

Abstract
Although Thr is equally represented as Ser in the human genome and is as good a nucleophile, it is never found in the active site of the large family of trypsin-like proteases that utilize the Asp/His/Ser triad. The molecular basis of the preference of Ser over Thr in the trypsin fold was investigated with X-ray structures of the thrombin mutant S195T free and bound to an irreversible active site inhibitor. In the free form, the methyl group of T195 is oriented toward the incoming substrate in a conformation seemingly incompatible with productive binding. In the bound form, the side chain of T195 reorients for efficient substrate acylation without causing steric clash within the active site. Rapid kinetics prove that this change is due to selection of an active conformation from a pre-existing ensemble of reactive and unreactive rotamers whose relative distribution determines the level of activity of the protease. Consistent with these observations, the S195T substitution is associated with a weak yet finite activity that enables identification of an unanticipated important role for S195 as the end-point of allosteric transduction in the trypsin fold. The S195T mutation abrogates the Na+-dependent enhancement of catalytic activity in thrombin, activated protein C and factor Xa, and significantly reduces the physiologically important allosteric effects of thrombomodulin on thrombin and of cofactor Va on factor Xa. The evolutionary selection of Ser over Thr in trypsin-like proteases was therefore driven by the need for high catalytic activity and efficient allosteric regulation.
Enzymes utilizing a catalytic Ser as nucleophile to broker cleavage of peptide bonds are widely distributed in nature and found in all kingdoms of cellular life as well as many viral genomes (1, 2). A typical genome contains 2–4% of genes encoding for proteolytic enzymes (3) and over one third of them are serine proteases. Nucleophilicity of the catalytic Ser is typically achieved through a catalytic triad of Asp/His/Ser residues, commonly referred to as the charge relay system (4, 5). The architecture of the triad first emerged almost 50 years ago from the structure of chymotrypsin (6): the carboxylate of D102 (chymotrypsinogen numbering) accepts a H-bond from the Nε1 atom of H57, which in turn accepts a H-bond at the Nε2 atom from the Oγ atom of S195. The H-bond between D102 and H57 facilitates the abstraction of the proton from S195 and generates a potent nucleophile (5, 7, 8). The Oγ atom of S195 attacks the carbonyl of the peptide substrate as a result of H57 acting as a general base (9). The oxyanion tetrahedral intermediate formed in the process is stabilized by the backbone N atoms of G193 and S195, which generate a positively charged pocket within the active site known as the oxyanion hole. H-bonding interactions in the oxyanion hole contribute between 1.5 to 3.0 kcal/mol to ground and transition state stabilization (10). Collapse of the tetrahedral intermediate generates the acyl-enzyme and stabilization of the newly created N-terminus is mediated by H57 (11). A water molecule then displaces the free polypeptide fragment and attacks the acyl-enzyme intermediate. The oxyanion hole stabilizes the second tetrahedral intermediate of the pathway and collapse of this intermediate liberates a new C-terminus in the substrate.
Combination of the Asp/His/Ser residues of the catalytic triad with the same or opposite handedness exists in at least four main protein folds, i.e., trypsin, subtilisin, prolyl oligopeptidase, and ClpP peptidase, vouching for four different evolutionary origins. Replacement of any member of the triad with other residues in subtilisin and trypsin largely compromises activity (7, 12, 13) and the effect is non-additive, indicating that the triad behaves as a highly cooperative unit (14). The catalytic S195 deserves special attention in the context of evolutionary origins. Ser is unique among amino acids because it is encoded by codons that cannot interconvert by single nucleotide replacement. Both AGY and TCN codons are documented (15, 16), indicating that the intermediates Thr and Cys along this transition should also be present in the protease family. Yet, only a few family members of viral origin use an active site thiol from a Cys residue (17) and no trypsin-like protease replaces Ser with Thr in the catalytic triad. The absence of Thr as a catalytic residue in trypsin-like proteases is intriguing because Thr is as reactive as a nucleophile as Ser, has the same occurrence in the human genome (18) and is used as an efficient nucleophile in other enzyme folds (19–21). Previous computational (22) and observational (23) studies have speculated that selection of Ser over Thr in the trypsin fold was driven by the steric clash caused by the methyl group within the active site, making Thr incompatible with catalytic activity. Functional support to this claim comes from the observation that the S195T mutant of trypsin is inactive and acquires minimal activity upon replacement of additional residues in the active site that relieve the steric clash (24). However, no structural evidence currently exists of the consequences of replacing Ser with Thr in the active site of a trypsin-like protease, nor is it known whether the S195T substitution has the same functional effect on other members of the trypsin family or causes additional perturbations.
