

Abstract
The vast majority of drugs entering human trials fail. This problem (called “attrition”) is widely recognized as a public health crisis, and has been discussed openly for the last two decades. Multiple recent reviews argue that animals may be just too different physiologically, anatomically, and psychologically from humans to be able to predict human outcomes, essentially questioning the justification of basic biomedical research in animals.
This review argues instead that the philosophy and practice of experimental design and analysis is so different in basic animal work and human clinical trials that an animal experiment (as currently conducted) cannot reasonably predict the outcome of a human trial. Thus, attrition does reflect a lack of predictive validity of animal experiments, but it would be a tragic mistake to conclude that animal models cannot show predictive validity.
A variety of contributing factors to poor validity are reviewed. The need to adopt methods and models that are highly specific (i.e., which can identify true negative results) in order to complement the current preponderance of highly sensitive methods (which are prone to false positive results) is emphasized. Concepts in biomarker-based medicine are offered as a potential solution, and changes in the use of animal models required to embrace a translational biomarker-based approach are outlined. In essence, this review advocates a fundamental shift, where we treat every aspect of an animal experiment that we can as if it was a clinical trial in a human population.
However, it is unrealistic to expect researchers to adopt a new methodology that cannot be empirically justified until a successful human trial. “Validation with known failures” is proposed as a solution. Thus new methods or models can be compared against existing ones using a drug that has translated (a known positive) and one that has failed (a known negative). Current methods should incorrectly identify both as effective, but a more specific method should identify the negative compound correctly. By using a library of known failures we can thereby empirically test the impact of suggested solutions such as enrichment, controlled heterogenization, biomarker-based models, or reverse-translated measures.
Keywords: animal biology, biomarker based, heterogenization, reverse-translated
Setting the Stage—Are Animal Researchers Creating a Future Without Animal Research?
Biomedical research at its core is ethically, societally, and scientifically justified by its ability to deliver real-world benefits to human health (Rollin 2006). Indeed, as researchers we assume that our work is meaningful, but the simple fact that roughly 90% of compounds entering human trials will fail should be a harsh reminder that, as Ioannidis bluntly pointed out, “most published research findings are false” (2005). While industry in general, and some academics in particular, have openly discussed the shockingly poor translation of basic animal work into human outcomes for some time (e.g., Cummings et al. 2014; Kola and Landis 2004; Paul et al. 2010; Tricklebank and Garner 2012), basic research in academia doggedly claims real meaning for human outcomes with almost every published result. However, as will be discussed, not all results are created equal(ly meaningful), and the creation of more or less meaningful results (i.e., the validity of the experiment) is entirely within the power of the experimenter. Yet few basic research articles will critically discuss limitations on the validity of the study, the likelihood that a result will translate to humans, or the steps taken in experimental design to maximize validity (how many mouse experiments, for instance, discuss whether an “anxiety-like” measure is actually clinically relevant, or how it was chosen over others to best match human symptoms?).
As a result, biomedical research is close to a tipping point with potentially dire consequences for the future, scope, and funding of basic animal research. Pharma has aggressively disinvested in animal research in the last few years (Hunter 2011), and the value of animal research for advancing human health is being questioned at the highest federal levels—for instance, the previous head of the NIH, Elias Zerhouni, has since argued that the NIH should shift emphasis to favor human work: “We all drank the Kool-Aid on that one, me included… The problem is that it [animal research] hasn't worked, and it's time we stopped dancing around the problem… We need to refocus and adapt new methodologies for use in humans to understand disease biology in humans” (McManus 2013). This should come as no surprise: the NIMH's 2008 strategic plan (National Institute of Mental Health 2008) mentions animal work a paltry two times in 38 pages; instead emphasizing the use of imaging, genomic, and other next-generation approaches to answer questions traditionally pursued in animals. Indeed, the NIMH's implementation and emphasis on Research Domain Criteria (RDoC) represent a clear implementation of this strategy, and a general example of the coming paradigm shift in biomedical research, from a reductionist animal-based “genomic and phenotyping” era to a human-based “biomarker and personalized medicine” era. Yet academic institutions continue to invest in increasingly elaborate and expensive infrastructure for animal studies (e.g., individually ventilated caging systems). These infrastructure investments are justified in terms of both need, and economic viability, by projections that an institution's research animal population census will grow, fuelled by increased funding support. While this may be reasonable in top-tier academic institutions, a systemic shortfall in funding for animal research would be catastrophic for most research universities. Pharma provides a cautionary tale: Kola and Landis pointed out in 2004 that Pharma's investment model was unrealistically unsustainable over the coming 5 to 10 years (Kola and Landis 2004), and, sure enough, Pharma has been forced to disinvest in animal research (Hunter 2011). Indeed a key strategy has been to shift the expense and risk of basic discovery into academia (Hunter 2011). Now that essentially the same forces come to bear on academic research, what can be done to prevent a similar contraction in the academic sector?
In theory, the answer is simple—we need to do a better job of producing animal results that translate to human outcomes. Similarly, the changes required are relatively simple, and this article outlines some of the systemic issues and potential solutions available to animal researchers. The real challenge, however, is changing the culture of biomedical research so that even small simple changes are adopted. The potential rewards are worth the effort. At the end of the day, the failure of animal results to translate is arguably the greatest laboratory animal welfare issue of our day and a source of many societal ills. Furthermore, real advances in personalized and biomarker-based research (the supposed panacea to the problems in animal research) will in truth be very hard to attain without animal models of disease development. It is hard to think of a better example where good-welfare-is-good-science, or good-welfare-is-good-business. If this problem can be solved, then the rewards for animal welfare, biomedical research, patient populations, return-on-the-funding-dollar, and society in general will be immeasurable.
Thus, this article is not intended primarily for researchers. The research community by and large has been unable or unwilling to heed the calls for changes in culture and practice in the literature (e.g., Andreatini and Bacellar 2000; Andrews and File 1993; Begley 2013; Begley and Ellis 2012; Brown and Wong 2007; Chesler et al. 2002; Crusio 2004; Crusio et al. 2009; Cummings et al. 2014; Geerts 2009; Gerlai 1996; Gerlai and Clayton 1999; Hay et al. 2014; Ioannidis 2005; Kola and Landis 2004; Mak et al. 2014; Nieuwenhuis et al. 2011; Paul et al. 2010; Peers et al. 2012; Prinz et al. 2011; Richter et al. 2009, 2010; Sena et al. 2010; Tricklebank and Garner 2012; van der Worp et al. 2010; Wolfer et al. 2002; Würbel 2000, 2001, 2002; Würbel and Garner 2007; Zahs and Ashe 2010). In particular, peer review can be a stifling force of inertia—innovative methods are less likely to be funded than established (but weaker) methods. Meanwhile, reviews or methods papers that challenge the status quo or point out that the Emperor has no clothes can be almost impossible to publish. In both cases, the risk and costs for young scientists are particular high, just at the time when they are most open to new ideas and at their most creative. Similarly, differential funding opportunities and success rates for grants proposing new work versus renewal of an existing program are a powerful disincentive to established researchers to adopt new methods or challenge the status quo (ironically at the time when they can be most influential). Researchers need to be given both the safe space in their careers to adopt new approaches, and the incentives to do so (Pusztai et al. 2013). For instance, multiple reviews of the systemic issues with knockout mice did nothing to change the culture (Crusio 2004; Wolfer et al. 2002), finally leading the premier publication for mouse behavioral genetics to enforce standards through editorial policy (Crusio et al. 2009). Similarly the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines seek to encourage standards (Kilkenny et al. 2010) incentivized by both institutions and journals. Thus, this review is targeted at other stakeholders with a vested interest in increasing the translational value of animal research (from patients, to funding bodies, to animal advocates, to university administrators, to Institutional Animal Care and Use Committee [IACUC] members, to journal editors, and beyond) in the hope of creating similar opportunities for a change in culture and practice.
Attrition—What Is It? What Causes It? And Why Should We Care?
The failure of drugs during the pipeline is termed “attrition” (Kola and Landis 2004) and can be subdivided into discovery or preclinical attrition (prior to human trials), and development or clinical attrition (during human trials) (Paul et al. 2010). Recent reviews of large-scale Food and Drug Administration (Kola and Landis 2004), international industry (Paul et al. 2010), and public (Hay et al. 2014) data sets indicate that clinical attrition rates range from approximately 80% to 97%, depending on the therapeutic area, with an average of approximately 90%.
Traditionally, attrition has been viewed as an issue with clinical trials, simply because this is when it is most visible and most costly. The most detailed economic data are provided by Paul and colleagues (2010) and provide an instructive window on the problem. Preclinical attrition rates are approximately 35% (i.e., one in three potential compounds will eventually enter human trials). However, these failures occur during relatively inexpensive phases of drug development and thus account for only 2% of the total cost of bringing a drug to market. By contrast, clinical attrition rates are approximately 90% (i.e., roughly one in nine compounds entering human trials will make it to market), but, because these phases of the pipeline are much more expensive, these failures account for 73% of the cost of bringing a drug to market (which totals approximately $1.8 billion in 2010 U.S. dollars) (Paul et al. 2010). These numbers underscore the huge societal impact of clinical attrition in particular—these costs can be diluted in a large patient population that will need the drug for a long time. Drugs for rare diseases, or for diseases with which patients are unlikely to seek help, and drugs that cure rather than manage diseases will be more expensive to the patient, and potentially too expensive to be worth development.
However, digging deeper into these data sets reveals that attrition is fundamentally an animal issue, not a human one. To understand why, we need to consider two sides of a coin: What are the identifiable reasons for failure in human trials? And what is the structure of decision making in the drug discovery pipeline itself? The drug discovery pipeline is traditionally divided into preclinical and clinical phases (which correspond to animal and human phases), but it can also be subdivided by the type of questions being asked. Early preclinical stages focus on potential efficacy (both in silico and in animals), followed by safety (in the last and most intensive and invasive use of animals and in the first “Phase I” human trials), followed by proof of efficacy (in Phase II and Phase III trials). This allows an estimate to be made of the reasons for clinical attrition—success during Phase I is roughly 50%, but the cumulative success from Phase II through to launch (i.e., when the efficacy of the drug is being assessed) is 20%. Accordingly, even small improvements in Phase II and Phase III success reduce the total cost of bringing a drug to market by 35% (Paul et al. 2010). Of course, efficacy is not an all-or-nothing phenomenon and drugs fail in these stages for other reasons. For example, bioavailability of the drug at the target may be very different in humans, limiting efficacy or narrowing the therapeutic window to the point where side effects become unmanageable; or drug–drug interactions may interfere with the study or preclude widespread use in a human population. Thus, the real issue is whether a new drug is efficacious enough given its cost of manufacture, the patient population, and the cost and efficacy of competing products. Nevertheless, taking very different approaches, Kola and Landis (2004) and Hay and colleagues (2014) identify lack of efficacy as the single largest reason for why a drug fails in human trials.
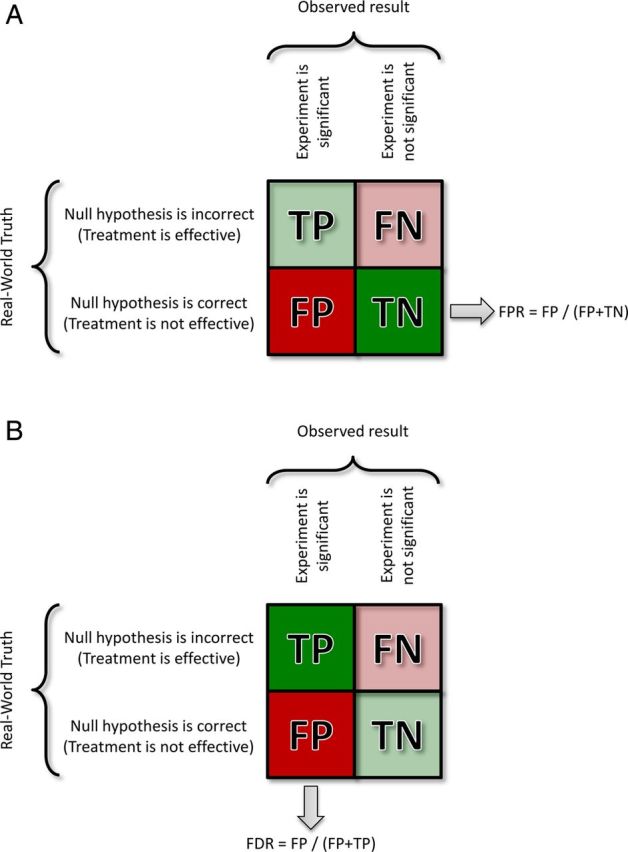
The other side of the coin is to think of the pipeline in terms of decision making. Thus the pipeline makes early go decisions on the basis of animal results, then turns its attention to making no-go decisions on the basis of safety, before finally assessing the predictive validity of an animal result (i.e., making a no-go decision) in the final and most expensive stages of the pipeline. From a statistical point of view, the initial stages of the pipeline emphasize sensitivity (making sure all hits are identified, at the cost of false positives), before emphasizing specificity (making sure that false positives are weeded out). (See Figure 1A for definitions.) From this perspective, attrition has a deceptively simple cure—moving the no-go decision making as early as possible in the pipeline. While this point has been made with respect to economics (Paul et al. 2010), it is just as important in terms of animal welfare. The scale and invasiveness of animal use in the pipeline peaks in preclinical toxicology. Thus every failure for lack of efficacy is a prima facie welfare issue through the needless use of animals in preclinical toxicology. If a highly specific no-go decision on the basis of efficacy could be inserted prior to this stage, then good-welfare-is-good-business: invasive animal use is massively reduced, as is attrition at the most costly stages of the pipeline.
