

Abstract
Biodegradable polymeric nanoparticles have the potential to be safer alternatives to viruses for gene delivery; however, their use has been limited by poor efficacy in vivo. In this work, we synthesize and characterize polymeric gene delivery nanoparticles and evaluate their efficacy for DNA delivery of herpes simplex virus type I thymidine kinase (HSVtk) combined with the prodrug ganciclovir (GCV) in a malignant glioma model. We investigated polymer structure for gene delivery in two rat glioma cell lines, 9L and F98, to discover nanoparticle formulations more effective than the leading commercial reagent Lipofectamine 2000. The lead polymer structure, poly(1,4-butanediol diacrylate-co-4-amino-1-butanol) end-modified with 1-(3-aminopropyl)-4-methylpiperazine, is a poly(β-amino ester) (PBAE) and formed nanoparticles with HSVtk DNA that were 138 ± 4 nm in size and 13 ± 1 mV in zeta potential. These nanoparticles containing HSVtk DNA showed 100% cancer cell killing in vitro in the two glioma cell lines when combined with GCV exposure, while control nanoparticles encoding GFP maintained robust cell viability. For in vivo evaluation, tumor-bearing rats were treated with PBAE/HSVtk infusion via convection-enhanced delivery (CED) in combination with systemic administration of GCV. These treated animals showed a significant benefit in survival (p = 0.0012 vs control). Moreover, following a single CED infusion, labeled PBAE nanoparticles spread completely throughout the tumor. This study highlights a nanomedicine approach that is highly promising for the treatment of malignant glioma.
Keywords: nanomedicine, brain tumor, DNA, gene therapy, polymer, nonviral gene delivery
Glioblastoma (GB) remains one of the most lethal cancers in humans with a median survival after maximal therapy of less than 2 years after first diagnosis.1−3 Despite improvements in the past few decades with intraoperative surgical techniques, chemotherapy, and radiation therapy, predictable curative treatment for GB does not exist yet. New insights into specific gene mutations and dysregulated signaling pathways of the pathogenesis of brain tumors4,5 have highlighted gene therapy as a potential approach for the treatment of GB. This approach is based on the local delivery of a vector or nanoparticle carrying genetic material to cause overexpression of a gene or replace a gene that is missing or under-expressed in order to kill cancer cells.6
Approaches to gene therapy for GB include the following : (1) delivery of suicide genes, which convert pro-drugs in situ and cause tumor cell death;7 (2) delivery of cytokine genes, which mobilize immune cells to fight the tumor;8,9 (3) delivery of tumor-suppressor genes, which induce apoptosis in tumor cells;10,11 and (4) delivery of conditionally replicating viruses to specifically lyse tumor cells while sparing normal tissue.12,13 Gene therapy has most often been performed using viral carriers. However, viruses pose significant safety risks due to their inherent toxicity, immunogenicity, and tumorigenicity.14
Nonviral gene delivery vectors have traditionally been unable to match the efficacy of viral gene delivery;15 however, they can be engineered to avoid the risks that viruses pose. Nonviral methods of gene delivery have recently expanded and several effective nanomaterials exist including lipid-based,16,17 polymeric,18−20 and inorganic21−23 nanoparticles, some of which have reached clinical trials.24 Successful DNA delivery can be achieved by designing materials that can overcome extra- and intracellular barriers.25−28 Cationic, primary amine-containing polymers such as poly(l-lysine) (PLL) can bind anionic DNA and compact it into positively charged nanoparticles. This protects the DNA and promotes cellular uptake via the electrostatic interaction between the cationic nanoparticle and anionic cell surface.28,29 Tertiary amine-containing polymers with high buffering capacities, such as poly(ethylenimine) (PEI), enable endocytosis and are then able to escape the endosome via the proton sponge mechanism.29 DNA release can be achieved by hydrolytic polymer degradation in the cytoplasm of the cell following escape from the endosome. Poly(β-amino ester)s (PBAEs) are a class of polymers that can be engineered to contain primary, secondary, and tertiary amines and hydrolytically cleavable ester bonds.30 These chemical properties enable effective DNA binding, endocytosis, endosomal escape, and intracellular DNA release within minutes to hours, all of which are prerequisite to nuclear uptake of the DNA31−33 PBAEs have previously been shown to be safe and effective DNA delivery vectors in vitro to several cell types and in vivo to retinal and brain tumor tissue.32−34 In previous studies, we have also shown that these polymers degrade quickly under physiological conditions, with a half-life of only a few hours.35 We believe that this is important both to minimize potential nanoparticle cytotoxicity as well as to ensure successful release of the DNA cargo.35 Interestingly, PBAEs can also be engineered to exhibit cell-type specificity and to selectively transfect tumor tissue while avoiding surrounding healthy tissue.34,36 These advantages make this class of polymers a promising option to use for the fabrication of polymeric gene delivery nanoparticles for the treatment of brain tumors. Convection-enhanced delivery (CED) has recently been shown to be effective for the delivery of polymeric nanoparticles encapsulating small molecule drugs, such as dithiazanine iodide, Doxil, and O6-benzylguanine, to brain tumors.37−39 Moreover, CED and gene therapy have been suggested as a promising combination for the treatment of glioma.40 Specifically, CED leads to better volume of distribution by maintaining a pressure gradient which enhances diffusion throughout the tumor mass.41 We hypothesized that intratumoral infusion via CED may represent an effective approach for the delivery of PBAE/DNA nanoparticles, as they are “soft” nanocomplexes which can be deformed and may more easily be convected though small spaces while encapsulating large DNA molecules.
The present study investigates the efficacy of PBAE nanoparticles for the intracellular delivery of the herpes simplex virus (HSV)-derived enzyme thymidine kinase (HSVtk), which acts as a suicide gene in an aggressive gliosarcoma model. Suicide therapy is based on the systemic delivery of an inactive prodrug with tumor-specific expression of a drug-activating enzyme (the suicide gene)42 in order to avoid toxicity in normal cells. The HSVtk-ganciclovir system has been previously used for gene therapy in several viral approaches such as with nonreplicating herpes virus or adenovirus.43−45 HSVtk catalyzes the phosphorylation of the cytotoxic nucleoside analogue ganciclovir (GCV) that can be incorporated into the DNA of actively proliferating cells, which disrupts DNA replication and halts cell division.46,47 Since the prodrug nucleosides are poor substrates for mammalian thymidine kinase, the toxic effect is initially restricted to cancer cells, as active GCV kills proliferating cells only.48 An attractive aspect of the HSVtk/GCV enzyme/prodrug system is that this therapy benefits from the phenomenon known as “bystander effect”, whereby even cancer cells that do not express HSVtk become sensitive to GCV due to the activation of GCV in neighboring transfected cancer cells.49,50
This work presents a biodegradable nanomedicine capable of effectively and selectively delivering DNA to malignant glioma in vivo. A library of PBAE nanoparticles was evaluated in vitro to optimize DNA delivery while minimizing toxicity. The optimal nanoparticle formulation was then physically characterized. We demonstrate that these nanoparticles can deliver an HSVtk transgene in vitro and initiate glioma cell killing via the local activation of GCV. We also demonstrate transfection of malignant gliomas in vivo. Using this nanoparticle-based therapy, we were able to statistically improve survival of rats with malignant glioma (Scheme 1). This work presents an exciting frontier in nanomedicine with significant potential to treat malignant gliomas.
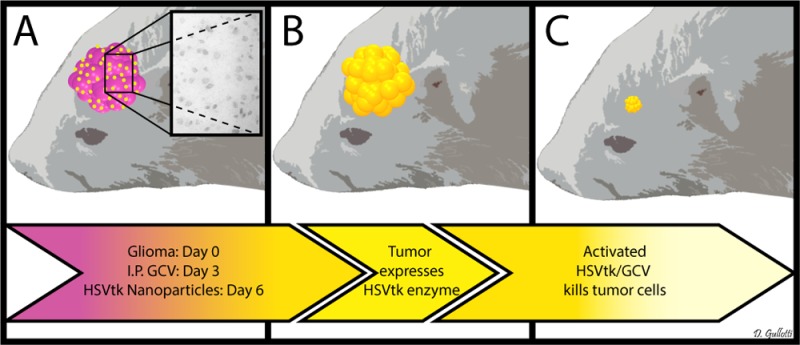
Scheme 1. Schematic representation of the in vivo study. The 9L bearing rats were treated with intraperitoneal administration of ganciclovir twice a day beginning on day 4 and then treated with a single CED infusion of PBAE/HSV-tk nanoparticles on day 6 (A). These treated animals showed a significant benefit in survival (p = 0.0012 vs control) (B–D).

Results and Discussion
PBAE-Based Nanoparticles Show High in Vitro Gene Delivery to Glioma Cells
A library of poly(β-amino ester)s (PBAEs) was synthesized following methods that we have previously described.31 Briefly, base monomers 1,4-butanediol diacrylate (B4) or 1,5-pentanediol diacrylate (B5) were polymerized via a Michael Addition reaction with side chain monomers 3-amino-1-propanol (S3), 4-amino-1-butanol (S4), or 5-amino-1-pentanol (S5) at ratios of either 1.05:1, 1.1:1, or 1.2:1 following Supporting Information Table S1, yielding acrylate-terminated polymers. These polymers were then end-capped with end-capping monomers pentane-1,3-diamine (E3), 2-((3-aminopropyl)amino)ethan-1-ol (E6), or 1-(3-aminopropyl)-4-methylpiperazine (E7). As an example, the polymer made from base monomer B4, side chain S4, and end-cap E7 is 1-(3-aminopropyl)-4-methylpiperazine end-modified poly(1,4-butanediol diacrylate-co-4-amino-1-butanol) and will be referred to as 447 for the duration of the manuscript. Figure 1 shows the monomer structures and the polymerization and end-capping reaction schemes. Gel permeation chromatography (GPC) was used to determine polymer size and polydispersity (Supporting Information Table S1). The Mn of the polymers varied from 3 to 16 kDa, with the average being 10 kDa. The PDI of the polymers varied from 1.74 to 8.07 among the polymer types, with the average PDI being 3.28. The lead polymer, 447, had a Mn of 11345 Da, a Mw of 36 814 Da, and a PDI of 3.25. It was chosen as the lead polymer based on its high gene delivery efficacy. It is possible that the proportion of polymer chains in the batch that are of relatively high molecular weight help in increasing the transfection. Some of our previous work has shown that increased PBAE polymer molecular weight can increase the binding affinity of the polymer with DNA and this can improve gene delivery efficacy.51 Polymer structure of polymer 447 was characterized via 1H NMR, and was shown to match previously described structures (Supporting Information Figure S1).52,53
Figure 1.

