

Abstract
Background:
There is preclinical synergism between taxanes and MK-2206. We aim to determine the maximum tolerated dose, safety, and activity of combining MK-2206 and paclitaxel in metastatic cancer.
Methods:
Patients received weekly doses of paclitaxel at 80mg/m2 on day 1, followed by MK-2206 orally on day 2 escalated at 90mg, 135mg, and 200mg. Treatment continued until progression, excessive toxicity, or patient request. Blood and tissue were collected for pharmacokinetic and pharmacodynamics markers. A cycle consisted of three weeks of therapy. Dose-limiting toxicity (DLT) was defined as unacceptable toxicity during the first cycle. All statistical tests were two-sided.
Results:
Twenty-two patients were treated, nine in dose escalation and 13 in dose expansion. Median age was 55 years. Median number of cycles was four. Dose escalation was completed with no DLT. CTCAE Grade 3 or higher adverse events were fatigue (n = 2), rash (n = 2), hyperglycemia (n = 1), and neutropenia (n = 7). Four patients in the expansion phase required MK-2206 dose reduction. Phase II recommended dose was established as paclitaxel 80mg/m2 weekly on day 1, and MK-2206 135mg weekly on day 2. Paclitaxel systemic exposure was similar in the presence or absence of MK-2206. Plasma MK-2206 concentrations were similar to data from previous phase I monotherapy. There was a statistically significant decrease in expression of pAKT S473 (P = .01) and pAKT T308 (P = .002) after therapy. PI3K/AKT/mTOR downregulation in tumor tissues and circulating markers did not correlate with tumor response or clinical benefit. There were five objective responses, and nine patients had stable disease.
Conclusion:
MK-2206 was well tolerated with paclitaxel. Preliminary antitumor activity was documented.
The PI3K/AKT/mTOR pathway is downstream of most growth factor tyrosine kinase receptors (TKRs) in cancer. It plays a key role in cell growth, protein translation, autophagy, metabolism, and cell survival (1,2). Pathway deregulation may occur through overexpression or activation of TKR, mutations and amplification of PIK3CA or AKT, and loss of negative regulators PTEN and INPP4B. Increased levels of phosphor-AKT and PTEN loss are poor outcome predictors (3). We showed that of 547 breast cancers tested, 117 (21.4%) had mutations in PIK3CA (4). In breast cancer cells, PTEN levels inversely correlated with AKT phosphorylation (5). Thus, PTEN-low tumors and PIK3CA mutant tumors may rely on AKT for oncogenic signaling. Therefore, AKT inhibitors may have a broader utility than TKR inhibitors.
Preclinical work with MK-2206 shows that many PIK3CA mutant and PTEN loss lines are sensitive (6). Loss of PTEN and PI3K signaling activation are associated with resistance to endocine therapy and trastuzumab (7–9). MK-2206 showed activity with improvement in breast cancer metastasis (10). In preclinical studies, MK-2206 demonstrated synergy with paclitaxel, and the combination had greater in vivo antitumor efficacy (6). Synergistic or additive inhibitory effects were also observed with docetaxel. Synergism was sequence-dependent and occurred when cells were treated with docetaxel followed by MK-2206 (11). Metabolism of MK-2206 in human liver is catalyzed by the cytochrome P450 3A4 isoenzyme (CYP3A4), as is docetaxel. In our previous phase I study using everolimus, there was a statistically significant pharmacokinetic (PK) interaction when combined with docetaxel, with severe adverse events (AEs) (12). Conversely, the same combination with paclitaxel had no PK interaction in our phase II neoadjuvant breast cancer trial (13).
The purpose of this study was to determine the MTD of the combination of weekly MK-2206 and paclitaxel (escalation) and to determine the safety and antitumor activity of the combination in metastatic breast cancer (expansion). Secondary objectives included PK of the combination, baseline molecular markers and pharmacodynamic markers in blood, and tumor tissue that may predict clinical activity.
Methods
The study was an open-label phase I study combining weekly paclitaxel with MK-2206 in advanced solid tumors with an expansion in advanced breast cancer (NCT01263145). Eligible patients had histologically confirmed metastatic tumors that had failed at least two therapy lines (escalation) and metastatic breast cancer that had progressed after maximum three therapy lines (expansion). Patients had measurable disease by Response Evaluation Criteria in Solid Tumors (RECIST) version 1.0 or evaluable disease (14), were age 18 years or older, had adequate organ function including HgbA1c under 8%, Eastern Cooperative Oncology Group (ECOG) performance status (PS) 0–2. Prior treatment with PI3K pathway inhibitors and paclitaxel for early disease was permitted. Patients were excluded if pregnant, breastfeeding, or taking CYP3A4 inducers or inhibitors. Washout period was 21 days. Radiographic evaluations were performed every nine weeks. The clinical trial was reviewed yearly and approved by institutional review boards. Patients provided written informed consent.
