

Abstract
JMJD3 (KDM6B) is an H3K27me3 demethylase and counteracts polycomb-mediated transcription repression. However, the function of JMJD3 in vivo is not well understood. Here we show that JMJD3 is highly expressed in cells of the chondrocyte lineage, especially in prehypertrophic and hypertrophic chondrocytes, during endochondral ossification. Homozygous deletion of Jmjd3 results in severely decreased proliferation and delayed hypertrophy of chondrocytes, and thereby marked retardation of endochondral ossification in mice. Genetically, JMJD3 associates with RUNX2 to promote proliferation and hypertrophy of chondrocytes. Biochemically, JMJD3 associates with and enhances RUNX2 activity by derepression of Runx2 and Ihh transcription through its H3K27me3 demethylase activity. These results demonstrate that JMJD3 is a key epigenetic regulator in the process of cartilage maturation during endochondral bone formation.
Keywords: JMJD3, RUNX2, chondrocyte, endochondral bone formation
Introduction
During development, endochondral bone formation is a process by which most skeletal elements of the body are formed (Karsenty, 2008; Yang, 2009; Long and Ornitz, 2013). Endochondral ossification starts from the differentiation of condensed mesenchymal cells to chondrocytes and the formation of cartilaginous templates. Subsequently, chondrocytes of the cartilage template undergo proliferation, hypertrophy, and finally are replaced by trabecular bone and bone marrow. Meanwhile, the perichondral mesenchymal cells flanking the hypertrophic chondrocytes differentiate into osteoblasts and form the periosteum or cortical bone (Kronenberg, 2003; Karsenty et al., 2009; Lefebvre and Bhattaram, 2010; Long and Ornitz, 2013). This process is in contrast to the intramembranous ossification, where mesenchymal cells directly differentiate into bone without an intermediate cartilage template (Karsenty et al., 2009; Lefebvre and Bhattaram, 2010; Long, 2012).
Many transcription factors are required for normal cartilage development and endochondral bone formation (Kronenberg, 2003; Karsenty et al., 2009; Lefebvre and Bhattaram, 2010; Long and Ornitz, 2013). Among them, RUNX2 is an essential regulator for chondrocyte maturation and endochondral bone formation. Runx2-null mice display severe chondrodysplasia with decreased proliferation and lack or delayed hypertrophy of chondrocytes (Inada et al., 1999; Kim et al., 1999; Ueta et al., 2001; Yoshida et al., 2004; Zhang et al., 2009; Takarada et al., 2013). Conversely, overexpression of Runx2 in chondrocytes induces premature hypertrophy of chondrocytes and acceleration of endochondral ossification (Takeda et al., 2001; Ueta et al., 2001).
In addition to transcription factors, gene transcription also relies on the state of histone modifications (Strahl and Allis, 2000; Kubicek and Jenuwein, 2004; Martin and Zhang, 2005; Greer and Shi, 2012). Tri-methylation of lysine 27 on histone H3 (H3K27me3) is a hallmark of transcriptional repression and is established by enhancer of zeste homolog 2 (EZH2), a core subunit of the polycomb repressive complex 2 (PRC2) (Czermin et al., 2002; Muller et al., 2002). The recent identification of the Jumonji domain-containing proteins UTX (KDM6A) and JMJD3 (KDM6B) as H3K27me3-specific demethylases, reveals that polycomb-mediated gene silencing is reversible and dynamically regulated (Agger et al., 2007; De Santa et al., 2007; Hong et al., 2007; Jepsen et al., 2007; Lan et al., 2007; Lee et al., 2007; Xiang et al., 2007). Genome-wide localization of H3K27me3 in embryonic stem cells (ESCs) reveals that H3K27me3 specifically resides at the promoters of key developmental regulators (Bernstein et al., 2006; Boyer et al., 2006; Bracken et al., 2006; Lee et al., 2006). Hence, H3K27me3 modification may play essential roles in embryo development and it is an impetus to study the function of JMJD3 in vivo. Indeed, Jmjd3-deficient mice were found to die shortly after birth with developmental defects in lung, macrophage, and neuron (Satoh et al., 2010; Burgold et al., 2012; Li et al., 2014; Park et al., 2014). These results illustrate the importance of JMJD3 during development.
Here, we observed that homozygous Jmjd3-deficient mice exhibited markedly reduced proliferation and hypertrophy of chondrocytes and a severe delay of endochondral ossification. JMJD3 genetically associated with RUNX2 to favor chondrocyte proliferation and maturation. JMJD3 biochemically interacted with and enhanced RUNX2 activity to favor the expression of Runx2 and Ihh. These results establish JMJD3 as a critical epigenetic factor for cartilage development and endochondral bone formation.
Results
JMJD3 was highly expressed in prehypertrophic and hypertrophic chondrocytes during endochondral bone formation
To investigate the role of JMJD3 in embryonic development, we performed a tissue survey on mouse embryo by immunohistochemistry. The survey indicated that JMJD3 was highly expressed in the nuclei of chondrocytes of embryonic vertebral body, compared with other tissues such as muscle, heart, and brain (Supplementary Figure S1A). Western blot analysis confirmed that JMJD3 was expressed in the nuclei of chondrocytes (Figure 1A). In view of this observation, we next analyzed the expression pattern of JMJD3 in mice at different developmental stages. Immunohistochemistry studies showed that JMJD3 was initially expressed weakly in the center of cartilaginous templates at embryonic day (E) 12.5 (Figure 1Ba). Subsequently, the expression of JMJD3 obviously increased in the prehypertrophic and hypertrophic chondrocytes of growth plates at E13.5 and E14.5 (Figure 1Bb–e). At E18.5, JMJD3 was mainly expressed in chondrocytes of the resting, prehypertrophic, and proximal hypertrophic zones, and weakly distributed in chondrocytes at proliferating and distal hypertrophic zones (Figure 1Bf–i). These results suggest that JMJD3 may modulate chondrocyte maturation during endochondral bone formation.
Figure 1.
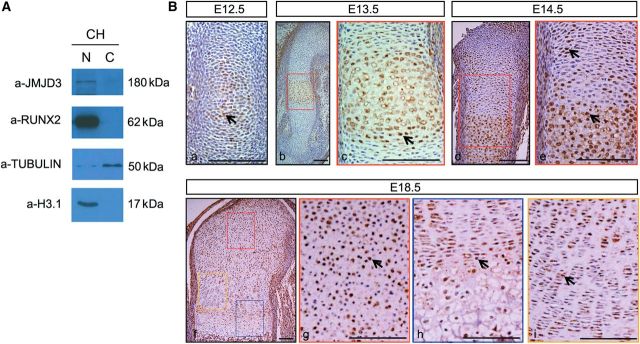
JMJD3 was highly expressed in prehypertrophic and hypertrophic chondrocytes. (A) Western blot analysis of JMJD3 in cytoplasmic (C) and nuclear (N) fractions of primary chondrocytes (CH) from E14.5 limbs. RUNX2 is a positive control. (B) Immunohistochemistry of JMJD3 at E12.5 (a), E13.5 (b, c), E14.5 (d, e), and E18.5 (f–i). WT humerus longitudinal sections counterstained with hematoxylin. Positive signal appears brown in the nuclear. The boxed regions in b, d, and f are magnified in c, e, g, h, and i with matched color rim, respectively. The black arrows indicate JMJD3-positive chondrocytes. Scale bar, 200 μm.
