

Abstract
Background
Distinct, partly competing, “waves” have been proposed to explain human migration in(to) today’s Island Southeast Asia and Australia based on genetic (and other) evidence. The paucity of high quality and high resolution data has impeded insights so far. In this study, one of the first in a forensic environment, we used the Ion Torrent Personal Genome Machine (PGM) for generating complete mitogenome sequences via stand-alone massively parallel sequencing and describe a standard data validation practice.
Results
In this first representative investigation on the mitochondrial DNA (mtDNA) variation of East Timor (Timor-Leste) population including >300 individuals, we put special emphasis on the reconstruction of the initial settlement, in particular on the previously poorly resolved haplogroup P1, an indigenous lineage of the Southwest Pacific region. Our results suggest a colonization of southern Sahul (Australia) >37 kya, limited subsequent exchange, and a parallel incubation of initial settlers in northern Sahul (New Guinea) followed by westward migrations <28 kya.
Conclusions
The temporal proximity and possible coincidence of these latter dispersals, which encompassed autochthonous haplogroups, with the postulated “later” events of (South) East Asian origin pinpoints a highly dynamic migratory phase.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-014-1201-x) contains supplementary material, which is available to authorized users.
Keywords: East Timor (Timor-Leste), Island Southeast Asia, Mitochondrial DNA, mtDNA haplogroup P, Human migration, First settlers, Population genetics, Forensic mtDNA analysis, Next generation sequencing, Ion Torrent PGM
Background
The Democratic Republic of Timor-Leste (East Timor) is located in the Lesser Sunda Islands (Nusa Tenggara) of Island (or Maritime) Southeast Asia (ISEA), between Mainland Southeast Asia (MSEA) and Australia, the Indian and the Pacific Ocean. It extends over the eastern part of Timor, the adjacent islands Ataúro and Jaco, and Oecusse, an exclave within western Timor (Indonesia) (Figure 1). Under Portuguese rule since the 16th century, East Timor declared its independence in 1975 and obtained it in 2002 after Indonesian occupation. The country has a population of ~1.1 million, a total area of 14,919 km2 and its highest peak reaches 2,963 meters. Thirty-two languages are spoken in the 13 districts; four working languages are used [1,2]. Archaeological, ethnographic, linguistic and genetic investigations of ISEA have outlined colonization on the crossroads of multiple migrations between today’s (S)EA mainland, Australia and the Pacific islands. The area of eastern Indonesia (including East Timor) has been described as a “melting pot” and a migratory “highway” based on genetic data [3-5]. Initial DNA studies have provided insights into East Timor’s complex composition [3,6-12].
Figure 1.
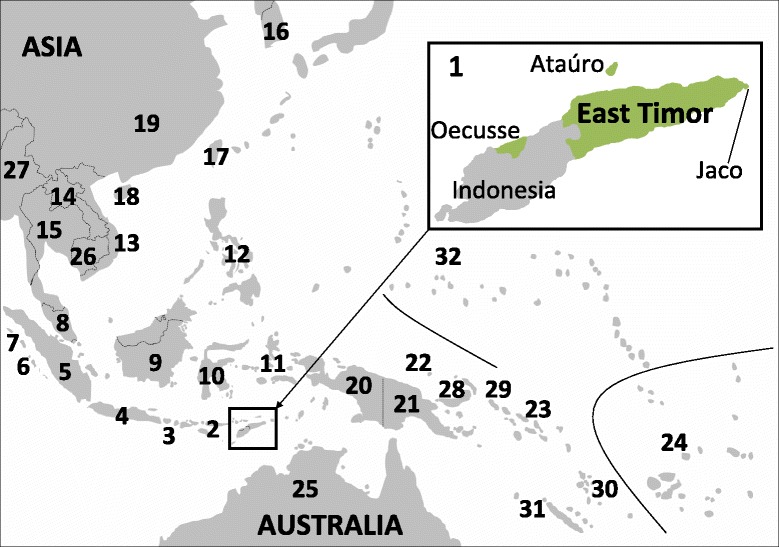
The geographic location of East Timor and the populations included in this study. 1 - East Timor, 2 - Nusa Tenggara, 3 - Bali, 4 - Java, 5 - Sumatra, 6 - Mentawai, 7 - Nias, 8 - Peninsular Malaysia, 9 - Borneo, 10 - Sulawesi, 11 - Moluccas, 12 - Philippines, 13 - Vietnam, 14 - Laos, 15 - Thailand, 16 - South Korea, 17 - Taiwan, 18 - Hainan, 19 - Mixed Han (China), 20 - WNG, 21 - PNG, 22 - Admiralty Islands, 23 - Solomon Islands, 24 – Polynesia/Fiji, 25 - Australia, 26 - Cambodia, 27 - Myanmar, 28 - New Britain and New Ireland (Bismarck Archipelago), 29 - Bougainville, 30 - Vanuatu, 31 - New Caledonia, 32 - Micronesia, 33 - New Zealand Maori (not on map). Populations 2–7, 9–11, and 20 are in Indonesia; 22, 28 and 29 in PNG. The population codes were retained throughout this study. For references see Additional file 11 where subpopulations are indicated with suffixes.
The routes and timing of human dispersal in the area remain to be fully clarified (cf. [13]). In any assumption, the influence of a changing climate needs to be considered. The sea level in ISEA rose after ~70 thousand years ago (kya), then sank from ~30 kya during the last glacial maximum (LGM) until ~18 kya, whereupon the landmasses of Sunda(land) (i.e. Sumatra, Java, Borneo, the Malay Peninsula and nearby islands) and Sahul (or Meganesia; i.e. Australia, New Guinea, Tasmania and nearby islands) became partly submersed again by the rising sea. Dramatic coastline shifts occurred ~14.5, 11.5, and 7.5 kya, creating the extant archipelago topography [4,5,14-16]. Timor was an island even during the LGM.
The pioneer migration out of Africa brought the first anatomically modern humans, likely hunter-gatherers and beachcombers able to perform (short) voyages, to Sahul ~60-40 kya [5,17-21]. The expansion from Sunda to Sahul might have occurred along northern (via Sulawesi) or southern Wallacea (via Nusa Tenggara and Timor) [5,14,15,20,22-28]. The so-called Austro-Melanesian, “negrito”, or Australoid first settlers are associated with non-Austronesian (i.e. Papuan) languages, spoken as far east as the Solomon Islands [15,24,29-31]. Their initial “fast train” migration likely left individuals carrying deep-rooting mtDNA founder haplotypes along its course. Autochthonous mtDNA lineages of ISEA, Melanesia and Australia support a long-term in situ development and date the first modern human arrival to >48 kya [3,5,14,26,28-30,32-34]. The presence of Upper Paleolithic settlers in the region is confirmed by findings in East Timor (>42 kya) [20,27], Borneo (~46 kya) [33], eastern Papua New Guinea (PNG) (~43-49 kya) [35], Melanesia and Australia (≥48 kya) [3,5,25,30,36]. Submergence could have caused a scarcity of sites (cf. [37,38]). Subsequent migrations led to admixture with or replacement of the present foragers and shaped the genetic diversity of ISEA. A major southward expansion out of Taiwan is postulated from mid-Holocene (~8-4 kya) linguistic and cultural changes associated with the spread of livestock domestication, agriculture and Austronesian (AN) speakers. Their arrival in eastern Indonesia (and Timor) is estimated to ~4 kya from Neolithic findings. This migration has found support by dispersal patterns of mtDNA lineages with ample basal diversity in Taiwan, and is connected to the East Asian (EA) proportion of the genetic pool in ISEA and AN speaking populations [3,5,15,24,28-30,39-42]. However, the so-called “out of Taiwan” (OOT) event has been questioned both in linguistics as single source of AN languages [30] and in genetics, since the model does not predict a direction and AN speakers share few mtDNA lineages despite matrilocality [5,43]. Moreover, archaeobotanic research indicates plant domestication in ISEA before this presumed advent [16]. A two-step arrival, ~60-40 kya and ~8-4 kya, does not appear to fully reflect the demographic history of the region [24,44]. MtDNA coalescence ages, phylogenetic and dispersal patterns that neither fit with long term in situ development >40 kya nor southward population expansions <8 kya indicate a broader timeframe and geographic origin of migrations [5,28,30]. Additional mid-Holocene expansions, labelled “express train” and “slow boat” [28,45], might have originated elsewhere, possibly within ISEA, MSEA or Near Oceania [3,5,24,28,30]. To account for these inconsistencies and to integrate the expected effects of a changing environment, such as facilitated migration by exposed and forced displacement by submersing land [15,24], a more comprehensive model needs to be outlined to explain the extant maternal genetic landscape of ISEA. The classical “waves” might rather represent longer periods of migration than short, distinct events. The second “wave” may be extended to a period of ongoing gene flow in the late Pleistocene and early Holocene between ~40/30-10/5 kya, with substantial, recurrent population expansions and shifts between Asia and Sahul and within ISEA, that may include the dispersals postulated from lineage-specific investigations [5,15,24,28,30,40,42,46]. These intermediate migrations are supported by archaeological, linguistic, Y-chromosomal and autosomal DNA evidence ([5,16,30]: “early train”). Further complexity might be added by gene flow from India into Australia ~4.2 kya [47] and “historic” movements from China, Arabia and India associated with trade and the spread of religions [3,5].
Even in the classical scenario, East Timor is of high genetic interest: it could have accommodated the “last step” before the colonization of Sahul, westward (back) migrations have been suggested, and the southward OOT migration could have extended there [3,5,27,28,40,48]. Nevertheless, mtDNA composition of its population is scarcely described: the available data comprise 38 hypervariable segment I (HVS-I) sequences [3] and 133 not individually reported haplotypes [10]. We here present the first representative complete mtDNA control region (CR) reference dataset for East Timor, comprising 324 country-wide samples (Additional files 1 and 2) sequenced according to highest forensic quality standards. Our aim was to explore the potential of this geographically restricted sample in providing insights into the history of (particularly the less investigated initial) human dispersal into and over the entire region. We thus also generated complete mitogenome sequences of 17 samples and fundamentally refined the phylogeny of mtDNA haplogroup P1 that appeared most promising for phylogeographic reconstructions. We used the Ion Torrent Personal Genome Machine (PGM) on an mtDNA population sample in a forensic environment as stand-alone approach for the first time; this study therefore significantly contributes towards the implementation of massively parallel sequencing (MPS) into high quality mtDNA typing routine.
Results and discussion
Ample mtDNA variation of the East Timor population
The diverse haplogroup spectrum of East Timor revealed in this first representative mtDNA study illustrates its position at the crossroads of several migrations between ISEA, Melanesia and Australia – and likely also the area’s rapid change from a large continental landmass to an archipelago [4]. We detected 164 different CR haplotypes in the 324 mtDNAs (disregarding cytosine indels around nps 16193, 309 and 573); 116 of these (35.8%) were unique in the dataset. Macro-haplogroup M comprised 50.3% of the samples in 14 haplogroups; macro-haplogroup N 49.7% in 22 haplogroups (Table 1). Most frequent were Q1 (14.2%) and M7c1 (13.9%). Using all information, 11.1% of the samples could be assigned to a terminal “twig” of the mtDNA Phylotree ([49], build 16) (10.8% based on CR). The most frequent CR haplotypes, relative to the revised Cambridge Reference Sequence (rCRS) [50], were 73G 146C 199C 263G 309.1C 315.1C 489C 523del 524del 16223T 16295T 16362C 16519C (haplogroup M7c1) and 73G 152C 249del 263G 309.1C 315.1C 521del 522del 523del 524del 16129A 16172C 16294T 16304C 16362C 16519C (haplogroup F1a4a1), with 4.0% each (Additional file 3). Figure 2 depicts the proportions of haplogroups, their phylogenetic relations and postulated geographic origin. The latter reflects influence from two geographic macro-regions: mainly (S)EA lineages were detected (56.5%; haplogroups B, D, F, M7, M10, M21, M71, M73, N21, R9). A considerable proportion indicated expansions of EA precursors in(to) ISEA and Polynesia (22.8%; haplogroups B4a1a1, E), followed by mtDNAs with indigenous Melanesian/eastern Indonesian/Near Oceanian origin (19.4%; haplogroups P, Q, R14). Admixture from outside eastern Eurasia could only be concealed among the unresolved M* and R* mitogenomes (1.2%). The 324 novel East Timor mtDNA haplotypes (307 CR sequences, 17 complete mitogenomes) are illustrated in Additional files 3 and 4, available in GenBank [KJ655583-KJ655889, KJ676774-KJ676790; http://www.ncbi.nlm.nih.gov/genbank] and included in the European DNA Profiling Group MtDNA Population Database (EMPOP) [EMP00534; http://empop.org] [51].
Table 1.