Study Therapy
MK-2206 was provided by Cancer Therapy Evaluation Program (CTEP), and paclitaxel was commercially available. Participants were considered for three dose-escalation levels and for a dose-expansion cohort once MTD was established. Paclitaxel was given at a fixed dose of 80mg/m2 intravenously (IV) weekly on day 1, and MK-2206 was escalated at 90mg, 135mg, and 200mg orally weekly on day 2. Premedication for paclitaxel consisted of dexamethasone 10mg on week 1, 4mg IV on week 2, and discontinued after if no infusion reaction occurred. Once MTD was reached, patients with metastatic breast cancer were treated. Cycle length was three weeks, and treatment was continued until disease progression, unacceptable toxicity, patient refusal, or physician’s decision.
Safety and Efficacy
Safety assessments were conducted at baseline, at weekly basis during the first cycle, then every cycle or earlier if toxicity occurred. Patients removed from study for AEs were followed until resolution or stabilization. Toxicity was graded according to National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE), version 4.0. A DLT was defined as any grade 3/4 nonhematologic toxicity, grade 3/4 rash or hyperglycemia lasting more than 72 hours, grade 4 febrile neutropenia that required hospitalization, and any grade 3 hematologic toxicity that required treatment delay beyond two weeks. If toxicity occurred, an appropriate treatment was used. If grade 3 or 4 toxicity occurred, drug was held until the toxicity was grade 1 or less. If grade 3 or 4 toxicity recurred, continuation of treatment was discussed with a decision made after consideration of relative risks and benefits. Therapy was restarted at a -1 dose level (MK-2206 of 135mg PO weekly); if a second grade 3 or 4 toxicity occurred, therapy was stopped. Paclitaxel doses were reduced by 25% (60mg/m2 IV weekly) after 12 weeks of therapy if patients develop grade 2 or greater neuropathy or nail changes.
All patients underwent anatomic imaging at presentation and every three cycles to establish response. A decrease in the size of the sum of the diameters of the index lesions of greater than or equal to 30% was considered a partial response (PR). Stable disease (SD) was considered if stability was documented at least once for four or more weeks. Clinical benefit was documented if PR or SD response lasted for 18 weeks (six cycles). Progression of disease (PD) was defined as 20% or greater increase of the index lesions or appearance of new lesions.
Pharmacokinetic Analysis
For paclitaxel, blood samples were drawn on days 1 and 15 of cycle 1 at predose, 10 minutes and 50 minutes following infusion, and then 10 minutes, 30 minutes, one hour, two hours, four hours, eight hours, and 24 hours after infusion completion. MK-2206 samples were collected on day 2 of cycle 1 at predose, one hour, two hours, four hours, six hours, eight hours, 24 hours, 72 hours, and 144 hours after MK-2206 dose; on day 16 of cycle 1 at predose, one hour, two hours, four hours, six hours, eight hours, 24 hours, 72 hours, and 144 hours after MK-2206 dose. Individual plasma concentration time data for paclitaxel and MK-2206 were used to generate PK parameter estimates using compartmental and noncompartmental methods utilizing WinNonLin Professional 5.3 (Pharsight Corp., St. Louis, MO). Peak plasma concentration (Cmax) and time to peak concentration (Tmax) were determined by data observation. Areas under the plasma concentration time curve (AUC) from zero to 24 hours postdose (AUC0-24), zero to 144 hours postdose (AUC0-144), and zero to infinity (AUC0-inf) were calculated by the linear trapezoidal method. Drug clearance (Cl) was determined by dose/AUC. Elimination half-life (t1/2) was calculated by 0.693/k, and apparent volume of distribution was calculated by Cl/k (15).
Biomarker Assessment
Fine-needle aspirations (FNAs) and core biopsies were obtained pretreatment and at two weeks. Samples were evaluated by reverse phase protein arrays (RPPA) to assess PI3K activation status. The antibodies used are listed in Supplementary Table 1 (available online). Core biopsies were assessed for PTEN and INPP4B expression by immunohistochemistry (IHC) and mutations at the PI3K pathway genes. Whole blood for pharmacodynamics assessments was collected at least at five time points: C1-D1 (pretreatment), C1-D2, C1-D3, C1-D5, C1-D8, C1-D15, C1-D16, C1-D17, C1-D19, and C2-D1 (prior to next dose). Peripheral blood mononuclear cells (PBMCs) and platelet-enriched plasma (PRP) were obtained and evaluated by RPPA. The full biomarker methodology including bioinformatics analysis is described in the Supplementary Methods (available online).
Statistical Analysis
For RPPA data analysis, paired t test was used to examine the difference between baseline and post-treatment samples by dose level (details are presented in the Supplementary Materials, available online). Two sample two-sided t tests were used to test the association between: 1) baseline protein expression, 2) pharmacodynamic changes (PDϪ), and 3) PI3K activity score with tumor response or clinical benefit. We defined PDϪ as the difference on protein expression after therapy exposure (PDϪ = post-treatment expression – pretreatment expression in tumor tissues and day 2-day 1, day 3-day1, and day 3-day 2 for circulating markers). PI3K activity score was defined as the sum of the phosphor-protein expression levels of AKT, mTOR, 4EBP1, S6K, and S6 (PI3K score = pS6-240/244 + pS6-S235/23(16)6 + pS6K-T389 + pmTOR-S2448 + p4EBP1-S65 + p4EBP1-T37/46 + pPRAS40-T246 + pAKT-S473 + pAKT-T308). For multiplicity adjustments, the Benjamini Hochberg (BH) (16) procedure was employed to control the False Discovery Rate (FDR). We used thresholds 0.3 or 0.1 for FDR to calibrate for the top proteins of interest. Fisher’s exact tests were performed to test the association of INPP4B, PTEN, or either protein presence or loss with tumor response or clinical benefit. A P value of less than .05 was considered statistically significant.