Dwarfism and skeletal developmental defects in Jmjd3–/– mice
To characterize the role of JMJD3 during skeletogenesis, we generated Jmjd3-deficient mice (Supplementary Figure S2A–G). Jmjd3 heterozygote (Jmjd3 +/−) mice were viable, fertile, and showed no visible phenotypic change. Crosses between Jmjd3+/− mice yielded Jmjd3−/− embryos in a predicated Mendelian ratio before birth with no enhanced perinatal death (Supplementary Table S1). Consistent with previous study (Satoh et al., 2010; Li et al., 2014), Jmjd3−/− mice died with cyanosis within 0.5–2 h after birth, with developmental defects of lung tissues (Supplementary Figure S3A). The prominent gross appearances of Jmjd3–/– embryos were dwarfism with thoracic kyphosis and short limbs (Figure 2A). To characterize the dwarfism phenotype, we weighed the embryos from E12.5 to E18.5. The weight of wild-type (WT) and Jmjd3–/– embryos was indistinguishable at E12.5. From E14.5 to E16.5, Jmjd3–/– embryos weighted less than WT littermates, though the difference is not statistical significant. This difference was even more striking at E18.5 and E19.5 (Figure 2B).
Figure 2.

Dwarfism and developmental defects of skeleton in Jmjd3−/− mice. (A) Gross appearance of Jmjd3+/+, Jmjd3+/−, and Jmjd3−/− littermates at E18.5. Scale bar, 1 mm. (B) Quantification of body weight of Jmjd3+/+, Jmjd3+/−, and Jmjd3−/− embryos at E12.5 (n = 4), E14.5 (n = 8), E16.5 (n = 6), E18.5 (n = 6), and E19.5 (n = 4). *P < 0.05. Error bar indicates the standard error (SE) of the mean. Statistical significance is determined by two-tailed Student's t-test. (C) Alizarin red (bone mineralization) and alcian blue (cartilage) stained skeleton of E18.5 WT (upper panels) and Jmjd3−/− (lower panels) littermates (a, b). Magnified images are shown for ribs and sternum (c, d), vertebrae (e, f), ilium (g, h), hindlimbs (i, j), forelimbs (k, l), and metacarpus (m, n). Red arrows indicate a delay in the formation of the primary ossification center of Jmjd3−/− skeletal elements. Scale bar, 1 mm. (D) Skeleton preparations of forelimbs of Jmjd3+/+ and Jmjd3−/− embryos were stained by alcian blue and/or alizarin red staining at E12.5 (a, b), E13.5 (c, d), E14.5 (e, f), and E16.5 (g, h). Red arrows indicate smaller and shorter humeri of Jmjd3−/− embryos. Scale bar, 1 mm. (E) Quantification of length of Jmjd3+/+ and Jmjd3−/− humeri at E12.5, E13.5, E14.5, and E16.5. * P < 0.05, ** P < 0.01. Error bar indicates the SE (n ≥ 4). Statistical significance is determined by two-tailed Student's t-test.
To examine whether developmental defects of skeleton was responsible for the dwarfism, thoracic kyphosis, and short limbs of Jmjd3–/– embryos at E18.5, we performed alcian blue and alizarin red staining of skeletal preparations. No obvious difference in number or position of skeletal elements between WT and Jmjd3+/− embryos was observed (Supplementary Figure S3B). However, Jmjd3−/− embryos exhibited clear dwarfism of all appendicular and axial skeletal elements (Figure 2C). These skeletal elements distinctively exhibited a lack or smaller size of mineralized bone tissue that should be stained in red, such as in sternum, spinal column, ilium, hindlimbs, forelimbs, and metacarpus (Figure 2Cc–n). These findings indicated that endochondral ossification in Jmjd3−/− embryos was severely delayed.
Given that JMJD3 was highly expressed in chondrocytes (Figure 1), we test whether abnormality of the cartilage development was involved in the delay of endochondral ossification in Jmjd3–/– embryos. We focused our analysis on the long bones of the limbs. In limbs, cartilage development starts at E12.5 when condensed mesenchymal cells begin to differentiate into chondrocytes (Kronenberg, 2003; Karsenty et al., 2009; Lefebvre and Bhattaram, 2010; Long and Ornitz, 2013). Therefore, we analyzed the skeletal preparation of limbs from E12.5 to E16.5. The results showed that the length of limbs was generally reduced in Jmjd3–/– embryos compared with WT littermates (Figure 2D and E). Quantitative analysis indicated that the length of Jmjd3−/− humeri mildly reduced by 11.2% at E12.5. Subsequently, the length of humeri decreased more severely by 18.4% at E13.5, 23.1% at E14.5, and 22.9% at E16.5 (Figure 2E). Accordingly, ossified size of humerus in Jmjd3−/− mice was obviously smaller than that in WT littermates at E16.5 (Figure 2Dh). Similar developmental defects of cartilage templates occurred in other skeletal elements, such as in hindlimbs and vertebrae (data not shown). These observations strongly indicated that cartilage developmental abnormalities were responsible, at least partially, for the delayed endochondral ossification in Jmjd3–/– embryos.
Reduced hypertrophy and proliferation of chondrocytes in Jmjd3−/− mice
Given that the length of cartilage templates was most reduced in E14.5 Jmjd3−/− embryos (Figure 2D and E), we performed histological examinations on embryos at this stage. Hematoxylin and eosin (H&E) staining showed that enlarged hypertrophic chondrocytes overtly presented at the center of WT femur, whereas small prehypertrophic and immature hypertrophic chondrocytes were still in Jmjd3−/− littermates (Figure 3Aa–d). Consistent with the histology observation, in situ hybridization showed an obviously narrower domain of Col10a1, a marker of chondrocyte hypertrophy, in the developing femur of Jmjd3−/− embryo than WT littermates (Figure 3Ae, f). In contrast, no staining difference in Col2a1, an early marker of chondrocytes differentiation, was detected between WT and Jmjd3–/– femurs (Figure 3Ag, h). These data indicated that chondrocyte hypertrophy was severely delayed in Jmjd3–/– embryos at this stage. Consistently, the signaling of Indian hedgehog (Ihh), a critical secretory factor from prehypertrophic chondrocytes that tightly controls chondrocyte proliferation and hypertrophy (St-Jacques et al., 1999), was still a whole domain in the middle part of Jmjd3–/– femur but was separated by hypertrophic chondrocytes into two parts in WT littermates at this stage (Figure 3Ai, j). In addition, the mildly weaker intensity of Col10a1 and Ihh staining was observed in Jmjd3–/– embryos compared with WT mice (Figure 3Af, j). Combined with previous observation that expression of JMJD3 increased in prehypertrophic and hypertrophic chondrocytes during endochondral bone formation (Figure 1B), we speculated that JMJD3 may positively regulate chondrocyte hypertrophy. Consistently with this conjecture, we observed that the domain of hypertrophic chondrocytes was reduced in Jmjd3–/– embryos compared with WT littermates by H&E staining at E16.5 (Figure 3Ak, l and B). In situ hybridization also showed that the domain and intensity of Col10a1 staining were obviously reduced in Jmjd3–/– embryos compared with WT littermates (Figure 3Am, n). In light of the investigations that an increase in the cellular volume of hypertrophic chondrocytes is a major determinant of the rate of longitudinal bone growth (Breur et al., 1991; Wilsman et al., 1996), we speculate that delayed chondrocyte hypertrophy is responsible for the remarkable shortening of long bones in E14.5 Jmjd3–/– embryos. Furthermore, the delayed hypertrophy of chondrocytes should be responsible for the retardation of endochondral ossification in Jmjd3–/– mice. In agreement with this speculation, von Kossa staining showed that less mineralization occurred in E16.5 Jmjd3–/– femur compared with WT littermates (Figure 3Ap).