MtDNA haplogroup frequencies in our East Timor sample
| Haplogroup | n | Frequency (%) |
|---|---|---|
| B4* | 4 | 1.2 |
| B4a1* | 18 | 5.6 |
| B4a1a1 | 16 | 4.9 |
| B4a1a3a | 5 | 1.5 |
| B4b1 | 8 | 2.5 |
| B4c1b2a2 | 9 | 2.8 |
| B4c2 | 2 | 0.6 |
| B5b* | 4 | 1.2 |
| B5b1c | 8 | 2.5 |
| D5b1c1 | 5 | 1.5 |
| D6a | 1# | 0.3 |
| E1a1a | 39 | 12.0 |
| E1a2 | 5 | 1.5 |
| E1b | 7 | 2.2 |
| E2 | 7 | 2.2 |
| F1a1a | 1 | 0.3 |
| F1a2 | 1 | 0.3 |
| F1a3a | 16 | 4.9 |
| F1a4a1 | 29 | 9.0 |
| F3b1a | 1 | 0.3 |
| M* | 2 | 0.6 |
| M7c1 | 45 | 13.9 |
| M10 | 1 | 0.3 |
| M21b | 1# | 0.3 |
| M71a2 | 1 | 0.3 |
| M73a | 2 | 0.6 |
| N21a | 2 | 0.6 |
| P1* | 2 | 0.6 |
| P1d | 7# | 2.2 |
| “P1e”§ | 5# | 1.5 |
| Q1 | 46 | 14.2 |
| Q3 | 1# | 0.3 |
| R* | 2 | 0.6 |
| R9c1* | 3 | 0.9 |
| R9c1a | 15 | 4.6 |
| R9c1b2 | 1 | 0.3 |
| R14 | 2 | 0.6 |
| Total | 324 | 100 |
#classified after complete mitogenome sequencing.
§postulated novel clade.
n - number of mtDNAs. Haplogroups according to [49], build 16.
Figure 2.

Schematic phylogenetic tree of all haplogroups observed in the 324 East Timor samples. Their phylogenetic relation and postulated geographic origin are indicated. The sizes of the circles correspond to haplogroup frequencies. Stem lengths are of no information content. The tree is rooted in the MRCA. All information available was used. Haplogroups are according to [49], build 16.
New insights from complete mitogenomes
Our analyses at the highest resolution substantially contributed to elucidate and refine the phylogeny of haplogroup P1. This is not “monotypic” as previously described ([49], build 16), which would be uncommon for a successful founder. The most parsimonious tree reconstructed from the available 20 (thereof 12 novel) mitogenomes ([52-55], this study) confirmed subclade P1d and added considerable internal variation to P1d1 with five additional basal branches (Figure 3, Additional file 5). Five East Timor samples and a PNG singleton [52] defined a novel branch characterized by the motif P1-152-13722-@16176 (relative to the rCRS [50]) that we tentatively called “P1e”, expanding the current mtDNA nomenclature [49]. The East Timor P1e quintet additionally shared the transition np 8286, resulting in a previously studied polycytosine stretch (e.g., [56]). Two mitogenomes [52,53] indicated additional basal P1 branches. The earlier proposed subclades P1a-c (cf. [48]) were defined partly by homoplasic markers and group with P1d and P1e lineages of our novel phylogeny. Multiple back-mutations of transitions currently defining haplogroup P1 ([49], build 16), viz. nps 212, 16176, and 16266, indicate the need for an updated nomenclature relying solely on the transition at np 16357 as CR marker.
Figure 3.
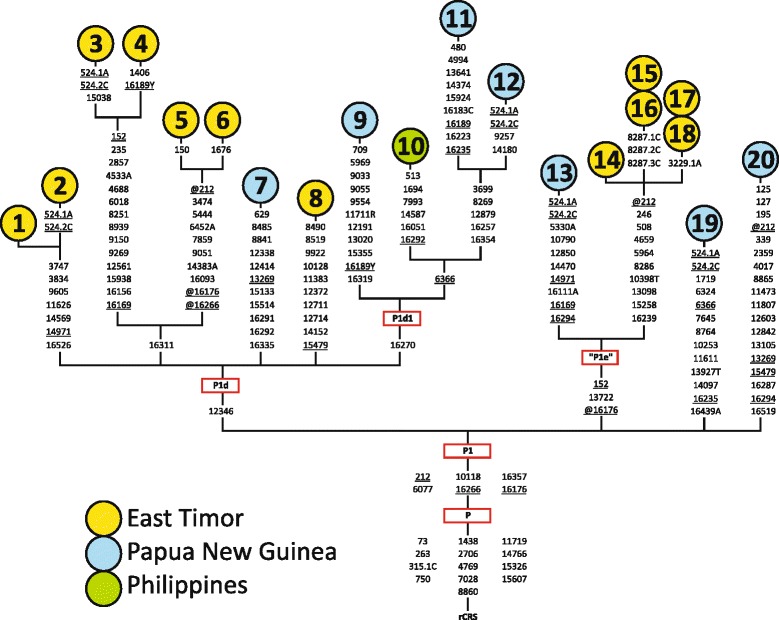
The refined phylogeny of haplogroup P1. The 12 novel completely sequenced haplogroup P1 mitogenomes from East Timor are shown together with the eight previously published. All differences are indicated (disregarding cytosine insertions after nps 309 and 16193) relative to the rCRS [50]. Haplogroups are according to [49], build 16. “P1e” is a novel suggestion. Bases are indicated according to the IUBMB nucleotide code. The prefix @ indicates the reversion of a mutation occurring earlier in the phylogeny. Underlined mutations are recurrent in the tree. For details and references, see Additional file 5.
The five completely sequenced non-P1 mitogenomes revealed novel lineages within haplogroups D6a, M21b, M73a, Q3, and R9c1b2 (Additional file 6).
The genetic position of East Timor
Based on HVS-I and HVS-II data, East Timor (29%) was amid the other island population in the unique haplotype proportions (8-47%, median 45.5%) and the intra-population random match probabilities (RMPs) (1:37–1:67, East Timor: 1:59). Mainland populations yielded higher proportions of unique haplotypes (median: 58.5%) and lower RMPs, likely due to smaller sample sets from islands, colonization bottlenecks and founder effects/isolation (cf. [41]). In total, 18.6% of the East Timor haplotypes (comprising 42.6% of the samples) were also found in other populations. The Philippine population shared 18.1% of its haplotypes (33.1% of its samples) with East Timor, followed by Taiwan (12.7|23.3%), all other populations were ≤8.6|6.3%. East Timor ranked second (10.70) in the intra-population numbers of mean pairwise differences (MPD) (9.08 (Philippines) - 11.88 (PNG)) (Table 2, Additional file 7). The inter-population MPD maxima of 13.52 (uncorrected) and 2.46 (corrected) derived from comparisons with the PNG sample. For East Timor, the inter-population MPD were found highest also with PNG and lowest with the Philippines. The pairwise FST values for East Timor indicate a pronounced genetic differentiation if compared with PNG (0.12) and minor ones with all other populations (≤0.05). The values including PNG were 0.12-0.20, while all other comparisons yielded ≤0.05. AMOVA revealed that the observed genetic variation was mainly attributable to variance within populations (95.1%) (Additional file 8).
Table 2.
Genetic diversity of the East Timor population and 10 surrounding populations using HVS-I and HVS-II
| Population | Reference | n a | hts b | Unique hts | MPD c | RMP d |
|---|---|---|---|---|---|---|
| 1A - East Timor | this study | 324 | 145 | 93 (29%) | 10.705 ± 4.886 | 0.017 |
| 8 - Peninsul. Malaysia* | [57] | 205 | 152 | 115 (56%) | 10.353 ± 4.743 | 0.009 |
| 12 - Philippines | [28] | 130 | 83 | 61 (47%) | 9.080 ± 4.206 | 0.023 |
| 13 - Vietnam | [58] | 187 | 153 | 134 (72%) | 10.104 ± 4.637 | 0.009 |
| 14 - Laos | [59] | 214 | 163 | 130 (61%) | 10.185 ± 4.670 | 0.009 |
| 15 - Thailand | [60] | 190 | 133 | 103 (54%) | 9.962 ± 4.576 | 0.012 |
| 16 - South Korea | [61] | 692 | 471 | 378 (54%) | 9.372 ± 4.309 | 0.005 |
| 17 - Taiwan | [45] | 640 | 122 | 51 (8%) | 9.642 ± 4.425 | 0.027 |
| 18 - Hainan | [62] | 290 | 137 | 74 (26%) | 10.315 ± 4.721 | 0.015 |
| 19 - Mixed Han (China) | [63] | 262 | 242 | 228 (87%) | 10.478 ± 4.792 | 0.005 |
| 21 - Papua New Guinea | [34,64] | 142 | 85 | 48 (34%) | 11.881 ± 5.408 | 0.016 |
*(Singapore).
anumber of mtDNAs.
bnumber of haplotypes.
cmean number of pairwise differences.
drandom match probability.
Reading frame: nps 16080–16193, 16194–16365, 73–300; total sample size: n = 3,276. For geographic locations, see Figure 1.
Restricting the comparisons to HVS-I to expand the population number, East Timor (15%) again positioned among the island populations (except Borneo) in the unique haplotype proportions (median: 14.5%). The values for mainland populations, except Australia and Peninsular Malaysia, were generally higher also here (median: 45.5%). East Timor yielded an RMP of 1:26, similar to other ISEA populations (Bali, Java, Sulawesi, and the Philippines) (Table 3), and 54.3% of its haplotypes (comprising 74.0% of the samples) were also found in the 24 surrounding populations. In general, I(S)EA and Oceanian populations shared greater proportions with East Timor than Mainland Asians. The Moluccas ranked highest (41.7%) in the shared haplotype proportions and second in the proportions of individuals, where Polynesia was highest (72.1%). This and similarly high values in other Oceanian populations were caused by the outstanding prevalence of shared B4a1(a1) haplotypes. The influence of greatly differing haplotype numbers and haplogroup spectra (see below) is also visible for Nusa Tenggara and New Guinea (NG) that shared rather low (~10%) haplotype proportions with East Timor compared to the other (geographically more distant) ISEA populations, but large sample proportions (49.6% resp. 24.9%). Australia ranked lowest in both aspects (4.0|9.6%) (Additional file 9). The highest numbers of intra-population MPD were obtained from NG (7.04), Polynesia was lowest (1.79). East Timor again ranked high (5.98), closest to Nusa Tenggara (5.82). The inter-population MPD maxima for HVS-I were 9.59 (uncorrected) and 5.18 (corrected). For East Timor, the lowest (uncorrected) number of inter-population MPD was calculated with Sulawesi (5.57) and the highest with WNG (8.16). Based on the corrected numbers, MPD of East Timor were lowest with the Moluccas (0.04) and Nusa Tenggara (0.07); the remaining values were more than double, up to 2.12 (Polynesia). AMOVA revealed that 87.6% of the variation was attributable to differences within populations (Additional file 10). We used the pairwise HVS-I FST values (discussed in Additional file 10) for an MDS analysis to depict genetic distances between populations (Figure 4). A limited genetic differentiation indicating high gene flow between most (S)EA populations was reflected by the resulting large main cluster. East Timor located in a separate cluster with the Nusa Tenggara and the Moluccan samples, slightly shifted towards the outlying “eastern” populations from NG and the Admiralties. Little gene flow was also indicated with the outlying populations from Polynesian and the Solomon Islands; this mirrored the distant position shown by the genetic indices. Closer to the main cluster, about equidistant to East Timor, were the ISEA outliers Nias, Java, Mentawai and, interestingly, also the Australian sample in the small range analyzed. All outlier islands were characterized by low haplotype diversity/high RMP (see above).
Table 3.
Genetic diversity of the East Timor population and 24 surrounding populations using HVS-I
| Population | Reference | n a | hts b | Unique hts | MPD c | RMP d |
|---|---|---|---|---|---|---|
| 1 - East Timor (pooled) | [3], this study | 362 | 94 | 52 (14%) | 5.976 ± 2.857 | 0.039 |
| 1A - East Timor | this study | 324 | 87 | 49 (15%) | 5.938 ± 2.841 | 0.039 |
| 2 - Nusa Tenggara | [3,5,24,65] | 1699 | 345 | 178 (10%) | 5.816 ± 2.783 | 0.017 |
| 3 - Bali | [5,24] | 570 | 138 | 71 (12%) | 5.643 ± 2.711 | 0.034 |
| 4 - Java | [5,24] | 97 | 49 | 31 (32%) | 5.570 ± 2.698 | 0.035 |
| 5 - Sumatra | [5,17,40] | 228 | 107 | 70 (31%) | 5.520 ± 2.664 | 0.023 |
| 6 - Mentawai | [5] | 128 | 16 | 4 (3%) | 4.687 ± 2.311 | 0.126 |
| 7 - Nias | [5,41] | 499 | 61 | 29 (6%) | 4.766 ± 2.334 | 0.153 |
| 8 - Peninsular Malaysia* | [17,57] | 470 | 158 | 98 (21%) | 6.350 ± 3.016 | 0.029 |
| 9 - Borneo | [24] | 157 | 102 | 77 (49%) | 5.345 ± 2.593 | 0.017 |
| 10 - Sulawesi | [5,24] | 437 | 153 | 108 (25%) | 4.804 ± 2.351 | 0.039 |
| 11 - Moluccas | [24,65] | 74 | 36 | 23 (31%) | 5.425 ± 2.643 | 0.055 |
| 12 - Philippines | [24,28] | 483 | 140 | 89 (18%) | 5.052 ± 2.457 | 0.035 |
| 13 - Vietnam | [58] | 187 | 119 | 91 (49%) | 5.445 ± 2.634 | 0.018 |
| 14 - Laos | [59] | 214 | 127 | 95 (44%) | 5.689 ± 2.737 | 0.018 |
| 15 - Thailand | [60] | 190 | 108 | 79 (42%) | 5.584 ± 2.694 | 0.02 |
| 16 - South Korea | [61] | 692 | 306 | 215 (31%) | 4.853 ± 2.370 | 0.019 |
| 17 - Taiwan (indigenous) | [24,45] | 718 | 86 | 35 (5%) | 5.197 ± 2.518 | 0.042 |
| 18 - Hainan | [62] | 293 | 99 | 45 (15%) | 5.338 ± 2.583 | 0.025 |
| 19 - Mixed Han (China) | [63] | 262 | 194 | 159 (61%) | 5.533 ± 2.668 | 0.008 |
| 20 - West New Guinea | [66] | 227 | 74 | 39 (17%) | 7.045 ± 3.320 | 0.046 |
| 21 - Papua New Guinea | [34,64] | 201 | 72 | 34 (17%) | 6.733 ± 3.188 | 0.046 |
| 22 - Admiralty Islands | [67,68] | 203 | 41 | 20 (10%) | 5.693 ± 2.739 | 0.152 |
| 23 - Solomon Islands | [29] | 703 | 102 | 41 (6%) | 3.660 ± 1.855 | 0.208 |
| 24 - Polynesia | [65,67] | 394 | 56 | 28 (7%) | 1.793 ± 1.038 | 0.340 |
| 25 - Australia (indigenous) | [34,69] | 146 | 50 | 22 (15%) | 4.555 ± 2.252 | 0.042 |
*including Singapore.
anumber of mtDNAs.
bnumber of haplotypes.
cmean number of pairwise differences.
drandom match probability.