Results
Patients Characteristics and Disposition
Supplementary Figure 1 (available online) summarizes the patient disposition. Twenty-two patients were enrolled, nine patients in dose escalation, and 13 patients in dose expansion. Three patients in the dose expansion were replaced because of lack of compliance (did not show up or receive any treatment), early progression (new metastasis documented within the first week of therapy) and severe therapy-related toxicities at first dose. Table 1 summarizes the clinical characteristics of all patients. Median age was 55 years (range 34–79 years). There were five men and 17 women. All patients but one had an ECOG PS of 0 or 1. In addition to breast cancer (n = 14), other primary tumors included colorectal (n = 2), ovarian (n = 1), endometrial (n = 1), ocular melanoma (n = 2), and squamous cell carcinoma of the head and neck (n = 2). Median previous therapy lines were 3 (range 1–10) for dose escalation and 1 (range 0–3) for expansion.
Table 1.
Patient and tumor characteristics*
| Characteristic | Patients (n = 22) | |
|---|---|---|
| Dose escalation | Dose expansion | |
| (n = 9) | (n = 13) | |
| No. (%) | No. (%) | |
| Age, y | ||
| Mean (range) | 52 (34–60) | 67 (34–79) |
| Sex | ||
| Male | 5 (55.6) | 0 (0.0) |
| Female | 4 (44.4) | 13 (100) |
| Race | ||
| African American | 0 | 2 (15.4) |
| White | 8 (88.9) | 10 (76.9) |
| Hispanic | 1 (11.1) | 1 (7.7) |
| ECOG performance status | ||
| 0 | 5 (55.5) | 10 (76.9) |
| 1 | 4 (44.4) | 2 (15.4) |
| 2 | 0 | 1 (7.7) |
| Primary site | ||
| Colorectal | 2 (22.2) | - |
| Breast | 1 (11.1) | 13 (100.0) |
| Ovary | 1 (11.1) | - |
| Ocular melanoma | 2 (22.2) | - |
| Head and neck | 2 (22.2) | - |
| Endometrial | 1 (11.1) | - |
| Number of therapies for metastatic cancer | ||
| 0 | 0 (0.0) | 3 (23.1) |
| 1 | 2 (22.2) | 5 (38.5) |
| 2 | 2 (22.2) | 1 (7.7) |
| 3 | 2 (22.2) | 4 (30.8) |
| ≥4 | 3 (33.3) | 0 (0.0) |
* ECOG = Eastern Cooperative Oncology Group.
Dose-Limiting Toxicities and Phase II Recommended Dose
There were no DLTs during the dose escalation. The MTD was paclitaxel 80mg/m2 IV weekly on day 1, and MK-2206 200mg orally weekly on day 2. However, four patients in the dose expansion required dose reductions of MK-2206 because of grade 2/3 rash. Therefore, the phase II recommended dose (P2RD) was defined as paclitaxel 80mg/m2 weekly on day 1 and MK-2206 135mg weekly on day 2 in every three-week cycle.
Safety and Tolerability
Patients in dose escalation received a median of eight cycles (range = 1–12), and patients in dose expansion received a median of three cycles (range: 0–10). Three patients in dose expansion did not complete one cycle, precluding efficacy assessment by protocol. Two of them discontinued therapy, one because of rapid progression, and one because of grade 4 toxicities attributed to study drugs. Paclitaxel and MK-2206 were tolerated well by most patients (Table 2), with most AEs being grade 1 to 2. Most common AEs included alopecia (n = 19, 90.4%), rash (n = 16, 76.2%), anemia (n = 16, 76.2%), dysgeusia (n = 5, 23.8%), fatigue (n = 16, 76.2%), neutropenia (n = 15, 71.4%), and hyperglycemia (n = 14, 66.6%). CTCAE Grade 3 or higher adverse events were fatigue (n = 2, 9.5%), rash (n = 2, 9.5%), hyperglycemia (n = 1, 4.8%), and neutropenia (n = 7, 33.5%). Serious AEs were considered related to MK-2206. They occurred in a single patient that developed grade 4 hyperglycemia and grade 4 rash after MK-2206 first dose and required admission to the hospital for supportive management.
Table 2.