Figure 3.

Severely reduced hypertrophy and proliferation of chondrocytes in Jmjd3−/− mice. (A) Representative H&E staining (a–d, k, l), in situ hybridization with probes of Col10a1 (e, f, m, n), Col2a1 (g, h), and Ihh (i, j), and Von Kossa staining (o, p) at E14.5 (a–j) and E16.5 (k–p) femurs of WT (upper) and Jmjd3−/− (lower) littermates. The boxed regions in a and b are magnified in c and d with matched color rim, respectively. Black arrows denote a delay of chondrocyte hypertrophy in Jmjd3−/− mice. Red arrows denote decreased mineralization in periosteum of Jmjd3−/− femur. Scale bar, 200 μm. (B) Quantification of the length of hypertrophic chondrocyte zones at E16.5 WT and Jmjd3−/− femur. * P < 0.05, two-tailed Student's t-test. Error bar indicates the SE (n = 3). (C) Immunohistochemistry of BrdU-labeled chondrocytes counterstained with hematoxylin in humeri sections of WT (upper) and Jmjd3−/− (lower) littermates at E14.5. The boxed regions in the left panels are magnified in the right panels with matched color rim, respectively. Arrows indicate BrdU-positive nuclei. Scale bar, 200 μm in a, b and 50 μm in c, d. (D) Quantification of proliferation rate in cartilage growth plate, represented by the percentage of BrdU-positive cells in WT and Jmjd3−/− humeri sections at E14.5. **P < 0.01, two-tailed Student's t-test. Error bar indicates the SE (n = 3).
To examine whether the severe dwarfism of long bones was also associated with abnormal proliferation of chondrocytes in E14.5 Jmjd3−/− mice, we performed in vivo BrdU incorporation and immunohistochemistry assays. The results revealed that the proliferative index of chondrocytes significantly decreased in cartilage growth plate of Jmjd3–/– embryos compared with WT littermates (Figure 3C and D). The result was confirmed by the immunohistochemistry investigation with an anti-Ki67 antibody (data not shown). We also performed in situ TUNEL assay, but did not detect any noticeable difference of apoptotic cells in the cartilage growth plates between WT and mutant samples at E14.5 and E16.5 (Supplementary Figure S4A and B). Therefore, in addition to delayed hypertrophy of chondrocytes, significantly decreased proliferation of chondrocytes was also responsible for the severe dwarfism in growth plates of E14.5 Jmjd3–/– embryos.
JMJD3 genetically cooperates with RUNX2 to regulate chondrocyte proliferation and hypertrophy during endochondral ossification
E14.5 Jmjd3−/− embryos displayed severe retardation of chondrocyte hypertrophy and decreased proliferation of chondrocytes, which is reminiscent of the function of RUNX2, a master regulator of chondrocyte maturation (Inada et al., 1999; Kim et al., 1999; Takeda et al., 2001; Ueta et al., 2001; Yoshida et al., 2004; Zhang et al., 2009; Takarada et al., 2013; Chen et al., 2014). Since JMJD3 is a H3K27 demethylase, we speculate that it may be a coactivator of RUNX2 in cartilage development. To address this possibility, we generated and analyzed Jmjd3+/−;Runx2+/− embryos. Examination of skeletal preparations showed that Jmjd3+/–;Runx2+/– embryos display significant delay in endochondral ossification than WT, Jmjd3+/–, or Runx2+/– mice at E15.5. This was evident when studying the development of ribs, limbs, and ilium (Figure 4Aa1–c4 and Ba, b). Alcian blue staining showed that chondrocyte hypertrophy was much delayed in the compound-heterozygous embryos, compared with WT or single-heterozygous littermates (Figure 4Ad1–d4 and Bc). In situ hybridization for Col10a1 also confirmed this finding (Figure 4Ae1–e4). In addition, KI67 staining by immunohistochemistry revealed that the proliferative index of chondrocytes most decreased in cartilage growth plate of compound-heterozygous embryos among all littermates (Figure 4Af1–g4 and Bd). Collectively, these results clearly demonstrate that JMJD3 genetically cooperates with RUNX2 to promote chondrocyte proliferation and hypertrophy during endochondral ossification, which supports the model that JMJD3 is a coactivator of RUNX2 in chondrocytes.
Figure 4.
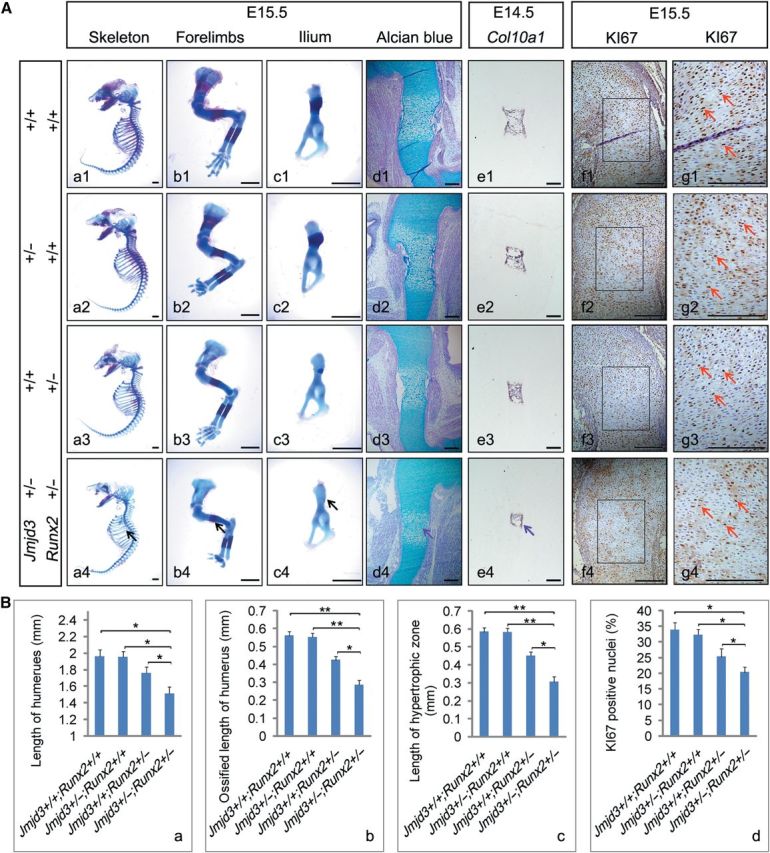
JMJD3 genetically cooperates with RUNX2 to regulate chondrocyte proliferation and hypertrophy during endochondral ossification. (A) Representative skeletal preparation (a1–c4), alcian blue staining (d1–d4), in situ hybridization with Col10a1 (e1–e4) probe, and immunohistochemistry of KI67 counterstained with hematoxylin (f1–f4) of Jmjd3+/+; Runx2+/+ (first row), Jmjd3+/–; Runx2+/+ (second row), Jmjd3+/+; Runx2+/– (third row), and Jmjd3+/–; Runx2+/– (bottom row) mice at E15.5 (a1–d4; f1–g4) and E14.5 (e1–e4). The boxed regions in f1–f4 are magnified in g1–g4, respectively. Black arrows indicate the most delayed bone ossification in ribs (a1–a4), forelimbs (b1–b4), and ilium (c1–c4) of Jmjd3+/–;Runx2+/– mice. Purple arrows indicate the shortest hypertrophic zone of chondrocytes in humeri of Jmjd3+/–;Runx2+/– mice at E15.5 (d4) and E14.5 (e4). Red arrows indicate KI67-positive nuclei (g1–g4). Scale bar, 1 mm in a1–c4 and 200 μm in d1–g4. (B) Quantification of full humerus length (a), length of ossified zone of humeri (b), length of hypertrophic zone of humeri (c), and proliferation rate in cartilage growth plate represented by the percentage of KI67-positive cells (d) of Jmjd3+/+; Runx2+/+, Jmjd3+/–; Runx2+/+, Jmjd3+/+; Runx2+/–, and Jmjd3+/–; Runx2+/– mice at E15.5. *P < 0.05, **P < 0.01. Two-tailed Student's t-test. Error bar represents the SE (n = 3).