Figure 1.

A Venn diagram representation of the effects of specificity and sensitivity on validity and false discovery. The figure illustrates an imaginary space of all the deficits we might want to detect. Within this space, deficits detected by the test and deficits actually relevant to the human disorder are shown as two separate areas. (A) Basic definitions. Deficits detected by the test are either: FP = False Positive (irrelevant to the disorder) or TP = True Positive (relevant to the disorder). Deficits relevant to the disorder but undetected by the test are FN = False Negative. Deficits irrelevant to the disorder and undetected by the test are TN = True Negative. Thus we are primarily concerned with the region of overlap (TP). (B) Representing validity. The reality of a behavioral test is that it will measure multiple behavioral domains, represented by the different lobes of the area representing the test. While a test may contain a lobe relevant to the disorder (TP), it will also contain lobes irrelevant to the disorder (FP). Thus FDR (False Discovery Rate) = FP / (FP + TP) is a useful proxy to validity—high false discovery rates indicate low validity (Richter et al. 2009). These FP domains, however, may be relevant to a different disorder, in which case the test may detect deficits relevant to both but cannot distinguish between them (i.e., it lacks discriminant validity). (C) Sensitivity and FDR. The immediate temptation is to increase the sensitivity of the test, but sensitivity is the proportion of all meaningful deficits detected = TP / (TP + FN) (i.e., it does not consider FP). Thus an indiscriminant expansion of the deficits detected by the test will increase sensitivity but at the expense of also increasing FDR and decreasing validity. This is one of the many problems with using multiple phenotyping tasks to test the same hypothesis (as illustrated by the expansion and addition of FP “lobes”). (D) Specificity is the proportion of all nonmeaningful deficits correctly identified = TN / (TN + FP). Thus specific tests minimize FP, and minimize FDR, but at the risk of increasing FN (i.e., though every result is much more believable, the cost is that we not be able to detect all relevant deficits). As argued in the text, the best strategy is to follow our existing overly sensitive phenotyping tests, with a round of highly specific tests, maximizing the chance of detecting all TP, but then quickly weeding out most FP.
The remainder of this review addresses the question of what this highly specific decision might look like in the absence of human trials. We will cover three topics: evidence that animal studies are the ultimate source of false positives and poor efficacy; reasons for the poor predictive validity of animal studies; and a new approach to animal models that resolves these problems, provides specificity, and supports the move to personalized medicine.
Animal Studies as the Ultimate Source of Attrition
The logic tying failures in clinical trials to basic research in animals is seductively straightforward. Every drug entering human trials, by definition, “worked” in an animal model in terms of both safety and efficacy, and efficacy is the primary reason why drugs fail in human trials. Thus, the primary reason for these failures can be traced back directly to false positives in animal models committing the pipeline to develop a drug that will ultimately fail. Straightforward data can be used to make this case. For instance, as reviewed by Zahs and Ashe (2010), over 200 different interventions have been reported to be effective in the APP mouse model of Alzheimer's disease, yet none has proven effective in human trials. Indeed the attrition rate for Alzheimer's drugs from 2002 to 2012 was 96.4% (Cummings et al. 2014). Similarly, approximately 500 compounds have been reported as effective in reducing the effects of acute ischemic stroke in animal models, yet only 2 have proven effective in humans (van der Worp et al. 2010).
However, both of these reviews (van der Worp et al. 2010; Zahs and Ashe 2010) offer a more nuanced view, questioning what the model(s) are actually modeling and whether this is truly relevant to human outcomes. In fact, a similar theme can be seen regardless of the particular focus of papers in the recent literature: from publication bias (Ioannidis 2005; Prinz et al. 2011; Sena et al. 2010); to adopting human clinical practices such as blinding (Kilkenny et al. 2009; Macleod et al. 2008; Muhlhausler et al. 2013) or matching (Hånell and Marklund 2014; Muhlhausler et al. 2013; Richter et al. 2009; Würbel and Garner 2007); to exposing problems in phenotyping and arguing for refined behavioral measures (Andreatini and Bacellar 2000; Arguello and Gogos 2006; Brown and Wong 2007; Gerlai and Clayton 1999; Insel 2007; Tricklebank and Garner 2012) including reverse translation (Garner et al. 2006, 2011; Harding et al. 2004); to discussing methodological and interpretational issues in genetically engineered mice (Crusio 2004; Crusio et al. 2009; Wolfer et al. 2002); to observations that unenriched animals are functionally immune suppressed enough to influence the model (Ader et al. 1991; Cao et al. 2010), or more generally that phenotypic plasticity in response to the environment might change experimental outcomes (Chesler et al. 2002), and that testing across diverse environments and biological backgrounds better matches human trials (Festing 2014; Mak et al. 2014; Richter et al. 2009, 2010, 2011; Wurbel 2002); to criticizing the inappropriate use of low-power naïve statistical approaches (Festing 2014; Peers et al. 2012; Prinz et al. 2011; Würbel and Garner 2007; Wurbel et al. 2013) particularly in basic research (Nieuwenhuis et al. 2011); to arguing that the model usefully recapitulates physiology without capturing predictive outcomes (Cummings et al. 2014; Zahs and Ashe 2010); even to fundamental biological issues with the models themselves (Begley and Ellis 2012; Geerts 2009; Mak et al. 2014).
Modeling the Disease, or Modeling the Model?
Thus all of these papers reach a general consensus—that when models fail to predict human outcomes, perhaps we are not really modeling the human disease, but just modeling the model. To illustrate this concept, consider four thought-experiments:
Is a knockout mouse based on a rare allele in humans a model of the disease, or a model of the subpopulation? For instance, Tourette syndrome and trichotillomania are generally considered to be a polygenic disorders where most risk alleles confer small, low-penetrance risk. However, a rare mutation in the SLITRK1 gene found in only a few individuals confers extremely high risk for both disorders (Abelson et al. 2005; Zuchner et al. 2006). Are the knockout mice inspired by these findings (Shmelkov et al. 2010), even though they show repetitive behavior, plausibly a general model of either disorder, or just a model of these rare human families?
Is a chemical lesion modeling terminal disease going to tell us about early disease processes? For instance, recent advances in Parkinson's disease show that at the point of diagnosis most patients have already lost over 50% of dopaminergic projections. In fact, we now recognize that there is an early, non-dopaminergic “pre-motor” phase to Parkinson's disease (Tolosa et al. 2007). Clearly we would like to be able to screen for these early stages and potentially slow or prevent progression of dopaminergic cell death. However, as our models are constrained to dopaminergic lesions, while they may model the terminal stages of Parkinson's disease, can they realistically help us understand, detect, or prevent the pre-motor onset of the disease in humans?
Can we realistically expect phenotyping to tell us about a specific human symptom? In humans, there is an entire discipline—neuropsychology—that describes the subtle differences in cognition that distinguish different disorders. Indeed, neuropsychology is a major driver of the new RDoC approach from the NIMH, and neuropsychology provides some of the best biomarkers in psychiatry (Garner et al. 2011; Holliday et al. 2005). For instance, the nature of memory deficits in Alzheimer's, versus normal aging, versus schizophrenia, versus traumatic brain injury, are fundamentally different—so is it really plausible that the Morris water maze (MWM) can meaningfully model, let alone distinguish, all of these human phenomena?
Is our animal population a realistic representation of human variability? Humans are variable. Indeed it is our variability in risk for illness and response to treatment that is the focus of modern medicine (Feinberg 2007; National Institute of Mental Health 2008). We would never perform a human drug trial in 42-year-old white males with identical educational levels, identical socioeconomic statuses, identical jobs, identical houses with identical (locked) thermostats, identical wives, identical diets, identical exercise regimes, in the same small town in Wisconsin, who all incidentally had the same grandfather. So can we realistically expect mice in exactly this kind of Stepford experiment to tell us anything about humans in general, or variability in risk or response in particular?
The answer to each of these thought-experiments is clearly “no,” but it is a qualified “no.” As many authors have argued, the secret to using animal models is to understand their limitations (e.g., Insel 2007). Thus all the thought-experiments above are not examples of bad models, but examples of the need to work within the limitations of a model or measure. This point is neatly illustrated by the difference between cardiovascular and cancer drug attrition. Cardiovascular drugs consistently have the lowest attrition, while cancer drugs consistently have one of the worst success rates (e.g., 20% versus 5%: Kola and Landis 2004). Authors in the cancer literature often blame a fundamental biological difference for this high attrition rate (Begley and Ellis 2012; Mak et al. 2014). Yet differences in heart physiology between humans and other animals are profound (and the impact on models has been intensively studied) (e.g., Vaidya et al. 1999; Wakimoto et al. 2001). Thus even major differences in animal versus human biology do not preclude a model from having strong predictive validity. Indeed, recognizing those differences may be key to ensuring translation, particularly in neuroscience, where general physiology can be quite different (e.g., in Alzheimer's disease: Kokjohn and Roher 2009; Zahs and Ashe 2010) and where simplistic approaches to behavioral phenotypes can be very misleading (see below).
Instead, this review proposes that the real difference between basic research in animal models and human clinical trials that ultimately causes attrition is the underlying axiomatic differences in the philosophy and methodology of animal versus human research. These differences are discussed in two steps. First, the following section emphasizes three deeply pervasive methodological issues that have received relatively little attention in the literature. These three issues can be thought of as logical traps that basic science has become increasingly blind to, and which should always be avoided. Then, a human-inspired approach to animal experiments is proposed, and the contingent changes in perception of what makes a good animal model are emphasized.
Three Traps That Have Become Business as Usual
Trap Number One: The Word “-like”
The word “-like” (as in “OCD-like” or “anxiety-like”) has become pervasive in behavioral neuroscience, but it represents an incredibly dangerous slip in logic. The trap is simple to understand: calling a measure “-like” does not make it so (that is an empirical issue of validity). Calling a measure “-like” is a rhetorical device that gives the measure a sheen of respectability and scientific caution, while in truth masking the fact that no attempt has been made to validate the measure or that it is being used despite being known to be invalid. In either case, this is simply bad science. The trap is that, although the initial “-like” may be well intentioned (as a kind of placeholder for the need to validate), with enough publications, the measure slips into perception as being validated. In other words, “-like” takes the place of empirical validation. This critique leads to two inevitable questions: why are “-like” measures so pervasive in neuroscience? And why do we think they have been validated (and is there evidence that they are invalid)?
Behavioral phenotyping and the pervasiveness of “-like.” The pervasiveness of the “-like” logical trap is tightly tied to the widespread use of behavioral phenotyping in behavioral neuroscience. As before, the argument here is not that behavioral phenotyping is inherently flawed but that it is the wrong tool to use if we want to predict human outcomes. Again the issue is one of sensitivity versus specificity (Figure 1A). Behavioral phenotyping tasks are designed to be overly sensitive (i.e., they hope to detect any relevant deficit at the risk of also detecting many false positives) (Figures 1B and C). This is a fine strategy for early stages of basic research, especially if the effect of a mutation or treatment is not known. However, without highly specific follow-up measures (which are not widely used) these false positives can never be weeded out, and thus phenotyping alone is a poor strategy for translational research (Figure 1D). That being said, why has phenotyping fallen into the “-like” trap?