Polymer synthesis scheme and monomer chemical structures. Base monomers (B) and side chain monomers (S) are polymerized, and polymers are then end-capped with end-capping monomers (E).
The in vitro DNA delivery efficacy of the PBAE library was assessed in 9L rat gliosarcoma (9L) and F98 rat glioma (F98) cell lines using plasmid DNA coding for green fluorescent protein (GFP). Cytotoxicity was measured using an MTS assay, and transfection was assessed using high-throughput flow cytometry and fluorescence microscopy. Nanoparticles were formed by mixing polymer and DNA at mass ratios of 30, 60, or 90 polymer-to-DNA (w/w) in aqueous conditions and delivering to the cells at a final DNA concentration of 5 ng/μL (Figure 2). Of the nanoparticle formulations tested on 9L cells, ten nanoparticle formulations enabled greater than 50% transfection with less than 20% toxicity (Figure 2A,C). In F98 cells, eight nanoparticle formulations enabled GFP expression in greater than 50% of cells while maintaining less than 20% toxicity (Figure 2B,D). Compared to Lipofectamine 2000, a leading commercially available nonviral transfection reagent, we found that three PBAE nanoparticle formulations had superior performance in 9L cells and 15 had superior performance in F98 cells. Interestingly, the polymer structures that were found to lead to the highest efficacy in one glioma cell type were not necessarily the optimal structures for the other glioma cell type. For example, 453 30 (w/w) transfected 68 ± 3% of F98s, but only 9 ± 1% of 9Ls. In contrast, 457 90 (w/w) shows some signs of potential cytotoxicity in F98, but has higher transfection (64 ± 4%) and no cytotoxicity in 9Ls. This potential cell-type specificity based on polymer structure is something that we have observed with other PBAE structures and other cell types, such as human endothelial cells.54,55 In considering polymer structure that makes up the nanoparticles, the E7 end-capping group 1-(3-aminopropyl)-4-methylpiperazine generally led to improved transfection compared to the E3 or E6 end-capping groups across the base polymers and glioma cell types evaluated. This is consistent with prior work done in other cell types, showing that the E7 end-cap is generally one of the most effective in our library.34,52,56 Among the non-E7 polymers, 536 60 (w/w) and 536 90 (w/w) nanoparticles had the most robust gene expression (respectively, 59 ± 5% and 65 ± 2% in 9L cells, and 73 ± 3% and 71 ± 5% in F98 cells).
Figure 2.

PBAE nanoparticles effectively transfect 9L and F98 malignant glioma cells in vitro. All polymers were screened at 30, 60, and 90 (w/w) delivering 0.6 μg of GFP DNA (A and B). Of the nanoparticles tested on 9L and F98 cells, three and 15 formulations, respectively, were found to deliver GFP DNA more effectively than commercially available transfection reagent Lipofectamine 2000 (*p < 0.05 versus Lipofectamine with 0.6 μg DNA via one-way ANOVA with Dunnett’s post-test). Fluorescence microscopy shows cells transfected with GFP using PBAE nanoparticles (C and D). Transfection efficacy was quantified using flow cytometry. Loss in metabolic activity was quantified using an MTS assay with colorimetric readout, measured by a multiplate reader.
Critically, certain polymeric nanoparticle formulations, such as 447 30 (w/w), transfected both glioma cells lines at similarly high levels and without any cytotoxicity. This result matches prior investigation of PBAEs56 that showed polymers of moderate hydrophobicity led to high transfection rates without significantly compromising cell viability. On the basis of the results of this study, we chose polymer 447 at 30 (w/w) as the optimal nanoparticle formulation for both 9L and F98 cells (77 ± 3% and 68 ± 1% transfection, respectively). Polymer 447 formed nanoparticles with GFP DNA through self-assembly that were 131 ± 3 nm in size and 15 ± 0.4 mV in zeta potential and with HSVtk DNA that were 138 ± 4 nm in size and 13 ± 1 mV in zeta potential (neither particle size nor zeta potential is statistically different between these two formulations). Even though the plasmid sizes were different, the total nucleic acid mass that was used to form the nanoparticles was the same and this led to nanoparticles with the same biophysical properties. This finding also matches our lab’s previous finding that DNA plasmid sequence and length does not affect the nanoparticle size or charge of these PBAE-based nanoparticles within the plasmid DNA size range of 2–26 kb.56 These nanoparticles also compared favorably to transfection with a leading commercially available nonviral transfection reagent, Lipofectamine 2000, which led to 52 ± 1% and 37 ± 4% transfection in 9L and F98 cells, respectively (see Supporting Information Table S2 for full statistical analysis on transfection efficacy).
The polymer that makes up these nanoparticles, 447, has also recently shown robust transfection of other cancer types, including human brain cancer cells.34 Although the physicochemical properties that govern the efficacy and activity of PBAE-based nanoparticles are currently under investigation and likely include the chemical structure and molecular weight of the polymer, the polymer–DNA binding strength, and the cellular uptake pathway of the nanoparticles,51,56,57 the exact mechanisms have not yet been fully elucidated. For example, we have seen that two cell types with very different transgene expression rates can show statistically similar nanoparticle uptake rates.31,34,58 Early work in our group also suggests that the particular cell uptake pathway can affect successful transfection rates with PBAE nanoparticles.57 Nonetheless, we have consistently observed certain trends across various cell types and culture systems. For instance, the polymers used here were synthesized using monomer ratios that yielded products with relatively high molecular weight, which we and others have shown to have a generally positive correlation with transfection efficacy,51,56,59 and the small molecules used for polymer library synthesis were chosen based on their transfection efficacy in other work.51,56,57
PBAE/HSVtk Nanoparticles and a Ganciclovir Prodrug Kill Glioma Cells in Vitro
We sought to examine the antitumor efficacy of the PBAE/HSVtk nanoparticles in vitro. Using the optimal nanoparticle formulation 447 at 30 (w/w), we delivered plasmid DNA encoding either GFP or HSVtk, and treated the cells with GCV at 0, 5, and 50 μg/mL. Viability was assessed quantitatively via cell counting in 9L and F98 cells (Figure 3B,D). We found that cancer cell death was dependent on the presence of both HSVtk and GCV, as HSVtk-transfected cells were viable when no GCV was present, while GFP-transfected cells were viable in spite of the presence of GCV. Specifically, we observed that the nanoparticle-mediated HSVtk/GCV-induced cytotoxicity was powerful, resulting in 106 ± 3% cell death of 9Ls and 96 ± 7% cell death of F98s at 5 μg/mL GCV when the cells were transfected with HSVtk versus GFP. At 50 μg/mL GCV, 104 ± 5% of 9L cells and 101 ± 2% of F98 cells were dead. These measurements of approximately 100% cell death are consistent with the complete cell death that was observed by microscopy (Figure 3A,C).
Figure 3.

PBAE delivery of HSVtk plasmid enables GCV-mediated killing of malignant glioma cells in vitro. 9L and F98 cells were transfected with plasmids encoding either GFP or HSVtk and then treated with 0, 5, or 50 μg/mL GCV prodrug. Cells treated with both HSVtk and GCV exhibit 100% cancer cell killing, measured by cell counting, versus GFP-transfected cells treated with GCV, showing that GCV-induced cell killing is dependent on the presence of HSVtk. Additionally, HSVtk without GCV does not kill cells.
Although the two plasmids used, GFP and HSVtk, are of different size and may result in slightly different transfection rates, the lack of difference in the physicochemical properties of nanoparticles formed with different types of DNA allows the use of the GFP plasmid as a nonfunctional control for the HSVtk plasmid. As both these types of nanoparticles are formed with the same total mass of polymer and vnucleic acid and have the same physicochemical properties, there is not expected to be a difference in polymer- or nanoparticle-induced toxicity between the two plasmids or their particle distribution. While the percent of cells transfected with GFP cannot be assumed to be exactly, the same as the percent of cells positive for HSVtk, the strong cytotoxic effect of GCV in HSVtk-transfected cells shows that this transfection, like the GFP transfection, is sufficient for a significant biological effect. These data suggest that nanoparticle-based delivery of the HSVtk gene enables local activation of GCV into a cell-killing drug, which is known to cause cell death in malignant glioma lines via activation of apoptosis.47,60 Moreover, the proportion of cells killed (approximately 100%) was greater than the transfection efficiency of the glioma cells (<80%), illustrating the strong bystander effect of the HSVtk/GCV therapeutic strategy. This is particularly important in the treatment of a tumor, as even if <100% of the cancer cells are transfected in vivo, the fraction of cancer cells that positively express HSVtk can lead to the apoptosis of neighboring untransfected tumor cells. This nanoparticle-mediated suicide gene therapy finding is consistent with studies done in other laboratories with an analogous HSVtk/GCV viral gene therapy strategy.61,62 Few previous studies have been conducted using nonviral HSVtk gene delivery in cancer models. Two examples in the literature include Neurotensin (NTS)-polyplex nanoparticles63 used to transfect triple negative breast cancer cells (MDA-MB-231), and poly(ethylene glycol)–poly(γ-benzyl-l-glutamate) (PEG–PBLG)64 nanoparticles used to transfect oral squamous cell carcinoma (Tca8113). The transfection efficacy with these nonviral nanoparticle systems was lower than in our present study, with only 18% and 30% transfected, respectively. In those cases, the efficacy of GCV treatment was shown to increase cancer cell death to 50% and 80% through the bystander mechanism. In contrast, our approach with PBAE nanoparticles led to 70% transfection efficacy with 100% cell death. To our knowledge, this is the first demonstration of significant efficacy and anticancer effects of PBAE/DNA delivery to treat brain cancer.