Reading frame: nps 16080–16180, 16195–16354; total sample size: n = 9,634. For geographic locations, see Figure 1.
Figure 4.
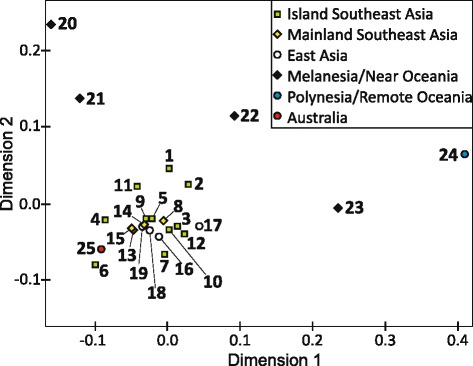
MDS plot displaying the genetic distances between East Timor and 24 surrounding populations. Based on pairwise F ST values in HVS-I (nps 16080–16180, 16195–16354). For population codes, see Figure 1. For details and references, see Table 3, Additional file 10.
Elucidating the initial settlement from its maternal footprints
Haplogroup P exhibited widespread distribution in both northern and southern Sahul (and beyond) as the only autochthonous lineage. In our efforts to reconstruct the initial migrations between Sunda and Sahul we therefore particularly focussed on this haplogroup. P generally occurred at frequencies <10%, but central and southern populations revealed proportions up to 56.3%. In most cases (also in East Timor), these mtDNAs completely or mainly consisted of P1 representatives; the highest P1 proportion was reached in PNG (43.8%). P1 lineages not further resolved by HVS-I (P1*) and the derived P1d1 mostly occurred together. The remaining P lineages were less widespread even if combined and always occurred in parallel with and in smaller proportions than P1 (when more than a P singleton was present), except in Australia (where P1 was virtually absent, see below) and the Philippines, as well as New Caledonia, the Admiralty Islands and the Kula Ring (where more unassigned P than P1 samples were found). PxP1 proportions >3.9% were only revealed in the Philippines, NG, Australia, and New Caledonia (up to 32.0%) (Figure 5, Additional file 11).
Figure 5.

Distribution of haplogroup P mtDNAs in East Timor and surrounding populations. (A) P1 and P1d1. These categories discernible with HVS-I data enabled equal treatment of all populations included. All other, including unassigned, P clades are combined into “PxP1”; (B) P1-P10. All available information was considered (reading frames varied). The total of haplogroup P has been set to 100% for every population to better depict (small) proportions. Inconclusive P samples were denoted as “P*”. Frequencies are indicated by colored circle segments. See legends for color codes. Population reference numbers located within a circle indicate that the analyzed haplogroup(s) were not found in this population. Populations: 1 - East Timor, 2 - Nusa Tenggara (2A - West Timor, 2B - Lembata, 2C - Sumba, 2D - Lombok, 2E - Flores, 2F - Solor, 2G - Adonara, 2H - Pantar, 2K - Alor), 3 - Bali, 4 - Java, 5 - Sumatra, 6 - Mentawai, 7 - Nias, 8 - Peninsular Malaysia, 9 - Borneo, 10 - Sulawesi, 11 - Moluccas (11A - Ternate, 11B – Ambon), 12 - Philippines, 13 - Vietnam, 14 - Laos, 15 - Thailand, 16 - South Korea, 17 - Taiwan, 18 - Hainan, 19 - Mixed Han (China), 20 - WNG (20A - highlands, 20B - lowlands, 20C - Southwest and Lowland Riverine), 21 – PNG (21A - pooled, 21B - Wewak, 21C - Bundi, 21D - Gidra, 21E - East Sepik, 21F- Kula Ring), 22 - Admiralty Islands, 23 - Solomon Islands, 24 - Polynesia/Fiji, 25 - Australia (25A - Northwest, Northern Territory, Far North Queensland, 25B - New South Wales, Paakintji, Ngiyambaa), 26 - Cambodia, 27 - Myanmar, 28 - New Britain and New Ireland, 29 - Bougainville, 30 - Vanuatu, 31 - New Caledonia, 32 - Micronesia, 33 - New Zealand Maori. For more information, see Additional file 11.
Both an eastern Indonesian and a Melanesian (Near Oceanian) origin for haplogroup P(1) have been proposed [3,28,48,70]. The high-resolution phylogeographic reconstruction using the 20 available complete mitogenomes already indicates that not even the basal P1 diversity is concentrated in eastern Indonesia (Figure 3): all sublineages were also found in NG, and samples from that island and the Philippines pointed out additional branches at all hierarchical levels [52-55]. A western Melanesian cradle is also supported by published CR data. Various P1 sublineages are dispersed in eastern Indonesia and beyond, but both the frequency and diversity peak lay in NG, whose central role was confirmed by the remaining P clades. Besides the more widespread P4a, NG shared P3 with Australian populations, and P9 only found in Taiwan and the Philippines (the latter based on a CR pattern, maintained until Phylotree’s build 15 [49]). The only P lineages not found in NG were P10 from the Philippines, and those restricted to Australia, viz. P4b and P5-8. Vice versa, P2 is only reported in NG, but has not been assessed in most other populations (Figure 5, Additional file 11).
The coalescent ages of the autochthonous Southwest Pacific haplogroup P fit intriguingly well with the archaeological dating of the first settlements in this area (~48-40 kya): it was estimated to 51.7-65.4 ky based on CR [5,34,64,71] and 54.8 ky using complete mitogenomes. The latter calculations yielded 41.3-53.0 ky for P2’10, P3, P4 and P6; estimates for the remaining clades were not made due to paucity of data [72]. Our maximum likelihood (ML) point estimate for haplogroup P1 considering the entire mtDNA molecule and all 20 sequences was 36.1 ky, P1d was dated at 31.9 ky, P1d1 at 28.1 ky and P1e at 27.4 ky. Rho (ρ) statistics yielded remarkably similar estimates completely overlapping with the ML ranges (Table 4). Thus, there is no indication for an age underestimation by ρ statistics, an effect noticed using low-resolution data [73]. The novel dating of P1 was consistent with results using fewer samples [72]. Partial mitogenome-based age reports for P1 were 28.0-53.8 ky [3,5,34,44,48,71]. It appears that haplogroup P and its sublineages arose in a successful period at the cusp of or shortly after the first arrival of anatomically modern humans, merely the most successful clade P1 occurred separately and later.
Table 4.
Molecular divergences and age estimates obtained by Maximum Likelihood and Rho statistics for haplogroup P1
| Haplogroup | N a | All nucleotide substitutions | |||||||
|---|---|---|---|---|---|---|---|---|---|
| ML b | S.E. c | T (ky) d | ΔT (ky) d | ρ | σ | T (ky) d | ΔT (ky) d | ||
| P1 | 20 | 12.8 | 1.2 | 36.1 | 3.7 | 12.5 | 1.4 | 35.3 | 4.3 |
| > P1d | 11 | 11.4 | 1.0 | 31.9 | 3.1 | 11.1 | 1.3 | 31.1 | 3.9 |
| >> P1d1 | 4 | 10.1 | 1.1 | 28.1 | 3.2 | 9.5 | 1.9 | 26.3 | 5.6 |
| > P1e | 6 | 9.9 | 1.6 | 27.4 | 4.8 | 9.7 | 2.7 | 26.9 | 8.1 |
anumber of complete mtDNA sequences.
bmaximum likelihood molecular divergence.
cstandard error.
dage estimates using the corrected molecular clock [74].
The P lineage spectrum in Australia is distinct from those from all other areas (as is possibly that of the Philippines and Taiwan). It is made up almost completely of the most ancient clades (P4b, P5-8; 48.0-52.9 ky); P3, rare and shared with NG at a very low level (≤1.7%), is also rather old (41.2 ky). Despite a singular report of the only other shared P lineage, the youngest and elsewhere predominant P1, this contradicts an origin of haplogroup P in central/southern Sahul (Australia) followed by (back-)migrations to northern Sahul (NG). Rather it favours a southward colonization prior to ~36 kya, when P1 developed, and little later north–south exchange even though a landbridge existed until ~8 kya [25,34,44,53,69,75-77]. Developing this scenario further, the more recent P clades, such as P1, would have arisen later during an “incubation” stage in northern Sahul from root P* mtDNA carriers left behind. Therefore, they were not or very limitedly represented on the forefront of settlers that proceeded southward and gave rise to Australian mtDNA diversity (cf. [78] for a similar scenario for South America). Our age estimates also contraindicate that haplogroup P1 is old but simply did not move on [77]. Intriguingly, the singular reported Australian P1 haplotype was revealed in a Northeast Aboriginal population considered closer to SEA Natives than other Australian groups in oral history and due to anthropological traits [77] (cf. the “negrito” ancestral connection hypothesis [31]). Apart from that, it could also derive from more recent, possibly individual, migration or displacement – a general caveat when equating lineage ages with timing of migration.
The synopsis of haplogroup P data indicates that eastern Indonesia, including East Timor, was not the cradle of this founder lineage, thus probably did not lie on the initial main route that likely led into northern Sahul, but was rather populated from there after haplogroup P1 (36.1 kya), P1d1 (28.1 kya), and likely even P1e (27.4 kya) (whose dispersal data are scant), had arisen. The landmass increase from ~30 kya (see above) could have triggered the demographic expansion at ~28-24 kya indicated in the Bayesian Skyline Plot (BSP) from western Melanesian P1 sequences (Figure 6), the time when all known P1 subclades emerged (29.7 ± 2.3 kya) (Table 4) - and could also have led to a geographic expansion that brought, among others, carriers of haplogroup P1 westward. The lack of PxP1 lineages in East Timor may be caused by the fact that a finite sample does not cover each and every rare lineage, or by drift, as they are found in and around eastern Indonesia [3,5,55]. For the same reasons - and the lack of mitogenomic data - we would not consider a more detailed dating of the westward movement reliable as it is based on the presence of only a P1e sublineage and the lack of P1d1 in East Timor as termini ante resp. post quem. The second most widespread P lineage, P4a, is much younger (18.6 ky) [72], absent from Australia, could also have arisen in northern Sahul, thus supporting an ongoing dynamic phase (Figure 5, Additional file 11).
Figure 6.
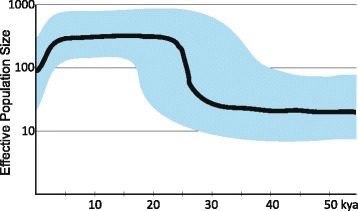
Bayesian Skyline Plot of mtDNA haplogroup P1. The hypothetical effective female population size is based on the complete P1 mitogenomes (Additional files 4 and 5) assuming a generation time of 25 years.
The distribution of haplogroups Q and Q1 resembled that of P and P1 to a very high degree. Q carriers were only absent in the southern- and northernmost populations included; the possible cradle of this indigenous haplogroup, again lay in NG: proportions reached >70%, and only there and in the Bismarck Archipelago, all three Q clades were found. Proportions >10% were revealed in the Moluccas (11.6%), East Timor (15.5%) and its northwest Nusa Tenggara neighbor islands (≤32.8%), as well as in western Melanesia (NG and its Northeast, ≤71.7%). Haplogroup Q proportions consisted predominantly of Q1, the most frequent “pioneer” lineage of eastern Indonesia in general [3] and East Timor, highly diverse even in the CR (Additional file 3). Q2 is widespread, and generally rare (except in the Bismarck Archipelago). Lineage Q3 was found on the latter, in this study (<1%), and in NG (≤14.9%), however, only one subclade carries HVS-I polymorphisms. A single Q sample was reported from Australia, a Q2 – again from the North [34] (Figure 7, Additional file 11). Haplogroup Q has been dated to 32.0-44.5 (even 74.6) ky using partial [5,34,44,64] and 37.5 ky using complete mitogenomes [72]. The age estimates for Q1, Q2, and Q3 from the latter study were 18.1, 28.7 and 31.0 ky, respectively, and widely ranged (2.9-48.0 ky) when partial mitogenomic data were employed [3,5,34,44,48,64,71].