Given their role in screening, behavioral phenotyping tasks are also designed to give quick and easy readouts, preferably using automated off-the-shelf equipment, which can be performed by researchers with no formal training in the behavioral sciences (Wahlsten 2001). The lack of formal training in ethology or psychology is the first reason why most tasks are referred to as “-like.” Validation itself is of little interest to users of phenotyping, instead the assumption is that the measure is as meaningful as any other assay that may be purchased by the lab.
While some phenotyping tasks are designed de novo, most are derived from ethology or experimental psychology. Given the emphasis on throughput, these tasks are then stripped of time-consuming elements, which are often essential controls; and meaningless but easily automated measures are added. For example, the open field test typical of phenotyping commits most of the errors warned against in the original literature (Archer 1973; Walsh and Cummins 1976). An important consequence of the oversimplification of such measures and the removal of controls is that phenotyping measures are extremely nonspecific and are sensitive to a wide range of confounding variables (Figure 1C). For instance, the single biggest predictor of performance in the MWM is the degree of retinal atrophy of the mouse, not memory (Brown and Wong 2007). That is, the MWM could just as well be interpreted as a measure of blindness not memory. Similarly, the best predictor of the tail-flick measure of pain sensitivity is the identity of the experimenter, not the genetics of the mouse (Chesler et al. 2002); and the elevated plus maze (EPM) shows many features that to an ethologist or psychologist suggest weak validity (Andreatini and Bacellar 2000; Andrews and File 1993). Furthermore, important ethological considerations are often overlooked. For instance, while the MWM makes sense for a marshland species like the Norway rat, the MWM for mice does not survive a thorough ethological analysis (Gerlai and Clayton 1999). However, as phenotyping often points out, such considerations are of little interest as long as the task can detect differences (Hossain et al. 2004), and are dismissed by using the word “-like.”
Worse still, the choice of these tasks is often based on simple word matches, unaware of the technical use of the word in the behavioral literature. For instance, “anxiety,” strictly defined, refers to the normal conflict of two motivations (hence the deliberate conflict of fear and exploration in most tests) (Gray 1987). This is just fundamentally different from the pathological anxiety that characterizes disorders such as obsessive-compulsive disorder (OCD) or generalized anxiety disorder, in which the essence of the pathology of the disorder is that anxiety is experienced when there is no conflict, or is experienced to an excessive degree in response to minor conflict (American Psychiatric Association 2013). As a result, the use of measures of normal anxiety to measure pathological anxiety is dubious at best. At the most extreme, researchers with no human clinical knowledge will latch onto completely superficial behavioral similarities, label them “-like”, and actually use behaviors in mice that would indicate differential diagnoses in humans. The best example of this is OCD, where any vaguely repetitive behavior in mice is referred to as “OCD-like.” Unfortunately, stereotypies, self-injury, and body-focused repetitive behavior disorders (e.g., trichotillomania) are all exclusionary differentials (i.e., if the presenting behavioral symptom is a stereotypy, a diagnosis of OCD is not possible) (American Psychiatric Association 2013). In fact, this author is unaware of any mouse model with “OCD-like” behavior where that behavior is not actually an exclusionary differential in humans. Such models may throw light on physiology common to repetitive behavior disorders (including OCD), but they cannot be a valid model of OCD specifically.
The misuse of barbering (fur and whisker pulling) neatly illustrates both points. It is widely misinterpreted as normal dominance behavior (i.e., a low level of barbering behavior is considered a social deficit) (Hånell and Marklund 2014), despite the fact that ethologists firmly debunked this interpretation many years ago (Garner et al. 2004a; Van de Weerd et al. 1992). At the same time, barbering is also widely used as an “OCD-like behavior” (e.g., Hill et al. 2007). Aside from the fact that it is farcical for a behavior to be both abnormal and normal at the same time, barbering is in fact a well-validated model of trichotillomania (Garner et al. 2004b, 2011), and trichotillomania is an exclusionary diagnosis for OCD (American Psychiatric Association 2013).
Have phenotyping measures actually been validated? The topic of validity and validation is complex (for in-depth dicussion of different kinds of validity, and how to test for them, see Tricklebank and Garner [2012], summarized in Table 1). Nevertheless, the most important thing to understand about validity is that it is completely distinct from reliability (Martin and Bateson 1986). Validity asks whether my measure or model actually means what it is supposed to, and what the limits of that inference might be—both questions being empirically answerable. Conversely, reliability asks whether my measure gives the same result under different circumstances (e.g., when it is repeated, or if two different raters score the same behavior). However, a completely reliable measure can be invalid. For example, a phrenologist could measure the size and location of bumps on your head on two different days and get the same results (test–retest reliability), or two phrenologists could measure your head and get the same results (inter-rater reliability), but these results say absolutely nothing about personality or criminality.
Table 1.
Three dimensions of validity (adapted from Tricklebank and Garner 2012). Validity can be thought of as three independent dimensions, and most tests of validity involve at least two of these dimensions (for instance, the failure of mouse models to predict human outcomes is a failure of predictive, external, convergent validity). For additional discussion, see (Campbell and Fiske 1959; Martin and Bateson 1986; Tricklebank and Garner 2012; Willner 1986; Würbel 2000)
| Dimension | Subtype | Definition and examples |
|---|---|---|
| Face v Construct v Predictive | Face | Does the measure or model appear outwardly similar to what it is supposed to measure or model in terms of behavior, phenomenology, epidemiology, etc.? (For example, does a fear measure resemble fear responses for the species? Does the animal behavior resemble the behavior seen in human patients?) |
| Construct | Does the measure or model involve the mechanism or processes that is supposed to measure or model (at physiological, neuropsychological, motivational levels, etc.)? (For example, can the measure actually access these processes? Is the methodology consistent with the theory behind the measure? Does an animal model involve the same physiology as the human measure or condition?) | |
| Predictive | Does the measure or model actually predict outcomes it is supposed to? (For example, does a behavioral stress measure predict stress hormone levels? Does an animal model predict human drug response? Does the animal model respond only to treatments that successfully treat human patients?) | |
| Internal v External | Internal | Are the methodology and results of the measure or model consistent with both the theory and existing data from the model system? (For example, is the methodology consistent with the mathematics describing the measured properties? Is the measure ecologically relevant to the test species? Does the measure agree with other measures of the same property in the same individuals?) |
| External | Are results from the measure or model broadly applicable? (For example, is the kind of fear measured in a fear test broadly applicable to the kind of fear being modeled in humans? Does the model give the consistent results across a range of environmental conditions that accurately reflect the range of environmental conditions experienced by human patients?) | |
| Convergent v Discriminant | Convergent | Does the measure or model show broad agreement with properties of the thing being measured, or properties of the human condition being modeled? (For example, are different measures of fear correlated? Does the model show similar behaviors to the human condition? Do drugs that treat human patients also treat model symptoms? Is the gene knocked out in the model also downregulated in human patients? Do mechanisms in the model mirror those in humans?) |
| Discriminant | Does the measure or model exclude alternative processes or differential diagnoses? (For example, is a fear measure clean, or is it correlated with measures of other behavioral traits? Does the model show behaviors, physiology, or symptoms atypical of the human conditions, or typical of a differential diagnosis to the human condition? Do drugs that fail to treat humans also fail to treat the model? Do all human patients show downregulation of the gene knocked out in the model, or only a subset? Do mechanisms that distinguish human disorders or subtypes also distinguish the animal models?) |
Given the central importance of validity to behavioral science, it is worrisome that few phenotyping tasks have been formally validated. Instead, the quality of behavioral measures in phenotyping is primarily assessed on whether they can discriminate between treatments or genotypes (e.g., Hossain et al. 2004); whether they are off the shelf, quick, and easy to perform (e.g., Hånell and Marklund 2014); and whether they are reliable (e.g., Wahlsten et al. 2003). Indeed classic validation papers often demonstrate nothing more than the sensitivity to distinguish mouse strains (e.g., Moy et al. 2004) or the reliability of the measure (e.g., Nadler et al. 2004). However, highly sensitive measures have an inherent tendency to decrease validity and increase false discovery rates (FDRs) (Figures 1B and C).
Most measures assume convergent face validity (i.e., they outwardly resemble the intended deficit being measured), but, as the examples above illustrate, this is a very weak and often misleading assumption. For instance, stereotypies lack face and construct validity as a measure of OCD, but realizing so requires knowledge of human symptomatology and diagnostics. Most also have accrued a level of convergent internal predictive validity, in that they respond to drugs that work in humans. The limitations of this form of validation should be obvious. First, human disorders are not diagnosed by drug response, and most psychoactive drugs have multiple (side) effects (in other words, although some simple tics in Tourette syndrome respond to haloperidol, not all behaviors in mice that respond to haloperidol are tics). Second, this confines us to find “more of the same” (i.e., just because known analgesics cause changes in the tail-flick test, it does not follow that an analgesic with a novel mode of action would) (Tricklebank and Garner 2012).
Normally in ethology or psychology, we would want to see stronger, quantifiable forms of validation. For instance, when multiple measures of anxiety are taken, one would expect them to correlate (internal, convergent, construct validity), but this is often not the case (e.g., Binder et al. 2004). Similarly, we would expect a measure not to be sensitive to alternative deficits (internal, discriminant, construct validity), but again this is often not the case (e.g., for the MWM: Brown and Wong 2007). More subtly, when we want to detect traits (e.g., pathological anxiety), the measure should not be fooled by states. That is, we want to detect stable features of the animal's behavior, not fleeting moment-to-moment changes, Again this is often not the case (e.g., for measures of anxiety and fear: Andreatini and Bacellar 2000; Andrews and File 1993; Miller et al. 2006). Finally, if a measure really is detecting pathology relevant to humans, then it should predict drugs that work (external, predictive, convergent validity) and drugs that do not work (external, predictive discriminant validity) in humans. However, the relatively high failure rate of central nervous system (CNS) drugs in human trials (Kola and Landis 2004) suggests that this, too, is not the case.
Thus “-like” is clearly not good enough. Validating existing phenotyping measures, however, is not a constructive exercise—we already know that most phenotyping measures are of limited validity. Furthermore, even a crude measure is useful for making an initial screen. Instead, a better strategy is to use phenotyping measures correctly (i.e., for initial screens) and complement them with new measures specifically designed to weed out the false positives injected by the use of phenotyping (Figure 1D). In other words, if we want animal results to predict human outcomes, then we need to measure the same clinical phenomena in animals that we do in humans (Garner et al. 2011; Insel 2007; Malkesman et al. 2009; Tricklebank and Garner 2012).
Trap Number Two: Going Fishing
The single most effective defense against false positives is having a strong specific hypothesis. This is for two reasons. First, the logic underlying the calculation of a p value requires a null hypothesis: p = the chance of observing an effect this large or greater, if we can assume that there is truly no effect. Therefore, without a null hypothesis, a p value is meaningless (see Figure 2A). Second, the overall chance of a false positive result increases the more tests are performed (a problem called multiplicity). For instance, if I perform five tests, the chance that at least one will be significant at p < 0.05 by chance alone is 23% (Grafen and Hails 2002). For this reason, we apply multiple-testing corrections when we are performing post hoc tests of a significant interaction in analysis of variance (ANOVA), for example, so that the overall probability of any of the multiple comparisons between means equals the “family level” limit of p < 0.05 with which the original F ratio was tested (Neter et al. 1996). A strong hypothesis predicts that a particular manipulation will have a particular effect in a particular measure, thus there is one test per hypothesis and the experiment is protected from multiplicity. The golden rule is that if I use multiple tests to assess the same hypothesis, then I must correct for multiple testing (Benjamini and Yekutieli 2001).
Figure 2.
The “confusion matrix” is used to describe the contributions of different results to the properties of measures or experiments, and the assumptions underlying their calculation (it is the mathematical representation of the Venn diagrams in Figure 1). In this case we consider experimental outcomes. Rows show the truth in the real world, and columns the outcome of an experiment. FP = False Positive; FN = False Negative; TP = True Positive; TN = True Negative.
(A) A p value is the probability, assuming that the null hypothesis is true (that there is no real treatment effect) of obtaining a result at least as large as the one observed. One useful way to think about this concept is to imagine that we knew for a fact that the null hypothesis is true and performed thousands of experiments. The resulting False Positive Rate (FPR) would be equal to the significance level chosen (i.e., if p < 0.05 is accepted as significant, then the FRP will = 5%). TP and FN are whited out because they are not considered in the logic of a p value. (B) When the null hypothesis is not known (and cannot be assumed), p values lose their meaning. This is typically the case in genomic studies and is argued here as also true for phenotyping. Instead all that is known is that positive results were observed. In this case, we can calculate a False Discovery Rate (FDR) as the proportion of positive findings expected to be False Positives. FN and TN are whited out because they are not considered in the logic of FDR. As with p values, a q value can be estimated for each result, or we can simply label all results less than a critical q value. In either case the q value represents the FDR at which the result ceases to be believable, so q < 0.1 means that a result is not believable once we are unwilling to tolerate a FDR of more than 10%. Like a p value, a result with a q < 0.1 (for instance) may still be a FP.