JMJD3 biochemically interacts with RUNX2
To further test the hypothesis that JMJD3 is a coactivator of RUNX2, we determine whether JMJD3 interacts with RUNX2 biochemically. In HEK293T cells co-expressing Flag-tagged JMJD3 and His-tagged RUNX2, immunoprecipitation of Flag-JMJD3 pulled down His-RUNX2 (Figure 5A). Immunoprecipitation of RUNX2 also pulled down endogenous JMJD3, or vice versa, in the primary culture of chondrocytes from E14.5 limbs (Figure 5B and C). Next, we mapped the domains of JMJD3 responsible for the interaction with RUNX2. Three Flag-tagged truncated JMJD3 proteins containing either the N-terminus (JMJD3-N, 1–562 aa), the middle part (JMJD3-M, 563–1162 aa), or the C-terminus (JMJD3-C, 1163–1641 aa) were co-expressed with His-RUNX2 in HEK293T cells, and immunoprecipitation of His-RUNX2 pulled down JMJD3-N and JMJD3-C, but not JMJD3-M (Figure 5D). We also determined the region of RUNX2 responsible for interaction with JMJD3 using the same strategy. Immunoprecipitation assays indicated that the N-terminus (RUNX2-N, 1–108 aa) and C-terminus of RUNX2 (RUNX2-C, 259–528 aa), but not the middle region of RUNX2 (RUNX2-M, 109–258 aa), interacted with JMJD3 (Figure 5E). Given that co-immunoprecipitation (CoIP) experiment can detect both directly and indirectly interacted proteins, we performed GST-pulldown experiment with purified recombinant proteins to determine whether JMJD3 can directly interact with RUNX2. The results indicated that JMJD3-N, but not JMJD3-C, directly interacted with RUNX2 (Figure 5F and Supplementary Figure S4C). Conversely, RUNX2-N, but not RUNX2-C, directly interacted with JMJD3-N (Figure 5G and Supplementary Figure S4D). In addition, to determine whether JMJD3 co-localizes with RUNX2 in vivo, we co-expressed Flag-tagged JMJD3 and His-Myc-tagged RUNX2 in HEK293T cells. Immunofluorescence under confocal microscope indicated that JMJD3 co-localized with RUNX2 in the nuclei of HEK293T cells (Supplementary Figure S4E). Consistently, confocal microscopy revealed that JMJD3 co-localized with RUNX2 in the nuclei of chondrocytes in the prehypertrophic zones of E14.5 tibia (Supplementary Figure S4Fe, c). We also observed that JMJD3 co-localized with RUNX2 in the nuclei of perichondrial osteoblasts (Supplementary Figure S4Fe, f). Thus, these results strongly suggest that JMJD3 interacts with RUNX2 in vivo and supports the hypothesis that JMJD3 is a coactivator of RUNX2.
Figure 5.
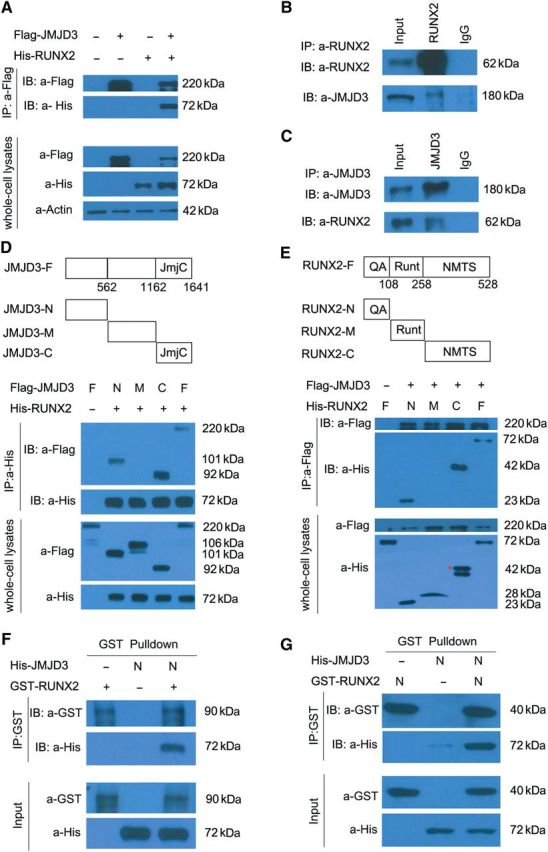
JMJD3 biochemically interacts with RUNX2. (A) Coimmunoprecipitation (Co-IP) of His-RUNX2 with Flag-JMJD3 in transiently transfected HEK293T cells. Immunoprecipitation (IP) was performed with an anti-Flag antibody, and RUNX2 proteins were detected by immunoblot (IB) analysis with anti-His antibody. IB of whole-cell lysates without immunoprecipitation was used to detect protein expression throughout experiments. (B) Co-IP of endogenous JMJD3 with RUNX2 in the primary culture of chondrocytes of E14.5 limbs. IP was performed with anti-RUNX2 antibody. JMJD3 proteins were detected by IB assay with anti-JMJD3 antibody. (C) Co-IP of endogenous RUNX2 with JMJD3 in the primary culture of chondrocytes from E14.5 embryos. IP was performed with anti-JMJD3 antibody, and RUNX2 proteins were detected by IB assay with anti-RUNX2 antibody. (D and E) Co-IP of constructs as indicated co-expressed in 293T cells. Cell extracts were immunoprecipitated with anti-His antibody (D) or anti-Flag antibody (E), followed by IB with an anti-Flag antibody and anti-His antibody. Red asterisk indicates the correct size of His-RUNX2-C proteins. JmjC, Jumonji C domain. QA, glutamine/alanine rich domain. Runt, runt homology domain. NMTS, nuclear matrix targeting signal. (F and G) GST-pulldown experiments with bacterially expressed GST-RUNX2 (F) or GST-RUNX2-N (G) and in vitro transcribed/translated JMJD3-N as indicated. The above experiments were repeated at least three times.