Figure 7.
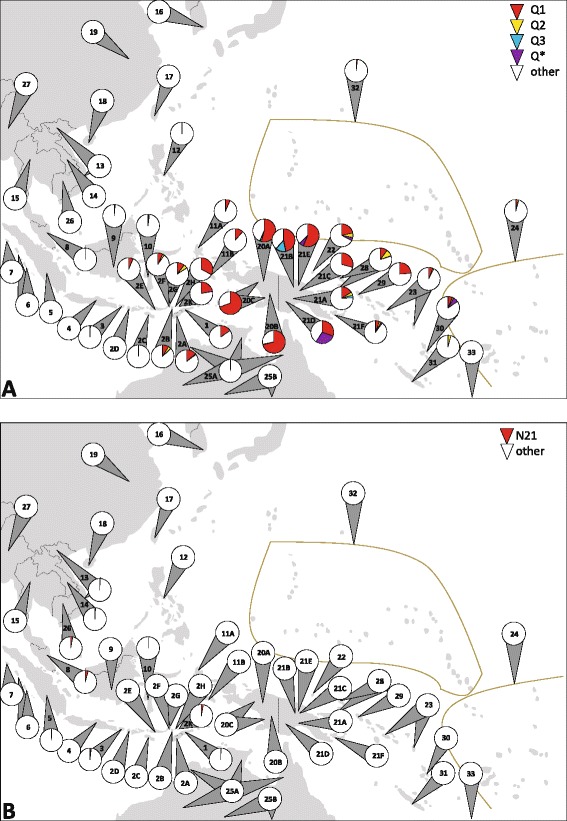
Distribution of mtDNA haplogroups Q and N21 in East Timor and surrounding populations. (A) Haplogroups Q1-Q3. Unassigned/inconclusive Q samples were denoted as “Q*”; (B) Haplogroup N21. All available information was considered (reading frames varied). Frequencies in populations are indicated by colored circle segments. See legends for color codes. Population reference numbers located within a circle indicate that the analyzed haplogroup(s) were not found in this population. For the populations included, see Figure 5. For details and references, see Additional file 11.
The complete absence of haplogroup Q carriers from Australia (except a single occurrence; possible explanations are discussed above) indicates that southern Sahul was populated before this lineage arose (37.5 kya), as we postulate for haplogroup P1. The outlined westward movements might also have caused the wide spread of the predominant Q1, the youngest Q subclade, after 18.1 kya - the timing we hypothesised from haplogroup P4a (18.6 kya) (Figure 7, Additional file 11). These coincidences support a common history of haplogroups P1, P4a and Q(1) and their spread probably even within the same group of settlers - separated from the events that led to the colonization of today’s Australia.
Westward movements after a stall in migration in northern Sahul are the most likely scenario that explains our findings (Figure 8). They have also been postulated in archaeology from old Australian vs. younger Timorese sites. Our genetic dating is however much younger than the first known settlements in Timor (>42 kya), but older than those of modern humans in Flores (<12 kya) [27]. A northward migration after an initial colonization of the South (>37.5 kya) appears less likely because of the complete lack of old Australian P lineages everywhere else. Several mtDNA genetic indices confirm that the relation between East Timor and NG is indeed not a recent one (see above). NG populations, due to their extremely high frequencies of haplogroups P and Q, appear generally far from all ISEA populations in these analyses. Still, East Timor yielded the least genetic differentiation from NG of all samples included, possibly because of its most proximal location in westward expansions (Figures 5 and 7, Additional files 8, 10 and 11). The “almost complete (female) isolation between the two regions” [48] is also seen in nuclear DNA investigations. These support a continuous population history for Sahul dating to probably ≥50 kya demonstrating a deep common origin of Australians and Papua New Guineans with little later migration [36,79]. The often depicted split of initial Sahul settlers into a northern and southern group after arrival on the continent, followed by isolation (e.g., [25,34,48]), would not explain the age gap in mtDNA lineages between South and North alone, unless an “incubatory” phase is taken into consideration. Separate settlement waves from Sunda to NG and to Australia [3] would also need to integrate the later development of P1 and Q from root haplotypes.
Figure 8.
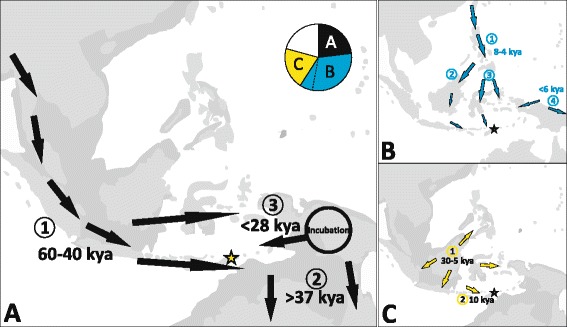
Migration routes and their contribution to the East Timor mtDNA pool. The major postulated migration events into ISEA and our novel findings are depicted. The asterisk highlights East Timor. The darker grey areas indicate the predicted late Pleistocene coastline. (A) The initial human settlement carrying haplogroups P, Q, N21 and others that arrived (1) between 60–40 kya. Our results indicate (2) a colonization of Australia (southern Sahul) before 37 kya and (3) an incubation period in northern Sahul (NG) followed by westward expansions after 28 kya; (B) The Holocene (1) southward out of Taiwan movement marked by haplogroups M7c1, D5, F1a3, F1a4 between 8–4 kya followed a (2) western or (3) eastern route (that we favour for East Timor), and (4) a local arisal, possibly connected to (1), of the “Polynesian motif” ~6 kya followed by west- and eastward migrations; (C) the postglacial expansion of haplogroup E (and others) (1) originating in eastern Sunda and a dispersal 30–5 kya that (2) reached eastern Indonesia ~10 kya. The inlay pie chart in (A) depicts the proportions of haplogroups associated with (A), (B) and (C) within the extant East Timor population. The dashed line separates the proportion of the “Polynesian motif”, as it has also been described to derive from a separate event. See text for details.
From the similarly small proportions of autochthonous haplogroups along both, we cannot favour a northern or southern route through Wallacea for the initial settlers [20,25,27]. The clustering of the Sulawesi and MSEA populations in the MDS plot rather derives from the footprints of later inputs in the mtDNA pool (Figure 4, Additional file 11).
Haplogroup N21, another postulated “first settlement” marker, found in proportions ≤4.2% in MSEA and ISEA, has been dated to 22.4 ky [72]. East Timor was its easternmost location. This evidence is not easily compatible with migration scenarios (Figure 7, Additional file 11). In total, the autochthonous lineages considered to have arisen in the course of the initial dispersal, i.e. P, Q, M21, M73, N21, R14, and possibly further unresolved M* and R* clades [3,5,17,24,28,34,40,55,80], account for 22.2% of our East Timor sample. This confirms previous estimates for ISEA [24].
Traces of later settlements
We do not find that “the mitochondrial genomes of Timorese women predominantly derive from Papuan progenitors” [4], but rather that additional migration after initial colonization is necessary to explain the predominant proportion of (S)EA lineages, mirrored by high haplotype sharing rates and relatively small genetic, despite large geographic, distance (Figure 2, Additional files 7, 8, 9 and 10). We investigated East Timor’s mtDNA composition (as a pooled sample from this study and [3]) in the light of four particular “later” migratory events postulated to have occurred between (S)EA and Melanesia during late Pleistocene and mid-Holocene in previous studies, often overlapping in terms of time and geography. When their contributions are merged, 52.3% of the East Timor population (53.3% of our sample) can be explained. Figure 8 depicts the migration routes we found to be relevant to the extant East Timorese mtDNA pool.
-
(i)
A Neolithic OOT expansion into ISEA in mid-Holocene ~8-4 kya [3,5,24]: its postulated mtDNA marker, the former haplogroup entity M7c3c (see Additional file 12), reached a frequency of 12.4% in East Timor (13.9% in our sample set). Also haplogroups D5 (1.4%), F1a3 (4.4%), F1a4 (8.6%), and F3b(1) (0.3%) have been associated with this migration [24,28,30,80]. Thus, 27.1% (our sample: 29.6%) of the East Timor mtDNAs could be related to this dispersal. A ~20% contribution for ISEA was previously estimated [24]. The OOT-related mtDNA haplogroups (as of published records) peak in the Philippines and eastern ISEA, but are almost absent from MSEA (except the basal D5), Melanesia, Polynesia, and Australia (Additional file 12): this migration might thus have ended in eastern Indonesia and not proceeded eastward as hypothesized [28] (see below). A western (Taiwan-Philippines-Borneo-Sumatra/Java) and an eastern (Taiwan-Philippines-Sulawesi/Moluccas) route into ISEA have been suggested [3,5,24,30,81], as well as several subevents via more than a single route [28]. Our results support an eastern route into East Timor: the combined marker haplogroups’ proportions were larger along this route (Additional files 11 and 12), the western route marker Y2 [24] was absent in East Timor, and the haplotype-based genetic parameters put East Timor closest to Nusa Tenggara (probably relevant in both routes), and Sulawesi and the Moluccas, proximal only on the eastern route. In the MDS plot, Sulawesi located at some distance from the cluster of East Timor-Nusa Tenggara-Moluccas, but closest to the Philippines and Taiwan, the postulated origin of the migration. Larger distances from the latter two, but similar ones to East Timor, were yielded by the “western route” populations (Figure 4, Additional files 7, 8, 9 and 10).
-
(ii)
A pre-mid-Holocene expansion in(to) ISEA and Polynesia [16,65,70]: marker haplogroup B4a1a1 probably originated in the vicinity of the Bismarck archipelago and might have reached Timor westward along a “voyaging corridor” [16,37]: the wide spread of this lineage (and AN languages) [56,82,83], highlights early navigating routes and capabilities. The so-called “Polynesian motif” (B4a1a1a until Phylotree [49], build 15) comprises 5.0% of the East Timor sample (additional 5.6% for B4a1*) (Table 1). Notably, B4a1a has also been considered an OOT marker [28,30]; hence, the “maternal” contribution of that event to East Timor would rise to 32.1% (34.5% in our sample). The two dispersals would be strongly interwoven, and eastern ISEA (East Timor) would not only represent the last section of a southward corridor (see above), but also the assumed bottleneck through which only few haplogroups made it to Melanesia and Polynesia [28,84,85].
-
(iii)
A major postglacial eastward expansion into ISEA in late Pleisto- and early Holocene ~30-5 kya ([15,24,30]: “early train”): accordingly, lineage E originated on the eastern Sunda coastline and during this expansion, caused by the rising sea, reached eastern Indonesia ~10 kya [15,16,24]. Carriers were widespread in ISEA and Near Oceania, with a focus off the former eastern rim of Sunda. The second highest frequency was observed in East Timor (19.3%). The second marker lineage, B4c2, was generally rare. Altogether, 19.9% of the East Timor dataset (18.5% of our sample) could be traced back to this Neolithic dispersal (Additional files 11 and 12). Remarkably, also haplogroup E has been linked to the OOT expansion [42].
-
(iv)
A Holocene arrival ~7 kya from MSEA to ISEA [24,40] may have played a minor role in populating its far East. Marker haplogroup F1a1*, frequent throughout eastern Indonesia, MSEA, and on Taiwan, is a singleton in the Philippines (contra OOT) and East Timor (0.3%), where the second marker haplogroup, N9a6, was absent (Additional files 11 and 12).
Conclusions
Our results from the oldest indigenous mtDNA lineages of the Southwest Pacific region indicate migratory events that commenced <28 (possibly <18) kya, more than 15–20 ky after the initial arrival to Sahul. We thus confer the extension of the rather strictly timed classical two stages (cf. [44]) also upon the “initial wave” of colonization and suggest a much broader timescale of events connected to indigenous haplogroups. Secondary expansions of initial settlers and westward migrations have been postulated before (cf. [27]) and dated at ~40 kya [48]. This is the first clear genetic indication of a temporal proximity and possible coincidence of dispersals which encompassed autochthonous haplogroups with postulated “later” events of (S)EA origin reconstructed from younger lineages. In our scenario, it seems now probable that the “initial” and “later” migration routes, or even the population groups themselves, were or became intertwined. Ongoing genetic (and linguistic) “diffusion” of humans (that at some point also brought P and Q carriers to islands east of NG) was likely facilitated once trade corridors were established [16,37,80,86]. The peak of Sahul’s surface area ~30-18 kya coincides with the highly dynamic migratory period we report and might have triggered it. Population dispersals (geographic expansions) do not necessarily also mean population expansion [4], but BSPs from thousands of samples in the HVS-I range (Indonesia) and tens of complete mitogenomes (Philippines and Malaysia) from mixed haplogroups have (remarkably similarly to our BSP from haplogroup P1; Figure 6) indicated a slowly growing population in the Pleistocene that peaked at 20–15 kya and declined thereafter [4,30,87]. This seems to pinpoint “substantial impact of common environmental forces” in ISEA [4].