PBAE/DNA Nanoparticles Can Be Lyophilized with No Change in Properties or Efficacy
Lyophilization of nanoparticles prior to in vivo administration has several benefits. Drying the nanoparticles enables subsequent reconstitution at a higher concentration, which was particularly beneficial for intracranial injections, in which volumes were limited due to size and pressure constraints within the brain. Lyophilized PBAE nanoparticles can also be stored for years without losing efficacy34,53 and are easier to administer, as the user simply needs to add water and inject. The amount of solutes added to the particles during formulation is easily adjustable to ensure that the resulting aqueous suspension is isotonic after adding water. To ensure that the 447 30 (w/w) nanoparticles did not lose efficacy after lyophilization, lyophilized particles were compared to freshly prepared particles. We compared the size and zeta potential of 447 30 (w/w) nanoparticles and found no significant difference between freshly prepared and lyophilized nanoparticles (Figure 4A). We also compared in vitro transfection of fresh versus lyophilized nanoparticles and found that measurements of transfection efficacy, percent of GFP-positive cells, and geometric mean fluorescence showed no statistical difference (p > 0.05) (Figure 4B). Finally, we imaged fresh and lyophilized nanoparticles via transmission electron microscopy (TEM), and found that nanoparticles from each batch were morphologically similar (Figure 4C,D).
Figure 4.

Nanoparticles maintain their physical characteristics and transfection capability following lyophilization. Fresh and lyophilized nanoparticles showed no statistical difference (p > 0.05) in their sizes and zeta potentials (A) and showed no statistical difference in percent transfection and geometric mean GFP in 9L cells (B). TEM imaging of fresh (C) and lyophilized (D) nanoparticles shows nanoparticles of the same size and morphology (scale bar = 100 nm).
PBAE Nanoparticles Are Safe To Deliver to the Brain
We assessed the safety of delivering PBAE/DNA nanoparticles to the brain in vivo in both wild type healthy rats (at day 3 and at day 60 post-nanoparticle infusion) and 9L tumor-bearing rats (at day 3 post-nanoparticle infusion). We used 25 μL as the infusion volume, which was previously shown to be safe.65 PBAE/GFP 447 30 (w/w) nanoparticles were injected in 10% (w/v) sucrose and were tested at 26 μg DNA with 780 μg PBAE. This dose was found to be safe and was used for subsequent in vivo studies. The 50 mg/kg GCV injected twice a day was well tolerated and was therefore chosen for all efficacy studies.
With the exception of animals euthanized at the early time point (day 3 post-nanoparticle infusion) for histopathological analysis, all nontumor-bearing animals survived until the end of the study (60 days post-nanoparticle infusion). No signs of neurotoxicity or paralysis were observed. The histopathology from day 3 (healthy wild type rats and 9L tumor-bearing rats) and day 60 (healthy wild type rats) showed no discernible signs of early or late tissue damage and did not include cytological changes, edema, gliosis, neuroinflammation, or necrosis (Supporting Information Figure S2). These data provide in vivo confirmation of one of the strengths of biodegradable PBAEs, a strong safety profile, even when used at high concentration.
Intratumoral Infusion of PBAE/GFP Nanoparticles Leads to Wide Distribution and Transfection in the Brain
The intratumoral distribution and transfection of PBAEs nanoparticles after local brain delivery was assessed. To evaluate the nanoparticle distribution and transfection through the tumor in vivo, we used nanoparticles containing Cy5-labeled GFP DNA and examined the distribution of Cy5+ and GFP+ cells (Figure 5A–D). On Day 7, 24 h post-nanoparticle infusion, the tumor mass was approximately 2 × 3 mm, extending from the cortex toward the ipsilateral caudate/putamen area and invading the left lateral ventricle (Figure 5A). Immunofluorescence staining showed high GFP signal in the corresponding area of the tumor. Specifically, 24 h after CED infusion of Cy5-labeled GFP DNA-containing nanoparticles, the nanoparticles transfected the tumor mass, while the normal brain remained GFP-negative (Figure 5B). Importantly, the nanoparticles transfected cells fairly homogeneously throughout the entire tumor mass, even distant from the site of infusion (Figure 5D–F).
Figure 5.

Local brain delivery of PBAE/GFP Nanoparticle via CED leads to effective tumor transfection in vivo. Coronal section of a 9L tumor bearing rat brain at 7 days post PBAE/GFP infusion showing the tumor region (A, scale bar = 2 mm). Fluorescence microscopy images show GFP+ transfected cells in the tumor area (B, scale bar = 2 mm). Enlarged area shows a wide distribution of GFP+ cells within the entire tumor area including the periphery (C, scale bar = 500 μm). The colocalization of GFP and Cy5 shows that the nanoparticles penetrate into the cells and successfully transfect them (D–F, scale bar: 50 μm). Re,: Cy5; green, GFP; blue, DAPI.
Limited tumor penetration and low transfection rates are major obstacles in nonviral gene therapy for the treatment of brain tumors.37 Transport depends on physio-anatomic barriers66 and the physicochemical characteristics of the particle, including its size, shape, and surface charge. In this study we show that CED positively affects the biodistribution of the PBAE nanoparticles compared to bolus injection (Supporting Information Figure S3). This is due to the creation of a positive pressure gradient, which creates bulk fluid movement in the brain interstitium. Tumor-bearing rats treated with a bolus injection of an equal amount of PBAE/DNA nanoparticles underwent brain analysis via immunofluorescence staining as well. Analysis showed that bolus injection led to transfection in the areas around the needle-path at the bottom of the tumor, with much of the core and margins without nanoparticle-mediated gene transfer. After CED infusion, on the other hand, the nanoparticle-mediated exogenous gene expression led to positive GFP signal across the whole tumor mass, from the core to the top, bottom, and margins. Moreover, the normalized GFP fluorescence per pixel of the tumor area (using ImageJ software) showed a 36% increase by CED infusion compared to bolus injection (15.8 ± 0.1 RFU vs 11.6 ± 0.2 RFU, vs 11.6 ± 0.2 RFU, p < 0.0001) These findings show for the first time that PBAE nanoparticles can penetrate the entirety of the tumor volume and therefore are promising for gene delivery when combined with CED.
PBAE/HSVtk Nanoparticle/GCV Treatment Leads to Prolonged Survival of 9L Gliosarcoma-Bearing Rats
After demonstrating the in vitro efficacy of PBAE/HSVtk nanoparticles in F98 and 9L glioma cells and the in vivo safety of PBAE nanoparticles for intratumoral transfection, we proceeded to test their in vivo therapeutic efficacy in 9L tumor-bearing rats (Scheme 1). 9L gliosarcoma is a highly aggressive syngeneic glioma model67,68 and is well-known as a challenging survival model due to its fast rate of growth. This model, although rodent-based, has achieved undisputed clinical relevance as it has been used as a preclinical model for multiple clinical trials in the investigation of novel chemo and immunotherapies, drug delivery strategies, and gene therapies.69−74 Animal survival after treatment with PBAE/HSVtk nanoparticles in combination with GCV was significantly longer compared to the untreated control group (p = 0.0012) (Figure 6). PBAE/HSVtk nanoparticles were also found to provide survival benefits compared to all the other control groups: GCV only (p = 0.0102), PBAE/GFP + GCV (p = 0.027), and HSVtk DNA+ GCV (p = 0.027).
Figure 6.
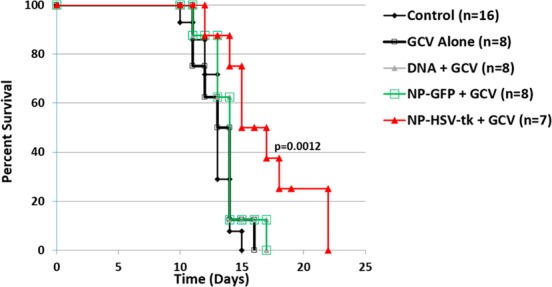
PBAE/HSVtk nanoparticles and ganciclovir (GCV) extend survival in a 9L glioasarcoma model. Kaplan–Meier plots of F344 rats that were implanted with 9L and either given no treatment (9L Control, n = 16); 50 mg/kg twice a day of systemic administration of GCV on days 4–13 (GCV Alone, n = 8); intracranial infusion of PBAE/GFP nanoparticles plus systemic administration of GCV (NP-GFP + GCV, n = 8); intracranial infusion of HSVtk DNA plus systemic administration of GCV (DNA + GCV n = 8); or intracranial infusion of PBAE/HSVtk nanoparticles plus systemic administration of GCV (NP-HSVtk + GCV, n = 8). The median survival of the group receiving PBAE/HSVtk nanoparticles in combination with GCV is significantly longer compared to that of the untreated control group (p = 0.0012).
Considering that 9L is characterized by an exponential-Gompertzian curve of growth after 6 days,67,68,75 such a statistically significant benefit in survival in the group treated with PBAE/HSVtk plus GCV is promising. The efficacy of the HSVtk/GCV paradigm has previously been shown in the 9L model using other strategies76,77 and the bystander effect and a resulting immune response can both contribute to the overall cytotoxicity of the PBAE/HSVtk GCV treatment.78,79 Due to the positive relationship between the bystander effect and the percent of HSVtk transfected cells,50 the improvement in survival observed here is highly indicative of strong PBAE transfection efficacy in vivo matching what was also observed in vitro. PBAE-based gene delivery has a potential role as alternative strategy to virus mediated gene therapy for glioblastoma.