In this study we report high resolution X-ray crystal structures of the S195T mutant of the clotting protease thrombin, free and bound to the irreversible inhibitor H-D-Phe-Pro-Arg-CH2Cl (PPACK). The structures document expected and unexpected consequences of replacing Ser with Thr as a nucleophile. Functionally, the mutation reduces but does not abrogate activity and reveals that S195 is needed to broker catalysis and allosteric regulation in the trypsin fold.
Materials and Methods
Reagents
Thrombin wild-type and mutant S195T were expressed as prethrombin-2 in E. coli, refolded and purified to homogeneity as previously described (25). Protein C was expressed, purified to homogeneity and activated as described elsewhere (26, 27). Factor X cDNA modified to include an epitope for the HPC4 antibody was cloned into a pENTR/D-TOPO entry vector and ultimately the pDEST40 expression vector (Life Technologies). The protein was expressed using HEK293 cells, purified using HPC4 affinity chromatography, activated using RVV-X (Haematologic Technologies) and further purified to homogeneity using a Q-Sepharose column.
Functional assays
The effect of Na+ on the hydrolysis of the chromogenic substrates H-D-Phe-Pro-Arg-p-nitroanilide (FPR) for thrombin, pyroGlu-Pro-Arg-p-nitroanilide for activated protein C, or H-D-Phe-Gly-Arg-p-nitroanilide for factor Xa was studied as detailed elsewhere (28, 29). The effect of thrombomodulin on the hydrolysis of protein C was studied using progress curves of hydrolysis of a substrate specific for activated protein C (30), or alternatively using a discontinuous assay where the reaction was stopped at different intervals with hirudin and the amount of activated protein C was estimated from a standard curve of activity (31). Prothrombin activation by factor Xa wild-type or mutant S195T was determined in the presence of 25 μM phospholipids, with and without 40 nM cofactor Va, as reported elsewhere (32). Stopped-flow fluorescence measurements of FPR binding to S195T thrombin were carried out with an Applied Photophysics SX20 spectrometer, using an excitation of 295 nm and a cutoff filter at 320 nm (29, 33). Samples of thrombin at a final concentration of 200 nM were mixed 1:1 with 60 μL solutions of the same buffer (0.1% PEG8000, 400 mM ChCl, 50 mM Tris, pH 8.0 at 15 °C) containing variable concentrations of FPR. Each trace was determined by averaging a minimum of eight traces. Final kobs values were determined by taking the average of three kobs values determined for each concentration during three independent titrations. Due to the low activity of the S195T mutant, no appreciable hydrolysis of FPR was detected over the time scale of the experiment.
X-ray studies
Crystallization of the thrombin mutant S195T free and bound to PPACK was achieved at 22 °C by the vapor diffusion technique using an Art Robbins Instruments Phoenix liquid handling robot and mixing equal volumes (0.3 μL) of protein and reservoir solution (Table 1). Crystals of S195T free were grown in less than two weeks in the presence of 10 mg/mL protein, 200 mM potassium formate, 21% PEG3350 and were cryoprotected prior to flash freezing in a solution of 200 mM potassium formate, 25% PEG3350, 25% glycerol. Crystals of S195T bound to PPACK were also grown in less than two weeks in the presence of 8.4 mg/mL protein, 100 mM MES, pH 6.5, 15% PEG6000, 5% MPD, 2 mM PPACK and were cryoprotected prior to flash freezing in a solution of 100 mM MES, pH 6.5, 20% PEG6000, 40% MPD. X-ray diffraction data were collected with a home source (Rigaku 1.2 kW MMX007 generator with VHF optics) Rigaku Raxis IV++ detector and were indexed, integrated, and scaled with the HKL2000 software package (34). The structures were solved by molecular replacement using MOLREP from the CCP4 suite (35) and the Protein Data Bank entry 1SHH for thrombin bound to PPACK (28). Refinement and electron density generation were performed with REFMAC5 from the CCP4 suite, and 5% of the reflections were randomly selected as a test set for cross validation. Model building and analysis of the structures were conducted with COOT (36). The structure of S195T bound to PPACK was also subject to a final round of refinement with PHENIX (37) and PDB_REDO (38). Ramachandran plots were calculated using PROCHECK (39). Statistics for data collection and refinement are summarized in Table 1. Atomic coordinates and structure factors have been deposited in the Protein Data Bank (accession codes 4RKJ for S195T free and 4RKO for S195T bound to PPACK).