Weaker hypotheses—for instance, that my manipulation will cause a difference in any of the measures taken—break this rule. The phenotyping philosophy of “any difference is interesting” is a perfect example. For instance, consider a phenotyping experiment that uses three measures of anxiety (open-field, EPM, and light–dark box), each with multiple submeasures: the number of comparisons used to test the single hypothesis (that anxiety differs by treatment) increases exponentially, and we can virtually guarantee that at least one will be significant by chance alone. The trap is to then cherry-pick the one apparently significant result (or, worse still, ignore conflicting results), which virtually guarantees a false positive (Begley 2013; Ioannidis 2005).
Thus the statistical issues facing phenotyping are much closer to genotyping (where we may be testing for differences in thousands of genes) than traditional hypothesis-led science. Given a weak hypothesis, p values become increasingly meaningless (because we are no longer safe in our assumption that the null hypothesis is true). Instead, now all we know for a fact is that we observed a positive result. In this case, we can still calculate a test—a q value, or the chance that the observed result(s) are a false discovery (see Figure 2B) (Benjamini and Yekutieli 2001).
Weak-hypothesis discovery techniques (like microarrays, RNA-seq, or phenotyping) are often unfairly characterized as fishing expeditions. This is unfair both to fishing and to discovery. At least a fisherman knows what species, sexes, or sizes she wants to catch, and can throw back the fish she does not want. In other words, she may use a sensitive method to catch all the available fish, but she then uses a specific method to screen out the ones she doesn't want. Naïve analysis of phenotyping or genotyping data has no such safety net—there is no way of picking out the real results from the false discoveries. To be fair, at least in genotyping we can use q values and FDR calculations to statistically isolate the results most likely to be true, and we can use biologically informed techniques (such as gene set enrichment analysis [GSEA]) to pick out the most biologically plausible results (Subramanian et al. 2005). There is no reason why we cannot apply the same logic to analysis of phenotyping data (and every reason to do so). At the very least, we should be testing phenotyping data with q values. This could be achieved by either figuring an FDR correction for the whole experiment, or perhaps with more focused hypotheses for each behavioral domain. This is a straightforward calculation and can be derived from the list of all observed p values relevant to a hypothesis (Benjamini and Yekutieli 2001). Similarly, the logic of GSEA can also be applied—if multiple tests are used to address the same hypothesis, then isolated results should be disbelieved, and results in agreement should carry more weight.
Trap Number Three: Confirmation is not Hypothesis Testing
The final trap is by no means confined to neuroscience. At its broadest, it can be described as mistaking a tool for a model. If we had a surgical lesion model (e.g., spared nerve injury), it would obviously be wrong to think of the scalpel used to perform the surgery as the model. However, we consistently mistake molecular scalpels in the form of knockout mice (for instance) as models. The fact that the tool is itself an animal is beside the point. This trap is triggered when we mistake confirmation for hypothesis testing. The difference should be obvious: we test hypotheses by falsifying them, but a confirmatory experiment does the exact opposite—it attempts to prove the original finding. As a result, a logical error in the interpretation of the original finding can be permutated through each confirmatory experiment. In the case of knockout mice, the pervasive mistake is to assume that the specific genetic lesion in the mouse is also the cause of the phenotype in humans (or wild-type mice). The specific genetic lesion (the genetic scalpel) may be rare or even impossible in the wild, and may bring about its effects through knock-on consequences on other molecular pathways. Table 2 illustrates this flawed logic using the example of knockout cars (where gas and brake pedals are removed), and the failure of confirmatory experiments to detect the false positive result. It shows the 1:1 correspondence to a series of experiments that lead to the biologically impossible implication that trichotillomania (which affects 3% to 4% of women) is caused by a spontaneous silencing of the HOXB8 gene in bone marrow white blood stem cells (Chen et al. 2010; Greer and Capecchi 2002).
Table 2.
Confirmatory experiments do not test for alternative physiological, biological, or psychological interpretations. The first column illustrates a series of experiments by an antique British sports car enthusiast. By failing to consider whole-car biology, the experimenter erroneously concludes that gas (accelerator) pedals are not only the cause of rapid aging that plagues these classic cars, but also the cause of accidents in general. Hopefully the flaws in the interpretation are glaring. The second column summarizes a series of experiments reporting the unexpected finding that Hoxb8 deletion leads to excessive grooming and self-injury (in addition to the expected skeletal abnormalities seen in these mice) (Greer and Capecchi 2002), and finally attributing the unexpected behavioral phenotype to selective Hoxb8 deletion in “hematopoietic” (bone marrow) derived cells (Chen et al. 2010). Note that the logical reasoning is identical for both series of experiments. In the case of the mice, the same lack of whole-animal thinking leads to a completely implausible model. It might well be the case that through a complex cascade of events this genetic scalpel influences the true disease process, but this mouse cannot represent a meaningful disease process in humans (i.e., it cannot be a model). For instance, it is implausible that a selective somatic mutation in bone marrow, or selective pathological downregulation, of HOXB8 occurs in 3.5% of women. What is particularly shocking about this example is that Chen et al. (2010) go as far as to imply that bone marrow transplant might be a possible therapy in humans
| Knockout antique British sports cars | Knockout mice |
|---|---|
|
|
|
|
|
|
|
|
|
|
In fact, barbering provides another very instructive example. Hill and colleaagues (2007) knocked out the gene coding for aromatase in order to engineer estrogen-deficient mice as a model of male-biased early-onset tic-like OCD. The logic being that this subset of OCD patients show evidence of lowered catechol-O-methyltransferase (COMT) activity, and the corresponding COMT gene is regulated by estrogen. Sure enough, male knockout mice showed increased barbering relative to wild-type, while female mice did not. Aside from the fact that barbering is not a model of OCD, what these authors failed to realize is that barbering severity and incidence is higher in females than in males (Garner et al. 2004b) and that an estrogen pulse specifically mediated by aromatase is required to masculinize the brain (Lephart 1996). Thus the male knockout mice had feminized brains, and, sure enough, are simply showing the same levels of barbering behavior as wild-type and knockout female mice. No amount of confirmatory experiments (just like with the Hoxb8 studies in Table 2) can overcome this basic biological error.
The trap here is that, while confirmatory experiments do rule out technical errors (like background strain effects), they are designed to check the veracity of a tool, not a model. However, if we want to avoid false positives in terms of translation to humans, we need to rule out biological, developmental, and psychological whole-animal alternative explanations. One of the simplest ways to do so is to model the development of disease itself. Again, this doesn't mean abandoning knockout mice—genetic scalpels are hugely useful for testing particular hypotheses, they just are a very poor choice for modeling the development of a disease in a wild-type population.
Solutions—Biomarkers and Preventative Personalized Medicine
The modern push toward personalized medicine is intimately tied to the idea of biomarkers (Gottesman and Gould 2003; Gould and Gottesman 2006; National Institute of Mental Health 2008). If we consider the development of a disorder from a risk allele, then we can follow disease development through a chain of genomic, physiological, biological, and psychological measures to the final symptomatology. Through development, these measures become less determinant (penetrant), increasingly complex, and may diverge into nonpathological subclinical traits, into different subtypes of a disorder, or even into different disorders. This building complexity reflects the increasing influence of epistatic, epigenetic, and environmental risk factors through development. Biomarkers are measurable obligate steps that development has to pass through. They are bottlenecks under the control of epigenetic or environmental factors (the term “endophenotype” refers to a biomarker that is an immediate determinant consequence of a gene or environmental event that triggers disease development). By understanding the factors regulating these bottlenecks we can potentially stop or strongly curtail disease development—the classic example in psychiatry being phenylketonuria (Diamond 1996). As a result, biomarkers are key to screening and also preventing disease, as well as personalizing treatment not to a patient's symptoms but to the developmental biology of those symptoms (Gottesman and Gould 2003).
If we are concerned with preventative and personalized medicine, then we clearly need animal models in which we can discover and follow biomarkers that predict onset, severity, or treatment response. In addition, models based on biomarkers resolve many of the problems discussed above. At its simplest, if a model is based on human biomarkers, then we no longer have to worry about whether a measure is “-like,” and, if that biomarker is a druggable target, then we have a direct readout of predictive efficacy. Two important caveats should be mentioned. First, the word “biomarker” is widely misused to mean any clinical sign (often biochemical) correlated with disease, in contrast to the strong causal definition of a biomarker in the original literature (Gottesman and Gould 2003; Gould and Gottesman 2006). Such correlational “biomarkers” may still be useful for model validation, or as measures of treatment response, or predictors of outcome. However, they are unlikely to be targets (other than for purely symptomatic treatment). Instead, true causal biomarkers are much more likely to be targets that can block disease pathogenesis itself. Second, reverse-translating biomarkers from humans to animals allows us to directly test the causality of a human biomarker. For instance, the limited causality seen in transgenic mouse models of Alzheimer's disease has been critical in rethinking the pathogenesis of the disease (Kokjohn and Roher 2009; Sabbagh et al. 2013; Zahs and Ashe 2010). Thus these caveats are, if anything, arguments for the power of the rigorous application of the original causative definition of a biomarker. Accordingly, several authors have argued for biomarker-based, spontaneous, or reverse-translated models (which are essentially synonymous) as solutions to the issue of predictive validity (e.g., Garner et al. 2011; Gould and Gottesman 2006; Insel 2007; Malkesman et al. 2009; Tricklebank and Garner 2012; Zahs and Ashe 2010). In some cases, this calls for a new model, but in reality it calls for a new approach to animal modeling (Insel 2007). As this review has emphasized, such a new approach can often involve repurposing existing models or technologies. Simply put, biomarkers emphasize that the “new medicine,” and hence the new kind of model, will be about the process of disease, not the pattern of symptoms.
What Does Modeling Process Rather Than Pattern Entail?
Spontaneous and Variable Models
At the most basic, if we want to study individual differences in disease, then we need models where individuals vary in severity or incidence, so that biomarkers can be manipulated. Existing spontaneous models (where animals are not treated, but naturally occurring individual differences are studied) provide a pool of candidates, such as the Ossabaw pig model of metabolic syndrome (Dyson et al. 2006), or the use of stereotypies in captive animals as a model of stereotypies in autism (Garner and Mason 2002; Garner et al. 2003, 2011; Lewis et al. 2006). This represents a fundamental shift from the conventional wisdom in induced models (i.e., a treatment induces the model versus control animals), where variability between animals is viewed as a nuisance to be controlled. If individual variability is the red meat of human medicine, then we must embrace and study the variability in animal models.
This point underlines the difference between a tool and a model discussed above. For instance, Hoxb8 mutant mice show 100% penetrance: all animals pull hair, both male and female (Greer and Capecchi 2002). Thus one of the most salient piece of disease biology in trichotillomania and in hair pulling in other animals (the strong female bias) (Dufour and Garner 2010) cannot be studied. Indeed, the biomarker concept can be viewed as fundamentally opposed to reductionist determinism. If the real nature of disease is the modulation of molecular events by the environment, then the further along disease development progresses the less determinant it becomes. Thus a good model will be one where genetics confers risk, not certainty, and the modulators of that risk can be studied.
Reverse Translation
Just because a model is spontaneous, does not mean that it is valid. For spontaneous models to be useful we need to be able to measure human biomarkers, using the same methodology, in the model; and novel biomarkers discovered in the model should be apparent in human patients (if they are not then the development of the disease and the human is critically different). This requirement actually leads to one of the most powerful features of a biomarker approach to modeling. It allows for rapid validation of the model, and for validation of findings from the model, without having to commit to a traditional drug development pipeline (Tricklebank and Garner 2012). Thus the first step is “reverse-translation”—that is, taking from humans highly specific biomarkers that are known to have clinical implications (in terms of disease development, prognosis, diagnosis, or drug response) and adapting them to animals. Insulin insensitivity in spontaneous models of metabolic syndrome is a simple example. In fact, psychiatry is replete with candidates, given the extensive literature tying particular symptoms to specific neuropsychological measures (and the RDoCs represent an evolution of this approach), which can often be reverse-translated as maze or operant tasks. For instance, neuropsychological measures of frontal cortex function that predict the particular kinds of repetitive behavior across diagnoses as broad as autism, schizophrenia, OCD, and trichotillomania (Garner 2006) have been reverse-translated for many species (Birrell and Brown 2000; Dias et al. 1996; Garner et al. 2006) and accordingly distinguish between the different kinds of repetitive behavior in mice (Garner et al. 2011). Other well-established reverse-translated paradigms include cognitive bias (relevant to depression) (Harding et al. 2004) and delay discounting (relevant to addiction and ADHD) (Oberlin and Grahame 2008).
