Chem. 276:14414-14419. [DOI] [PubMed] [Google Scholar]
- 20.Harada, H., B. Becknell, M. Wilm, M. Mann, L. J. Huang, S. S. Taylor, J. D. Scott, and S. J. Korsmeyer. 1999. Phosphorylation and inactivation of BAD by mitochondria-anchored protein kinase A. Mol. Cell. 3:413-422. [DOI] [PubMed] [Google Scholar]
- 21.Holzberg, D., C. G. Knight, O. Dittrich-Breiholz, H. Schneider, A. Dorrie, E. Hoffmann, K. Resch, and M. Kracht. 2003. Disruption of the c-JUN-JNK complex by a cell-permeable peptide containing the c-JUN delta domain induces apoptosis and affects a distinct set of interleukin-1-induced inflammatory genes. J. Biol. Chem. 278:40213-40223. [DOI] [PubMed] [Google Scholar]
- 22.Jakobi, R., E. Moertl, and M. A. Koeppel. 2001. p21-activated protein kinase gamma-PAK suppresses programmed cell death of BALB3T3 fibroblasts. J. Biol. Chem. 276:16624-16634. [DOI] [PubMed] [Google Scholar]
- 23.Jiang, K., B. Zhong, C. Ritchey, D. L. Gilvary, E. Hong-Geller, S. Wei, and J. Y. Djeu. 2003. Regulation of Akt-dependent cell survival by Syk and Rac. Blood 101:236-244. [DOI] [PubMed] [Google Scholar]
- 24.Joneson, T., and D. Bar-Sagi. 1999. Suppression of Ras-induced apoptosis by the Rac GTPase. Mol. Cell. Biol. 19:5892-5901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim, R., K. Tanabe, Y. Uchida, M. Emi, H. Inoue, and T. Toge. 2002. Current status of the molecular mechanisms of anticancer drug-induced apoptosis. The contribution of molecular-level analysis to cancer chemotherapy. Cancer Chemother. Pharmacol. 50:343-352. [DOI] [PubMed] [Google Scholar]
- 26.Lee, Y., and E. Shacter. 1997. Bcl-2 does not protect Burkitt's lymphoma cells from oxidant-induced cell death. Blood 89:4480-4492. [PubMed] [Google Scholar]
- 27.Lizcano, J. M., N. Morrice, and P. Cohen. 2000. Regulation of BAD by cAMP-dependent protein kinase is mediated via phosphorylation of a novel site, Ser155. Biochem. J. 349:547-557. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Mira, J. P., V. Benard, J. Groffen, L. C. Sanders, and U. G. Knaus. 2000. Endogenous, hyperactive Rac3 controls proliferation of breast cancer cells by a p21-activated kinase-dependent pathway. Proc. Natl. Acad. Sci. USA 97:185-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Missy, K., P. Van, V., P. Raynal, C. Viala, G. Mauco, M. Plantavid, H. Chap, and B. Payrastre. 1998. Lipid products of phosphoinositide 3-kinase interact with Rac1 GTPase and stimulate GDP dissociation. J. Biol. Chem. 273:30279-30286. [DOI] [PubMed] [Google Scholar]
- 30.Motoyama, A. B., and N. E. Hynes. 2003. BAD: a good therapeutic target? Breast Cancer Res. 5:27-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murga, C., M. Zohar, H. Teramoto, and J. S. Gutkind. 2002. Rac1 and RhoG promote cell survival by the activation of PI3K and Akt, independently of their ability to stimulate JNK and NF-κB. Oncogene 21:207-216. [DOI] [PubMed] [Google Scholar]
- 32.Nobes, C. D., P. Hawkins, L. Stephens, and A. Hall. 1995. Activation of the small GTP-binding proteins Rho and Rac by growth factor receptors. J. Cell Sci. 108:225-233. [DOI] [PubMed] [Google Scholar]
- 33.Nunez, G., and L. del Peso. 1998. Linking extracellular survival signals and the apoptotic machinery. Curr. Opin. Neurobiol. 8:613-618. [DOI] [PubMed] [Google Scholar]
- 34.Pervaiz, S., J. Cao, O. S. Chao, Y. Y. Chin, and M. V. Clement. 2001. Activation of the RacGTPase inhibits apoptosis in human tumor cells. Oncogene 20:6263-6268. [DOI] [PubMed] [Google Scholar]
- 35.Ridley, A. J. 2001. Cyclin' round the cell with Rac. Dev. Cell. 1:160-161. [DOI] [PubMed] [Google Scholar]
- 36.Ruggieri, R., Y. Y. Chuang, and M. Symons. 2001. The small GTPase Rac suppresses apoptosis caused by serum deprivation in fibroblasts. Mol. Med. 7:293-300. [PMC free article] [PubMed] [Google Scholar]
- 37.Sahai, E., and C. J. Marshall. 2002. RHO-GTPases and cancer. Nat. Rev. Cancer 2:133-142. [DOI] [PubMed] [Google Scholar]
- 38.Schnelzer, A., D. Prechtel, U. Knaus, K. Dehne, M. Gerhard, H. Graeff, N. Harbeck, M. Schmitt, and E. Lengyel. 2000. Rac1 in human breast cancer: overexpression, mutation analysis, and characterization of a new isoform, Rac1b. Oncogene 19:3013-3020. [DOI] [PubMed] [Google Scholar]
- 39.Schurmann, A., A. F. Mooney, L. C. Sanders, M. A. Sells, H. G. Wang, J. C. Reed, and G. M. Bokoch. 2000. p21-activated kinase 1 phosphorylates the death agonist Bad and protects cells from apoptosis. Mol. Cell. Biol. 20:453-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taghiyev, A. F., N. V. Guseva, H. Harada, C. M. Knudson, O. W. Rokhlin, and M. B. Cohen. 2003. Overexpression of BAD potentiates sensitivity to tumor necrosis factor-related apoptosis-inducing ligand treatment in the prostatic carcinoma cell line LNCaP. Mol. Cancer Res. 1:500-507. [PubMed] [Google Scholar]
- 41.Tsujimoto, Y. 2003. Cell death regulation by the Bcl-2 protein family in the mitochondria. J. Cell Physiol. 195:158-167. [DOI] [PubMed] [Google Scholar]
- 42.Yang, E., J. Zha, J. Jockel, L. H. Boise, C. B. Thompson, and S. J. Korsmeyer. 1995. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell 80:285-291. [DOI] [PubMed] [Google Scholar]
- 43.Yang, F. C., R. Kapur, A. J. King, W. Tao, C. Kim, J. Borneo, R. Breese, M. Marshall, M. C. Dinauer, and D. A. Williams. 2000. Rac2 stimulates Akt activation affecting BAD/Bcl-XL expression while mediating survival and actin function in primary mast cells. Immunity 12:557-568. [DOI] [PubMed] [Google Scholar]
- 44.Zha, J., H. Harada, E. Yang, J. Jockel, and S. J. Korsmeyer. 1996. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell 87:619-628. [DOI] [PubMed] [Google Scholar]
- 45.Zhang, B., Y. Gao, S. Y. Moon, Y. Zhang, and Y. Zheng. 2001. Oligomerization of Rac1 GTPase mediated by the carboxyl-terminal polybasic domain. J. Biol. Chem. 276:8958-8967. [DOI] [PubMed] [Google Scholar]
- 46.Zhang, B., Y. Zhang, and E. Shacter. 2003. Caspase 3-mediated inactivation of Rac GTPases promotes drug-induced apoptosis in human lymphoma cells. Mol. Cell. Biol. 23:5716-5725. [DOI] [PMC free article] [PubMed] [Google Scholar]