JMJD3 cooperates with RUNX2 to regulate Runx2 and Ihh transcription
To determine whether JMJD3 modulates RUNX2 transcriptional activity on the promoters of their co-target genes, we first analyzed expression of chondrocyte-specific genes in WT and Jmjd3−/− embryos. RT-qPCR showed that the mRNA levels of Runx2, Ihh, Col10a1, and MMP13 in limb chondrocytes were reduced in E14.5 Jmjd3–/– embryos compared with WT mice (Figure 6A). Similar results were observed in primarily cultured WT and Jmjd3–/– limb chondrocytes or Jmjd3 knockdown chondrocytes of E14.5 embryos (Supplementary Figure S5Aa, b). Since RUNX2 has been shown to bind and activate its own promoter via a positive feedback loop (Ducy et al., 1999), and Ihh is a target gene of RUNX2 (Yoshida et al., 2004), we tested whether JMJD3 directly regulates the transcription of the two genes.
Figure 6.
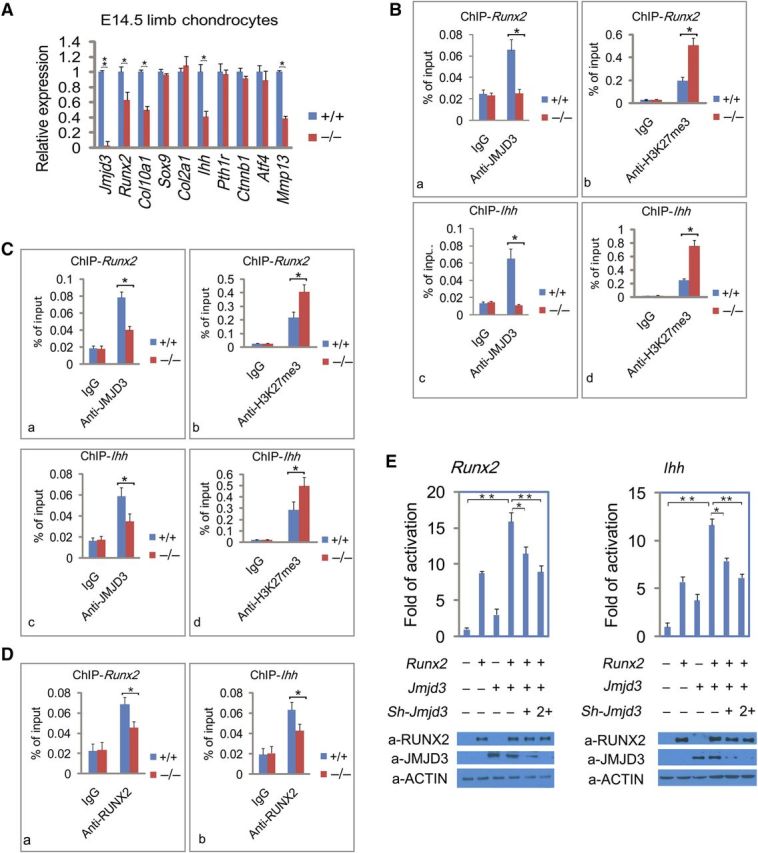
JMJD3 cooperates with RUNX2 to regulate Runx2 and Ihh transcription. (A) RT-qPCR analysis to determine the expression levels of chondrocyte-specific genes relative to Gapdh in limb chondrocytes of WT and Jmjd3–/– mice at E14.5. (B) ChIP-qPCR assays with antibodies specific for JMJD3 (a, c) or H3K27me3 (b, d,) at the promoters of Runx2 (a, b) and Ihh (c, d) in WT and Jmjd3−/− primary chondrocytes. (C) ChIP-qPCR assays with antibodies specific for JMJD3 (a, c) or H3K27me3 (b, d) at the promoters of Runx2 (a, b) and Ihh (c, d) in WT and Runx2−/− primary chondrocytes. (D) ChIP-qPCR assays with antibodies specific for RUNX2 at the promoters of Runx2 (a) and Ihh (c) in WT and Jmjd3−/− primary chondrocytes. (E) Luciferase reporter assays. ATDC5 chondrocytes were transfected with the pGL3-basic reporter containing 2 kb Runx2 promoter (a) or 2 kb Ihh promoter (b) with or without lentiviral expression vectors encoding Runx2, Jmjd3, or Jmjd3-shRNA (sh-Jmjd3) as indicated. The basal luciferase activity for each reporter was calculated as 1 in y-axis. Western blots show levels of expressed proteins in the lysates of transfected cells (bottom). The above experiments were repeated three times. Positions of primers (B, D, and E) in the promoters of Runx2 and Ihh for ChIP-qPCR experiments were showed in Supplementary Figure S5Ba–c. Signals were shown as the percentage of the input. * P < 0.05, ** P < 0.01, two-tailed Student's t-test. Error bar represents the SE of three independent experiments.
We first examined whether JMJD3 directly bound to the promoters of Runx2 and Ihh by chromatin immunoprecipitation (ChIP) followed by qPCR analyses in ATDC5 chondrocytes and primary chondrocytes. Indeed, JMJD3 was revealed to be strongly recruited to the promoters of the two genes (Supplementary Figures S5Ba, b and S6Ba, c). Consistent with the biochemical role of JMJD3 as H3K27 demethylase, the levels of H3K27me3 at the promoters of Runx2 and Ihh markedly increased in Jmjd3−/− versus WT primary chondrocytes (Figure 6Bb, d). In contrast, no H3K27me3 was detected on the promoter of the housekeeping gene Gapdh in primary chondrocytes of WT and Jmjd3−/− mice (Supplementary Figure S5Bc). Therefore, these results clearly demonstrated that JMJD3 directly derepressed Runx2 and Ihh transcription in chondrocytes by erasure of H3K27me3 with its H3K27me3 demethylase activity. Our result was further supported by a recent report that silencing of JMJD3 expression increased the level of H3K27me3 on the promoter regions of Runx2 in MC3T3-E1 osteoblasts (Yang et al., 2013a). In addition, to test whether JMJD3 is the only enzyme to erase H3K27me3 at the promoters of these genes, we performed ChIP-qPCR assays with mouse embryonic stem (ES) cells as a positive control. The results showed that H3K27me3 level in ES cells was slightly higher than that in JMJD3-null primary chondrocytes, but obviously higher than that in WT chondrocytes (Supplementary Figure S5Ca, b). These results indicated that JMJD3 might be a major eraser of H3K27me3 at the promoters of Runx2 and Ihh during chondrocyte maturation.
In order to test whether JMJD3 binding to target genes is reliant on RUNX2 during chondrocyte maturation, we examined JMJD3 occupancies at promoters of Runx2 and Ihh genes in WT and Runx2−/− primary chondrocytes by ChIP-qPCR assays. The results showed that the level of JMJD3 was obviously reduced at the promoters of Runx2 and Ihh genes in Runx2−/− compared with WT chondrocytes (Figure 6Ca, c). Consistently, the levels of H3K27me3 at the promoters of Runx2 and Ihh markedly increased in Runx2−/− versus WT chondrocytes (Figure 6Cb, d). These results suggest that RUNX2 can recruit JMJD3 to target genes during chondrocyte maturation. In addition, to test whether JMJD3 can affect binding of RUNX2 to target genes, we performed ChIP-qPCR with anti-RUNX2 antibody in WT and Jmjd3−/− primary chondrocytes. The results showed that RUNX2 level was reduced at the promoters of Runx2 and Ihh genes in Jmjd3−/− chondrocytes compared with WT chondrocytes. These findings suggest that JMJD3 can also affect RUNX2 binding to its target genes (Figure 6D).