HSVtk/GCV therapy has previously been used in brain tumor treatment clinical trials with retroviral80−82 and adenoviral83 vectors with limited success in progression-free and overall survival. This has been attributed to poor distribution of the carrier and limited delivery of HSVtk into the tumor. The HSVtk/GCV system has also been used to investigate the efficacy of nonviral vectors in Phase I–II studies conducted with cationic liposomal vectors, which showed, via positron emission tomography with a 124I-labeled specific substrate for HSV-1, presence of thymidine kinase in one out of five patients.84 A subsequent study using liposomes had encouraging results by showing reduction of the tumor volume mass in two out of eight patients.85 Yet, nonviral gene therapy is generally considered less efficient than viral gene therapy.86 While siRNA delivery nanomedicine approaches have been progressing rapidly, including approaches such as spherical nucleic acids for the treatment of glioblastoma,87 DNA delivery has been more challenging. This is likely due to in large part to the much larger size of DNA as a nanomedicine drug cargo and the need for it to be delivered into nucleus rather than just into the cytoplasm.84 Due to the wide tumor penetration, high exogenous gene expression, and bystander effect capabilities, the PBAE/HSVtk nanoparticles may be able to overcome the previously encountered limitations and be a promising new nanomedicine for brain cancer.
Conclusions
In this work, we have developed a new gene transfer nanomedicine that causes the expression of suicide gene herpes simplex virus type I thymidine kinase (HSVtk) within brain tumor cells in vitro and in vivo. Biodegradable nanoparticles were synthesized, characterized, and utilized to treat malignant glioma using convection-enhanced delivery (CED). The lead polymer structure, poly(β-amino ester) 1-(3-aminopropyl)-4-methylpiperazine end-modified poly(1,4-butanediol diacrylate-co-4-amino-1-butanol), formed DNA nanoparticles with suitable biophysical properties for intracellular delivery and a wide biodistribution when injected intracranially. CED led to improved levels of tumor transfection. These PBAE/HSVtk nanoparticles combined with systemic administration of ganciclovir as a prodrug led to a significant increase in survival in a 9L glioma model (p = 0.0012 vs control). Our results provide the first demonstration of a successful nonviral nanomedicine method for HSVtk/GCV treatment of brain cancer.
Materials and Methods
Polymer Materials
Monomers used for polymer synthesis were purchased as follows: 1,4-butanediol diacrylate (B4; Alfa Aesar, Ward Hill, MA); 1,5-pentanediol diacrylate (B5; Monomer-Polymer and Dajac Laboratories, Trevose, PA); 3-amino-1-propanol (S3; Alfa Aesar); 4-amino-1-butanol (S4; Alfa Aesar); 5-amino-1-pentanol (S5; Alfa Aesar); pentane-1,3-diamine (E3; TCI America); 2-(3-aminopropylamino)ethanol (E6; Sigma-Aldrich); 1-(3-aminopropyl)-4-methylpiperazine (E7; Alfa Aesar). Lipofectamine 2000 was purchased from Invitrogen (Carlsbad, CA) and used according to manufacturer’s instructions. The pEGFP-N1 plasmid (GFP) was purchased from Elim Biopharmaceuticals and amplified by Aldevron (Fargo, ND). Herpes simplex virus type 1-derived thymidine kinase (HSVtk) gene was cloned into the pcDNA3.1 vector (Life Technologies) according to the manufacturer’s protocols. Label IT Tracker Cy5 Kit was purchased from Mirus Bio (Madison, WI). For staining, 2-(4-amidinophenyl)-1H -indole-6-carboxamidine (DAPI) was purchased from Life Technologies, propidium iodide (PI) was purchased from Invitrogen (Carlsbad, CA), and the conjugated antibody anti-Ki67 Alexa Fluor 647 (rabbit anti-rat 1:50) was purchased from Cell Signaling Technology (Beverly, MA). CellTiter 96 AQueous One MTS assay was purchased from Promega (Fitchburg, WI). Ganciclovir was purchased from both Invivogen (San Diego, CA) and Euroasian Chemicals PVT LTD (Lower Parel, Mumbai, India).
Polymer Synthesis
PBAEs were synthesized using a two-step reaction (Figure 1) in a manner similar to Bhise et al.(88) Base monomers B4, B5, or B6 were each polymerized by Michael Addition of one of the side chain monomers S3, S4, or S5 at ratios following Supporting Information Table S1, for 24 h at 90 °C in the absence of solvent. Monomer acrylate-to-amine molar ratios used for synthesis ranged from 1.2:1 to 1.05:1. For the second step of synthesis, the diacrylate-terminated base polymers (B-S) were dissolved in anhydrous tetrahydrofuran (THF, Sigma) at 100 mg/mL and combined with 0.2 M amine-containing small molecules (E3, E6, or E7) as polymer endcapping groups. The reaction was conducted for 1 h at room temperature while shaking. Polymers were then purified to remove excess monomer via precipitation in diethyl ether. The ether was decanted to collect polymer, the polymer was washed again with ether, the ether was decanted, and then the polymer was allowed to dry under vacuum for 48 h. The neat polymers were then dissolved in dimethyl sulfoxide (DMSO) at 100 mg/mL and stored at −20 °C in small aliquots to limit freeze–thaw cycles. The molecular weight and polydispersities of the polymers were determined by gel permeation chromatography (GPC; Waters, Milford, MA) in BHT-stabilized tetrahydrofuran with 5% DMSO and 1% piperidine. Number-averaged and weight-averaged molecular weights (Mn and Mw, respectively) were measured using polystyrene standards (Supporting Information Table S1). Purity of the leading polymer, 1-(3-aminopropyl)-4-methylpiperazine end-modified poly(1,4-butanediol diacrylate-co-4-amino-1-butanol) (447), was confirmed by 1H NMR spectra (Supporting Information Figure S1).
Preparation of Cy5-Labeled DNA
GFP DNA was labeled with Cy5 using the Label IT Tracker Cy5 Kit following manufacturer instructions. The amount of Cy5 labeling was measured to be approximately one dye molecule per 205 base pairs.
Cell Culture
F98 glioma cells were provided from R. Barth (Ohio State University, Columbus, OH) and the 9L gliosarcoma line was obtained from the Brain Tumor Research Center (University of California, San Francisco, CA). Cells were grown in high glucose Dulbecco’s modified Eagle’s medium (DMEM; Gibco Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich), 0.8 mM l-glutamine, and 1% penicillin–streptomycin (Gibco Life Technologies, Carlsbad, CA). Cells were cultured at 37 °C in a humidified incubator with 5% CO2.
In Vitro DNA Delivery to Glioma Cells Utilizing PBAE Nanoparticles
One day prior to transfection, 9L and F98 cells were plated in 96-well cell culture plates at a cell density of 10 000 cells/well in 100 μL of complete medium, and allowed to adhere at 37 °C overnight. For nanoparticle preparation, following previously reported protocols,34,88 GFP DNA was diluted to 60 μg/mL in 25 mM sodium acetate pH 5 buffer (NaAc). PBAEs were diluted from their stock solutions in DMSO in 25 mM NaAc and added to DNA solutions at PBAE/DNA mass ratios (w/w) of 30, 60, or 90. Polymers were added to DNA 1:1 (v/v), mixed via pipetting, and incubated at room temperature for 10 min to allow for self-assembly, which has previously been shown to be sufficient time for DNA binding to the polymer.56 Nanoparticles (20 μL) in NaAc were added directly to the cells that were in 100 μL/well of cell culture medium in 96-well plates (final DNA concentration of 600 ng/well). Nanoparticles were incubated with the cells for 2 h at 37 °C, after which the media was removed and replaced with fresh, complete media. Four replicates were evaluated for each transfection condition. For lyophilized nanoparticle formation and evaluation (Figure 4), nanoparticles were initially formed as described above, including allowing 10 min for self-assembly. Subsequently, d-sucrose was added as a cryoprotectant to a final concentration of 30 mg/mL sucrose, the nanoparticles were frozen at −80 °C, and they were lyophilized as we have recently described.53 Lyophilized nanoparticles were stored at −20 °C until use and were then reconstituted in sterile water and used at the same concentration as freshly prepared nanoparticles.
Evaluation of Transfection by Flow Cytometry and Fluorescence Microscopy
Transfection efficacy was evaluated by measuring the percentage of cells expressing the exogenously delivered GFP DNA. Fluorescence microscopy images were obtained using a Zeiss Axio observer A1 microscope with a Zeiss Axiocam MRm camera using AxioVision Release 4.8.2 software. Transfection efficacy was evaluated by microscopy after 48 h at 5× magnification.
Flow cytometry was completed 48 h following transfection using an Intellicyt high-throughput autosampler attached to an BD Accuri C6 flow cytometer (emission filter: 530/30 nm). Hypercyt software was used to assign events to each well and FlowJo 7 software (Treestar) was used to analyze the flow cytometry results. Plates were prepared for flow cytometry by trypsinization using 30 μL of 0.05% Trypsin–EDTA, followed by addition of 170 μL of a solution of 2% FBS in PBS. Samples were transferred to round-bottom 96-well plates and centrifuged, and 170 μL of volume was removed. Cells were then resuspended via pipetting and loaded onto the Hypercyt autosampler.
Measurement of Cytotoxicity
Nonspecific cell toxicity was defined as the loss of metabolic activity in each well following transfection. Cell toxicity was determined at 24 h post-transfection using a CellTiter 96 AQueous One MTS assay following manufacturer’s instructions. A BioTek Synergy 2 Microplate Reader was used to read absorbance at 490 nm, and cell toxicity was determined by normalizing the metabolic activity values of treated wells to untreated wells.