Table 1.
Crystallographic data for thrombin mutant S195T.
| free | PPACK-bound | |
|---|---|---|
| Buffer | 200 mM K formate | 100 mM MES, pH 6.5 |
| PEG | 3350 (21%) | 6000 (15%) |
| PDB ID | 4RKJ | 4RKO |
|
| ||
| Data collection: | ||
| Wavelength (Å) | 1.54 | 1.54 |
| Space group | P21212 | P21 |
| Unit cell dimensions (Å) | a=61.4, b=91.1, c=50.5 | a=44.6, b=73.3, c=48.7, β=113.4° |
| Molecules/asymmetric unit | 1 | 1 |
| Resolution range (Å) | 40-1.7 | 40-1.85 |
| Observations (unique) | 247223 (31895) | 83278 (22779) |
| Completeness (%) | 99.8 (98.8) | 92.0 (86.8) |
| Rsym (%) | 8.6 (56.1) | 8.1 (51.8) |
| I/σ(I) | 20.3 (2.9) | 13.4 (1.8) |
|
| ||
| Refinement: | ||
| Resolution (Å) | 40-1.7 | 40-1.85 |
| Rcryst, Rfree | 0.16, 0.21 | 0.17, 0.20 |
| Reflections (working/test) | 30132/1605 | 20421/1157 |
| Protein atoms | 2319 | 2358 |
| Solvent molecules | 229 | 145 |
| PPACK/Na+ | - | 1/1 |
| Rmsd bond lengthsa (Å) | 0.006 | 0.008 |
| Rmsd anglesa (°) | 1.1 | 1.3 |
| Rmsd ΔB (Å2) (mm/ms/ss)b | 2.38/2.54/3.43 | 2.75/3.15/4.24 |
| <B> protein (Å2) | 34.9 | 39.5 |
| <B> solvent (Å2) | 47.2 | 41.5 |
| <B> PPACK/Na+ (Å2) | - | 32.7/32.2 |
|
| ||
| Ramachandran plot: | ||
| Most favored(%) | 100.0 | 100.0 |
Root-mean-squared deviation (Rmsd) from ideal bond lengths and angles and Rmsd in B-factors of bonded atoms.
mm, main chain-main chain; ms, main chain-side chain; ss, side chain-side chain.
Analysis of PDB structures of trypsin-like proteases and zymogens
Structures with a resolution <2.0 Å from the trypsin Pfam family (PF00089) were obtained from the Protein Data Bank (PDB) and analyzed. The structurally analogous residue corresponding to H57 and S195 were determined by superpositioning of each structure against a reference molecule (PDB ID 1SHH) using the program FAST (40). Structures with mutations at either H57 or S195 were excluded from analysis resulting in a total of 565 independent chains. No structure of a trypsin-like protease carrying T195 was detected in the PDB. The structures were then assessed to determine if the active site was occupied either by residues from other chains (including symmetry mates) or the presence of a ligand or other heteroatom resulting in 175 chains in this group. For all 565 chains the dihedral angle (C-Cα-Cβ-Oγ) of the residue corresponding to S195 was calculated and adjusted to the range 0–360°. The H-bonding distance between S195 and H57 was measured from the Nε2 atom of H57 to the Oγ atom of S195.