Treatment-related adverse events
| Drug-related adverse events | Grades | Level 1 (n = 3) | Level 2 (n = 3) | Level 3 (n = 3) | Expansion (n = 12) | Total (n = 21) |
|---|---|---|---|---|---|---|
| Acne face/scalp | All 3–4 |
0 0 |
1 0 |
0 0 |
0 0 |
1 0 |
| Alopecia | All 3–4 |
2 0 |
3 0 |
2 0 |
12 0 |
19 0 |
| Anemia | All 3–4 |
1 0 |
2 0 |
2 0 |
11 1 |
16 1 |
| Anorexia | All 3–4 |
1 0 |
3 0 |
1 0 |
2 0 |
7 0 |
| Arthralgia | All 3–4 |
1 0 |
1 0 |
0 0 |
0 0 |
2 0 |
| Deep vein thrombosis | All 3–4 |
0 1 |
0 0 |
0 0 |
0 0 |
0 1 |
| Dry skin | All 3–4 |
0 0 |
1 0 |
2 1 |
0 0 |
3 1 |
| Diarrhea | All 3–4 |
0 0 |
0 0 |
2 0 |
0 0 |
2 0 |
| Dysgeusia | All 3–4 |
2 0 |
0 0 |
1 0 |
2 0 |
5 0 |
| Fatigue | All 3–4 |
1 1 |
3 1 |
2 0 |
10 0 |
16 2 |
| Fever | All 3–4 |
0 0 |
0 0 |
0 0 |
1 0 |
1 0 |
| Flu-like symptoms | All 3–4 |
0 0 |
1 0 |
1 0 |
4 0 |
6 0 |
| Hyperglycemia | All 3–4 |
1 0 |
2 0 |
2 0 |
9 1 |
14 1 |
| Hypertriglyceridemia | All 3–4 |
2 0 |
0 0 |
0 0 |
0 0 |
2 0 |
| Mucositis | All 3–4 |
2 0 |
1 0 |
2 0 |
5 0 |
10 0 |
| Myalgias | All 3–4 |
0 0 |
2 0 |
0 0 |
2 0 |
4 0 |
| Nail changes | All 3–4 |
4 0 |
3 1 |
1 0 |
1 0 |
9 1 |
| Nausea | All 3–4 |
2 0 |
2 0 |
1 0 |
6 0 |
11 0 |
| Neuropathy | All 3–4 |
2 0 |
3 0 |
1 0 |
9 2 |
15 2 |
| Neutropenia | All 3–4 |
0 0 |
3 0 |
0 1 |
12 6 |
15 7 |
| Prolonged QTc | All 3–4 |
1 0 |
0 0 |
0 0 |
0 0 |
1 0 |
| Pruritus | All 3–4 |
0 0 |
1 0 |
1 0 |
0 0 |
2 0 |
| Rash | All 3–4 |
2 0 |
3 0 |
3 0 |
8 2 |
16 2 |
Pharmacokinetics
Plasma concentration time profiles for paclitaxel on C1D1 and C1D15 following 80mg/m2 IV infusion over one hour on a weekly schedule in combination with 90mg or 135mg MK-2206 orally were similar with polyphasic elimination after achieving similar peak concentrations at the end of drug administration (Supplementary Figure 2, available online). Following a noncompartmental analysis to fit the concentration time data, all PK parameters estimated were not statistically significantly different between days or doses across both agents (Table 3). After two weekly doses of 200mg MK-2206 on C1-D2 and C1-D9, mean paclitaxel exposure on C1-D15 measured by both AUC0-24 and AUC0-inf increased by 30% to 40% in addition to a more than two-fold increase in mean peak concentration when compared with 135mg MK-2206. These PK changes in paclitaxel on C1-D15 are most likely caused by the increased dose of MK-2206 but the interpatient variability is high. In published data, a mean paclitaxel Cmax of 2000–2500ng/mL was observed and AUC did not exceed 5000 hr•ng/mL (17–19). There were no changes in paclitaxel terminal half-life. MK-2206 PK on a weekly administration schedule at all dose levels when combined with paclitaxel was characterized by a relatively slow absorption (mean Tmax range = 4..8-6..8 hours) with a subtle biphasic elimination (Supplementary Figure 3, available online) leading to a mean terminal half-life of 50 to 60 hours, consistent with previous single-agent data (10). Peak concentration and systemic exposure of MK-2206 were dose dependent. Mean peak concentrations and AUC0-144hr at 90mg and 135mg dose levels following the third dose at C1-D16 were similar to single agent data after steady state was reached (10). At a 200mg dose, mean systemic exposure over 144 hours at C1-D2 (24,245 nM•hr) was similar to previous single agent data for 300mg weekly (AUC0-168hr, 27,700 nM•hr) that led to three out of three patients with grade 3 skin rash. Less than a 2.5-fold increase in drug accumulation was observed based on AUC0-24 and AUC0-inf systemic exposure comparison between C1-D2 and C1-D16, which is similar to published data (10,15,19).
Table 3.