To test whether JMJD3 can functionally cooperate with RUNX2 to regulate Runx2 and Ihh transcription, we performed luciferase reporter assays in HEK293 cells. While JMJD3 alone only slightly potentiated Runx2 and Ihh transcription, it obviously synergized with RUNX2 to promote the expression of the two genes, which was reduced by shRNA against Jmjd3 in a dose-dependent manner (Supplementary Figure S5Da, b). Similar results were observed in ATDC5 chondrocyte cell line (Figure 6E). These findings strongly demonstrated that JMJD3 could modulate transcriptional activity of RUNX2 and facilitate the expression of their co-target genes.
Discussion
Chondrocyte proliferation and hypertrophy are essential for endochondral bone formation (Karsenty, 2008; Yang, 2009; Long and Ornitz, 2013). Dysregulation of these processes will lead to severe chondrodysplasia and skeleton malformation diseases (Kornak and Mundlos, 2003; Baldridge et al., 2010; Warman et al., 2011). Many transcription factors and signaling pathways have been demonstrated to orchestrate these processes (Kronenberg, 2003; Karsenty et al., 2009; Yang, 2009; Baldridge et al., 2010; Lefebvre and Bhattaram, 2010; Long and Ornitz, 2013). However, the function of epigenetic regulators in these processes has not been well established. Recently, histone deacetylase HDAC4 and H3K9 methyltransferase ESET were revealed to inhibit the chondrocyte hypertrophy in vivo, though their roles in chondrocyte proliferation were not described (Vega et al., 2004; Yang et al., 2013b). In addition, histone lysine demethylase PHF2 was showed to be recruited by ARID5B to promote chondrogenesis (Hata et al., 2013). Here, we demonstrate that JMJD3 promotes chondrocyte proliferation and hypertrophy because it is a coactivator of RUNX2. Indeed, JMJD3 is most highly expressed in prehypertrophic and hypertrophic chondrocytes as RUNX2 does (Vega et al., 2004). Jmjd3-mutant mice display severely decreased proliferation and delayed hypertrophy of chondrocytes. Genetic and biochemical means reveal that JMJD3 cooperates with RUNX2 to promote chondrocyte proliferation and hypertrophy. Our results establish the critical role of JMJD3 in chondrocyte proliferation and hypertrophy and extend the knowledge of epigenetic regulators in regulation of cartilage development.
Recent work by an independent group has demonstrated that Jmjd3-deleted mice die shortly after birth without hypoplasia of lung and dwarfism (Burgold et al., 2012). This in vivo evidence would seem to argue against our and other findings (Satoh et al., 2010; Li et al., 2014). We carefully analyzed this discrepancy and found that Burgold et al. (2012) actually trapped only one of the two promoters of Jmjd3 (De Santa et al., 2007). Therefore, the phenotypes of their Jmjd3 knockout mice were closely resemble those of our Jmjd3+/− mice, which had no dwarfism and hypoplasia of the lung.
Although our results do not exclude by any means the possibility that JMJD3 also regulates other important processes during endochondral bone formation, such as differentiation of condensed mesenchymal cells to chondrocytes, significantly reduced length of long bones in Jmjd3−/− embryos at E14.5–E16.5 implicates that JMJD3 may play more critical role in regulation of proliferation and hypertrophy of chondrocytes at these stages (Figure 2D and E). This speculation is also supported by the observation that the remarkably reduced proliferation and delayed hypertrophy of chondrocytes occur in Jmjd3+/−;Runx2+/− mice compared with WT, Jmjd3+/–, or Runx2+/– littermates. The data also reveal that RUNX2 is a major target of JMJD3 during chondrocyte maturation, though we do not exclude the possibility that JMJD3 also regulates other important transcription factors in this process. In addition, abnormity of osteoblast differentiation should also account for the marked retardation of endochondral ossification in Jmjd3−/− mice. This was confirmed by our observation that osteoblast differentiation was also delayed in Jmjd3−/− and Jmjd3+/−;Runx2+/−mice compared with WT, Jmjd3+/–, or Runx2+/– mice (unpublished data). The results were consistent with a recent report that JMJD3 was required for the differentiation of MC3T3E1 to osteoblasts through Runx2 (Yang et al., 2013a). Therefore, these data suggest that JMJD3 may regulate both chondrocyte maturation and osteoblast differentiation through RUNX2.
RUNX2 transcriptional activity is mediated by multiple domains during skeletogenesis. The conserved Runt domain is essential for RUNX2 to selectively bind to specific cis-acting elements of target genes. The N-terminus containing a short stretch of glutamine/alanine (QA) repeats and the C-terminal containing the nuclear matrix targeting signal (NMTS) domain are required for maximal transcriptional activity of RUNX2 by interacting with chromatin remodeling factors and epigenetic modifiers (Schroeder et al., 2005; Ziros et al., 2008; Chen et al., 2014). Previously, HDAC4 was revealed to interact with Runt domain and inhibited the transcriptional activity of RUNX2 and chondrocyte hypertrophy (Vega et al., 2004). Here, we demonstrated that JMJD3 physically interacted with the N-terminus of RUNX2 and thus enhanced RUNX2 transcriptional activity during chondrocyte maturation. Since JMJD3 cannot directly bind to DNA sequence, it is conceived that RUNX2 may recruit JMJD3 to target genes to promote chondrocyte maturation. This suggestion was confirmed by ChIP-qPCR assays, which showed that the level of JMJD3 at the promoters of Runx2 and Ihh genes was obviously decreased in Runx2−/− compared with WT chondrocytes (Figure 6Ca, c). It is interesting that JMJD3 deletion also affected binding of RUNX2 to the promoters of its target genes (Figure 6D). This may be associated with more condensed chromatin at the promoters of target genes resulted from JMJD3 deletion. Therefore, impairment of RUNX2 binding to target genes by JMJD3 deletion may be a mechanism responsible for the decreased proliferation and hypertrophy of chondrocytes in Jmjd3−/− mice.
The obviously decreased mRNA level of Runx2 and Ihh and accordingly elevated level of H3K27me3 on the two promoters in Jmjd3–/– mice indicate that JMJD3 regulates the expression of the two genes with its H3K27me3 demethylase activity. The physical interaction between JMJD3 and RUNX2 also implicates that JMJD3 may cooperate with RUNX2 to regulate transcription of more target genes during chondrocyte maturation. In view that JMJD3 regulates gene transcription during cell differentiation in demethylase-dependent and/or demethylase-independent manners (Agger et al., 2007; De Santa et al., 2007; Miller et al., 2010; Chen et al., 2012; Shpargel et al., 2014), we are not able to rule out the possibility that JMJD3 also regulates the transcription of these genes in a demethylase-independent manner during chondrocyte maturation. Future studies should aim at addressing this question by using JMJD3 enzyme-dead knock-in mice.