Evaluation of HSVtk/GCV-Induced Cytotoxicity in 9L and F98 Glioma Cell Lines
Cells were transfected as described above using polymer 447 at 30 (w/w), using either HSVtk or GFP DNA (n = 4). GCV was added to the cell culture medium at 24 h post-transfection at concentrations from 0 to 50 μg/mL and replenished every 2 days. At 5 days post-transfection, cells were stained with PI, fixed, and then stained with DAPI. Each well was photographed at 5× magnification to capture PI and DAPI fluorescence as well as a brightfield image using a Zeiss Axio observer A1 microscope with a Zeiss Axiocam MRm camera using AxioVision Release 4.8.2 software. Cells positive for PI and/or DAPI were counted using ImageJ v1.47 software. PI cell counts were subtracted from DAPI cell counts to remove dead cells from the total cell count, and then the total cell count of each well was normalized to that of untreated wells.
Nanoparticle Physical Characterization
The hydrodynamic radius and zeta potential of the leading nanoparticle formulation, 447 30 (w/w), was determined via dynamic light scattering (DLS) using a Malvern Zetasizer NanoZS (Malvern Instruments, Malvern, U.K.). Fresh and lyophilized nanoparticles were formed as previously described for transfection, and then diluted into PBS at a 1:6 (v/v) dilution in order to better approximate the physiological salt concentration and pH that particles would experience in cell culture or in an organism. To measure particle size, the intensity-weighted Z-average of the particle diameter is reported in nm. Zeta potentials were analyzed using the Smoluchowski model and average electrophoretic mobilities were measured at 25 °C at pH 7.4.
Transmission Electron Microscopy
Transmission electron microscopy (TEM) was used to image 447 30 (w/w) nanoparticles using a Philips/FEI BioTwin CM120 transmission electron microscope. Nanoparticles were formed as described for transfection, and 5 μL of the nanoparticle solution was loaded onto a carbon-coated copper TEM grid and allowed to dry completely prior to imaging.
Animals
Female F344 rats, weighing 125–175 g each (Harlan Bioproducts, Indiana, IN), were housed in standard facilities and were provided with ad libitum access to food and water. The policies and guidelines of the Johns Hopkins University Animal Care and Use Committee were strictly followed throughout the study.
Tumor Implantation
F344 rats were intracranially implanted with 9L gliosarcoma, which was maintained and passaged every 2–3 weeks. For surgical intracranial implantation, the tumor was removed from the carrier animal, cut into 2 mm3 pieces, and placed in sterile 0.9% saline on ice as previously described.89 Rats were anesthetized with an intraperitoneal injection of 3–5 mL/kg of a stock solution containing ketamine HCl (75 mg/kg; 100 mg/mL), xylazine (7.5 mg/kg; 100 mg/mL), and ethanol (14.25%) in sterile 0.9% NaCl. For the orthotopic tumor inoculation, the head was shaved and prepared with alcohol and Prepodyne solution (DeLaval, Inc.). To expose the sagittal and coronal sutures, a midline scalp incision was made. A small hole was made using an electric drill in the skull centered 3 mm lateral to the sagittal suture and 5 mm posterior to the coronal suture. The superior sagittal sinus was carefully avoided. Under microscopic magnification, a dural opening and then a cortical opening were made. A small area of cortex and white matter was resected. Once hemostasis was achieved, a single tumor piece (2 mm3) was placed into the resection cavity. The skin was then closed with surgical staples. All surgical procedures were performed using standard sterile surgical technique.
In Vivo Nanoparticle Administration
Under full anesthesia, 6 days after tumor inoculation, the original incision was opened and the original burr hole was located. For Convection-Enhanced Delivery (CED), a 25-gauge needle was stereotactically placed at a depth of 3 mm into the rat striatum. The infusion was performed using an UltraMicroPump (UMP3) with SYS-Micro4 Controller (World Precision Instruments, Inc., Sarasota, FL) at a rate of 1 μL/min for 25 min. After the injection, the needle was maintained in the cortex for another 5 min to avoid backflow. Following needle removal, the incision was stapled and the animal was allowed to awaken and recover. Bolus administration delivered the same volume of nanoparticles by manual injection as previously described.89
Throughout the study, three types of DNA were complexed with 447 30 (w/w) to form nanoparticles and used in vivo in lyophilized form: (1) GFP, (2) Cy5-labeled GFP, and (3) HSVtk. The nanoparticles were stored in −20 °C and were resuspended in water prior to injection. The particles were injected both by bolus (manual injection) or infused by CED. The two methods of nanoparticle administration were compared using 12 tumor-bearing rats infused with PBAE/GFPs 6 days after tumor implantation. The animals were euthanized at 24 h postinfusion and the brains were fixed in formalin for imaging. The efficacy study was performed using CED.
In Vivo Safety Studies
The safety of intracranial injection of PBAEs was evaluated. PBAE 447/GFP DNA nanoparticles (26 μg DNA/780 μg polymer) were infused in a volume of 25 μL using CED in six wild type healthy rats and three 9L tumor-bearing rats for a total number of 9 rats. All rats were observed daily for any signs of neurotoxicity. Three animals from each group were euthanized 3 days after nanoparticle infusion, while the remaining nontumor-bearing animals were observed for 60 days after nanoparticle infusion and then euthanized. Subsequently, their brains were harvested and placed in formalin for histopathological analysis. Specifically, the brains were cut in 3 slices of 2 mm: one centered on the site of the injection and the other two centered 2 mm anteriorly and posteriorly from it.
The safety of systemically injected ganciclovir was assessed in 6 rats for 10 days: 3 rats were treated with intraperitoneal administration of 50 mg/kg once a day and the other 3 with 50 mg/kg twice a day for 10 days. Rats were evaluated daily for signs of pain and distress, including ruffled fur, dehydration, hunched position, weakness, lethargy, immobility, lack of coordination, labored respiration, or cyanosis according to the Johns Hopkins Animal Care and Use Guidelines. At the end of the study, the brains were fixed, sectioned, and processed for light microscopic analysis to determine histopathology and to evaluate tissue damage.
Efficacy Studies
For intracranial implantation, 48 F344 rats (24 rats for the first study and 24 for the second) were anesthetized and received tumor as described above. Six days after tumor implantation, when tumor area was approximately 2 × 3 mm2, the rats were randomized into the following groups: Control group, which received an intracranial infusion of 25 μL of saline by CED (n = 16); GCV group which received intraperitoneal administration of 50 mg/kg of GCV twice a day (n = 8); NP-GFP + GCV group, which received intracranial infusion of PBAE/GFP nanoparticles plus intraperitoneal administration of GCV (n = 8); DNA + GCV group, which received intracranial infusion of HSVtk DNA plus intraperitoneal administration of GCV (n = 8); and the NP-HSVtk + GCV group, which received intracranial infusion of PBAE/HSVtk nanoparticles plus intraperitoneal administration of GCV (n = 8).
GCV was administered for 10 days, from Day 4 to Day 13, i.e. starting 4 days after tumor implantation and 2 days before PBAE/HSVtk infusion due to the aggressive nature of the 9L glioma, as described previously.48 The animals were monitored daily and assessed for neurological impairment. The animals were perfused as they became moribund and brains were placed in formalin for histological analysis.
Brain Imaging
Immunofluorescence staining of the in vivo transfection was assessed. A total of 6 rats were used for imaging in vivo transfection 24 hr post-infusion via CED. Animals were infused at 6 days after tumor implantation with Cy5-labeled GFP DNA utilizing the DNA/polymer ratio used for the efficacy study. A comparison of nanoparticle delivery via bolus injection and CED infusion was subsequently conducted. The image analysis was performed with ImageJ as previously described90 and the whole tumor was considered as the region of interest (ROI). The boundaries of the tumor area were determined by a neuropathologist via blind analysis of H&E and DAPI stained slides. The corrected total cell fluorescence (CTCF) intensity of the whole tumor area, both after CED infusion and after bolus injection, was compared and calculated as follows: integrated density – (area of selected cell × mean fluorescence of background readings), normalized by the number of pixels in the tumor area. To evaluate PBAE distribution between the two administration methods and how this might affect transfection efficacy, 12 9L tumor-bearing rats were divided into two groups: a CED group (n = 6) and an intracranial bolus injection group (n = 6). Cy5 labeled GFP-nanoparticles were infused via CED infusion or bolus injection on Day 6 and the rats were sacrificed 24 h later. The animals were anesthetized and perfused with 0.1 M phosphate-buffered saline (PBS), followed by 4% paraformaldehyde in 0.1 M PBS solution. The brain was immediately removed from the skull and transferred to 4% paraformaldehyde at 4 °C for 24 h.
Immunohistochemistry
The brains, fixed in 4% paraformaldehyde at 4 °C for at least 24 h, were cryoprotected by sinking in 30% sucrose in 0.1 M PBS for 3 days and then embedded in Optimal Cutting Temperature compound (OCT) compound. Cryosection slides were prepared at 10 μm using a Leica CM1905 cryostat. The slices were imaged to assess transfection by confocal laser scanning microscope (Leica TCS SP5Microsystems) and captured with Axiovision software (Axiovision Rel 4.9).
Data Analysis and Statistical Methods
Transfection efficacy of PBAE nanoparticles was compared to Lipofectamine 2000 using One-way ANOVA and Dunnett posttests (GraphPad Prism 5.0). Comparisons of fresh and lyophilized nanoparticles were conducted using two-tailed t tests (GraphPad Prism 5.0). The statistical analysis for survival was completed using Kaplan–Meier survival plots (GraphPad Prism 5.0), and the survival curves were compared using the Log-rank (Mantel-Cox) test. p < 0.05 was considered statistically significant in all cases.