Mean pharmacokinetic parameters estimated for paclitaxel and MK-2206
| MK-2206 parameters | 90mg Qweekly | 135mg Qweekly | 200mg Qweekly | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C1D2 (n = 3) |
SD | C1D16 (n = 3) |
SD | C1D2 (n = 3) |
SD | C1D16 (n = 3) | SD | C1D2 (n = 16) |
SD | C1D16 (n = 10) |
SD | |
| AUC0-24, nM.hr | 2589.8* | N/A | 3206.9 | 1273.2 | 3757.9 | 2043.0 | 5427.7 | 3957.2 | 6724.6 | 3250.0 | 6427.5 | 2264.2 |
| AUC0-144, nM.hr | N/A | N/A | N/A | N/A | N/A | N/A | 19169.5 | 13590.2 | 24244.8 | 10906.5 | 24050.5 | 7290.7 |
| AUC0-inf, nM.hr | 11597.6* | N/A | 22335.9 | 25829.5 | 9803.9 | 5575.4 | 23955.2 | 16600.7 | 30954.7 | 13187.5 | 28546.8 | 10317.1 |
| Cmax, nM | 123.7 | 25.9 | 167.7 | 68.7 | 224.7 | 129.8 | 307.8 | 221.2 | 392.1 | 168.1 | 357.6 | 171.1 |
| t1/2, hr† | 56.9* | N/A | 49.9 | 28.3 | 20.3‡ | 6.5 | 29.6 (60.6)‡ | 26.8 (13.2) | 56.2 | 11.0 | 60.5 | 11.9 |
| Tmax, hr† | 6.5 | 1.0 | 5.1 | 1.4 | 5.5 | 2.1 | 4.8 | 1.5 | 5.1 | 3.9 | 6.8 | 4.1 |
| Cl, L/hr | 19.0* | N/A | 21.4 | 15.1 | 40.3 | 22.9 | 17.9 | 8.9 | 18.3 | 7.2 | 19.2 | 6.7 |
| Vz, L | 1563.0* | N/A | 1399.9 | 345.5 | 1132.3 | 325.9 | 1628.0 | 918.0 | 1512.0 | 586.4 | 1736.3 | 804.4 |
| Paclitaxel parameters | C1D1 (n = 3) |
SD | C1D15 (n = 3) |
SD | C1D1 (n = 3) |
SD | C1D15 (n = 3) | SD | C1D1 (n = 15) |
SD | C1D15 (n = 10) |
SD |
| AUC0-24, hr.ng/mL | 6780.1 | 2414.1 | 5036.2 | 421.5 | 5364.2 | 772.9 | 5633.1 | 1022.1 | 19008.1 | 32538.1 | 7739.9 | 6215.0 |
| AUC0-inf, hr.ng/mL | 7375.6 | 2881.2 | 5639.4 | 432.8 | 5933.4 | 592.1 | 6239.8 | 1304.2 | 19644.3 | 32574.6 | 8331.4 | 6294.1 |
| Cmax, ng/mL | 4323.3 | 620.0 | 2408.9 | 593.8 | 3247.1 | 1207.6 | 3170.3 | 642.0 | 18465.6 | 36675.6 | 6536.6 | 9193.8 |
| t1/2, hr | 6.4 | 2.8 | 10.7 | 2.9 | 10.6 | 2.2 | 8.9 | 5.0 | 9.9 | 1.7 | 9.8 | 2.0 |
| Cl, L/hr/m2 | 12.0 | 4.3 | 14.2 | 1.0 | 13.6 | 1.4 | 13.2 | 3.1 | 10.7 | 5.5 | 12.1 | 4.4 |
| Vz, L/m2 | 99.7 | 25.6 | 222.2 | 70.6 | 209.7 | 64.0 | 155.3 | 67.0 | 154.2 | 88.4 | 173.1 | 74.3 |
* n =1
† Harmonic mean ± pseudo standard deviation is reported. AUC = area under the curve.
‡ t1/2 using four- to 24-hour postdose timepoints reported. Value in parentheses for C1D16 includes 72- and 144-hour time points.
Antitumor Activity
From the 21 patients who received therapy, there were five PR and nine SD. Three responses occurred in the expansion cohort and two in dose-escalation group, one in a patient with metastatic breast cancer and the other one in a patient with metastatic colorectal cancer. SD was documented in three patients in expansion cohort and in six patients in the dose-escalation cohort: two squamous cell carcinomas of the head and neck, two ocular melanomas, one ovarian, and one endometrial cancer. Maximum percentage of disease change by RECIST in patients with at least one follow-up scan is presented in Figure 1.
Figure 1.
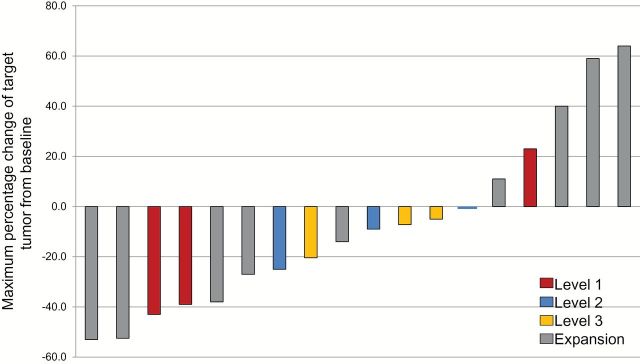
Maximum percentage change of target tumor from baseline by Response Evaluation Criteria in Solid Tumors in patients with at least one follow-up scan.