As an H3K27me3 demethylase, JMJD3 counteracts the transcription repression by PRC2 complex and biochemically interacts with the core subunits of trithorax complex, such as WDR5, RBBP5, ASH2L, and BRG1 (De Santa et al., 2007; Miller et al., 2010). These results suggest that JMJD3 should belong to the trithorax complex. Therefore, our results suggest that PRC2 and trithorax proteins may antagonistically regulate the RUNX2 activity and result in the precise timing of chondrocyte hypertrophy during endochondral bone formation. Consistent with this model, high level of H3K27me3 was found on the promoters of RUNX2 target genes, such as Runx2 and Ihh, in the present study. Based on these results, we hypothesize that, to avoid premature differentiation of chondrocytes, PRC2 proteins should maintain the repression of these genes by negatively regulating RUNX2 activity. Upon differentiation, JMJD3 removes H3K27me3 and facilitates RUNX2 to promote the transcription of target genes, which results in the promotion of chondrocyte hypertrophy (Figure 7A and B). Furthermore, in view that HDAC4 and ESET negatively mediate chondrocyte hypertrophy through inhibiting RUNX2 (Vega et al., 2004; Yang et al., 2013b), we propose that chondrocyte hypertrophy is dynamically regulated by various antagonistic epigenetic regulators in many aspects or levels through RUNX2.
Figure 7.
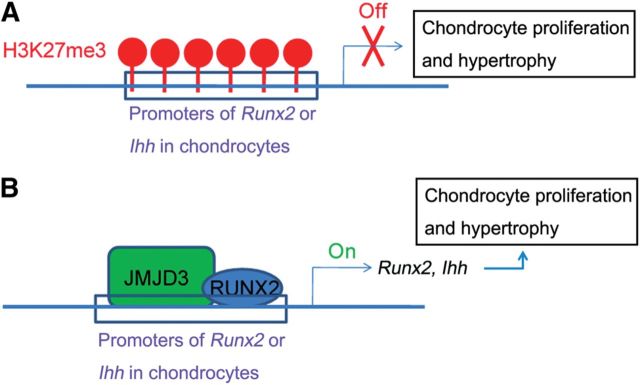
Schematic representation illustrates that JMJD3 cooperates with RUNX2 to regulate proliferation and hypertrophy of chondrocytes. (A) H3K27me3 modification maintains the transcription repression of RUNX2 target gene Runx2 and Ihh in chondrocytes. (B) JMJD3 erases the H3K27me3 modification and facilitates RUNX2 to regulate the transcription of Runx2 and Ihh in chondrocytes.
In summary, our results provide the first in vivo evidence that histone demethylase JMJD3 acts as a key coactivator of RUNX2 and promotes chondrocyte proliferation and hypertrophy during endochondral ossification. Further studies are needed to elucidate in greater detail the physiological and pathological roles of JMJD3 in skeletogenesis.
Materials and methods
Materials, methods, and associated references are described in Supplementary Materials and methods.
Supplementary material
Supplementary material is available at Journal of Molecular Cell Biology online.
Funding
This work was supported by the National Natural Science Foundation of China (91219304), National Basic Research Program of China (2010CB529705, 2011CB510103, 2014CB943100), and the Council of Shanghai Municipal Government for Science and Technology.
Conflict of interest: none declared.
Supplementary Material
Acknowledgements
We wish to thank Prof. Weiguo Zou and Xiaoyan Ding (Shanghai Institute of Biochemistry and Cell Biology) for scientific input, Brendan Lee (Baylor College of Medicine) for sharing the probes, and all CDC lab members for discussion and technical assistances.
References
- Agger K., Cloos P.A., Christensen J., et al. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature. 2007;449:731–734. doi: 10.1038/nature06145. [DOI] [PubMed] [Google Scholar]
- Baldridge D., Shchelochkov O., Kelley B., et al. Signaling pathways in human skeletal dysplasias. Annu. Rev. Genomics Hum. Genet. 2010;11:189–217. doi: 10.1146/annurev-genom-082908-150158. [DOI] [PubMed] [Google Scholar]
- Bernstein B.E., Mikkelsen T.S., Xie X., et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- Boyer L.A., Plath K., Zeitlinger J., et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349–353. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- Bracken A.P., Dietrich N., Pasini D., et al. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 2006;20:1123–1136. doi: 10.1101/gad.381706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breur G.J., VanEnkevort B.A., Farnum C.E., et al. Linear relationship between the volume of hypertrophic chondrocytes and the rate of longitudinal bone growth in growth plates. J. Orthop. Res. 1991;9:348–359. doi: 10.1002/jor.1100090306. [DOI] [PubMed] [Google Scholar]
- Burgold T., Voituron N., Caganova M., et al. The H3K27 demethylase JMJD3 is required for maintenance of the embryonic respiratory neuronal network, neonatal breathing, and survival. Cell Rep. 2012;2:1244–1258. doi: 10.1016/j.celrep.2012.09.013. [DOI] [PubMed] [Google Scholar]
- Chen S., Ma J., Wu F., et al. The histone H3 Lys 27 demethylase JMJD3 regulates gene expression by impacting transcriptional elongation. Genes Dev. 2012;26:1364–1375. doi: 10.1101/gad.186056.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Ghori-Javed F.Y., Rashid H., et al. Runx2 regulates endochondral ossification through control of chondrocyte proliferation and differentiation. J. Bone Miner. Res. 2014;29:2653–2665. doi: 10.1002/jbmr.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czermin B., Melfi R., McCabe D., et al. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell. 2002;111:185–196. doi: 10.1016/s0092-8674(02)00975-3. [DOI] [PubMed] [Google Scholar]
- De Santa F., Totaro M.G., Prosperini E., et al. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell. 2007;130:1083–1094. doi: 10.1016/j.cell.2007.08.019. [DOI] [PubMed] [Google Scholar]
- Ducy P., Starbuck M., Priemel M., et al. A Cbfa1-dependent genetic pathway controls bone formation beyond embryonic development. Genes Dev. 1999;13:1025–1036. doi: 10.1101/gad.13.8.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer E.L., Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012;13:343–357. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata K., Takashima R., Amano K., et al. Arid5b facilitates chondrogenesis by recruiting the histone demethylase Phf2 to Sox9-regulated genes. Nat. Commun. 2013;4:2850. doi: 10.1038/ncomms3850. [DOI] [PubMed] [Google Scholar]
- Hong S., Cho Y.W., Yu L.R., et al. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc. Natl Acad. Sci. USA. 2007;104:18439–18444. doi: 10.1073/pnas.0707292104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inada M., Yasui T., Nomura S., et al. Maturational disturbance of chondrocytes in Cbfa1-deficient mice. Dev. Dyn. 1999;214:279–290. doi: 10.1002/(SICI)1097-0177(199904)214:4<279::AID-AJA1>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Jepsen K., Solum D., Zhou T., et al. SMRT-mediated repression of an H3K27 demethylase in progression from neural stem cell to neuron. Nature. 2007;450:415–419. doi: 10.1038/nature06270. [DOI] [PubMed] [Google Scholar]
- Karsenty G. Transcriptional control of skeletogenesis. Annu. Rev. Genomics Hum. Genet. 2008;9:183–196. doi: 10.1146/annurev.genom.9.081307.164437. [DOI] [PubMed] [Google Scholar]
- Karsenty G., Kronenberg H.M., Settembre C. Genetic control of bone formation. Annu. Rev. Cell Dev. Biol. 2009;25:629–648. doi: 10.1146/annurev.cellbio.042308.113308. [DOI] [PubMed] [Google Scholar]