In 1869, AR Wallace noted that the Timorese appeared closer to inhabitants of the Moluccas and NG than SEA and speculated about admixture [88]. Almost one and a half century later, we tried to shed light on these relations by characterizing the mtDNA composition of East Timorese. The blurring of older “footprints” by subsequent migrations, and the paucity of available (founder lineage) mitogenomic data may have confounded our assumptions on dispersal and time estimates. In any event, the migration history (to be) reconstructed from mtDNA is only the “female side” of the medal, but “necessarily coupled” to the history of the entire population [4]. Other genetic markers (cf. [36,86,89]) will support or modify our proposals, but contribute to the complete picture only together with non-genetic disciplines, such as archaeology (cf. [90]) and linguistics - particularly, since the geographically overlapping AN and Papuan language families are thought to be separable in time; with the reservation of difficulties in speaker affiliation and language grouping (cf. [3,29,59]). All-embracing studies on human colonization will need to consider also other hominin species or populations [20,91-93].
Methods
Sample collection
Three hundred and twenty-four randomly selected East Timor residents (175 female, 149 male) voluntarily donated buccal swabs under informed consent. The study was approved by the Universidade Nacional Timor Lorosa'e. For most individuals, maternal origin within East Timor could be verified over three generations; for 36 donors, information was incomplete but indicated East Timor origin. Their birthplaces comprised all 13 districts. One donor was born in Flores (Indonesia) with a maternal line traceable to East Timor. Five donors indicated maternal origin in West Timor, one in Java (both Indonesia), four could not provide any information. All were included in this study to reflect the extant population (Additional files 1 and 2).
MtDNA sequence data generation and interpretation
DNA was extracted using standard protocols [94]. The forensic quality CR (nps 16024–16569, 1–576) Sanger type-sequencing (STS) protocols included amplification of a single segment, redundant sequencing coverage, independent data inspection and final validation to ensure precise base calling [95]. Contiguous sequences were aligned [96] with respect to the rCRS [50] using Sequencher v5.0 (GeneCodes Corporation, Ann Arbor, MI, USA) and assigned to mtDNA haplogroups according to Phylotree [49], build 16, aided by the EMMA software package [97]. In order to define their phylogenetic position, five mtDNAs were subjected to entire mitogenome STS following forensic protocols [98]. To resolve the still poorly described phylogeny of haplogroup P1, also three P1 samples were included in these analyses.
Emerging MPS solutions now offer a more accessible option to yield highly desired full mitogenome data [99], but investigations benefiting from phylogenetic knowledge found published data inappropriate for the quality needs of the field requiring highest accuracy [100] - just as STS flaws attracted attention in the last decade (e.g., [101]). Few studies applied MPS on mtDNA in a forensic environment [102-106]. In this study, we used the Ion Torrent PGM under forensic quality control for the first time on a (small) population sample by stand-alone approach for complete mitogenome MPS on the eight samples described above and nine additional bona fide P1 samples. We used Ion PGM Sequencing 200 Kit v.2 chemistry on an Ion 316 chip, and applied strict quality control according to a preceding validation study of the PGM in forensic mtDNA sequencing, that suggested MPS, with appropriate care, as a valuable alternative also in a forensic environment [106]. Raw data were inspected twice using independent software, mirroring the gold standard in STS (see above). The latter was applied ex post to clarify remaining discordance. Phylogenetic plausibility checks were performed on the dataset.
Phylogenetic reconstructions, age and demographic estimates
The most parsimonious phylogenetic trees were manually reconstructed from the 20 haplogroup P1 sequences, as well as the five non-P1 complete mitogenomes to depict their position among published sequences [17,28,44,52-55,80,87,107-116]. To obtain ML molecular divergences for haplogroup P1 PAML 4.4 [117] was used, assuming the HKY85 mutation model with γ-distributed rates, as previously suggested [118]. The ML estimates were compared with those obtained from the averaged distance (ρ) of a clade’s haplotypes to the respective root haplotype, accompanied by a heuristic estimate of the standard error (σ) calculated from an estimate of the genealogy. All calculations were performed on the complete mtDNA haplotypes. Mutational distances were converted into years using a corrected molecular clock [74]. Concerns about the estimation accuracy via ρ statistics have been raised considering the small and hypervariable CR [73]; recent studies based on entire mitogenomes found good concordance between ML and ρ results (e.g., [38,119]).
The P1 complete mitogenome dataset was analyzed with BEAST v.1.7 [120] to obtain a BSP of the effective female population size. We used the HKY substitution model (γ-distributed rates) and a relaxed molecular clock (lognormal in distribution across, and uncorrelated between branches) for 5,000,000 iterations, drew samples every 10,000 Markov chain Monte Carlo steps, and visualized the output using Tracer v.1.5 [120] assuming a generation time of 25 years, as in [4,119].
Analysis of maternal relatedness
To avoid a biased representation of lineages by closely related individuals, donors with identical mitogenomes were inspected for maternal relatedness. Also those that only varied in polycytosine- and (AC)n-stretch lengths were considered, as such differences can result from analysis and detection conditions [100,121-123]. After typing 15 autosomal STR loci and the amelogenin length polymorphism, pedigree construction, and likelihood ratio (LR) calculation using reported STR allele frequencies [7] [correcting the 10.2 allele frequency of D18S51 to 0.0 (L Souto, pers. comm.)], no donor pair revealed close maternal relatedness (i.e., mother-child and sibling constellations) applying a cut-off LR of 1,000 [124,125] (data not shown).
Forensic and population genetic parameters of East Timor and surrounding populations
The mtDNA composition of East Timor was compared to populations from surrounding areas by calculating haplotype-based forensic and population genetic intra- and interpopulation parameters, taking the entire quality-checked datasets into account. Samples of identical geographic origin were merged from different publications to better reflect true variation and reduce any effects of error in small samples (cf. [24]) after careful inspection of metadata to avoid multiple representations of individuals. We determined the proportions of unique haplotypes, RMPs (as sum of squared mtDNA haplotype frequencies), counted MPD within populations and between populations (both uncorrected and corrected, i.e. reduced by the mean of MPD observed within the two populations compared), performed an analysis of molecular variance (AMOVA) using ARLEQUIN v.3.5.1.2 [126] and generated an FST distance matrix for Multidimensional Scaling (MDS) analysis with the R software package [127] function cmdscale(). All sequences were trimmed to a greatest common range: (i) eleven populations (3,276 samples) from (S)EA and Melanesia [28,34,45,57-64] were included based on HVS-I and -II data (nps 16080–16193, 16194–16365, 73–300); (ii) another analysis covered 25 populations (9,634 individuals) from (S)EA, Melanesia, Polynesia, and Australia [3,5,17,24,28,29,34,40,41,45,57-69] in an HVS-I range (nps 16080–16180, 16195–16354). See Figure 1, Tables 2 and 3 for details.
Phylogeographic investigations
To assess the distribution of selected marker mtDNA haplogroups, frequency values in surrounding populations were collected from published records after phylogenetic inspection [3,5,17,24,28-30,34,40,41,44,45,48,52,53,55,57-69,75-77,80,86,115,116,128-141]. Data were mostly restricted and sample sizes often very small. As outlined, they were merged in case of identical geographic origin. To analyze the dispersal of haplogroup P(1) in greater detail, we distinguished categories discernible in HVS-I: P1*, P1d1, and the “non-P1” P samples (PxP1). We chose the greatest common (i.e. available, in many cases) sequenced fragment for an unbiased picture and assumed haplogroup P1 status when a minimal pattern of [73G 263G 315.1C] 16357C, relative to the rCRS [50], was present, as done before [3], because the remaining current CR markers ([49], build 16) were not found reliable (see above). The transition at np 16357 is a phylogenetic marker of 17 additional mtDNA haplogroups ([49], build 16). All are clearly discernible from P1 by their HVS-I pattern and/or are not expected to occur in the investigated area. Supporting our assumptions, all haplotypes in EMPOP [51] (v2.3, release 11, n = 34,617) that carried a private transition at np 16357 and could be mistaken for P1 fell into the latter category. In further analyses, we looked at the dispersal of the remaining P and other marker lineages, inasmuch they were detectable from available data. We labelled P and Q mtDNAs not assigned to a specific lineage due to lack of information as “P*” and “Q*”.
To assess and depict the impact from surrounding regions on the mtDNA pool, the East Timor haplogroups were classified according to their geographic origin as per previous reports, where terminology and assignments were overlapping and contradictory at times [3,5,15,16,24,28-30,32,34,40,70,80].
Availability of supporting data
The sequence data generated are available in GenBank [KJ655583-KJ655889, KJ676774-KJ676790; http://www.ncbi.nlm.nih.gov/genbank] and included in EMPOP [EMP00534; http://empop.org].
Acknowledgements
The authors are most grateful to the donors who participated in this study and thank the Universidade Nacional de Timor Lorosa'e (UNTL) for this cooperation, the staff at the Portuguese Embassy at Díli, Timor-Leste and João Souto Miranda for local support, Helena Moreira and Filipa Tavares (Department of Biology, University of Aveiro, Campus de Santiago, Aveiro, Portugal) for preparing the sampling material, the latter and Daniela Niederwieser (Institute of Legal Medicine, Medical University of Innsbruck, Innsbruck, Austria) for STR analyses, and Mannis van Oven (Department of Forensic Molecular Biology, Erasmus MC University Medical Center Rotterdam, Rotterdam, The Netherlands) for giving advice on mtDNA haplogroup nomenclature. This work was supported by the Foundation of Science and Technology Portugal (FCT); the Programa Operacional Temático Factores de Competitividade (COMPETE), co-funded by the European Community Fund FEDER with the Project PTDC/CS-ANT/108558/2008; the FCT fellowship SFRH/BD/63165/2009; the Austrian Science Fund (FWF) [L397] and [P22880-B12]; the European Union Seventh Framework Programme (FP7/2007-2013) under grant agreement n° 285487; the intramural funding program of the Medical University Innsbruck for young scientists MUI-START, Project 2013042025 and the Theodor Körner Fonds zur Förderung von Wissenschaft und Kunst.
Abbreviations
- AMOVA
Analysis of molecular variance
- AN
Austronesian
- BSP
Bayesian Skyline Plot
- codR
Coding region of the mtDNA
- CR
Control region of the mtDNA
- EA
East Asia(n)
- EMPOP
European DNA Profiling Group MtDNA Population Database
- HVS
Hypervariable segment of the mtDNA
- ISEA
Island SEA
- ky
Thousand years
- kya
ky ago
- MDS
Multidimensional scaling
- MPD
Mean pairwise differences
- MPS
Massively parallel sequencing
- MRCA
Most recent common ancestor
- MSEA
Mainland SEA
- mtDNA
Mitochondrial DNA
- NG
New Guinea(n)
- np
Nucleotide pair
- OOT
“out of Taiwan” (dispersal)
- PNG
Papua NG
- rCRS
Revised Cambridge Reference Sequence
- RMP
Random match probability
- (S)EA
(South) EA
- STR
Short tandem repeat
- STS
Sanger-type sequencing
- WNG
West NG
Additional files
Origin of the 324 East Timor sample donors. East Timor and its 13 districts are shown. The diameter of every circle is proportional to the number of samples deriving from each district (see legend). The asterisk indicates the capital, Díli. The Díli district includes Ataúro island. The striped area is part of Indonesia. For a detailed list, see Additional file 2.
Detailed origin of the 324 East Timor sample donors. The district of origin is listed for each donor. n.a. = not available.
CR profiles, haplogroups and GenBank accession numbers for the 324 East Timor samples. Haplogroups according to [49], build 16. Differences are relative to the rCRS [50]. Bases are indicated according to the IUBMB nucleotide code. The prefix @ indicates the reversion of a mutation occurring earlier in the phylogeny. Differences indicating potentially novel, not (yet) defined haplogroups are indicated as suffixes.
Mutational profiles, haplogroups and GenBank accession codes of the 17 novel complete East Timor mitogenomes. Haplogroups according to [49], build 16. Differences are relative to the rCRS [50]. Bases are indicated according to the IUBMB nucleotide code.
List of the complete haplogroup P1 mitogenomes depicted in Figure 3 .
Novel non-P1 complete mitogenomes from East Timor. The five East Timor samples are shown in the context of published sequences. Color codes indicate the country of origin. All identified differences are relative to the rCRS [50], disregarding cytosine insertions after nps 16193 and 309. Haplogroups according to [49], build 16. Bases are indicated according to the IUBMB nucleotide code. The prefix @ indicates the reversion of a mutation occurring earlier in the phylogeny. Underlined mutations are recurrent in the tree. References and GenBank accession numbers are indicated for each sample. Samples marked with an asterisk are available from GenBank only [Pradutkanchana 2010, unpublished data].
Shared haplotypes between East Timor and 10 surrounding populations based on HVS-I and HVS-II.
Intra-population comparisons for East Timor and 10 surrounding populations based on HVS-I and HVS-II. Part A: Design and results of AMOVA. Part B: Mean pairwise differences. Part C: F ST comparisons.
Shared haplotypes between East Timor and 24 surrounding populations based on HVS-I.
Intra-population comparisons for East Timor and 24 surrounding populations based on HVS-I. Part A: Design and results of AMOVA. Part B: Mean pairwise differences. Part C: F ST comparisons.
Frequency of selected mtDNA haplogroups in East Timor and surrounding populations. For a graphical representation of frequencies, see Figures 5 and 7 and Additional file 12.