Pharmacodynamics and Biomarker Analysis
Tumor Tissues
Samples for RPPA were available from 21 patients at baseline and 16 patients post-treatment. Outcome information was available in 18 patients. There was a statistically significant difference between baseline and post-treatment expression of pAKT-S473 (P = .01) and pAKT-T308 (P = .002) (Figure 2A). However, at a false discovery rate (FDR) threshold of 0.1, there were no statistically significant differences in protein expression from baseline to post-treatment in any dose levels. At baseline level, no protein was found differentially expressed for tumors that had responses vs no responses. Two proteins were found differentially expressed among patients who had clinical benefit vs no benefit: pPKCα-S657 (P < .001) and VEGFR2 (P = .003). There was a statistically significant association of pharmacodynamic changes (PDϪ) of p27 with response (P < .001). There were no PDϪ associated with clinical benefit. There were no statistically significant associations between baseline PI3K activity score with response (means -19.0 vs -20.5 for response and nonresponse, respectively, P = .70) or clinical benefit (Means -19.9 vs -20.7 for benefit vs no benefit, respectively, P = .63) (Figure 2B). When assessing PI3K/ activation score from baseline to post-treatment, there was no association of pathway downregulation and response or clinical benefit (P = .28 and >.99, respectively) (Supplementary Tables 2 and 3, available online). Lastly, at an FDR threshold of 0.1, PDϪ changes were found not to be dose dependent.
Figure 2.
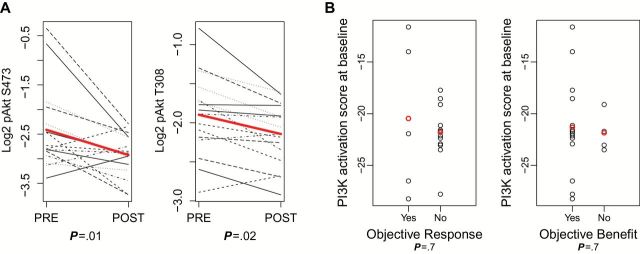
A) Differences between baseline and post-treatment expression of pAKT-S473 (P = .01), and pAKT-T308 (P = .003) with two-sided t test. B) Baseline PI3K activity scores showing no associations with tumor response (means -20.464 vs -21.801 for response and nonresponse, respectively, P = .7), or clinical benefit (Means -21.311 vs -21.846 for benefit vs no benefit, respectively, P = .07). Analysis was done with two-sided t test, P < .05 was considered statistically significant.
Baseline tissues for IHC were available in 16 patients. PTEN loss was documented in three tumors and INPP4B loss in nine tumors. Two tumors had both PTEN and INPP4B loss. There was no association of INPP4B, PTEN or either protein loss with response (P > .99 for all cases), or clinical benefit (P > .99, .45, and >.99, respectively).
Adequate quality DNA for sequencing was available in 10 patients. A PIK3CA E545K mutation was detected in a patient with head and neck cancer who had SD for 10 weeks. No AKT or PTEN mutations were found.
Circulating Markers
Whole blood was collected on day 1 (pretreatment), day 2 (post-paclitaxel but pre-MK-2206) and day 3 (post-paclitaxel and MK-2206). Blood was available in 17 patients, and outcome information was available in 15. Comparisons were made from day 2 to day 1, day 3 to day 1, and day 3 to day 2.
In PRPs, at an FDR threshold of 0.1, there was a statistically significant decrease of pAKT-S473, pAKT-T308, and p70S6K-T389, (P < .001 in all) from day 2 to day 3 (Figure 3A). There was no association of PDϪ (day 2-day 1, day 3-day 1 or day 3-day 2) with tumor response. There were no statistically significant associations between baseline PI3K activity score with tumor response (Means -8.6 vs -9.3 for response and nonresponse, respectively, P = .61). When assessing changes in PI3K activation score from baseline to post-treatment, there was a statistically significant decrease in activity score from day 2 to day 3 (means -8.9 vs -11.4, P < .001), consistent with inhibition of PI3K signaling. However, there was no association of pathway downregulation and tumor response at any time interval (P > .2 in all) (Supplementary Tables 2–4, available online). Lastly, at an FDR threshold of 0.3, pAKT inhibition was dose dependent at day 3 (pAKT-S473: P = .003, pAKT-T308: P = .02), at PDϪ from day 1 to day 3 (pAKT-S473: P = .014, pAKT T308: P = .01), and from day 2 to day 3 (pAKT S473: P = .02, pAKT-T308: P = .01) (Figure 3B). Associations with clinical benefit were not possible because of limited patient numbers. Data on PBMCs and PRP showed similar results, but AKT inhibition was more apparent in PRPs.
Figure 3.

A) Decline of pAKT-T308 and pAKT-S473 in platelets on day 3, after treatment with paclitaxel + MK-2206 (MK-2206 administered D2). B) Dose dependence of pAKT levels at day 3 (D3) or by measuring pharmacodynamic (PD) changes from day 2 to day 3 (D3-D2) or from day 1 to day 3 (D3-D1).
Discussion
We completed a phase I trial of paclitaxel in combination with MK-2206. We found that the RP2D was paclitaxel 80mg/m2 weekly on day 1 and MK-2206 135mg weekly on day 2. Grade 3 or higher adverse events were fatigue, rash, hyperglycemia, and neutropenia. Toxicity was less than expected possible from concomitant steroid use, and except for cytopenias related to chemotherapy most clinically significant AEs were consistent and milder to what has been previously reported with MK-2206 monotherapy or in combination with trastuzumab (10,20). We saw no PK interactions, paclitaxel exposure was similar in the presence or absence of MK-2206, and plasma MK-2206 concentrations appeared similar to data from phase 1 monotherapy (10), supporting our taxane selection avoiding possible CYP3A4 interactions (12).