- Kim I.S., Otto F., Zabel B., et al. Regulation of chondrocyte differentiation by Cbfa1. Mech. Dev. 1999;80:159–170. doi: 10.1016/s0925-4773(98)00210-x. [DOI] [PubMed] [Google Scholar]
- Kornak U., Mundlos S. Genetic disorders of the skeleton: a developmental approach. Am. J. Hum. Genet. 2003;73:447–474. doi: 10.1086/377110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronenberg H.M. Developmental regulation of the growth plate. Nature. 2003;423:332–336. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- Kubicek S., Jenuwein T. A crack in histone lysine methylation. Cell. 2004;119:903–906. doi: 10.1016/j.cell.2004.12.006. [DOI] [PubMed] [Google Scholar]
- Lan F., Bayliss P.E., Rinn J.L., et al. A histone H3 lysine 27 demethylase regulates animal posterior development. Nature. 2007;449:689–694. doi: 10.1038/nature06192. [DOI] [PubMed] [Google Scholar]
- Lee T.I., Jenner R.G., Boyer L.A., et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301–313. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.G., Villa R., Trojer P., et al. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science. 2007;318:447–450. doi: 10.1126/science.1149042. [DOI] [PubMed] [Google Scholar]
- Lefebvre V., Bhattaram P. Vertebrate skeletogenesis. Curr. Top. Dev. Biol. 2010;90:291–317. doi: 10.1016/S0070-2153(10)90008-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Wang H.Y., Chepelev I., et al. Stage-dependent and locus-specific role of histone demethylase Jumonji D3 (JMJD3) in the embryonic stages of lung development. PLoS Genet. 2014;10:e1004524. doi: 10.1371/journal.pgen.1004524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long F. Building strong bones: molecular regulation of the osteoblast lineage. Nat. Rev. Mol. Cell Biol. 2012;13:27–38. doi: 10.1038/nrm3254. [DOI] [PubMed] [Google Scholar]
- Long F., Ornitz D.M. Development of the endochondral skeleton. Cold Spring Harb. Perspect. Biol. 2013;5:a008334. doi: 10.1101/cshperspect.a008334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin C., Zhang Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005;6:838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- Miller S.A., Mohn S.E., Weinmann A.S. Jmjd3 and UTX play a demethylase-independent role in chromatin remodeling to regulate T-box family member-dependent gene expression. Mol. Cell. 2010;40:594–605. doi: 10.1016/j.molcel.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller J., Hart C.M., Francis N.J., et al. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell. 2002;111:197–208. doi: 10.1016/s0092-8674(02)00976-5. [DOI] [PubMed] [Google Scholar]
- Park D.H., Hong S.J., Salinas R.D., et al. Activation of Neuronal gene expression by the JMJD3 demethylase is required for postnatal and adult brain neurogenesis. Cell Rep. 2014;8:1290–1299. doi: 10.1016/j.celrep.2014.07.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T., Takeuchi O., Vandenbon A., et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat. Immunol. 2010;11:936–944. doi: 10.1038/ni.1920. [DOI] [PubMed] [Google Scholar]
- Schroeder T.M., Jensen E.D., Westendorf J.J. Runx2: a master organizer of gene transcription in developing and maturing osteoblasts. Birth Defects Res. C Embryo Today. 2005;75:213–225. doi: 10.1002/bdrc.20043. [DOI] [PubMed] [Google Scholar]
- Shpargel K.B., Starmer J., Yee D., et al. KDM6 demethylase independent loss of histone H3 lysine 27 trimethylation during early embryonic development. PLoS Genet. 2014;10:e1004507. doi: 10.1371/journal.pgen.1004507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Jacques B., Hammerschmidt M., McMahon A.P. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999;13:2072–2086. doi: 10.1101/gad.13.16.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl B.D., Allis C.D. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- Takarada T., Hinoi E., Nakazato R., et al. An analysis of skeletal development in osteoblast-specific and chondrocyte-specific runt-related transcription factor-2 (Runx2) knockout mice. J. Bone Miner. Res. 2013;28:2064–2069. doi: 10.1002/jbmr.1945. [DOI] [PubMed] [Google Scholar]
- Takeda S., Bonnamy J.P., Owen M.J., et al. Continuous expression of Cbfa1 in nonhypertrophic chondrocytes uncovers its ability to induce hypertrophic chondrocyte differentiation and partially rescues Cbfa1-deficient mice. Genes Dev. 2001;15:467–481. doi: 10.1101/gad.845101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueta C., Iwamoto M., Kanatani N., et al. Skeletal malformations caused by overexpression of Cbfa1 or its dominant negative form in chondrocytes. J. Cell Biol. 2001;153:87–100. doi: 10.1083/jcb.153.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega R.B., Matsuda K., Oh J., et al. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell. 2004;119:555–566. doi: 10.1016/j.cell.2004.10.024. [DOI] [PubMed] [Google Scholar]
- Warman M.L., Cormier-Daire V., Hall C., et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am. J. Med. Genet. A. 2011;155A:943–968. doi: 10.1002/ajmg.a.33909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilsman N.J., Farnum C.E., Leiferman E.M., et al. Differential growth by growth plates as a function of multiple parameters of chondrocytic kinetics. J. Orthop. Res. 1996;14:927–936. doi: 10.1002/jor.1100140613. [DOI] [PubMed] [Google Scholar]
- Xiang Y., Zhu Z., Han G., et al. JMJD3 is a histone H3K27 demethylase. Cell Res. 2007;17:850–857. doi: 10.1038/cr.2007.83. [DOI] [PubMed] [Google Scholar]
- Yang Y. Skeletal morphogenesis during embryonic development. Crit. Rev. Eukaryot. Gene Expr. 2009;19:197–218. doi: 10.1615/critreveukargeneexpr.v19.i3.30. [DOI] [PubMed] [Google Scholar]
- Yang D., Okamura H., Nakashima Y., et al. Histone demethylase Jmjd3 regulates osteoblast differentiation via transcription factors Runx2 and osterix. J. Biol. Chem. 2013a;288:33530–33541. doi: 10.1074/jbc.M113.497040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Lawson K.A., Teteak C.J., et al. ESET histone methyltransferase is essential to hypertrophic differentiation of growth plate chondrocytes and formation of epiphyseal plates. Dev. Biol. 2013b;380:99–110. doi: 10.1016/j.ydbio.2013.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida C.A., Yamamoto H., Fujita T., et al. Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes Dev. 2004;18:952–963. doi: 10.1101/gad.1174704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Xiao Z., Luo J., et al. Dose-dependent effects of Runx2 on bone development. J. Bone Miner. Res. 2009;24:1889–1904. doi: 10.1359/JBMR.090502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziros P.G., Basdra E.K., Papavassiliou A.G. Runx2: of bone and stretch. Int. J. Biochem. Cell Biol. 2008;40:1659–1663. doi: 10.1016/j.biocel.2007.05.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.