Acknowledgments
This work was supported in part by the NIH (1R01EB016721). K.L.K. also thanks the NIH Cancer Nanotechnology Training Center (R25CA153952) at the JHU Institute for Nanobiotechnology for fellowship support and S.Y.T. thanks the NSF for fellowship support. Laboratory support from Joshua and Genine Fidler is also gratefully acknowledged. The authors would also like to thank Dr. Tara L. Deans for her assistance with the molecular biology components of this work.
Supporting Information Available
Polymer characterization via GPC, polymer characterization via1H NMR, additional statistical analysis, safety data, and convection enhanced delivery data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
× These authors contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Chaichana K. L.; Zadnik P.; Weingart J. D.; Olivi A.; Gallia G. L.; Blakeley J.; Lim M.; Brem H.; Quinones-Hinojosa A. Multiple Resections for Patients with Glioblastoma: Prolonging Survival. J. Neurosurg. 2013, 118, 812–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGirt M. J.; Chaichana K. L.; Gathinji M.; Attenello F. J.; Than K.; Olivi A.; Weingart J. D.; Brem H.; Quinones-Hinojosa A. R. Independent Association of Extent of Resection with Survival in Patients with Malignant Brain Astrocytoma. J. Neurosurg. 2009, 110, 156–162. [DOI] [PubMed] [Google Scholar]
- Stupp R.; Mason W. P.; van den Bent M. J.; Weller M.; Fisher B.; Taphoorn M. J. B.; Belanger K.; Brandes A. A.; Marosi C.; Bogdahn U.; et al. Radiotherapy Plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [DOI] [PubMed] [Google Scholar]
- Mao H.; LeBrun D. G.; Yang J.; Zhu V. F.; Li M. Deregulated Signaling Pathways in Glioblastoma Multiforme: Molecular Mechanisms and Therapeutic Targets. Cancer Invest. 2012, 30, 48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohgaki H.; Kleihues P. Genetic Pathways to Primary and Secondary Glioblastoma. Am. J. Pathol. 2007, 170, 1445–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowska A.; Nandhu M. S.; Behera P.; Chiocca E. A.; Viapiano M. S. Strategies in Gene Therapy for Glioblastoma. Cancers 2013, 5, 1271–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarogoulidis P.; Darwiche K.; Sakkas A.; Yarmus L.; Huang H.; Li Q.; Freitag L.; Zarogoulidis K.; Malecki M. Suicide Gene Therapy for Cancer—Current Strategies. J. Genet. Syndr. Gene Ther. 2013, 9, 16849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada H.; Villa L.; Attanucci J.; Erff M.; Fellows W. K.; Lotze M. T.; Pollack I. F.; Chambers W. H. Cytokine Gene Therapy of Gliomas: Effective Induction of Therapeutic Immunity to Intracranial Tumors by Peripheral Immunization with Interleukin-4 Transduced Glioma Cells. Gene Ther. 2001, 8, 1157–1166. [DOI] [PubMed] [Google Scholar]
- Okada H.; Pollack I. F. Cytokine Gene Therapy for Malignant Glioma. Expert Opin. Biol. Ther. 2004, 4, 1609–1620. [DOI] [PubMed] [Google Scholar]
- Lang F. F.; Bruner J. M.; Fuller G. N.; Aldape K.; Prados M. D.; Chang S.; Berger M. S.; McDermott M. W.; Kunwar S. M.; Junck L. R.; et al. Phase I Trial of Adenovirus-Mediated p53 Gene Therapy for Recurrent Glioma: Biological and Clinical Results. J. Clin. Oncol. 2003, 21, 2508–2518. [DOI] [PubMed] [Google Scholar]
- Wang T. J.; Huang M. S.; Hong C. Y.; Tse V.; Silverberg G. D.; Hsiao M. Comparisons of Tumor Suppressor p53, p21, and p16 Gene Therapy Effects on Glioblastoma Tumorigenicity in Situ. Biochem. Biophys. Res. Commun. 2001, 287, 173–180. [DOI] [PubMed] [Google Scholar]
- Ning J.; Wakimoto H.. Oncolytic Herpes Simplex Virus-Based Strategies: Toward a Breakthrough in Glioblastoma Therapy. Front. Microbiol. 2014, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markert J. M.; Medlock M. D.; Rabkin S. D.; Gillespie G. Y.; Todo T.; Hunter W. D.; Palmer C. A.; Feigenbaum F.; Tornatore C.; Tufaro F.; et al. Conditionally Replicating Herpes Simplex Virus Mutant, G207 for the Treatment of Malignant Glioma: Results of a Phase I Trial. Gene Ther. 2000, 7, 867–874. [DOI] [PubMed] [Google Scholar]
- Thomas C. E.; Ehrhardt A.; Kay M. A. Progress and Problems with the Use of Viral Vectors for Gene Therapy. Nat. Rev. Genet. 2003, 4, 346–358. [DOI] [PubMed] [Google Scholar]
- Pack D. W.; Hoffman A. S.; Pun S.; Stayton P. S. Design and Development of Polymers for Gene Delivery. Nat. Rev. Drug Discovery 2005, 4, 581–593. [DOI] [PubMed] [Google Scholar]
- Akinc A.; Zumbuehl A.; Goldberg M.; Leshchiner E. S.; Busini V.; Hossain N.; Bacallado S. A.; Nguyen D. N.; Fuller J.; Alvarez R.; et al. A Combinatorial Library of Lipid-Like Materials for Delivery of RNAi Therapeutics. Nat. Biotechnol. 2008, 26, 561–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple S. C.; Akinc A.; Chen J.; Sandhu A. P.; Mui B. L.; Cho C. K.; Sah D. W.; Stebbing D.; Crosley E. J.; Yaworski E.; et al. Rational Design of Cationic Lipids for siRNA Delivery. Nat. Biotechnol. 2010, 28, 172–176. [DOI] [PubMed] [Google Scholar]
- Breunig M.; Hozsa C.; Lungwitz U.; Watanabe K.; Umeda I.; Kato H.; Goepferich A. Mechanistic Investigation of Poly (Ethylene Imine)-Based siRNA Delivery: Disulfide Bonds Boost Intracellular Release of the Cargo. J. Controlled Release 2008, 130, 57–63. [DOI] [PubMed] [Google Scholar]
- Matsumoto S.; Christie R. J.; Nishiyama N.; Miyata K.; Ishii A.; Oba M.; Koyama H.; Yamasaki Y.; Kataoka K. Environment-Responsive Block Copolymer Micelles with a Disulfide Cross-Linked Core for Enhanced siRNA Delivery. Biomacromolecules 2009, 10, 119–127. [DOI] [PubMed] [Google Scholar]
- van der Aa L. J.; Vader P.; Storm G.; Schiffelers R. M.; Engbersen J. F. J. Optimization of Poly(Amido Amine)S as Vectors for siRNA Delivery. J. Controlled Release 2011, 150, 177–186. [DOI] [PubMed] [Google Scholar]
- Derfus A. M.; Chen A. A.; Min D. H.; Ruoslahti E.; Bhatia S. N. Targeted Quantum Dot Conjugates for siRNA Delivery. Bioconjugate Chem. 2007, 18, 1391–1396. [DOI] [PubMed] [Google Scholar]
- Elbakry A.; Zaky A.; Liebl R.; Rachel R.; Goepferich A.; Breunig M. Layer-by-Layer Assembled Gold Nanoparticles for siRNA Delivery. Nano Lett. 2009, 9, 2059–2064. [DOI] [PubMed] [Google Scholar]
- Kakizawa Y.; Furukawa S.; Ishii A.; Kataoka K. Organic-Inorganic Hybrid-Nanocarrier of siRNA Constructing through the Self-Assembly of Calcium Phosphate and PEG-Based Block Aniomer. J. Controlled Release 2006, 111, 368–370. [DOI] [PubMed] [Google Scholar]
- Forbes D. C.; Peppas N. A. Oral Delivery of Small RNA and DNA. J. Controlled Release 2012, 162, 438–435. [DOI] [PubMed] [Google Scholar]
- Curiel D. T.; Agarwal S.; Wagner E.; Cotten M. Adenovirus Enhancement of Transferrin Polylysine-Mediated Gene Delivery. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 8850–8854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midoux P.; Monsigny M. Efficient Gene Transfer by Histidylated Polylysine/pDNA Complexes. Bioconjugate Chem. 1999, 10, 406–411. [DOI] [PubMed] [Google Scholar]
- Gary D. J.; Puri N.; Won Y. Y. Polymer-Based siRNA Delivery: Perspectives on the Fundamental and Phenomenological Distinctions from Polymer-Based DNA Delivery. J. Controlled Release 2007, 121, 64–73. [DOI] [PubMed] [Google Scholar]
- Luo D.; Saltzman W. M. Synthetic DNA Delivery Systems. Nat. Biotechnol. 2000, 18, 33–37. [DOI] [PubMed] [Google Scholar]
- Boussif O.; Lezoualc’h F.; Zanta M. A.; Mergny M. D.; Scherman D.; Demeneix B.; Behr J.-P. A Versatile Vector for Gene and Oligonucleotide Transfer into Cells in Culture and in Vivo: Polyethylenimine. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 7297–7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn D. M.; Langer R. Degradable Poly(B-Amino Esters): Synthesis, Characterization, and Self-Assembly with Plasmid DNA. J. Am. Chem. Soc. 2000, 122, 10761–10768. [Google Scholar]