Reverse-translated tasks have an inherent construct validity. However, physiological, behavioral, and psychological differences between animals and humans can always still impact the predictive validity of a model (e.g., Zahs and Ashe 2010). Thus the validity of reverse-translated tasks or biomarkers should still be tested empirically. For instance, reverse-translated biomarkers should be specific predictors of the same symptoms in animals as in humans and distinguish between similar symptoms (Garner et al. 2011). They should respond to the same manipulations or drugs, again showing both sensitivity and specificity (Helms et al. 2006); and mice bred or engineered for a particular symptomatic state should also show changes in the biomarkers characteristic of at-risk human populations (Oberlin and Grahame 2008). They can also be validated using known failures (see below).
Adopting Human Clinical Trial Designs
As discussed above, we need to use experimental designs that study individual variability, rather than control it. Variability is inescapable, we can try to control it, but this is a losing proposition. For instance, mice vary systematically in their level of anxiety, abnormal behavior, and immune suppression according to their position in the cage rack (Ader et al. 1991; Garner et al. 2004a). In fact, female non-obese diabetic (NOD) mice are sufficiently anxious and immune suppressed that they show a delay in the onset of Type I (autoimmune) diabetes when housed higher on the cage rack (Ader et al. 1991). Some profound effects on experimental outcomes simply cannot be controlled—for instance, the identity of the experimenter (Chesler et al. 2002).
Furthermore, when an experiment is controlled to an arbitrary narrow environment, we lose the ability to infer anything about the generality of the result (Festing 2014; Fisher 1935; Würbel 2000), and the risk of false discoveries increases by an order of magnitude (Richter et al. 2009) (this point is developed in the first example below). The alternative approach is to measure sources of variability and to control for them in the statistical analysis (e.g., including control variables, perhaps for motivational level or general learning ability, measured elsewhere in the task). We can also adapt the classic solution in human trials of sampling across a diverse population and “matching” placebo- and drug-treated individuals (for instance), which is a special case of a “randomized block design” (Würbel and Garner 2007). In epidemiology, this concept is called stratifying (Woodward 1999). In agricultural statistics, it is called blocking (i.e., the subplots within one field are matched; Grafen and Hails 2002). In all cases, the idea is the same—we don't even need to know how or why pairs (or groups) of individuals are similar, we just need to put together pairs of individuals we expect to be more similar to each other than to other individuals (Festing 2014; Grafen and Hails 2002). In the case of mice, so many factors cluster on cage that we can treat cage as our matched pair (or block), if the treatment is applied to individual mice; or to adjacent pairs of cages on the rack if the treatment is applied to the cage (Würbel and Garner 2007). The impact is profound, reducing sample size by up to two orders of magnitude (see Figure 4). Yet the real surprise is not that a simple solution exists, but that such a fundamental cornerstone of experimental design in every other area of biological research is bizarrely absent from preclinical and basic work in animal models (Festing 2014).
Figure 3.

The biomarker model of disease development. As disease develops, physiological measures and ultimate symptomatology become increasingly complex and less determinant, reflecting the influence of genetic and environmental interactions with the underlying genotype (or original cause of the disease). Development may diverge and terminate before true disease onsets (yielding subclinical traits) or may diverge into closely related disorders. Biomarkers are bottlenecks in disease development, which can be measured, and ideally can also be manipulated by their environmental risk factors. Entirely determinant biomarkers of a genotype (or other immediate cause of disease), are called endophenotypes (Garner et al. 2011; Gottesman and Gould 2003; Gould and Gottesman 2006).
Figure 4.

The fallacy of arguing that “human designs” (i.e., controlled heterogenized designs) are less powerful than traditional neuroscience methods. Using the observed variance components in the data from a complex reverse-translated paradigm (Garner et al. 2006), the impact of different analytic approaches are simulated at different sample sizes, and the resulting p values plotted. The analysis can block (match) by Cage (C+), it can optionally include Sex if this variable is of experimental interest (S+), and Internal controls can be included (I+). Thus C+S+I+ is equivalent to a human matched pairs design with statistical (rather than experimental) matching for an internal control such as IQ. C-S-I- is a naïve t-test. (A) The observed effect size of +33.2%. (B-D) Simulated effect sizes of +10%, +20%, and +40% are shown. Note that the sample size required to achieve significance with the human design is extremely stable, essentially as a result of Mead's rule (Mead 1988). Note that blocking for biologically quantifiable (I+) sources of error, and unquantifiable but assignable (C+), are both required to maximize power. Note that for C+ analyses (where cage is nested in Sex), S+ and S- analyses produce identical results (as shown by the overlay of the dashed and solid lines of each color, and as would be expected from the partitioning of variance in a nested design). For C- analyses, including Sex in the analysis provides a slight benefit (reflecting the minimal impact of Sex on this particular data set). Thus an additional advantage of including Cage in particular is that it accounts for variables such as Sex, without having to worry about the magnitude of their impact on the data (Würbel and Garner 2007).
Enrichment and Home Cage Measures
We need models where the biological and environmental “background” on which the disease process develops is like humans. Animals are fundamentally designed to control stressors that they care about. Indeed, animals that cannot control (through behavior or physiology) even innocuous stressors can show catastrophic changes in biology (Weiss 1971). Thus we can define stress as the state in which an animal can safely control a stressor, and distress as the state in which an animal can control a stressor only by negatively impacting another biological system (Moberg 2000). For instance, if mice find standard housing conditions aversively cold (Gaskill et al. 2009), they can use nesting material to control this stressor (Gaskill et al. 2012, 2013a); and, without nesting material, they are demonstrably distressed as their reproductive output suffers (Gaskill et al. 2013b, 2013c). Thus the fundamental argument for enrichment is that an animal is actually abnormal without it.
We do not have to look far to find evidence that this might affect experimental outcomes. For instance, social housing has profound impacts on cancer models. Rats housed singly have massively increased risk and severity of mammary tumors (Hermes et al. 2009). Even in socially housed rats, those with stronger social relationships live longer and have smaller tumor burdens (Yee et al. 2008), while mice housed in groups are more responsive to chemotherapy (Kerr et al. 1997). But these effects are not confounds—they actually mimic the profound impact that social support has on disease progression in humans (Hermes et al. 2009; Kerr et al. 1997; Kroenke et al. 2006; Yee et al. 2008). Nonsocial enrichment also has profound effects—again enriched mice show lower mortality and slower tumor growth than standard housed mice, specifically because standard housed animals are sufficiently stressed to be immune suppressed (Cao et al. 2010). Indeed, the serum from enriched animals is able to suppress cancer cell growth in vitro (Cao et al. 2010). Thus perhaps a fundamental contributor to the poor predictive validity of cancer models (Begley and Ellis 2012; Mak et al. 2014) is not that the biology of the disease is different in humans and animals, but that the psychology is. In other words, animals in barren, uncontrollable environments are models of chronically stressed, socially isolated, and immune-suppressed humans; but, if we want good models of most human cancer patients receiving physical and social supportive care, then we need to think carefully about the social and physical enrichment of these animals.
Similarly, in neuroscience, we can consider the role that stressors may play in changing results. The most obvious stressor is handling by the experimenter themselves. For instance, the identity of the experimenter is a much better predictor of tail-flick pain sensitivity than the strain (i.e., genetics) of the mouse (Chesler et al. 2002); to the point that the mere odor of male experimenters is enough to cause a stress-induced analgesic effect in mice (Sorge et al. 2014). Similarly, handling affects performance in the EPM so profoundly that it completely corrupts the ability of the task to detect drug effects. Rats naïve to handling can show an effect of anxiolytic drugs, but not anxiogenic drugs, while rats habituated to handling show an effect of anxiogenic drugs, but not anxiolytic drugs (Andrews and File 1993). As most phenotyping tasks involve extensive handling, it is hard to see how the impact of this stressor can be eliminated. Again, the best solution may be to add a new round of specific measures taken in undisturbed animals in their home cage. Ethological observation (see www.mousebehavior.org for a guide to mouse ethology) is an obvious first step—for example, the use of spontaneous stereotypies as models of stereotypies in autism (Garner et al. 2011; Lewis et al. 2006). More complex cognitive tasks can be implemented using automated apparatuses, such as touchscreens for primates (Dias et al. 1996) or automated mazes attached to the home cage for rodents (Pioli et al. 2014). (Indeed, in this latter example, the automated maze was much more robust than the hand-run version of the same task in the same mice.)
Validating Animal Discoveries Without a Human Drug Trial
Biomarkers allow several strategies for making quick go/no-go decisions early in the pipeline (and thus to shift attrition to less economically and ethically less costly stages). Again, the idea is to introduce a specific biomarker-based stage to make early no-go decisions after the initial highly sensitive phenotyping stage identifies a potential compound. First, because reverse-translated biomarkers (e.g., insulin resistance) are direct measures of the mechanism of disease, a novel drug should affect the biomarker as well as other phenotypes in the model. Drugs that affect phenotypes without affecting biomarkers may not be treating the underlying disease and thus would be less likely to translate to humans, and can be abandoned earlier in the pipeline. Second, reverse-translated biomarkers allow us to follow disease development in animals from conception to death (while this is rarely the case in humans) and identify earlier novel biomarkers that lead to the known human (i.e., reverse-translated) biomarkers. These novel biomarkers not only provide druggable targets but also allow for rapid validation. Instead of performing a clinical trial, a short study can test for novel biomarkers found in the mouse and, if they are not found in humans, then a quick no-go decision can be made for compounds targeting these biomarkers early in the pipeline (Tricklebank and Garner 2012).
Validating Changes in Practice with Known Failures
Finally, perhaps the most important validation that can be performed is to compare a specific reverse-translated measure against a sensitive phenotyping measure, and to test whether they can correctly identify placebo treatment, treatment with a known effective drug, and treatment with a known failed drug. The specific test should correctly label the known ineffective drug as a negative, while the phenotyping measure should identify it falsely as a positive. Both should identify the known effective drug; and both should identify the control. This simple experiment resolves the “better the devil we know” catch-22, that it is completely unrealistic to expect industry or academia to adopt a new method or model that cannot be empirically justified (by a successful translation) until years down the pipeline in human trials. In fact, this strategy can be used to test the effectiveness of any of the changes in practice discussed above (for instance, we could test whether enriched mice would have correctly identified recent failures in cancer drug discovery).
Example 1: The Power of Reverse-Translated Biomarkers in a “Human” Design
The house mouse is an extraordinary animal in one particular aspect. Unlike other commensal species (a commensal lives in a niche created by humans), such as the pigeon, rat, or Rhesus macaque, mice have successfully followed us around the globe and colonized almost every environment humans have ever created (Latham and Mason 2004; Silver 1995). Unlike humans, they have done so without the benefit of language, or tools, or clothing. Instead they rely on phenotypic plasticity—or the ability of the same genome to express different highly adaptive phenotypes in different environments (Miner et al. 2005). Phenotypic plasticity is the ultimate expression of epigenetic regulation, and biomarkers represent the mechanism through which the environment can produce plastic phenotypes. Indeed, this connection is central to a biomarker-based approach to preventative, personalized medicine (Feinberg 2007). Thus mice are arguably the best model of humans because they are so like us in the one way that really matters (i.e., phenotypic plasticity) and because, as they have co-evolved with us, they are likely to respond to similar environments in similar ways, and thus share similar connections between phenotypic plasticity and disease.
When we standardize an experiment by trying to make everything the same (as is typical in basic science), we are in fact making an explicit assumption that phenotypic plasticity does not exist (Richter et al. 2009). For instance, in examining influences on the tail-flick test of pain sensitivity, Chesler and colleagues (2002) found that 27% of variability in response could be attributed to genotype (the treatment), 42% to the test environment, and 18% to phenotypic plasticity. If ignored, the 42% attributable to environment simply adds noise to the experiment (but does not change relative ranking of genotypes). This is the component that can be controlled by standardizing the environment, and thus would increase test sensitivity. However, if we choose one arbitrary environment, then the 18% due to phenotypic plasticity becomes indistinguishable from the true treatment effect, potentially generating a false positive result (i.e., a result only true in one arbitrary environment, not the general population). Thus, standardizing through homogenization not only decreases specificity and increases false discovery from phenotypic plasticity, but the increased sensitivity further adds to the risk of false discovery in general. This phenomenon is referred to as the “standardization fallacy” (Würbel 2000) and was warned against by the father of modern biostatistics (Fisher 1935). Indeed, it is the fundamental reason why almost all other fields of biological research (including human trials) examine a controlled heterogenous population and standardize statistically, as discussed above.