Dispersal of selected mtDNA haplogroups in East Timor and surrounding populations. (A) Haplogroups related to the “out of Taiwan” dispersal. (B) Haplogroups B4c2 and E; (C) Haplogroup F1a1. All available information was considered (reading frames varied between publications). Frequencies in populations are indicated by colored circle segments. See legends for color codes. Population reference numbers located within a circle indicate that the analyzed haplogroup(s) were not found in this population. For the populations included, see Figure 5. For details and references, see Additional file 11.
Footnotes
Sibylle M Gomes and Martin Bodner contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SMG conducted experiments, interpreted results, retrieved and quality-checked data, accomplished calculations, prepared tables and figures, drafted and helped writing the manuscript. MB retrieved and quality-checked data, accomplished calculations, prepared tables and figures, performed experiments, interpreted results, drafted, wrote, edited and revised the manuscript. LS designed and coordinated the project, collected the samples. BZ, GH and CS conducted experiments, interpreted results. AWR retrieved, edited and quality-checked sequence data. AA, AO and AT edited data, performed calculations, interpreted results, and contributed to the manuscript. FCR coordinated the project. WP coordinated the mtDNA analyses, supervised experiments, validated results, and contributed to writing the manuscript. All authors approved the final manuscript.
Contributor Information
Sibylle M Gomes, Email: sibylle.marcialgomes@ua.pt.
Martin Bodner, Email: martin.bodner@i-med.ac.at.
Luis Souto, Email: lsouto@ua.pt.
Bettina Zimmermann, Email: bettina.zimmermann@i-med.ac.at.
Gabriela Huber, Email: gabriela.e.huber@i-med.ac.at.
Christina Strobl, Email: christina.strobl@i-med.ac.at.
Alexander W Röck, Email: a.w.roeck@gmail.com.
Alessandro Achilli, Email: alessandro.achilli@unipg.it.
Anna Olivieri, Email: anna.olivieri@unipv.it.
Antonio Torroni, Email: antonio.torroni@unipv.it.
Francisco Côrte-Real, Email: fgoncalves@fmed.uc.pt.
Walther Parson, Email: walther.parson@i-med.ac.at.
References
- 1.National Statistics Directorate (NSD) [Timor-Leste], Ministry of Finance [Timor-Leste], and ICF Macro . Timor-Leste Demographic and Health Survey 2009–10. Díli: NSD and ICF Macro; 2010. [Google Scholar]
- 2.National Statistics Directorate (NSD) [Timor-Leste] and United Nations Population Fund (UNFPA). Population and Housing Census of Timor-Leste, 2010, Volume 3: Social and Economic Characteristics. NSD and UNFPA. 2011. https://www.mof.gov.tl/wp-content/uploads/2011/06/Publication-3-English-Web.pdf. Accessed 4 Feb 2015.
- 3.Mona S, Grunz KE, Brauer S, Pakendorf B, Castrì L, Sudoyo H, et al. Genetic admixture history of Eastern Indonesia as revealed by Y-Chromosome and mitochondrial DNA analysis. Mol Biol Evol. 2009;26:1865–77. [DOI] [PubMed]
- 4.Guillot EG, Tumonggor MK, Lansing S, Sudoyo H, Cox MP. Climate change influenced female population sizes through time across the Indonesian Archipelago. Hum Biol. 2013;85:135–152. doi: 10.3378/027.085.0306. [DOI] [PubMed] [Google Scholar]
- 5.Tumonggor MK, Karafet TM, Hallmark B, Lansing JS, Sudoyo H, Hammer MF, et al. The Indonesian archipelago: an ancient genetic highway linking Asia and the Pacific. J Hum Genet. 2013;58:165–73. [DOI] [PubMed]
- 6.Souto L, Gusmão L, Amorim A, Ferreira E, Côrte-Real F, Vieira DN, et al. POP-Gene TIMOR: first forensic DNA marker study of East-Timor people. Int Congr Ser. 2004;1261:201–3.
- 7.Souto L, Alves C, Gusmão L, Ferreira E, Amorim A, Côrte-Real F, et al. Population data on 15 autosomal STRs in a sample from East Timor. Forensic Sci Int. 2005;155:77–80. [DOI] [PubMed]
- 8.Souto L, Gusmão L, Amorim A, Côrte-Real F, Vieira DN. Y-STR haplotype diversity in distinct linguistic groups from East Timor. Am J Hum Biol. 2006;18:691–701. doi: 10.1002/ajhb.20553. [DOI] [PubMed] [Google Scholar]
- 9.Souto L, Gusmão L, Ferreira E, Amorim A, Côrte-Real F, Vieira DN. Y-chromosome STR haplotypes in East Timor: Forensic evaluation and population data. Forensic Sci Int. 2006;156:261–265. doi: 10.1016/j.forsciint.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 10.Souto L, Rocha AM, Pires A, Ferreira E, Kayser M, Amorim A, et al. Mitochondrial DNA variability in populations from East Timor (Timor Leste). Int Congr Ser. 2006;1288:115–7.
- 11.Souto L, Tavares F, Moreira H, Côrte-Real F. Evaluation of the PowerPlex Fusion System in a sample from East Timor. Forensic Sci Int Genet Suppl Ser. 2013;4:e105–e106. doi: 10.1016/j.fsigss.2013.10.054. [DOI] [Google Scholar]
- 12.Santos C, Phillips C, Fondevila M, Porras-Hurtado L, Carracedo Á, Souto L, et al. A study of East Timor variability using the SNPforID 52-plex SNP panel. Forensic Sci Int Genet. 2011;5:e25–6. [DOI] [PubMed]
- 13.Cox MP. The genetic environment of Melanesia: Clines, clusters and contact. In: Koven TV, editor. Population Genetics Research Progress. Hauppage: Nova Science Publishers; 2008. pp. 45–83. [Google Scholar]
- 14.Richards M, Bandelt HJ, Kivisild T, Oppenheimer S. A Model for the Dispersal of Modern Humans out of Africa. In: Bandelt HJ, Macaulay V, Richards M, editors. Human Mitochondrial DNA and the Evolution of Homo sapiens. Berlin and Heidelberg: Springer; 2006. pp. 225–265. [Google Scholar]
- 15.Soares P, Trejaut JA, Loo JH, Hill C, Mormina M, Lee CL, et al. Climate change and postglacial human dispersals in Southeast Asia. Mol Biol Evol. 2008;25:1209–18. [DOI] [PubMed]
- 16.Soares P, Rito T, Trejaut J, Mormina M, Hill C, Tinkler-Hundal E, et al. Ancient voyaging and Polynesian origins. Am J Hum Genet. 2011;88:239–47. [DOI] [PMC free article] [PubMed]
- 17.Macaulay V, Hill C, Achilli A, Rengo C, Clarke D, Meehan W, et al. Single, rapid coastal settlement of Asia revealed by analysis of complete mitochondrial genomes. Science. 2005;308:1034–6. [DOI] [PubMed]
- 18.Mellars P. A new radiocarbon revolution and the dispersal of modern humans in Eurasia. Nature. 2006;439(7079):931–935. doi: 10.1038/nature04521. [DOI] [PubMed] [Google Scholar]
- 19.Mellars P. Going east: new genetic and archaeological perspectives on the modern human colonization of Eurasia. Science. 2006;313(5788):796–800. doi: 10.1126/science.1128402. [DOI] [PubMed] [Google Scholar]
- 20.O’Connor S. New evidence from East Timor contributes to our understanding of earliest modern human colonisation east of the Sunda Shelf. Antiquity. 2007;81:523–535. doi: 10.1017/S0003598X00095569. [DOI] [Google Scholar]
- 21.Balme J. Of boats and string: The maritime colonisation of Australia. Quat Int. 2013;285:68–75. doi: 10.1016/j.quaint.2011.02.029. [DOI] [Google Scholar]
- 22.Birdsell JB. The recalibration of a paradigm for the first peopling of greater Australia. In: Allen J, Golson J, Jones R, editors. Sunda and Sahul: Prehistoric Studies in Southeast Asia, Melanesia and Australia. London: Academic Press; 1977. pp. 113–167. [Google Scholar]
- 23.Merriwether DA, Hodgson JA, Friedlaender FR, Allaby R, Cerchio S, Koki G, et al. Ancient mitochondrial M haplogroups identified in the Southwest Pacific. Proc Natl Acad Sci U S A. 2005;102:13034–9. [DOI] [PMC free article] [PubMed]
- 24.Hill C, Soares P, Mormina M, Macaulay V, Clarke D, Blumbach PB, et al. A mitochondrial stratigraphy for Island Southeast Asia. Am J Hum Genet. 2007;80:29–43. [DOI] [PMC free article] [PubMed]
- 25.Oppenheimer S. The great arc of dispersal of modern humans: Africa to Australia. Quat Int. 2009;202:2–13. doi: 10.1016/j.quaint.2008.05.015. [DOI] [Google Scholar]
- 26.Oppenheimer S. A single southern exit of modern humans from Africa: Before or after Toba? Quat Int. 2012;258:88–99. doi: 10.1016/j.quaint.2011.07.049. [DOI] [Google Scholar]
- 27.O’Connor S, Barham A, Spriggs M, Veth P, Aplin K, St Pierre E. Cave archaeology and sampling issues in the tropics: a case study from Lene Hara Cave, a 42,000 year old occupation site in East Timor, Island Southeast Asia. Aust Archaeol. 2010;71:29–40. [Google Scholar]
- 28.Tabbada KA, Trejaut J, Loo JH, Chen YM, Lin M, Mirazón-Lahr M, et al. Philippine mitochondrial DNA Diversity: a populated viaduct between Taiwan and Indonesia? Mol Biol Evol. 2010;27:21–31. [DOI] [PubMed]
- 29.Delfin F, Myles S, Choi Y, Hughes D, Illek R, van Oven M, et al. Bridging near and remote Oceania: mtDNA and NRY variation in the Solomon Islands. Mol Biol Evol. 2012;29:545–64. [DOI] [PubMed]
- 30.Jinam TA, Hong LC, Phipps ME, Stoneking M, Ameen M, Edo J, et al. Evolutionary history of continental Southeast Asians: “early train” hypothesis based on genetic analysis of mitochondrial and autosomal DNA data. Mol Biol Evol. 2012;29:3513–27. [DOI] [PubMed]
- 31.Endicott P. Introduction: revisiting the “negrito” hypothesis: a transdisciplinary approach to human prehistory in southeast Asia. Hum Biol. 2013;85(1–3):7–20. doi: 10.3378/027.085.0301. [DOI] [PubMed] [Google Scholar]
- 32.Metspalu M, Kivisild T, Bandelt HJ, Richards M, Villems R. The Pioneer Settlement of Modern Humans in Asia. In: Bandelt HJ, Macaulay V, Richards M, editors. Human Mitochondrial DNA and the Evolution of Homo sapiens. Berlin and Heidelberg: Springer; 2006. pp. 181–199. [Google Scholar]
- 33.Barker G, Barton H, Bird M, Daly P, Datan I, Dykes A, et al. The ‘human revolution’ in lowland tropical Southeast Asia: the antiquity and behavior of anatomically modern humans at Niah Cave (Sarawak, Borneo). J Hum Evol. 2007;52:243–61. [DOI] [PubMed]
- 34.Hudjashov G, Kivisild T, Underhill PA, Endicott P, Sanchez JJ, Lin AA, et al. Revealing the prehistoric settlement of Australia by Y chromosome and mtDNA analysis. Proc Natl Acad Sci U S A. 2007;104:8726–30. [DOI] [PMC free article] [PubMed]
- 35.Summerhayes GR, Leavesley M, Fairbairn A, Mandui H, Field J, Ford A, et al. Human adaptation and plant use in highland New Guinea 49,000 to 44,000 years ago. Science. 2010;330(6000):78–81. [DOI] [PubMed]
- 36.Rasmussen M, Guo X, Wang Y, Lohmueller KE, Rasmussen S, Albrechtsen A, et al. An Aboriginal Australian genome reveals separate human dispersals into Asia. Science. 2011;334:94–8. [DOI] [PMC free article] [PubMed]
- 37.Terrell JE, Welsch RL. Lapita and the temporal geography of prehistory. Antiquity. 1997;71:548–572. [Google Scholar]
- 38.Bodner M, Perego UA, Huber G, Fendt L, Röck AW, Zimmermann B, et al. Rapid coastal spread of First Americans: Novel insights from South America’s Southern Cone mitochondrial genomes. Genome Res. 2012;22:811–20. [DOI] [PMC free article] [PubMed]
- 39.Spriggs M. Chronology of the Neolithic Transition in Island Southeast Asia and the Western Pacific: a view from 2003. Rev Archaeol. 2003;24:57–80. [Google Scholar]
- 40.Hill C, Soares P, Mormina M, Macaulay V, Meehan W, Blackburn J, et al. Phylogeography and ethnogenesis of Aboriginal Southeast Asians. Mol Biol Evol. 2006;23:2480–91. [DOI] [PubMed]
- 41.van Oven M, Hämmerle JM, van Schoor M, Kushnick G, Pennekamp P, Zega I, et al. Unexpected island effects at an extreme: reduced Y chromosome and mitochondrial DNA diversity in Nias. Mol Biol Evol. 2011;28:1349–61. [DOI] [PubMed]
- 42.Ko AM, Chen CY, Fu Q, Delfin F, Li M, Chiu HL, et al. Early Austronesians: into and out of Taiwan. Am J Hum Genet. 2014;94(3):426–36. [DOI] [PMC free article] [PubMed]
- 43.Jordan FM, Gray RD, Greenhill SJ, Mace R. Matrilocal residence is ancestral in Austronesian societies. Proc R Soc B. 2009;276:1957–1964. doi: 10.1098/rspb.2009.0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Friedlaender JS, Friedlaender FR, Hodgson JA, Stoltz M, Koki G, Horvat G, et al. Melanesian mtDNA complexity. PLoS One. 2007;2:e248. [DOI] [PMC free article] [PubMed]
- 45.Trejaut JA, Kivisild T, Loo JH, Lee CL, He CL, Hsu CJ, et al. Traces of archaic mitochondrial lineages persist in Austronesian-speaking Formosan populations. PLoS Biol. 2005;3:e247. [DOI] [PMC free article] [PubMed]
- 46.Oppenheimer S, Richards M. Fast trains, slow boats, and the ancestry of the Polynesian islanders. Science Progress. 2001;84:157–181. doi: 10.3184/003685001783238989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pugach I, Delfin F, Gunnarsdóttir E, Kayser M, Stoneking M. Genome-wide data substantiate Holocene gene flow from India to Australia. Proc Natl Acad Sci U S A. 2013;110:1803–1808. doi: 10.1073/pnas.1211927110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Friedlaender J, Schurr T, Gentz F, Koki G, Friedlaender F, Horvat G, et al. Expanding Southwest Pacific mitochondrial haplogroups P and Q. Mol Biol Evol. 2005;22:1506–17 [Erratum Mol Biol Evol 2005, 22:2313]. [DOI] [PubMed]