Results
The consequences of replacing Ser with Thr as an active site nucleophile in the trypsin fold are revealed directly by the crystal structures of the S195T mutant of the clotting protease thrombin free and bound to PPACK. The structure in the free form, solved at 1.7 Å resolution (Table 1), shows the active site in the open E conformation (41, 42), with a Cα-Cα distance between the two conserved residues G193 and G216 of 10.3 Å and the side chain of W215 pointing away from the active site entrance (Figure 1A). The rmsd between the structure of S195T free and the E form of wild-type (28) is only 0.28 Å. The substituted T195 is clearly visible in the density map and has no influence on critical interactions within the active site, such as the H-bond between the catalytic D102 and H57 or the ion-pair between the N-terminal residue I16 and D194 needed to correctly fold the oxyanion hole upon the zymogen to protease transition (5). However, the methyl group of T195 points to the solvent and toward an incoming substrate with which it would be in direct steric clash, and sits in the position typically occupied by the Oγ atom of S195 (28, 43). The Oγ atom of T195 points to the opposite direction relative to the methyl group, toward the neighbor C42-C58 disulfide bond that remains 3.1 Å away, but makes a strong H-bonding interaction (2.9 Å) with the Nε2 atom of H57. The position of the H atom on the Nε2 atom of H57 estimated after optimization with PROPKA (44) at pH 7.0 is within 1.95 Å from the Oγ atom of T195 at an H-bond acceptor angle of 91.5°, consistent with the geometry expected of a strong H-bond. The molecular defect of T195 as a nucleophile in the trypsin fold is not due to lack of H-bonding interaction with H57, but rather to an “unreactive” rotamer that positions the methyl group in direct clash with the incoming substrate.
Figure 1.

Active site conformations of the thrombin mutant S195T free (A) or bound to PPACK (B,C) as determined by x-ray crystallography (Table 1). The side chain of T195 positions the methyl group toward an incoming substrate and the Oγ atom in the opposite direction but 3.1 Å away from the C42-C58 disulfide bond. This defective conformation of the nucleophile is corrected upon binding of PPACK (B, yellow sticks) that makes all the expected contacts with residues within the active site. The PPACK-bound conformation (C, PPACK removed for clarity) has an orientation of the Oγ atom of T195 compatible with substrate acylation. Shown are relevant H-bonding interactions. PPACK makes the expected contacts with the N atoms of T195 and G193 in the oxyanion hole, the N and O atoms of G216 and the carboxylate of D189, as well as a strong edge-to-face interaction with W215 in the aryl binding site. The presence of T195 does not alter the critical H-bonding interactions between D102 and H57, or the ion-pair between D194 and the N-terminus I16 that stabilizes the active site and oxyanion hole. There is no steric clash in the two orientations of T195 with residues within the active site. The distance of the methyl group from H57 is 4.1 Å in the free form (A) and 3.2 Å in the PPACK-bound form (C). Electron density (green mesh) is a 2F0-Fc map contoured at 1.0 σ.
Previous studies also speculated that activity in the S195T mutant would require a rotation of the T195 side chain impeded by the ensuing steric clash of the methyl group with the C42-C58 disulfide bond or the catalytic H57 (22–24). However, removal of the C42-C58 disulfide bond with Ala and Val substitutions confers on the S195T mutant of trypsin a significant level of activity only toward ester substrates (24). The mobility of T195 within the active site is directly documented by the structure of the S195T mutant bound to the irreversible inhibitor PPACK (Figure 1B) solved at 1.85 Å resolution (Table 1). A rotation of the side chain of T195 brings the Oγ atom in covalent interaction with PPACK as seen in the wild-type (28, 43) and repositions the methyl group to the back of the active site cavity next to the C42-C58 disulfide bond, but without causing any appreciable steric clash. Overall, the structure is practically identical (rmsd=0.25 Å) to that of wild-type bound to PPACK in the Na+-bound form (28). The inhibitor binds to the active site with all expected H-bonding interactions with D189, the oxyanion hole and the backbone O and N atoms of G216, as well as with a strong edge-to-face interaction with W215 (Figure 1B). Comparison of the free (Figure 1A) and PPACK-bound structures (Figure 1C) proves that the side chain of T195 may assume at least two different orientations within the active site without encroaching on the catalytic H57 or the C42-C58 disulfide bond. This observation bears on the role of S195 as a nucleophile.