Phenotypic plasticity is clearly widespread in behavioral neuroscience—both when identical experiments are performed in different laboratories (Crabbe et al. 1999; Richter et al. 2009), and even when repeated in the same laboratory (Chesler et al. 2002; Richter et al. 2010). Furthermore, when the impact of phenotypic plasticity and standardization were formally examined by Richter and colleagues (2009), as expected, attempting to standardize experiments through homogenization artificially inflated false discovery rates by nearly an order of magnitude, yet this problem can be avoided by adopting the human designs discussed above (Richter et al. 2009, 2010, 2011). A common objection to these and other papers from Würbel's group is that the cost of these advanced experimental designs is a loss of power (e.g., van der Staay et al. 2010; van der Staay and Steckler 2002). However, this is based on the strange assumption that one would incorrectly analyze such “human designs” with naïve t-tests.
Figure 4 illustrates this fallacy (and the general cost of using naïve univariate tests of any kind). In this figure, data are taken from a complex reverse-translated measure in our own lab (Garner et al. 2006), and experiments of various sizes simulated given the observed variance components in the original data. Figure 4 shows the effects of sample size, effect size, and different analytical models on the observed p value. These data were chosen to illustrate the fact that any lab, and reverse-translated measures, are just as susceptible to phenotypic plasticity. The only difference is how the data are analyzed. Note that they also illustrate the importance of internal controls (which are generally lost in phenotyping measures) and that including planned variability (in this case sex) does not affect power if properly analyzed. The most important point to take away is that human designs are remarkably insensitive to effect size. Whereas a t-test may need thousands of animals to detect a 10% effect size, the required sample size for a properly analyzed experiment barely increases. This reflects the successful partitioning of variance in such designs and is a good example of Mead's rule for estimating power in factorial designs (Mead 1988). Thus the standard counter-argument from traditional neuroscience is a straw man—a properly analyzed human design is inherently more powerful and more specific than a naïve phenotyping and t-test approach.
Example 2: A Biomarker-Based, Reverse-Translated Model
To illustrate the application of the ideas in this review, our work developing barbering in mice as a model of trichotillomania is briefly reviewed. Trichotillomania, or compulsive hair pulling, affects between 3% and 4% of women (Christenson and Mansueto 1999; Christenson et al. 1991), making it one of the most common disorders in women. Hair pulling in animals is also extremely common, and in mice the behavior is called “barbering” (Dufour and Garner 2010). We first used behavioral epidemiology to identify risk factors for barbering and to test for similarities to trichotillomania (specifically, a female bias, onset with sexual maturity, exacerbation by reproductive events, and exacerbation by stress) (Garner et al. 2004a, 2004b). This pattern of risk factors limits the range of potential human disorders to a small handful. We also excluded alternative explanations such as any connection to social dominance (Garner et al. 2004a). We then reverse translated neuropsychological biomarkers that distinguish between different repetitive behavior disorders in humans and specifically tested for biomarkers of OCD and autism, finding a pattern of biomarkers that matches only trichotillomania (Garner et al. 2011). Having validated the model, we tested a neutraceutical treatment mimicking the action of a selective serotonin reuptake inhibitor (SSRI), which in fact increased both prevalence and severity of the behavior (Dufour et al. 2010). We followed development through puberty, identifying a number of predictive biomarkers (Hess et al. 2008), all of which were pointing to a central role of metabolically derived oxidative stress. Meanwhile, Grant and colleagues (2009) reported that N-acetyl-cysteine (NAC), a compound with minimal psychoactive properties, alleviates symptoms in nearly 60% of patients. NAC, is the rate-limiting precursor to the brain's defense against oxidative stress. Accordingly, we found 10-fold increases in biomarkers of oxidative stress in barbering mice (Vieira et al. 2011) and have shown that NAC can both prevent and cure barbering in mice (Vieira et al. 2013). Thus, adopting a biomarker-based approach allowed us to validate a model, identify the underlying disease process, identify predictive biomarkers that could be used to screen young girls, and identify an innocuous neutraceutical preventative treatment.
The next step is to implement exactly the no-go decision-making step that was argued for at the start of the review. Now that the mouse model has identified novel biomarkers that are themselves druggable targets, we can go back to humans and test for these novel biomarkers. If present, we have identified a key part of the disease biology in humans; if not, then we (like most animal models) will have only modeled the model. Either answer is valuable.
Conclusions
The central argument of this review is that, if we want animal models to translate to human outcomes, then we need to start performing animal experiments as if they were human trials. A variety of pitfalls in current status quo methodology were emphasized, and key elements of a biomarker-based approach to animal models were emphasized. At the end of the day, the real challenge is to persuade researchers to adopt new methodologies on the basis of a leap of faith that doing so will improve human outcomes over a decade in the future. Unsurprisingly, few researchers have been willing to take such a risk. Therefore the most important idea in this review is that of “validation using known failures.” Every potential refinement to current practice (from enrichment, to human experimental design, to reverse-translated measures) can be empirically assessed by testing whether they would have correctly identified a failed compound as a true negative in animals, when traditional methods generate a false positive. Aggressively testing for ways to improve human translation is arguably one of the most important things the biomedical research community needs to do. For every moment we delay, patients will continue to suffer, and unimaginable amounts of money and animals are needlessly wasted.
Acknowledgments
Many of the ideas in this review developed during discussion with many colleagues, in particular: Hanno Würbel, Mark Tricklebank, Jeffrey Alberts, Michael Festing (not least for pointing out the connection to Fisher's original writings), Karen Parker, Amy Lossie, and Edmond Pajor. This work was supported in part by the National Institute Of Neurological Disorders And Stroke of the National Institutes of Health under Award Number R21NS088841R21NS088841. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- Abelson JF, Kwan KY, O'Roak BJ, Baek DY, Stillman AA, Morgan TM, Mathews CA, Pauls DL, Rašin M-R, Gunel M, Davis NR, Ercan-Sencicek AG, Guez DH, Spertus JA, Leckman JF, Dure LS, Kurlan R, Singer HS, Gilbert DL, Farhi A, Louvi A, Lifton RP, Šestan N, State MW. Sequence variants in SLITRK1 are associated with Tourette's Syndrome. Science. 2005;310:317–320. doi: 10.1126/science.1116502. [DOI] [PubMed] [Google Scholar]
- Ader DN, Johnson SB, Huang SW, Riley WJ. Group-size, cage shelf level, and emotionality in nonobese diabetic mice - Impact on onset and incidence of IDDM. Psychosom Med. 1991;53:313–321. doi: 10.1097/00006842-199105000-00005. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders: DSM-5. Washington DC: American Psychiatric Association; 2013. [Google Scholar]
- Andreatini R, Bacellar LFS. Animal models: Trait or state measure? The test-retest reliability of the elevated plus-maze and behavioral despair. Prog Neuropsychopharmacol Biol Psychiatry. 2000;24:549–560. doi: 10.1016/s0278-5846(00)00092-0. [DOI] [PubMed] [Google Scholar]
- Andrews N, File SE. Handling history of rats modified behavioural effects of drugs in the elevated plus-maze test of anxiety. Eur J Pharmacol. 1993;235:109–112. doi: 10.1016/0014-2999(93)90827-5. [DOI] [PubMed] [Google Scholar]
- Archer J. Tests for emotionality in rats and mice - Review. Anim Behav. 1973;21:205–235. doi: 10.1016/s0003-3472(73)80065-x. [DOI] [PubMed] [Google Scholar]
- Arguello PA, Gogos JA. Modeling madness in mice: One piece at a time. Neuron. 2006;52:179–196. doi: 10.1016/j.neuron.2006.09.023. [DOI] [PubMed] [Google Scholar]
- Begley CG. Six red flags for suspect work. Nature. 2013;497:433–434. doi: 10.1038/497433a. [DOI] [PubMed] [Google Scholar]
- Begley CG, Ellis LM. Drug development: Raise standards for preclinical cancer research. Nature. 2012;483:531–533. doi: 10.1038/483531a. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Annals Stat. 2001;29:1165–1168. [Google Scholar]
- Binder E, Droste SK, Ohl F, Reul JMHM. Regular voluntary exercise reduces anxiety-related behaviour and impulsiveness in mice. Behav Brain Res. 2004;155:197–206. doi: 10.1016/j.bbr.2004.04.017. [DOI] [PubMed] [Google Scholar]
- Birrell JM, Brown VJ. Medial frontal cortex mediates perceptual attentional set shifting in the rat. J Neurosci. 2000;20:4320–4324. doi: 10.1523/JNEUROSCI.20-11-04320.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RE, Wong AA. The influence of visual ability on learning and memory performance in 13 strains of mice. Learn Memory. 2007;14:134–144. doi: 10.1101/lm.473907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DT, Fiske DW. Convergent and discriminant validation by the multitrait-multimethod matrix. Psychol Bull. 1959;56:81–105. [PubMed] [Google Scholar]
- Cao L, Liu XL, Lin EJD, Wang CS, Choi EY, Riban V, Lin B, During MJ. Environmental and genetic activation of a brain-adipocyte BDNF/Leptin axis causes cancer remission and inhibition. Cell. 2010;142:52–64. doi: 10.1016/j.cell.2010.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SK, Tvrdik P, Peden E, Cho S, Wu S, Spangrude G, Capecchi MR. Hematopoietic origin of pathological grooming in Hoxb8 mutant mice. Cell. 2010;141:775–785. doi: 10.1016/j.cell.2010.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler EJ, Wilson SG, Lariviere WR, Rodriguez-Zas SL, Mogil JS. Identification and ranking of genetic and laboratory environment factors influencing a behavioral trait, thermal nociception, via computational analysis of a large data archive. Neurosci Biobehav Rev. 2002;26:907–923. doi: 10.1016/s0149-7634(02)00103-3. [DOI] [PubMed] [Google Scholar]
- Christenson GA, Mansueto CS. Trichotillomania: Descriptive characteristics and phenomenology. In: Stein DJ, Christenson GA, Hollander E, editors. Trichotillomania. Washington, DC: American Psychiatric Press, Inc; 1999. pp. 1–41. [Google Scholar]
- Christenson GA, Pyle RL, Mitchell JE. Estimated Lifetime Prevalence of Trichotillomania in College-Students. J Clin Psychiatr. 1991;52:415–417. [PubMed] [Google Scholar]
- Crabbe JC, Wahlsten D, Dudek BC. Genetics of mouse behavior: interactions with laboratory environment. Science. 1999;284:1670–1672. doi: 10.1126/science.284.5420.1670. [DOI] [PubMed] [Google Scholar]
- Crusio WE. Flanking gene and genetic background problems in genetically manipulated mice. Biol Psychiatry. 2004;56:381–385. doi: 10.1016/j.biopsych.2003.12.026. [DOI] [PubMed] [Google Scholar]
- Crusio WE, Goldowitz D, Holmes A, Wolfer D. Standards for the publication of mouse mutant studies. Genes Brain Behav. 2009;8:1–4. doi: 10.1111/j.1601-183X.2008.00438.x. [DOI] [PubMed] [Google Scholar]
- Cummings J, Morstorf T, Zhong K. Alzheimer's disease drug-development pipeline: few candidates, frequent failures. Alzheimer's Res Ther. 2014;6:37. doi: 10.1186/alzrt269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond A. Evidence for the importance of dopamine for prefrontal cortex functions early in life. Philos Trans R Soc Lond B Biol Sci. 1996;351:1483–1493. doi: 10.1098/rstb.1996.0134. discussion 1494. [DOI] [PubMed] [Google Scholar]
- Dias R, Robbins TW, Roberts AC. Dissociation in prefrontal cortex of affective and attentional shifts. Nature. 1996;380:69–72. doi: 10.1038/380069a0. [DOI] [PubMed] [Google Scholar]
- Dufour BD, Adeola O, Cheng HW, Donkin SS, Klein JD, Pajor EA, Garner JP. Nutritional up-regulation of serotonin paradoxically induces compulsive behavior. Nutr Neurosci. 2010;13:256–264. doi: 10.1179/147683010X12611460764688. [DOI] [PubMed] [Google Scholar]