Weekly paclitaxel is an effective therapy in metastatic breast cancer with response rates (RR) of 21% to 42% according to therapy line (21–25). However, when looking at third line setting, RR decrease to 15% with 33% SD (22). There is no phase II data on single agent MK-2206. In the phase I study there were no responses, but 18% of patients had SD. In our population, the combination resulted in a meaningful risk ratio of 24%, with 43% of patients experiencing SD. This was the selected regimen for I SPY 2 (Investigation of Serial Studies to Predict Your Therapeutic Response with Imaging and Molecular Analysis 2) (26). A recently reported phase I study of MK-2206 plus carboplatin and paclitaxel, docetaxel, or erlotinib reported early evidence of antitumor activity in head and neck cancer, with paclitaxel used in a three-week schedule (27).
In preclinical work, the effect of MK-2206 with/without paclitaxel on cell signaling, cell growth, and survival in vitro and in vivo in cells correlated with different PIK3CA and PTEN status. We found that MK-2206 had antitumor activity alone and in combination with paclitaxel, and that activity could be greater in PTEN loss or PIK3CA mutants (6). Work from Beaver, et al. showed that PIK3CA, but not AKT1 mutations, increased sensitivity to GDC-0941 (PI3K inhibitor) and MK-2206 (28). In our clinical trial, we found a statistically significant difference between baseline and post-treatment pAKT-S473 and pAKT-T308 expression, demonstrating that MK-2206 inhibited AKT phosphorylation consistent with decreased activity at biologically achieved levels. However, PI3K downregulation in tumor tissues, PBMCs, and platelets was not statistically significantly different between patients who had response or stable disease compared with patients who progressed. Thus, extent of pathway inhibition was not a predictor of outcome, but rather tumors may differ in the extent they rely on PI3K signaling. Further studies with adequate power are needed to determine whether even greater pathway inhibition could further improve antitumor efficacy of AKT inhibitors. There was no association of INPP4B or PTEN loss with tumor response or stable disease. We found a single PIK3CA mutation. In the MK-2206/trastuzumab phase I study, all 37 patients had baseline analysis of circulating DNA for PIK3CA mutations. Only three PIK3CA mutations were found, but their analysis did not confirm the hypothesis that tumors with PIK3CA mutations are more sensitive to MK-2206 (20). At this point, our data does not support patient selection by the PI3K aberrations explored. The correlative work of I SPY2 results may show other predictors. Other molecular aberrations or a systems biology approach should be considered for optimal biomarker discovery.
Our study had some limitations. We studied weekly dosing of MK2206, as we expected that weekly dosing would be more tolerable than daily dosing, but we cannot exclude the possibility that other intermittent schedules such as every other day or every third day may not be tolerable and more efficacious. Further, we have not explored the safety, tolerability of different timing of delivery. In the dose expansion, we included tumors of different histologic types and genomic backgrounds; this and escalating doses limit our ability to assess efficacy. Although we were successful in getting pretreatment and on-treatment biopsies, the sample size and number of responders limit our ability to make significant correlations with outcome. Our biomarker analysis has shown inhibition of Akt phosphorylation, but whether this is sufficient target inhibition requires more study.
Our results show evidence of antitumor activity of the combination of paclitaxel and MK-2206 in solid tumors and in breast cancer, and the combination was well tolerated. Paclitaxel did not affect the PK profile of MK-2206 at 135mg/week, suggesting that this AKT inhibitor can be safely combined with paclitaxel. Our data does not support patient selection by the PI3K aberrations explored, however this analysis was limited by sample size.
Funding
This work was supported in part by National Cancer Institute 1K23CA121994 (AMG), ASCO Career Development Award (AMG), Komen for the Cure Catalyst Award KG090341 (AMG), American Cancer Society Research Scholar Grant 121329-RSG-11-187-01-TBG (AMG), SUC2-AACR-DT0209 01(AMG, GBM, FMB), National Cancer Institute U01 CA062461-18 (RK), and the Commonwealth Foundation for Cancer research (AMG).
Supplementary Material
Results of this study were presented at the 2012 American Association of Cancer Research Annual Meeting.
The study funders had no role in design of the study, the collection, analysis, or interpretation of the data, the writing of the manuscript, nor the decision to submit the manuscript for publication.
We thank Sha Huang for pharmacokinetic sample analysis of paclitaxel; we thank Pharmacokinetics, Pharmacodynamics, and Drug Metabolism at Merck Research Laboratories-Oncology for pharmacokinetic sample analysis of MK-2206 and Lisa Norberg for all pharmacokinetic data organization, modeling, and analysis.