There is observational evidence that S195 itself is highly mobile within the active site. Analysis of 565 high resolution (<2.0 Å) structures currently deposited in the PDB documents a Gaussian distribution for both the H-bonding distance between the Oγ atom of S195 and the Nε2 atom of the catalytic H57 (Figure 2A) and the dihedral C-Cα-Cβ-Oγ angle of S195 (Figure 2B). There is no significant correlation (r=0.51) between the two quantities (Figure 2C), indicating that a stable H-bond between S195 and H57 can be established in multiple orientations of S195 within the active site. The dihedral angle of 305° observed with T195 in the free form (Figure 2B) lies outside of the Gaussian distribution of S195 and is clearly incompatible with a productive conformation of the nucleophile (Figure 1A), yet the H-bonding interaction with H57 remains strong. The relatively large set of dihedral angle/H-bonding arrangements available to S195 should be compatible with multiple levels of substrate binding and acylation, creating the potential for fine tuning catalytic activity with minimal perturbation of the structure (45).
Figure 2.
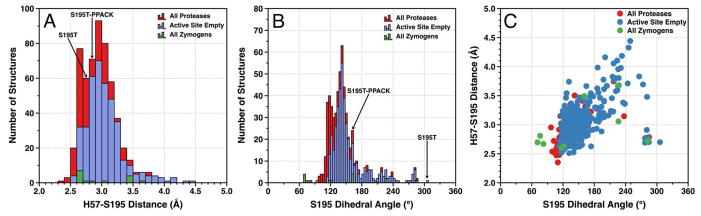
Distribution of the S195-H57 H-bonding distances (A) and dihedral angles (C-Cα-Cβ-Oγ) (B) of residue S195 in a total of 565 high resolution (<2.0 Å) structures of trypsin-like proteases and zymogens deposited in the PDB. The H-bonding distance between S195 and H57 features a Gaussian distribution with values of μ=3.1 Å and σ=0.3 Å barely affected by the presence of ligands in the active site (μ=3.0 Å, σ=0.3 Å). The S195 residue adopts a Gaussian distribution of dihedral angles with μ=156° and σ=37°, which is affected minimally by the presence of ligands within the active site (μ=148°, σ=36°). H-bonding distances and dihedral angles are also reported for the few zymogen strcutures documented in the PDB for the sake of completeness. The sample is however too small for statistical comparisons. (C) There is no correlation (r=0.51 or less) between the dihedral angle of S195 and its H-bonding distance from H57 in all three data sets.
The structure of the S195T mutant reported here is the first and only structure of a trypsin-like protease carrying Thr as a nucleophile, which makes it difficult to extend the observations made on S195 (Figure 2). If the conformation documented in the crystal structure of the S195T mutant in the free form is a genuine representation of an energetically stable rotamer of the nucleophile, it may be expected that the Gaussian distribution accessible to T195 is shifted toward rotamers with higher values of the dihedral angle (Figure 2B) that are unfavorable to substrate binding and catalysis. Importantly, binding of PPACK corrects this defect and shifts the dihedral angle toward a value (160°) that is closer to the mean (148°) of the S195 distribution (Figure 2B) without causing any significant steric clash within the active site. A facile conclusion suggested by the crystal structures is that binding of substrate induces a shift in the rotamer of T195 to facilitate acylation, but rapid kinetics reveal a different scenario. The chromogenic substrate FPR is the cleavable analog of PPACK and binds to the S195T mutant in the ms time scale over which no significant cleavage by the enzyme takes place (Figure 3A). The single exponential relaxation associated with the binding interaction (kobs) is independent of FPR concentration (Figure 3B). Induced fit requires the value of kobs to increase hyperbolically with the ligand concentration, while selection from a pre-existing ensemble is more versatile and gives rise to values of kobs that increase with, decrease with or are independent of ligand concentration (46). The data in Figure 3 provide unequivocal evidence that binding of FPR to the S195T mutant of thrombin occurs by selecting a productive conformation of the nucleophile from a pre-existing ensemble of reactive and unreactive rotamers.
Figure 3.