- Dufour BD, Garner JP. An ethological analysis of barbering behavior. In: Kalueff A, Bergner C, LaPorte J, editors. Neurobiology of Grooming Behavior. Cambridge, UK: Cambridge University Press; 2010. [Google Scholar]
- Dyson MC, Alloosh M, Vuchetich JP, Mokelke EA, Sturek M. Components of metabolic syndrome and coronary artery disease in female Ossabaw swine fed excess atherogenic diet. 2006. pp. 35–45. Comp Med 56. [PubMed]
- Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- Festing MFW. Randomized block experimental designs can increase the power and reproducibility of laboratory animal experiments. ILAR J. 2014;55:472–476. doi: 10.1093/ilar/ilu045. [DOI] [PubMed] [Google Scholar]
- Fisher RA. Edinburgh, London: Oliver and Boyde; 1935. The design of experiments. [Google Scholar]
- Garner JP. Ch 5. Perseveration and stereotypy - systems-level insights from clinical psychology. In: Rushen J, Mason G, editors. Stereotypic Animal Behaviour: Fundamentals and Applications to Welfare. 2nd ed. Wallingford, England, UK: CABI; 2006. pp. 121–152. [Google Scholar]
- Garner JP, Dufour B, Gregg LE, Weisker SM, Mench JA. Social and husbandry factors affecting the prevalence and severity of barbering (‘whisker trimming’) by laboratory mice. Appl Anim Behav Sci. 2004a;89:263–282. [Google Scholar]
- Garner JP, Mason GJ. Evidence for a relationship between cage stereotypies and behavioural disinhibition in laboratory rodents. Behav Brain Res. 2002;136:83–92. doi: 10.1016/s0166-4328(02)00111-0. [DOI] [PubMed] [Google Scholar]
- Garner JP, Meehan CL, Mench JA. Stereotypies in caged parrots, schizophrenia and autism: evidence for a common mechanism. Behav Brain Res. 2003;145:125–134. doi: 10.1016/s0166-4328(03)00115-3. [DOI] [PubMed] [Google Scholar]
- Garner JP, Thogerson CM, DuFour B, Würbel H, Murray JD, Mench JA. Reverse-translational biomarker validation of abnormal repetitive behaviors in mice: an illustration of the 4Ps modeling approach. Behav Brain Res. 2011;219:189–196. doi: 10.1016/j.bbr.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner JP, Thogerson CM, Würbel H, Murray JD, Mench JA. Animal neuropsychology: Validation of the intra-dimensional extra-dimensional set shifting task in mice. Behav Brain Res. 2006;173:53–61. doi: 10.1016/j.bbr.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Garner JP, Weisker SM, Dufour B, Mench JA. Barbering (fur and whisker trimming) by laboratory mice as a model of human trichotillomania and obsessive-compulsive spectrum disorders. Comp Med. 2004b;54:216–224. [PubMed] [Google Scholar]
- Gaskill BN, Gordon CJ, Pajor EA, Lucas JR, Davis JK, Garner JP. Heat or insulation: Behavioral titration of mouse preference for warmth or access to a nest. PLoS ONE. 2012;7:e32799. doi: 10.1371/journal.pone.0032799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaskill BN, Gordon CJ, Pajor EA, Lucas JR, Davis JK, Garner JP. Impact of nesting material on mouse body temperature and physiology. Physiol Behav. 2013a;110–111:87–95. doi: 10.1016/j.physbeh.2012.12.018. [DOI] [PubMed] [Google Scholar]
- Gaskill BN, Pritchett-Corning KR, Gordon CJ, Pajor EA, Lucas JR, Davis JK, Garner JP. Energy reallocation to breeding performance through improved nest building in laboratory mice. PLoS ONE. 2013b;8:e74153. doi: 10.1371/journal.pone.0074153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaskill BN, Rohr SA, Pajor EA, Lucas JR, Garner JP. Some like it hot: Mouse temperature preferences in laboratory housing. Appl Anim Behav Sci. 2009;116:279–285. [Google Scholar]
- Gaskill BN, Winnicker C, Garner JP, Pritchett-Corning KR. The naked truth: Breeding performance in nude mice with and without nesting material. Appl Anim Behav Sci. 2013c;143:110–116. [Google Scholar]
- Geerts H. Of mice and men: bridging the translational disconnect in CNS drug discovery. CNS Drugs. 2009;23:915–926. doi: 10.2165/11310890-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Gerlai R. Gene-targeting studies of mammalian behavior - Is it the mutation or the background genotype. Trends Neurosci. 1996;19:177–181. doi: 10.1016/s0166-2236(96)20020-7. [DOI] [PubMed] [Google Scholar]
- Gerlai R, Clayton NS. Analysing hippocampal function in transgenic mice: An ethological perspective. Trends Neurosci. 1999;22:47–51. doi: 10.1016/s0166-2236(98)01346-0. [DOI] [PubMed] [Google Scholar]
- Gottesman II, Gould TD. The endophenotype concept in psychiatry: Etymology and strategic intentions. Am J Psychiatry. 2003;160:636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- Gould TD, Gottesman II. Psychiatric endophenotypes and the development of valid animal models. Genes Brain Behav. 2006;5:113–119. doi: 10.1111/j.1601-183X.2005.00186.x. [DOI] [PubMed] [Google Scholar]
- Grafen A, Hails R. Modern Statistics for the Life Sciences. Oxford & New York: Oxford University Press; 2002. [Google Scholar]
- Grant JE, Odlaug BL, Kim SW. N-Acetylcysteine, a glutamate modulator, in the treatment of trichotillomania a double-blind, placebo-controlled study. Arch Gen Psychiatry. 2009;66:756–763. doi: 10.1001/archgenpsychiatry.2009.60. [DOI] [PubMed] [Google Scholar]
- Gray JA. The Psychology of Fear and Stress. Cambridge; New York: Cambridge University Press; 1987. [Google Scholar]
- Greer JM, Capecchi MR. Hoxb8 is required for normal grooming behavior in mice. Neuron. 2002;33:23–34. doi: 10.1016/s0896-6273(01)00564-5. [DOI] [PubMed] [Google Scholar]
- Hånell A, Marklund N. Structured evaluation of rodent behavioral tests used in drug discovery research. 2014. Front Behav Neurosci 8. [DOI] [PMC free article] [PubMed]
- Harding EJ, Paul ES, Mendl M. Animal behavior - Cognitive bias and affective state. Nature. 2004;427:312. doi: 10.1038/427312a. [DOI] [PubMed] [Google Scholar]
- Hay M, Thomas DW, Craighead JL, Economides C, Rosenthal J. Clinical development success rates for investigational drugs. Nat Biotech. 2014;32:40–51. doi: 10.1038/nbt.2786. [DOI] [PubMed] [Google Scholar]
- Helms CM, Reeves JM, Mitchell SH. Impact of strain and D-amphetamine on impulsivity (delay discounting) in inbred mice. Psychopharmacol. 2006;188:144–151. doi: 10.1007/s00213-006-0478-0. [DOI] [PubMed] [Google Scholar]
- Hermes GL, Delgado B, Tretiakova M, Cavigelli SA, Krausz T, Conzen SD, McClintock MK. Social isolation dysregulates endocrine and behavioral stress while increasing malignant burden of spontaneous mammary tumors. Proc Natl Acad Sci U S A. 2009;106:22393–22398. doi: 10.1073/pnas.0910753106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess SE, Lossie AC, Meisel RL, Franklin M, Garner JP. The role of estrogen in the onset and performance of plucking behavior in a mouse model of trichotillomania. 2008. Proceedings of the 38th Annual Meeting of the Society for Neuroscience.
- Hill RA, McInnes KJ, Gong ECH, Jones MEE, Simpson ER, Boon WC. Estrogen deficient male mice develop compulsive behavior. Biol Psychiatry. 2007;61:359–366. doi: 10.1016/j.biopsych.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Holliday J, Tchanturia K, Landau S, Collier D, Treasure J. Is impaired set-shifting an endophenotype of anorexia nervosa? Am J Psychiatry. 2005;162:2269–2275. doi: 10.1176/appi.ajp.162.12.2269. [DOI] [PubMed] [Google Scholar]
- Hossain SM, Wong BKY, Simpson EM. The dark phase improves genetic discrimination for some high throughput mouse behavioral phenotyping. Genes Brain Behav. 2004;3:167–177. doi: 10.1111/j.1601-183x.2004.00069.x. [DOI] [PubMed] [Google Scholar]
- Hunter J. Challenges for pharmaceutical industry: new partnerships for sustainable human health. Philos Trans R Soc Lond A. 2011;369:1817–1825. doi: 10.1098/rsta.2010.0377. [DOI] [PubMed] [Google Scholar]
- Insel TR. From animal models to model animals. Biol Psychiatry. 2007;62:1337–1339. doi: 10.1016/j.biopsych.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Ioannidis JPA. Why most published research findings are false. PLoS Med. 2005;2:696–701. doi: 10.1371/journal.pmed.0020124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr LR, Grimm MS, Silva WA, Weinberg J, Emerman JT. Effects of social housing condition on the response of the Shionogi mouse mammary carcinoma (SC115) to chemotherapy. Cancer Res. 1997;57:1124–1128. [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8:e1000412. doi: 10.1371/journal.pbio.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Parsons N, Kadyszewski E, Festing MFW, Cuthill IC, Fry D, Hutton J, Altman DG. Survey of the quality of experimental design, statistical analysis and reporting of research using animals. PLoS ONE. 2009;4:e7824. doi: 10.1371/journal.pone.0007824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokjohn TA, Roher AE. Amyloid precursor protein transgenic mouse models and Alzheimer's disease: understanding the paradigms, limitations, and contributions. Alzheimer's & dementia : the journal of the Alzheimer's Association. 2009;5:340–347. doi: 10.1016/j.jalz.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004;3:711–716. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- Kroenke CH, Kubzansky LD, Schernhammer ES, Holmes MD, Kawachi I. Social networks, social support, and survival after breast cancer diagnosis. J Clin Oncol. 2006;24:1105–1111. doi: 10.1200/JCO.2005.04.2846. [DOI] [PubMed] [Google Scholar]
- Latham N, Mason G. From house mouse to mouse house: the behavioural biology of free-living Mus musculus and its implications in the laboratory. Appl Anim Behav Sci. 2004;86:261–289. [Google Scholar]
- Lephart ED. A review of brain aromatase cytochrome P450. Brain Research Brain. Res Rev. 1996;22:1–26. [PubMed] [Google Scholar]
- Lewis MH, Presti MF, Lewis KB, Turner CA. Ch 7. The neurobiology of stereotypy I: Environmental complexity. In: Rushen J, Mason G, editors. Stereotypic Animal Behaviour: Fundamentals and Applications to Welfare. 2nd ed. Wallingford, England, UK: CABI; 2006. [Google Scholar]
- Macleod MR, van der Worp HB, Sena ES, Howells DW, Dirnagl U, Donnan GA. Evidence for the efficacy of NXY-059 in experimental focal cerebral ischaemia is confounded by study quality. Stroke. 2008;39:2824–2829. doi: 10.1161/STROKEAHA.108.515957. [DOI] [PubMed] [Google Scholar]
- Mak IW, Evaniew N, Ghert M. Lost in translation: animal models and clinical trials in cancer treatment. Am J Translat Res. 2014;6:114–118. [PMC free article] [PubMed] [Google Scholar]
- Malkesman O, Austin DR, Chen G, Manji HK. Reverse translational strategies for developing animal models of bipolar disorder. Dis Models Mech. 2009;2:238–245. doi: 10.1242/dmm.001628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin P, Bateson P. Measuring Behaviour: An Introductory Guide. Cambridge, England UK: Cambridge University Press; 1986. [Google Scholar]
- McManus R. Ex-Director Zerhouni Surveys Value of NIH Research. NIH Record. 2013;LXV:1. ,6,5. [Google Scholar]
- Mead R. The design of experiments : statistical principles for practical applications. Cambridge UK; New York: Cambridge University Press; 1988. [Google Scholar]
- Miller KA, Garner JP, Mench JA. Is fearfulness a trait that can be measured with behavioural tests? A validation of four fear tests for Japanese quail. Anim Behav. 2006;71:1323–1334. [Google Scholar]
- Miner BG, Sultan SE, Morgan SG, Padilla DK, Relyea RA. Ecological consequences of phenotypic plasticity. Trends Ecol Evol. 2005;20:685–692. doi: 10.1016/j.tree.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Moberg GP. Biological response to stress: implications for animal welfare. In: Moberg GP, Mench JA, editors. The biology of animal stress: basic principles and implications for animal welfare. Wallingford, UK: CABI; 2000. pp. 1–22. [Google Scholar]
- Moy SS, Nadler JJ, Perez A, Barbaro RP, Johns JM, Magnuson TR, Piven J, Crawley JN. Sociability and preference for social novelty in five inbred strains: an approach to assess autistic-like behavior in mice. Genes Brain Behav. 2004;3:287–302. doi: 10.1111/j.1601-1848.2004.00076.x. [DOI] [PubMed] [Google Scholar]
- Muhlhausler BS, Bloomfield FH, Gillman MW. Whole Animal Experiments Should Be More Like Human Randomized Controlled Trials. PLoS Biol. 2013;11:e1001481. doi: 10.1371/journal.pbio.1001481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadler JJ, Moy SS, Dold G, DT, Simmons N, Perez A, Young NB, Barbaro RP, Piven J, Magnuson TR, Crawley JN. Automated apparatus for quantitation of social approach behaviors in mice. Genes Brain Behav. 2004;3:303–314. doi: 10.1111/j.1601-183X.2004.00071.x. [DOI] [PubMed] [Google Scholar]
- National Institute of Mental Health. The National Institute of Mental Health Strategic Plan. 2008. Available online (www.nimh.nih.gov/about/strategic-planning-reports/index.shtml. ).