References
- 1. Hennessy BT, Smith DL, Ram PT, et al. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4(12):988–1004. [DOI] [PubMed] [Google Scholar]
- 2. Meric-Bernstam F, Gonzalez-Angulo AM. Targeting the mTOR signaling network for cancer therapy. J Clin Oncol. 2009;27(13):2278–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Saal LH, Holm K, Maurer M, et al. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res. 2005;65(7):2554–2559. [DOI] [PubMed] [Google Scholar]
- 4. Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68(15):6084–6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vasudevan KM, Barbie DA, Davies MA, et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell. 2009;16(1):21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sangai T, Akcakanat A, Chen H, et al. Biomarkers of response to Akt inhibitor MK-2206 in breast cancer. Clin Cancer Res. 2012;18(20):5816–5828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Miller TW, Perez-Torres M, Narasanna A, et al. Loss of Phosphatase and Tensin homologue deleted on chromosome 10 engages ErbB3 and insulin-like growth factor-I receptor signaling to promote antiestrogen resistance in breast cancer. Cancer Res. 2009;69(10):4192–4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nagata Y, Lan KH, Zhou X, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6(2):117–127. [DOI] [PubMed] [Google Scholar]
- 9. Berns K, Horlings HM, Hennessy BT, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12(4):395–402. [DOI] [PubMed] [Google Scholar]
- 10. Yap TA, Yan L, Patnaik A, et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J Clin Oncol. 2011;29(35):4688–4695. [DOI] [PubMed] [Google Scholar]
- 11. Hirai H, Sootome H, Nakatsuru Y, et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther. 2010;9(7):1956–1967. [DOI] [PubMed] [Google Scholar]
- 12. Moulder S, Gladish G, Ensor J, et al. A phase 1 study of weekly everolimus (RAD001) in combination with docetaxel in patients with metastatic breast cancer. Cancer. 2012;118(9):2378–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gonzalez-Angulo AM, Akcakanat A, Liu S, et al. Open-label randomized clinical trial of standard neoadjuvant chemotherapy with paclitaxel followed by FEC versus the combination of paclitaxel and everolimus followed by FEC in women with triple receptor-negative breast cancerdagger. Ann Oncol. 2014;25(6):1122–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92(3):205–216. [DOI] [PubMed] [Google Scholar]
- 15. MK-2206 Investigator’s Brochure. 6 ed. Release Date: February 16, 2013. [Google Scholar]
- 16. Benjamini Y, Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J Royal Stat Soc Series B-Methodological. 1995;57(1):289–300. [Google Scholar]
- 17. Chu Q, Mita A, Forouzesh B, et al. Phase I and pharmacokinetic study of sequential paclitaxel and trabectedin every 2 weeks in patients with advanced solid tumors. Clin Cancer Res. 2010;16(9):2656–2665. [DOI] [PubMed] [Google Scholar]
- 18. Bailey HH, Alberti DB, Thomas JP, et al. Phase I trial of weekly paclitaxel and BMS-214662 in patients with advanced solid tumors. Clin Cancer Res. 2007;13(12):3623–3629. [DOI] [PubMed] [Google Scholar]
- 19. Naoki K, Kunikane H, Fujii T, et al. Dose-escalating and pharmacokinetic study of a weekly combination of paclitaxel and carboplatin for inoperable non-small cell lung cancer: JCOG 9910-DI. Jpn J Clin Oncol. 2009;39(9):569–575. [DOI] [PubMed] [Google Scholar]
- 20. Hudis C, Swanton C, Janjigian YY, et al. A phase 1 study evaluating the combination of an allosteric AKT inhibitor (MK-2206) and trastuzumab in patients with HER2-positive solid tumors. Breast Cancer Res. 2013;15(6):R110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Seidman AD, Hudis CA, Albanell J, et al. Dose-dense therapy with weekly 1-hour paclitaxel infusions in the treatment of metastatic breast cancer. J Clin Oncol. 1998;16(10):3353–3361. [DOI] [PubMed] [Google Scholar]
- 22. Perez EA, Vogel CL, Irwin DH, et al. Multicenter phase II trial of weekly paclitaxel in women with metastatic breast cancer. J Clin Oncol. 2001;19(22):4216–4223. [DOI] [PubMed] [Google Scholar]
- 23. Gori S, Mosconi AM, Basurtol C, et al. Weekly paclitaxel in metastatic breast cancer patients: a phase II study. Tumori. 2002;88(6):470–473. [DOI] [PubMed] [Google Scholar]
- 24. Sato K, Inoue K, Saito T, et al. Multicenter phase II trial of weekly paclitaxel for advanced or metastatic breast cancer: the Saitama Breast Cancer Clinical Study Group (SBCCSG-01). Jpn J Clin Oncol. 2003;33(8):371–376. [DOI] [PubMed] [Google Scholar]
- 25. Lombardi D, Crivellari D, Scuderi C, et al. Long-term, weekly one-hour infusion of paclitaxel in patients with metastatic breast cancer: a phase II monoinstitutional study. Tumori. 2004;90(3):285–288. [DOI] [PubMed] [Google Scholar]
- 26. I-SPY2 Clinical Trials. https://www.ispy2.org Accessed November 23, 2013. [Google Scholar]
- 27. Molife LR, Yan L, Vitfell-Rasmussen J, et al. Phase 1 trial of the oral AKT inhibitor MK-2206 plus carboplatin/paclitaxel, docetaxel, or erlotinib in patients with advanced solid tumors. J Hematol Oncol. 2014;7(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Beaver JA, Gustin JP, Yi KH, et al. PIK3CA and AKT1 mutations have distinct effects on sensitivity to targeted pathway inhibitors in an isogenic luminal breast cancer model system. Clin Cancer Res. 2013;19(19):5413–5422. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.