- 49.van Oven M, Kayser M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum Mutat. 2009;30:E386–E394. doi: 10.1002/humu.20921. [DOI] [PubMed] [Google Scholar]
- 50.Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet. 1999;23:147. doi: 10.1038/13779. [DOI] [PubMed] [Google Scholar]
- 51.Parson W, Dür A. EMPOP-A forensic mtDNA database. Forensic Sci Int Genet. 2007;1:88–92. doi: 10.1016/j.fsigen.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 52.Ingman M, Kaessmann H, Pääbo S, Gyllensten U. Mitochondrial genome variation and the origin of modern humans. Nature. 2000;408:708–713. doi: 10.1038/35047064. [DOI] [PubMed] [Google Scholar]
- 53.Ingman M, Gyllensten U. Mitochondrial genome variation and evolutionary history of Australian and New Guinean Aborigines. Genome Res. 2003;13:1600–1606. doi: 10.1101/gr.686603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hartmann A, Thieme M, Nanduri LK, Stempfl T, Moehle C, Kivisild T, et al. Validation of microarray-based resequencing of 93 worldwide mitochondrial genomes. Hum Mutat. 2009;30:115–22. [DOI] [PubMed]
- 55.Delfin F, Min-Shan Ko A, Li M, Gunnarsdóttir ED, Tabbada KA, Salvador JM, et al. Complete mtDNA genomes of Filipino ethnolinguistic groups: a melting pot of recent and ancient lineages in the Asia-Pacific region. Eur J Hum Genet. 2014;22:228–37. [DOI] [PMC free article] [PubMed]
- 56.Handoko HY, Lum JK, Rismalia G, Kartapradja H. Length variations in the COII-tRNALys intergenic region of mitochondrial DNA in Indonesian populations. Hum Biol. 2001;73:205–223. doi: 10.1353/hub.2001.0018. [DOI] [PubMed] [Google Scholar]
- 57.Wong HY, Tang JS, Budowle B, Allard MW, Syn CK, Tan-Siew WF, et al. Sequence polymorphism of the mitochondrial DNA hypervariable regions I and II in 205 Singapore Malays. Leg Med (Tokyo). 2007;9:33–7. [DOI] [PubMed]
- 58.Irwin JA, Saunier JL, Strouss KM, Diegoli TM, Sturk KA, O’Callaghan JE, et al. Mitochondrial control region sequences from a Vietnamese population sample. Int J Legal Med. 2008;122:257–9. [DOI] [PubMed]
- 59.Bodner M, Zimmermann B, Röck A, Kloss-Brandstätter A, Horst D, Horst B, et al. Southeast Asian diversity: first insights into the complex mtDNA structure of Laos. BMC Evol Biol. 2011;11:49. [DOI] [PMC free article] [PubMed]
- 60.Zimmermann B, Bodner M, Amory S, Fendt L, Röck A, Horst D, et al. Forensic and phylogeographic characterization of mtDNA lineages from northern Thailand (Chiang Mai). Int J Legal Med. 2009;123:495–501. [DOI] [PubMed]
- 61.Lee HY, Yoo JE, Park MJ, Chung U, Shin KJ. Mitochondrial DNA control region sequences in Koreans: identification of useful variable sites and phylogenetic analysis for mtDNA data quality control. Int J Legal Med. 2006;120:5–14. doi: 10.1007/s00414-005-0005-6. [DOI] [PubMed] [Google Scholar]
- 62.Peng MS, He JD, Liu HX, Zhang YP. Tracing the legacy of the early Hainan Islanders- a perspective from mitochondrial DNA. BMC Evol Biol. 2011;11:46. doi: 10.1186/1471-2148-11-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yao YG, Kong QP, Bandelt HJ, Kivisild T, Zhang YP. Phylogeographic differentiation of mitochondrial DNA in Han Chinese. Am J Hum Genet. 2002;70:635–651. doi: 10.1086/338999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee EJ, Koki G, Merriwether DA. Characterization of population structure from the mitochondrial DNA vis-à-vis language and geography in Papua New Guinea. Am J Phys Anthropol. 2010;142:613–624. doi: 10.1002/ajpa.21284. [DOI] [PubMed] [Google Scholar]
- 65.Kayser M, Brauer S, Cordaux R, Casto A, Lao O, Zhivotovsky LA, et al. Melanesian and Asian origins of Polynesians: mtDNA and Y chromosome gradients across the Pacific. Mol Biol Evol. 2006;23:2234–44. [DOI] [PubMed]
- 66.Tommaseo-Ponzetta M, Attimonelli M, De Robertis M, Tanzariello F, Saccone C. Mitochondrial DNA variability of West New Guinea populations. Am J Phys Anthropol. 2002;117:49–67. doi: 10.1002/ajpa.10010. [DOI] [PubMed] [Google Scholar]
- 67.Ohashi J, Naka I, Tokunaga K, Inaoka T, Ataka Y, Nakazawa M, et al. Brief Communication: Mitochondrial DNA variation suggests extensive gene flow from Polynesian ancestors to indigenous Melanesians in the Northwestern Bismarck Archipelago. Am J Phys Anthropol. 2006;130:551–6. [DOI] [PubMed]
- 68.Kayser M, Choi Y, van Oven M, Mona S, Brauer S, Trent RJ, et al. The impact of the Austronesian expansion: evidence from mtDNA and Y chromosome diversity in the Admiralty Islands of Melanesia. Mol Biol Evol. 2008;25:1362–74. [DOI] [PubMed]
- 69.van Holst Pellekaan SM, Frommer M, Sved JA, Boettcher B. Mitochondrial control-region sequence variation in Aboriginal Australians. Am J Hum Genet. 1998;62:435–449. doi: 10.1086/301710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kayser M. The human genetic history of Oceania: near and remote views of dispersal. Curr Biol. 2010;20:R194–R201. doi: 10.1016/j.cub.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 71.Forster P, Torroni A, Renfrew C, Röhl A. Phylogenetic star contraction applied to Asian and Papuan mtDNA evolution. Mol Biol Evol. 2001;18:1864–1881. doi: 10.1093/oxfordjournals.molbev.a003728. [DOI] [PubMed] [Google Scholar]
- 72.Behar DM, van Oven M, Rosset S, Metspalu M, Loogväli EL, Silva NM, et al. A “Copernican” reassessment of the human mitochondrial DNA tree from its root. Am J Hum Genet. 2012;90:675–84. [DOI] [PMC free article] [PubMed]
- 73.Cox MP. Accuracy of molecular dating with the Rho statistic: Deviations from coalescent expectations under a range of demographic models. Hum Biol. 2008;80:335–357. doi: 10.3378/1534-6617-80.4.335. [DOI] [PubMed] [Google Scholar]
- 74.Soares P, Ermini L, Thomson N, Mormina M, Rito T, Röhl A, et al. Correcting for purifying selection: an improved human mitochondrial molecular clock. Am J Hum Genet. 2009;84:740–59. [DOI] [PMC free article] [PubMed]
- 75.van Holst Pellekaan SM, Ingman M, Roberts-Thomson J, Harding RM. Mitochondrial genomics identifies major haplogroups in Aboriginal Australians. Am J Phys Anthropol. 2006;131:282–294. doi: 10.1002/ajpa.20426. [DOI] [PubMed] [Google Scholar]
- 76.Huoponen K, Schurr TG, Chen Y, Wallace DC. Mitochondrial DNA variation in an aboriginal Australian population: evidence for genetic isolation and regional differentiation. Hum Immunol. 2001;62(9):954–969. doi: 10.1016/S0198-8859(01)00294-4. [DOI] [PubMed] [Google Scholar]
- 77.McAllister P, Nagle N, Mitchell RJ. The Australian Barrineans and their relationship to Southeast Asian Negritos: an investigation using mitochondrial genomics. Hum Biol. 2013;85:485–502. doi: 10.3378/027.085.0322. [DOI] [PubMed] [Google Scholar]
- 78.de Saint Pierre M, Gandini F, Perego UA, Bodner M, Gómez-Carballa A, Corach D, et al. Arrival of Paleo-Indians to the southern cone of South America: new clues from mitogenomes. PLoS One. 2012;7(12):e51311. [DOI] [PMC free article] [PubMed]
- 79.McEvoy BP, Lind JM, Wang ET, Moyzis RK, Visscher PM, van Holst Pellekaan SM, et al. Whole-genome genetic diversity in a sample of Australians with deep Aboriginal ancestry. Am J Hum Genet. 2010;87(2):297–305. [DOI] [PMC free article] [PubMed]
- 80.Peng MS, Quang HH, Dang KP, Trieu AV, Wang HW, Yao YG, et al. Tracing the Austronesian footprint in Mainland Southeast Asia: a perspective from mitochondrial DNA. Mol Biol Evol. 2010;27:2417–30. [DOI] [PubMed]
- 81.Spriggs M. Out of Asia: The spread of Southeast Asian Pleistocene and Neolithic maritime cultures in Island Southeast Asia and the western Pacific. In: O’Connor S, Veth P, editors. East of Wallace’s Line: studies of past and present maritime cultures of the Indo-Pacific region. Rotterdam: A.A. Balkema; 2000. pp. 51–75. [Google Scholar]
- 82.Razafindrazaka H, Ricaut FX, Cox MP, Mormina M, Dugoujon JM, Randriamarolaza LP, et al. Complete mitochondrial DNA sequences provide new insights into the Polynesian motif and the peopling of Madagascar. Eur J Hum Genet. 2010;18:575–81. [DOI] [PMC free article] [PubMed]
- 83.Gonçalves VF, Stenderup J, Rodrigues-Carvalho C, Silva HP, Gonçalves-Dornelas H, Líryo A, et al. Identification of Polynesian mtDNA haplogroups in remains of Botocudo Amerindians from Brazil. Proc Natl Acad Sci U S A. 2013;110:6465–9. [DOI] [PMC free article] [PubMed]
- 84.Spriggs M. The Neolithic and Austronesian expansion within Island Southeast Asia and into the Pacific. In: Chiu S, Sand C, editors. From Southeast Asia to the Pacific: Archaeological Perspectives on the Austronesian Expansion and the Lapita Cultural Complex. Taipei: Academia Sinica; 2007. pp. 104–125. [Google Scholar]
- 85.Pawley A. Prehistoric Migration and Colonisation Processes in Oceania: A view from Historical Linguistics and Archaeology. In: Lucassen J, Lucassen L, Manning P, editors. Migration History in World History: Multidisciplinary Approaches. Leiden: Brill Academic Publishers; 2010. pp. 77–112. [Google Scholar]
- 86.van Oven M, Brauer S, Choi Y, Ensing J, Schiefenhövel W, Stoneking M, et al. Human genetics of the Kula Ring: Y-chromosome and mitochondrial DNA variation in the Massim of Papua New Guinea. Eur J Hum Gen. 2014;22:1393–403. [DOI] [PMC free article] [PubMed]
- 87.Gunnarsdóttir ED, Li M, Bauchet M, Finstermeier K, Stoneking M. High-throughput sequencing of complete human mtDNA genomes from the Philippines. Genome Res. 2011;21:1–11. doi: 10.1101/gr.107615.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wallace AR. The Malay Archipelago: The land of the orang-utan, and the bird of paradise. A narrative of travel, with studies of man and nature. London: Macmillan and Co; 1869. [Google Scholar]
- 89.Veeramah KR, Hammer MF. The impact of whole-genome sequencing on the reconstruction of human population history. Nat Rev Genet. 2014;15:149–162. doi: 10.1038/nrg3625. [DOI] [PubMed] [Google Scholar]
- 90.Davidson I. Peopling the last new worlds: The first colonisation of Sahul and the Americas. Quat Int. 2013;285:1–19. doi: 10.1016/j.quaint.2012.09.023. [DOI] [Google Scholar]
- 91.Brown P, Sutikna T, Morwood MJ, Soejono RP, Jatmiko, Saptomo EW, et al. A new small-bodied hominin from the Late Pleistocene of Flores, Indonesia. Nature. 2004;431:1055–61. [DOI] [PubMed]
- 92.Krause J, Fu Q, Good JM, Viola B, Shunkov MV, Derevianko AP, et al. The complete mitochondrial DNA genome of an unknown hominin from southern Siberia. Nature. 2010;464:894–7. [DOI] [PMC free article] [PubMed]
- 93.Reich D, Patterson N, Kircher M, Delfin F, Nandineni MR, Pugach I, et al. Denisova admixture and the first modern human dispersals into Southeast Asia and Oceania. Am J Hum Genet. 2011;89:516–28. [DOI] [PMC free article] [PubMed]
- 94.Walsh PS, Metzger DA, Higuchi R. Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques. 1991;10:506–513. [PubMed] [Google Scholar]
- 95.Parson W, Bandelt HJ. Extended guidelines for mtDNA typing of population data in forensic science. Forensic Sci Int Genet. 2007;1:13–19. doi: 10.1016/j.fsigen.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 96.Bandelt HJ, Parson W. Consistent treatment of length variants in the human mtDNA control region: a reappraisal. Int J Legal Med. 2008;122:11–21. doi: 10.1007/s00414-006-0151-5. [DOI] [PubMed] [Google Scholar]
- 97.Röck AW, Dür A, van Oven M, Parson W. Concept for estimating mitochondrial DNA haplogroups using a maximum likelihood approach (EMMA) Forensic Sci Int Genet. 2013;7(6):601–609. doi: 10.1016/j.fsigen.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fendt L, Zimmermann B, Daniaux M, Parson W. Sequencing strategy for the whole mitochondrial genome resulting in high quality sequences. BMC Genomics. 2009;10:139. doi: 10.1186/1471-2164-10-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Irwin JA, Parson W, Coble MD, Just RS. MtGenome reference population databases and the future of forensic mtDNA analysis. Forensic Sci Int Genet. 2011;5:222–225. doi: 10.1016/j.fsigen.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 100.Bandelt HJ, Salas A. Current next generation sequencing technology may not meet forensic standards. Forensic Sci Int Genet. 2012;6(1):143–145. doi: 10.1016/j.fsigen.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 101.Salas A, Carracedo A, Macaulay V, Richards M, Bandelt HJ. A practical guide to mitochondrial DNA error prevention in clinical, forensic, and population genetics. Biochem Biophys Res Commun. 2005;335:891–899. doi: 10.1016/j.bbrc.2005.07.161. [DOI] [PubMed] [Google Scholar]
- 102.Mikkelsen M, Rockenbauer E, Wächter A, Fendt L, Zimmermann B, Parson W, et al. Application of full mitochondrial genome sequencing using 454 GS FLX pyrosequencing. Forensic Sci Int Genet Suppl Ser. 2009;2:518–9.