- Neter J, Kutner MH, Nachtsheim CJ, Wasserman W. Applied Linear Statistical Models. Chicago: Irwin; 1996. [Google Scholar]
- Nieuwenhuis S, Forstmann BU, Wagenmakers E-J. Erroneous analyses of interactions in neuroscience: a problem of significance. Nat Neurosci. 2011;14:1105–1107. doi: 10.1038/nn.2886. [DOI] [PubMed] [Google Scholar]
- Oberlin BG, Grahame NJ. Selection for high alcohol preference in mice reliably causes greater impulsivity during a delay discounting task. Alcohol Clin Exp Res. 2008;32:16A. [Google Scholar]
- Paul SM, Mytelka DS, Dunwiddie CT, Persinger CC, Munos BH, Lindborg SR, Schacht AL. How to improve R&D productivity: the pharmaceutical industry's grand challenge. Nat Rev Drug Discov. 2010;9:203–214. doi: 10.1038/nrd3078. [DOI] [PubMed] [Google Scholar]
- Peers IS, Ceuppens PR, Harbron C. In search of preclinical robustness. Nat Rev Drug Discov. 2012;11:733–734. doi: 10.1038/nrd3849. [DOI] [PubMed] [Google Scholar]
- Pioli EY, Gaskill BN, Gilmour G, Tricklebank MD, Dix SL, Bannerman D, Garner JP. An automated maze task for assessing hippocampus-sensitive memory in mice. Behav Brain Res. 2014;261:249–257. doi: 10.1016/j.bbr.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz F, Schlange T, Asadullah K. Believe it or not: how much can we rely on published data on potential drug targets? Nat Rev Drug Discov. 2011;10:712. doi: 10.1038/nrd3439-c1. [DOI] [PubMed] [Google Scholar]
- Pusztai L, Hatzis C, Andre F. Reproducibility of research and preclinical validation: problems and solutions. Nat Rev Clin Oncol. 2013;10:720–724. doi: 10.1038/nrclinonc.2013.171. [DOI] [PubMed] [Google Scholar]
- Richter SH, Garner JP, Auer C, Kunert J, Wurbel H. Systematic variation improves reproducibility of animal experiments. Nat Methods. 2010;7:167–168. doi: 10.1038/nmeth0310-167. [DOI] [PubMed] [Google Scholar]
- Richter SH, Garner JP, Wurbel H. Environmental standardization: cure or cause of poor reproducibility in animal experiments? Nat Methods. 2009;6:257–261. doi: 10.1038/nmeth.1312. [DOI] [PubMed] [Google Scholar]
- Richter SH, Garner JP, Zipser B, Lewejohann L, Sachser N, Touma C, Schindler B, Chourbaji S, Brandwein C, Gass P, van Stipdonk N, van der Harst J, Spruijt B, Võikar V, Wolfer DP, Würbel H. Effect of population heterogenization on the reproducibility of mouse behavior: A multi-laboratory study. PLoS ONE. 2011;6:e16461. doi: 10.1371/journal.pone.0016461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollin BE. Science and Ethics. Cambridge; New York: Cambridge University Press; 2006. [Google Scholar]
- Sabbagh JJ, Kinney JW, Cummings JL. Animal systems in the development of treatments for Alzheimer's disease: challenges, methods, and implications. Neurobiol Aging. 2013;34:169–183. doi: 10.1016/j.neurobiolaging.2012.02.027. [DOI] [PubMed] [Google Scholar]
- Sena ES, van der Worp HB, Bath PMW, Howells DW, Macleod MR. Publication bias in reports of animal stroke studies leads to major overstatement of efficacy. 2010. p. e1000344. PLoS Biol 8. [DOI] [PMC free article] [PubMed]
- Shmelkov SV, Hormigo A, Jing D, Proenca CC, Bath KG, Milde T, Shmelkov E, Kushner JS, Baljevic M, Dincheva I, Murphy AJ, Valenzuela DM, Gale NW, Yancopoulos GD, Ninan I, Lee FS, Rafii S. Slitrk5 deficiency impairs corticostriatal circuitry and leads to obsessive-compulsive-like behaviors in mice. Nat Med. 2010;16:598–602. doi: 10.1038/nm.2125. 591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver LM. Mouse genetics: concepts and applications. New York: Oxford University Press; 1995. [Google Scholar]
- Sorge RE, Martin LJ, Isbester KA, Sotocinal SG, Rosen S, Tuttle AH, Wieskopf JS, Acland EL, Dokova A, Kadoura B, Leger P, Mapplebeck JC, McPhail M, Delaney A, Wigerblad G, Schumann AP, Quinn T, Frasnelli J, Svensson CI, Sternberg WF, Mogil JS. Olfactory exposure to males, including men, causes stress and related analgesia in rodents. Nat Methods. 2014;11:629–632. doi: 10.1038/nmeth.2935. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolosa E, Compta Y, Gaig C. The premotor phase of Parkinson's disease. Parkinsonism Rel Disord. 2007;13:S2–S7. doi: 10.1016/j.parkreldis.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Tricklebank MD, Garner JP. The possibilities and limitations of animal models for psychiatric disorders. In: Rankovic Z, Bingham M, Nestler EJ, Hargreaves R, editors. Drug Discovery for Psychiatric Disorders. The Royal Society of Chemistry. Vol 28. 2012. pp. 534–556. [Google Scholar]
- Vaidya D, Morley GE, Samie FH, Jalife J. Reentry and fibrillation in the mouse heart: A challenge to the critical mass hypothesis. Circul Res. 1999;85:174–181. doi: 10.1161/01.res.85.2.174. [DOI] [PubMed] [Google Scholar]
- Van de Weerd HA, Vandenbroek FAR, Beynen AC. Removal of vibrissae in male mice does not influence social dominance. Behav Processes. 1992;27:205–208. doi: 10.1016/0376-6357(92)90177-F. [DOI] [PubMed] [Google Scholar]
- van der Staay FJ, Arndt SS, Nordquist RE. The standardization-generalization dilemma: a way out. Genes Brain Behav. 2010;9:849–855. doi: 10.1111/j.1601-183X.2010.00628.x. [DOI] [PubMed] [Google Scholar]
- van der Staay FJ, Steckler T. The fallacy of behavioral phenotyping without standardisation. Genes Brain Behav. 2002;1:9–13. doi: 10.1046/j.1601-1848.2001.00007.x. [DOI] [PubMed] [Google Scholar]
- van der Worp HB, Howells DW, Sena ES, Porritt MJ, Rewell S, O'Collins V, Macleod MR. Can Animal Models of Disease Reliably Inform Human Studies? PLoS Med. 2010;7:e1000245. doi: 10.1371/journal.pmed.1000245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira G, Lossie AC, Ajuwon K, Garner JP. Is hair and feather pulling a disease of oxidative stress? 2011. p. 57. Proceedings of the 45th International Congress of the International Society for Applied Ethology, Indianapolis, IN, USA.
- Vieira G, Mudla A, Garner JP, Lossie AC. Preventing, treating, and predicting Trichotillomania in a mouse model: A fundamental role for biomarkers of oxidative stress. 2013. 20th Annual National Conference on Hair Pulling & Skin Picking Disorders, Newark, NJ. [DOI] [PMC free article] [PubMed]
- Wahlsten D. Standardizing tests of mouse behavior: reasons, recommendations, and reality. Physiol Behav. 2001;73:695–704. doi: 10.1016/s0031-9384(01)00527-3. [DOI] [PubMed] [Google Scholar]
- Wahlsten D, Rustay NR, Metten P, Crabbe JC. In search of a better mouse test. Trends Neurosci. 2003;26:132–136. doi: 10.1016/S0166-2236(03)00033-X. [DOI] [PubMed] [Google Scholar]
- Wakimoto H, Maguire CT, Kovoor P, Hammer PE, Gehrmann J, Triedman JK, Berul CI. Induction of atrial tachycardia and fibrillation in the mouse heart. Cardiovasc Res. 2001;50:463–473. doi: 10.1016/s0008-6363(01)00264-4. [DOI] [PubMed] [Google Scholar]
- Walsh RN, Cummins RA. Open-field test: A critical-review. Psychol Bull. 1976;83:482–504. [PubMed] [Google Scholar]
- Weiss JM. Effects of coping behavior with and without a feedback signal on stress pathology in rats. J Comp Physiolog Psychol Vol. 1971;77:22–30. doi: 10.1037/h0031581. [DOI] [PubMed] [Google Scholar]
- Willner P. Validation criteria for animal models of human mental disorders: Learned helplessness as a paradigm case. Prog Neuropsychopharmacol Biol Psychiatry. 1986;10:677–690. doi: 10.1016/0278-5846(86)90051-5. [DOI] [PubMed] [Google Scholar]
- Wolfer DP, Crusio WE, Lipp HP. Knockout mice: simple solutions to the problems of genetic background and flanking genes. Trends Neurosci. 2002;25:336–340. doi: 10.1016/s0166-2236(02)02192-6. [DOI] [PubMed] [Google Scholar]
- Woodward M. Epidemiology: Study Design and Data Analysis. Boca Raton, FL: Chapman & Hall/CRC Press; 1999. [Google Scholar]
- Würbel H. Behaviour and the standardization fallacy. Nat Genet. 2000;26:263. doi: 10.1038/81541. [DOI] [PubMed] [Google Scholar]
- Würbel H. Ideal homes? Housing effects on rodent brain and behaviour. Trends Neurosci. 2001;24:207–211. doi: 10.1016/s0166-2236(00)01718-5. [DOI] [PubMed] [Google Scholar]
- Würbel H. Behavioral phenotyping enhanced - beyond (environmental) standardization. Genes Brain Behav. 2002;1:3–8. doi: 10.1046/j.1601-1848.2001.00006.x. [DOI] [PubMed] [Google Scholar]
- Würbel H, Garner JP. Refinement of rodent research though environmental enrichment and systematic randomization. NC3Rs. 2007;9:1–9. [Google Scholar]
- Würbel H, Richter SH, Garner JP. Reply to: “Reanalysis of Richter et al. (2010) on reproducibility”. Nat Methods. 2013;10:374. doi: 10.1038/nmeth.2446. [DOI] [PubMed] [Google Scholar]
- Yee JR, Cavigelli SA, Delgado B, McClintock MK. Reciprocal affiliation among adolescent rats during a mild group stressor predicts mammary tumors and lifespan. Psychosom Med. 2008;70:1050–1059. doi: 10.1097/PSY.0b013e31818425fb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahs KR, Ashe KH. ‘Too much good news’ - are Alzheimer mouse models trying to tell us how to prevent, not cure, Alzheimer's disease? Trends Neurosci. 2010;33:381–389. doi: 10.1016/j.tins.2010.05.004. [DOI] [PubMed] [Google Scholar]
- Zuchner S, Cuccaro ML, Tran-Viet KN, Cope H, Krishnan RR, Pericak-Vance MA, Wright HH, Ashley-Koch A. SLITRK1 mutations in Trichotillomania. Mol Psychiatry. 2006;11:888–889. doi: 10.1038/sj.mp.4001898. [DOI] [PubMed] [Google Scholar]