- Green J. J.; Langer R.; Anderson D. G. A Combinatorial Polymer Library Approach Yields Insight into Nonviral Gene Delivery. Acc. Chem. Res. 2008, 41, 749–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunshine J.; Green J. J.; Mahon K. P.; Yang F.; Eltoukhy A. A.; Nguyen D. N.; Langer R.; Anderson D. G. Small-Molecule End-Groups of Linear Polymer Determine Cell-Type Gene-Delivery Efficacy. Adv. Mater. 2009, 21, 4947–4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunshine J. C.; Sunshine S. B.; Bhutto I.; Handa J. T.; Green J. J. Poly(B-Amino Ester)-Nanoparticle Mediated Transfection of Retinal Pigment Epithelial Cells in Vitro and in Vivo. PLoS One 2012, 7, e37543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero-Cázares H.; Tzeng S. Y.; Young N. P.; Abutaleb A. O.; Quiñones-Hinojosa A.; Green J. J. Biodegradable Polymeric Nanoparticles Show High Efficacy and Specificity at DNA Delivery to Human Glioblastoma in Vitro and in Vivo. ACS Nano 2014, 8, 5141–5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunshine J. C.; Peng D. Y.; Green J. J. Uptake and Transfection with Polymeric Nanoparticles Are Dependent on Polymer End-Group Structure, but Largely Independent of Nanoparticle Physical and Chemical Properties. Mol. Pharmaceutics 2012, 9, 3375–3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozielski K. L.; Tzeng S. Y.; Mendoza B. A. H. d.; Green J. J. Bioreducible Cationic Polymer-Based Nanoparticles for Efficient and Environmentally Triggered Cytoplasmic siRNA Delivery to Primary Human Brain Cancer Cells. ACS Nano 2014, 8, 3232–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Patel T. R.; Sirianni R. W.; Strohbehn G.; Zheng M. Q.; Duong N.; Schafbauer T.; Huttner A. J.; Huang Y.; Carson R. E.; et al. Highly Penetrative, Drug-Loaded Nanocarriers Improve Treatment of Glioblastoma. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 11751–11756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito R.; Bringas J. R.; McKnight T. R.; Wendland M. F.; Mamot C.; Drummond D. C.; Kirpotin D. B.; Park J. W.; Berger M. S.; Bankiewicz K. S. Distribution of Liposomes into Brain and Rat Brain Tumor Models by Convection-Enhanced Delivery Monitored with Magnetic Resonance Imaging. Cancer Res. 2004, 64, 2572–2579. [DOI] [PubMed] [Google Scholar]
- Stephen Z. R.; Kievit F. M.; Veiseh O.; Chiarelli P. A.; Fang C.; Wang K.; Hatzinger S. J.; Ellenbogen R. G.; Silber J. R.; Zhang M. Redox-Responsive Magnetic Nanoparticle for Targeted Convection-Enhanced Delivery of O 6-Benzylguanine to Brain Tumors. ACS Nano 2014, 8, 10383–10395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juratli T. A.; Schackert G.; Krex D. Current Status of Local Therapy in Malignant Gliomas—A Clinical Review of Three Selected Approaches. Pharmacol. Ther. 2013, 139, 341–358. [DOI] [PubMed] [Google Scholar]
- Allard E.; Passirani C.; Benoit J.-P. Convection-Enhanced Delivery of Nanocarriers for the Treatment of Brain Tumors. Biomaterials 2009, 30, 2302–2318. [DOI] [PubMed] [Google Scholar]
- Freeman S. M.; Abboud C. N.; Whartenby K. A.; Packman C. H.; Koeplin D. S.; Moolten F. L.; Abraham G. N. The “Bystander Effect”: Tumor Regression When a Fraction of the Tumor Mass Is Genetically Modified. Cancer Res. 1993, 53, 5274–5283. [PubMed] [Google Scholar]
- Boucher P. D.; Ruch R. J.; Shewach D. S. Differential Ganciclovir-Mediated Cytotoxicity and Bystander Killing in Human Colon Carcinoma Cell Lines Expressing Herpes Simplex Virus Thymidine Kinase. Hum. Gene. Ther. 1998, 9, 801–814. [DOI] [PubMed] [Google Scholar]
- DiMaio J. M.; Clary B. M.; Via D. F.; Coveney E.; Pappas T. N.; Lyerly H. K. Directed Enzyme Pro-Drug Gene Therapy for Pancreatic Cancer in Vivo. Surgery 1994, 116, 205–213. [PubMed] [Google Scholar]
- Martuza R. L.; Malick A.; Markert J. M.; Ruffner K. L.; Coen D. M. Experimental Therapy of Human Glioma by Means of a Genetically Engineered Virus Mutant. Science 1991, 252, 854–856. [DOI] [PubMed] [Google Scholar]
- Rubsam L. Z.; Boucher P. D.; Murphy P. J.; KuKuruga M.; Shewach D. S. Cytotoxicity and Accumulation of Ganciclovir Triphosphate in Bystander Cells Cocultured with Herpes Simplex Virus Type 1 Thymidine Kinase-Expressing Human Glioblastoma Cells. Cancer Res. 1999, 59, 669–675. [PubMed] [Google Scholar]
- Tomicic M. T.; Thust R.; Kaina B. Ganciclovir-Induced Apoptosis in Hsv-1 Thymidine Kinase Expressing Cells: Critical Role of DNA Breaks, Bcl-2 Decline and Caspase-9 Activation. Oncogene 2002, 21, 2141–2153. [DOI] [PubMed] [Google Scholar]
- Amano S.; Gu C.; Koizumi S.; Tokuyama T.; Namba H. Timing of Ganciclovir Administration in Glioma Gene Therapy Using HSVtk Gene-Transduced Mesenchymal Stem Cells. Cancer Genomics Proteomics 2011, 8, 245–250. [PubMed] [Google Scholar]
- Denning C.; Pitts J. D. Bystander Effects of Different Enzyme-Prodrug Systems for Cancer Gene Therapy Depend on Different Pathways for Intercellular Transfer of Toxic Metabolites, a Factor That Will Govern Clinical Choice of Appropriate Regimes. Hum. Gene. Ther. 1997, 8, 1825–1835. [DOI] [PubMed] [Google Scholar]
- Mesnil M.; Yamasaki H. Bystander Effect in Herpes Simplex Virus-Thymidine Kinase/Ganciclovir Cancer Gene Therapy: Role of Gap-Junctional Intercellular Communication1. Cancer Res. 2000, 60, 3989–3999. [PubMed] [Google Scholar]
- Bishop C. J.; Ketola T.-M.; Tzeng S. Y.; Sunshine J. C.; Urtti A.; Lemmetyinen H.; Vuorimaa-Laukkanen E.; Yliperttula M.; Green J. J. The Effect and Role of Carbon Atoms in Poly(B-Amino ester)s for DNA Binding and Gene Delivery. J. Am. Chem. Soc. 2013, 135, 6951–6957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunshine J. C.; Akanda M. I.; Li D.; Kozielski K. L.; Green J. J. Effects of Base Polymer Hydrophobicity and End-Group Modification on Polymeric Gene Delivery. Biomacromolecules 2011, 12, 3592–3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzeng S. Y.; Guerrero-Cázares H.; Martinez E. E.; Sunshine J. C.; Quiñones-Hinojosa A.; Green J. J. Non-Viral Gene Delivery Nanoparticles Based on Poly(B-Amino Esters) for Treatment of Glioblastoma. Biomaterials 2011, 32, 5402–5410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green J. J. 2011 Rita Schaffer Lecture: Nanoparticles for Intracellular Nucleic Acid Delivery. Ann. Biomed. Eng. 2012, 40, 1408–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmueli R. B.; Sunshine J. C.; Xu Z.; Duh E. J.; Green J. J. Gene Delivery Nanoparticles Specific for Human Microvasculature and Macrovasculature. Nanomedicine 2012, 8, 1200–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzeng S. Y.; Green J. J. Subtle Changes to Polymer Structure and Degradation Mechanism Enable Highly Effective Nanoparticles for siRNA and DNA Delivery to Human Brain Cancer. Adv. Healthcare Mater. 2013, 2, 468–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.; Sunshine J. C.; Green J. J. Differential Polymer Structure Tunes Mechanism of Cellular Uptake and Transfection Routes of Poly(Beta-Amino Ester) Polyplexes in Human Breast Cancer Cells. Bioconjugate Chem. 2014, 25, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzeng S. Y.; Higgins L. J.; Pomper M. G.; Green J. J. Student Award Winner in the Ph.D. Category for the 2013 Society for Biomaterials Annual Meeting and Exposition, April 10–13, 2013, Boston, Massachusetts: Biomaterial-Mediated Cancer-Specific DNA Delivery to Liver Cell Cultures Using Synthetic Poly(Beta-Amino ester)s. J. Biomed. Mater. Res., Part A 2013, 101, 1837–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltoukhy A. A.; Siegwart D. J.; Alabi C. A.; Rajan J. S.; Langer R.; Anderson D. G. Effect of Molecular Weight of Amine End-Modified Poly(Beta-Amino ester)s on Gene Delivery Efficiency and Toxicity. Biomaterials 2012, 33, 3594–3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Lin J.; Chu J.; Ma Y.; Zeng S.; Luo Q. Activation of Caspase-3 Noninvolved in the Bystander Effect of the Herpes Simplex Virus Thymidine Kinase Gene/Ganciclovir (HSV-tk/GCV) System. J. Biomed. Opt. 2008, 13, 031209. [DOI] [PubMed] [Google Scholar]
- Immonen A.; Vapalahti M.; Tyynela K.; Hurskainen H.; Sandmair A.; Vanninen R.; Langford G.; Murray N.; Yla-Herttuala S. Advhsv-Tk Gene Therapy with Intravenous Ganciclovir Improves Survival in Human Malignant Glioma: A Randomised, Controlled Study. Mol. Ther. 2004, 10, 967–972. [DOI] [PubMed] [Google Scholar]