- 103.Mikkelsen M, Frank-Hansen R, Hansen AJ, Morling N. Massively parallel pyrosequencing of the mitochondrial genome with the 454 methodology in forensic genetics. Forensic Sci Int Genet. 2014;12:30–37. doi: 10.1016/j.fsigen.2014.03.014. [DOI] [PubMed] [Google Scholar]
- 104.Holland MM, McQuillan MR, O’Hanlon KA. Second generation sequencing allows for mtDNA mixture deconvolution and high resolution detection of heteroplasmy. Croat Med J. 2011;52:299–313. doi: 10.3325/cmj.2011.52.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Loreille O, Koshinsky H, Fofanov VY, Irwin JA. Application of next generation sequencing technologies to the identification of highly degraded unknown soldiers’ remains. Forensic Sci Int Genet Suppl Ser. 2011;3:e540–e541. doi: 10.1016/j.fsigss.2011.10.013. [DOI] [Google Scholar]
- 106.Parson W, Strobl C, Huber G, Zimmermann B, Gomes SM, Souto L, et al. Evaluation of next generation mtGenome sequencing using the Ion Torrent Personal Genome Machine (PGM). Forensic Sci Int Genet. 2013;7:543–9. [DOI] [PMC free article] [PubMed]
- 107.Tanaka M, Cabrera VM, González AM, Larruga JM, Takeyasu T, Fuku N, et al. Mitochondrial genome variation in Eastern Asia and the peopling of Japan. Genome Res. 2004;14:1832–50. [DOI] [PMC free article] [PubMed]
- 108.Dancause KN, Chan CW, Arunotai NH, Lum JK. Origins of the Moken Sea Gypsies inferred from mitochondrial hypervariable region and whole genome sequences. J Hum Genet. 2009;54:86–93. doi: 10.1038/jhg.2008.12. [DOI] [PubMed] [Google Scholar]
- 109.Ueno H, Nishigaki Y, Kong QP, Fuku N, Kojima S, Iwata N, et al. Analysis of mitochondrial DNA variants in Japanese patients with schizophrenia. Mitochondrion. 2009;9:385–93. [DOI] [PubMed]
- 110.Reiff DM, Spathis R, Chan CW, Vilar MG, Sankaranarayanan K, Lynch D, et al. Inherited and somatic mitochondrial DNA mutations in Guam amyotrophic lateral sclerosis and parkinsonism-dementia. J Neurol Sci. 2011;32:883–92. [DOI] [PubMed]
- 111.Scholes C, Siddle K, Ducourneau A, Crivellaro F, Järve M, Rootsi S, et al. Genetic diversity and evidence for population admixture in Batak Negritos from Palawan. Am J Phys Anthropol. 2011;146:62–72. [DOI] [PubMed]
- 112.Zheng HX, Yan S, Qin ZD, Wang Y, Tan JZ, Li H, et al. Major population expansion of East Asians began before Neolithic time: evidence of mtDNA genomes. PLoS One. 2011;6:e25835. [DOI] [PMC free article] [PubMed]
- 113.Liu J, Wang LD, Sun YB, Li EM, Xu LY, Zhang YP, et al. Deciphering the signature of selective constraints on cancerous mitochondrial genome. Mol Biol Evol. 2012;29:1255–61. [DOI] [PubMed]
- 114.Zhang AM, Jia X, Guo X, Zhang Q, Yao YG. Mitochondrial DNA mutation m.10680G >A is associated with Leber hereditary optic neuropathy in Chinese patients. J Transl Med. 2012;10:43. doi: 10.1186/1479-5876-10-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhang X, Qi X, Yang Z, Serey B, Sovannary T, Bunnath L, et al. Analysis of mitochondrial genome diversity identifies new and ancient maternal lineages in Cambodian aborigines. Nat Commun. 2013;4:2599. [DOI] [PubMed]
- 116.Summerer M, Horst J, Erhart G, Weißensteiner H, Schönherr S, Pacher D, et al. Large-scale mitochondrial DNA analysis in Southeast Asia reveals evolutionary effects of cultural isolation in the multi-ethnic population of Myanmar. BMC Evol Biol. 2014;14:17. [DOI] [PMC free article] [PubMed]
- 117.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24(8):1586–91. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 118.Perego UA, Achilli A, Angerhofer N, Accetturo M, Pala M, Olivieri A, et al. Distinctive Paleo-Indian migration routes from Beringia marked by two rare mtDNA haplogroups. Curr Biol. 2009;19:1–8. [DOI] [PubMed]
- 119.Olivieri A, Pala M, Gandini F, Hooshiar Kashani B, Perego UA, Woodward SR, et al. Mitogenomes from two uncommon haplogroups mark Late Glacial/Postglacial expansions from the Near East and Neolithic dispersals within Europe. PLoS ONE. 2013;8:e70492. [DOI] [PMC free article] [PubMed]
- 120.Drummond AJ, Suchard MA, Dong X, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lutz-Bonengel S, Sänger T, Pollak S, Szibor R. Different methods to determine length heteroplasmy within the mitochondrial control region. Int J Legal Med. 2004;118:274–281. doi: 10.1007/s00414-004-0457-0. [DOI] [PubMed] [Google Scholar]
- 122.Salas A, Bandelt HJ, Macaulay V, Richards MB. Phylogeographic investigations: The role of trees in forensic genetics. Forensic Sci Int. 2007;168:1–13. doi: 10.1016/j.forsciint.2006.05.037. [DOI] [PubMed] [Google Scholar]
- 123.Berger C, Hatzer-Grubwieser P, Hohoff C, Parson W. Evaluating sequence-derived mtDNA length heteroplasmy by amplicon size analysis. Forensic Sci Int Genet. 2011;5:142–145. doi: 10.1016/j.fsigen.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Egeland T, Mostad PF, Mevåg B, Stenersen M. Beyond traditional paternity and identification cases: selecting the most probable pedigree. Forensic Sci Int. 2000;110:47–59. doi: 10.1016/S0379-0738(00)00147-X. [DOI] [PubMed] [Google Scholar]
- 125.Bodner M, Irwin JA, Coble MD, Parson W. Inspecting close maternal relatedness: Towards better mtDNA population samples in forensic databases. Forensic Sci Int Genet. 2011;5:138–141. doi: 10.1016/j.fsigen.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10:564–567. doi: 10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- 127.The R Software Package. [http://cran.r-project.org]
- 128.Vigilant L, Stoneking M, Harpending H, Hawkes K, Wilson AC. African populations and the evolution of human mitochondrial DNA. Science. 1991;253(5027):1503–1507. doi: 10.1126/science.1840702. [DOI] [PubMed] [Google Scholar]
- 129.Redd AJ, Takezaki N, Sherry ST, McGarvey ST, Sofro AS, Stoneking M. Evolutionary history of the COII/tRNALys intergenic 9 base pair deletion in human mitochondrial DNAs from the Pacific. Mol Biol Evol. 1995;12(4):604–615. doi: 10.1093/oxfordjournals.molbev.a040240. [DOI] [PubMed] [Google Scholar]
- 130.Sykes B, Leiboff A, Low-Beer J, Tetzner S, Richards M. The origins of the Polynesians: an interpretation from mitochondrial lineage analysis. Am J Hum Genet. 1995;57(6):1463–1475. [PMC free article] [PubMed] [Google Scholar]
- 131.Lum JK, Cann RL, Martinson JJ, Jorde LB. Mitochondrial and nuclear genetic relationships among Pacific Island and Asian populations. Am J Hum Genet. 1998;63(2):613–624. doi: 10.1086/301949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Redd AJ, Stoneking M. Peopling of Sahul: mtDNA variation in aboriginal Australian and Papua New Guinean populations. Am J Hum Genet. 1999;65(3):808–828. doi: 10.1086/302533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lum JK, Cann RL. MtDNA lineage analyses: origins and migrations of Micronesians and Polynesians. Am J Phys Anthropol. 2000;113(2):151–168. doi: 10.1002/1096-8644(200010)113:2<151::AID-AJPA2>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 134.Friedlaender JS, Gentz F, Green K, Merriwether DA. A cautionary tale on ancient migration detection: mitochondrial DNA variation in Santa Cruz Islands, Solomon Islands. Hum Biol. 2002;74(3):453–71. doi: 10.1353/hub.2002.0029. [DOI] [PubMed] [Google Scholar]
- 135.Black ML, Dufall K, Wise C, Sullivan S, Bittles AH. Genetic ancestries in northwest Cambodia. Ann Hum Biol. 2006;33(5–6):620–627. doi: 10.1080/03014460600882561. [DOI] [PubMed] [Google Scholar]
- 136.Ricaut FX, Thomas T, Arganini C, Staughton J, Leavesley M, Bellatti M, et al. Mitochondrial DNA variation in Karkar Islanders. Ann Hum Genet. 2008;72:349–67. [DOI] [PubMed]
- 137.Vilar MG, Kaneko A, Hombhanje FW, Tsukahara T, Hwaihwanje I, Lum JK. Reconstructing the origin of the Lapita Cultural Complex: mtDNA analyses of East Sepik Province, PNG. J Hum Genet. 2008;53(8):698–708. doi: 10.1007/s10038-008-0301-3. [DOI] [PubMed] [Google Scholar]
- 138.Nur Haslindawaty AR, Panneerchelvam S, Edinur HA, Norazmi MN, Zafarina Z. Sequence polymorphisms of mtDNA HV1, HV2, and HV3 regions in the Malay population of Peninsular Malaysia. Int J Legal Med. 2010;124:415–426. doi: 10.1007/s00414-010-0469-x. [DOI] [PubMed] [Google Scholar]
- 139.Benton M, Macartney-Coxson D, Eccles D, Griffiths L, Chambers G, Lea R. Complete mitochondrial genome sequencing reveals novel haplotypes in a Polynesian population. PLoS One. 2012;7(4):e35026. doi: 10.1371/journal.pone.0035026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hwa HL, Ko TM, Chen YC, Lin CY, Huang YH, Tseng LH, et al. Sequence polymorphisms of mtDNA HV1, HV2 and HV3 regions in eight population groups living in Taiwan. Aust J Forensic Sci. 2012;44:243–52.
- 141.Heyer E, Georges M, Pachner M, Endicott P. Genetic diversity of four Filipino negrito populations from Luzon: comparison of male and female effective population sizes and differential integration of immigrants into Aeta and Agta communities. Hum Biol. 2013;85(1–3):189–208. doi: 10.3378/027.085.0310. [DOI] [PubMed] [Google Scholar]