- Moolten F. L. Drug-Sensitivity (Suicide) Genes for Selective Cancer-Chemotherapy. Cancer Gene Ther. 1994, 1, 279–287. [PubMed] [Google Scholar]
- Castillo-Rodriguez R. A.; Arango-Rodriguez M. L.; Escobedo L.; Hernandez-Baltazar D.; Gompel A.; Forgez P.; Martinez-Fong D. Suicide HSVtk Gene Delivery by Neurotensin-Polyplex Nanoparticles via the Bloodstream and GCV Treatment Specifically Inhibit the Growth of Human Mda-Mb-231 Triple Negative Breast Cancer Tumors Xenografted in Athymic Mice. PLoS One 2014, 9, e97151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D.; Wang A.; Huang H.; Chen Y. PEG-Pblg Nanoparticle-Mediated Hsv-Tk/GCV Gene Therapy for Oral Squamous Cell Carcinoma. Nanomedicine 2008, 3, 813–821. [DOI] [PubMed] [Google Scholar]
- Bernal G. M.; LaRiviere M. J.; Mansour N.; Pytel P.; Cahill K. E.; Voce D. J.; Kang S.; Spretz R.; Welp U.; Noriega S. E. Convection-Enhanced Delivery and in Vivo Imaging of Polymeric Nanoparticles for the Treatment of Malignant Glioma. Nanomedicine 2014, 10, 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodworth G. F.; Dunn G. P.; Nance E. A.; Hanes J.; Brem H.. Emerging Insights into Barriers to Effective Brain Tumor Therapeutics. Front. Oncol. 2014, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salhotra A.; Lal B.; Laterra J.; Sun P. Z.; van Zijl P. C.; Zhou J. Amide Proton Transfer Imaging of 9L Gliosarcoma and Human Glioblastoma Xenografts. NMR Biomed. 2008, 21, 489–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Tryggestad E.; Wen Z.; Lal B.; Zhou T.; Grossman R.; Wang S.; Yan K.; Fu D. X.; Ford E.; et al. Differentiation between Glioma and Radiation Necrosis Using Molecular Magnetic Resonance Imaging of Endogenous Proteins and Peptides. Nat. Med. 2011, 17, 130–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimeco F.; Rhines L. D.; Hanes J.; Tyler B. M.; Brat D.; Torchiana E.; Guarnieri M.; Colombo M. P.; Pardoll D. M.; Finocchiaro G. Paracrine Delivery of Il-12 against Intracranial 9L Gliosarcoma in Rats. J. Neurosurg. 2000, 92, 419–427. [DOI] [PubMed] [Google Scholar]
- Recinos V. R.; Tyler B. M.; Bekelis K.; Sarah Brem Sunshine B.; Vellimana A.; Li K. W.; Brem H. Combination of Intracranial Temozolomide with Intracranial Carmustine Improves Survival When Compared with Either Treatment Alone in a Rodent Glioma Model. Neurosurgery 2010, 66, 530–537. [DOI] [PubMed] [Google Scholar]
- Rhines L. D.; Sampath P.; DiMeco F.; Lawson H. C.; Tyler B. M.; Hanes J.; Olivi A.; Brem H. Local Immunotherapy with Interleukin-2 Delivered from Biodegradable Polymer Microspheres Combined with Interstitial Chemotherapy: A Novel Treatment for Experimental Malignant Glioma. Neurosurgery 2003, 52, 872–880. [DOI] [PubMed] [Google Scholar]
- Tamargo R. J.; Myseros J. S.; Epstein J. I.; Yang M. B.; Chasin M.; Brem H. Interstitial Chemotherapy of the 9L Gliosarcoma: Controlled Release Polymers for Drug Delivery in the Brain. Cancer Res. 1993, 53, 329–333. [PubMed] [Google Scholar]
- Walter K. A.; Cahan M. A.; Gur A.; Tyler B.; Hilton J.; Colvin O. M.; Burger P. C.; Domb A.; Brem H. Interstitial Taxol Delivered from a Biodegradable Polymer Implant against Experimental Malignant Glioma. Cancer Res. 1994, 54, 2207–2212. [PubMed] [Google Scholar]
- Weingart J. D.; Sipos E. P.; Brem H. The Role of Minocycline in the Treatment of Intracranial 9L Glioma. J. Neurosurg. 1995, 82, 635–640. [DOI] [PubMed] [Google Scholar]
- Stojiljkovic M.; Piperski V.; Dacevic M.; Rakic L.; Ruzdijic S.; Kanazir S. Characterization of 9L Glioma Model of the Wistar Rat. J. Neuro-Oncol. 2003, 63, 1–7. [DOI] [PubMed] [Google Scholar]
- Barba D.; Hardin J.; Ray J.; Gage F. H. Thymidine Kinase-Mediated Killing of Rat Brain Tumors. J. Neurosurg. 1993, 79, 729–735. [DOI] [PubMed] [Google Scholar]
- Miletic H.; Fischer Y.; Litwak S.; Giroglou T.; Waerzeggers Y.; Winkeler A.; Li H.; Himmelreich U.; Lange C.; Stenzel W. Bystander Killing of Malignant Glioma by Bone Marrow-Derived Tumor-Infiltrating Progenitor Cells Expressing a Suicide Gene. Mol. Ther. 2007, 15, 1373–1381. [DOI] [PubMed] [Google Scholar]
- Sandmair A.-M.; Turunen M.; Tyynelä K.; Loimas S.; Vainio P.; Vanninen R.; Vapalahti M.; Bjerkvig R.; Jänne J.; Ylä-Herttuala S. Herpes Simplex Virus Thymidine Kinase Gene Therapy in Experimental Rat Bt4c Glioma Model: Effect of the Percentage of Thymidine Kinase-Positive Glioma Cells on Treatment Effect, Survival Time, and Tissue Reactions. Cancer Gene Ther. 2000, 7, 413–421. [DOI] [PubMed] [Google Scholar]
- Sanson M.; Marcaud V.; Robin E.; Valéry C.; Sturtz F.; Zalc B. Connexin 43-Mediated Bystander Effect in Two Rat Glioma Cell Models. Cancer Gene Ther. 2002, 9, 149–155. [DOI] [PubMed] [Google Scholar]
- Rainov N. G. A Phase III Clinical Evaluation of Herpes Simplex Virus Type 1 Thymidine Kinase and Ganciclovir Gene Therapy as an Adjuvant to Surgical Resection and Radiation in Adults with Previously Untreated Glioblastoma Multiforme. Hum. Gene Ther. 2000, 11, 2389–2401. [DOI] [PubMed] [Google Scholar]
- Shand N.; Weber F.; Mariani L.; Bernstein M.; Gianella-Borradori A.; Long Z.; Sorensen A. G.; Barbier N. A Phase 1–2 Clinical Trial of Gene Therapy for Recurrent Glioblastoma Multiforme by Tumor Transduction with the Herpes Simplex Thymidine Kinase Gene Followed by Ganciclovir. Gli328 European-Canadian Study Group. Hum. Gene Ther. 1999, 10, 2325–2335. [DOI] [PubMed] [Google Scholar]
- Harsh G. R.; Deisboeck T. S.; Louis D. N.; Hilton J.; Colvin M.; Silver J. S.; Qureshi N. H.; Kracher J.; Finkelstein D.; Chiocca E. A.; et al. Thymidine Kinase Activation of Ganciclovir in Recurrent Malignant Gliomas: A Gene-Marking and Neuropathological Study. J. Neurosurg. 2000, 92, 804–811. [DOI] [PubMed] [Google Scholar]
- Trask T. W.; Trask R. P.; Aguilar-Cordova E.; Shine H. D.; Wyde P. R.; Goodman J. C.; Hamilton W. J.; Rojas-Martinez A.; Chen S. H.; Woo S. L.; et al. Phase I Study of Adenoviral Delivery of the HSV-tk Gene and Ganciclovir Administration in Patients with Current Malignant Brain Tumors. Mol. Ther. 2000, 1, 195–203. [DOI] [PubMed] [Google Scholar]
- Jacobs A.; Voges J.; Reszka R.; Lercher M.; Gossmann A.; Kracht L.; Kaestle C.; Wagner R.; Wienhard K.; Heiss W. D. Positron-Emission Tomography of Vector-Mediated Gene Expression in Gene Therapy for Gliomas. Lancet 2001, 358, 727–729. [DOI] [PubMed] [Google Scholar]
- Reszka R. C.; Jacobs A.; Voges J. Liposome-Mediated Suicide Gene Therapy in Humans. Methods Enzymol. 2005, 391, 200–208. [DOI] [PubMed] [Google Scholar]
- Pulkkanen K. J.; Yla-Herttuala S. Gene Therapy for Malignant Glioma: Current Clinical Status. Mol. Ther. 2005, 12, 585–598. [DOI] [PubMed] [Google Scholar]
- Jensen S. A.; Day E. S.; Ko C. H.; Hurley L. A.; Luciano J. P.; Kouri F. M.; Merkel T. J.; Luthi A. J.; Patel P. C.; Cutler J. I.; et al. Spherical Nucleic Acid Nanoparticle Conjugates as an RNAi-Based Therapy for Glioblastoma. Sci. Transl. Med. 2013, 5, 209ra152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhise N. S.; Gray R. S.; Sunshine J. C.; Htet S.; Ewald A. J.; Green J. J. The Relationship between Terminal Functionalization and Molecular Weight of a Gene Delivery Polymer and Transfection Efficacy in Mammary Epithelial 2-D Cultures and 3-D Organotypic Cultures. Biomaterials 2010, 31, 8088–8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler B.; Wadsworth S.; Recinos V.; Mehta V.; Vellimana A.; Li K.; Rosenblatt J.; Do H.; Gallia G. L.; Siu I. M.; et al. Local Delivery of Rapamycin: A Toxicity and Efficacy Study in an Experimental Malignant Glioma Model in Rats. Neuro. Oncol. 2011, 13, 700–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess A.; Vigneron S.; Brioudes E.; Labbe J. C.; Lorca T.; Castro A. Loss of Human Greatwall Results in G2 Arrest and Multiple Mitotic Defects Due to Deregulation of the Cyclin B-Cdc2/Pp2a Balance. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 12564–12569. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.