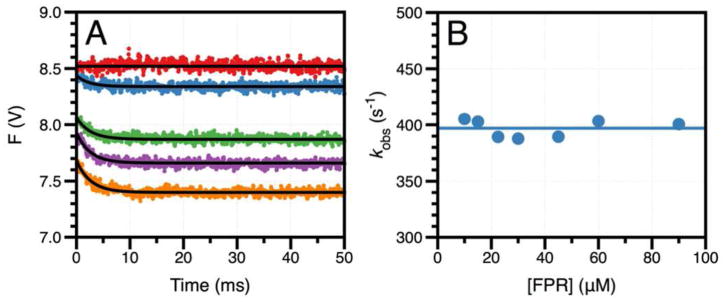
(A) Rapid kinetic traces of FPR binding to thrombin mutant S195T in the 0–50 ms timescale. Shown are the traces obtained at 0 (red circles), 10 (cyan circles), 30 (green circles), 45 (magenta circles), and 60 (orange circles) μM FPR. Solid lines were drawn according to a single exponential function. Experimental conditions are: 0.1% PEG8000, 400 mM ChCl, 50 mM Tris, pH 8.0 at 15 °C. (B) Values of kobs obtained from the analysis of the rapid kinetic traces according to a single exponential function. The solid horizontal line drawn at 400±10 s−1 is the average of all kobs values for FPR binding. The lack of dependence of kobs on FPR concentration is unequivocal evidence of a pre-existing ensemble of inactive and active conformations with FPR selecting the active one for binding (46). The value of kobs=400±10 s−1 is a measure of the rate of transition from the inactive to the active conformation. Because the kinetics refer to a pre-existing ensemble, this asymptotic value of kobs also coincides with the koff for FPR dissociation from the enzyme (46).
Consistent with the observations from X-ray structural biology and rapid kinetics, the S195T substitution brings about significant functional perturbations of thrombin and a >2,000-fold drop in the value of kcat/Km relative to wild-type due to 60-fold increase in Km and 40-fold decrease in kcat, with no evidence of changes in the underlying kinetic mechanism of hydrolysis. The energetic cost of rotating T195 within the active site for optimal acylation and binding is reflected in the perturbed values of Km and kcat. That is consistent with the properties of other site-specific perturbations of the enzyme affecting residues in the primary specificity site (47) or the oxyanion hole (10) for which naturally occurring mutations exist in the trypsin family. Absolute selection of Ser over Thr as the nucleophile in the trypsin fold was therefore driven by additional needs. Indeed, an important and completely unanticipated consequence of the S195T substitution is the resulting perturbation of allosteric effects. Trypsin-like proteases carrying Tyr or Phe at position 225 are endowed with Na+-dependent enhancement of catalytic activity (48). The Na+ binding site is located >15 Å away from residues of the catalytic triad and nestles between the 186- and 220- loops (28, 49) that also control the primary specificity of the enzyme (50). The Na+ effect is allosteric and influences the catalytic activity of highly specific proteases involved in blood coagulation and the complement system (16, 51). Binding of Na+ increases kcat/Km for the hydrolysis of FPR by thrombin nearly 25-fold and is abrogated by the S195T mutation (Figure 4A). The Kd for the thrombin-Na+ interaction is unaffected by the S195T substitution (Figure 4B), proving that S195 is involved exclusively in the allosteric transduction of Na+ binding into enhanced catalytic activity. This long-range communication is likely mediated by residues D221 in the Na+ loop and N143 next to the oxyanion hole, as identified by previous mutagenesis studies (28, 52). The S195T mutation also perturbs a physiologically important allosteric pathway. Binding of thrombomodulin to exosite I located >15 Å away from the active site (53, 54) enhances activity toward the anticoagulant protein C >2,000-fold (55). The effect is significantly reduced by the S195T mutation without appreciable perturbation of the binding affinity of the cofactor (Figure 4C). The intriguing functional consequences of the S195T mutation extend to other members of the trypsin fold. The S195T mutation abrogates the Na+ effect in activated protein C and factor Xa (Figure 5) and significantly reduces the physiologically important allosteric effect of cofactor Va on factor Xa (56) from 3,200-fold to 200-fold (Table 2). Altogether, these functional data prove that S195 not only ensures optimal catalytic activity but also functions as the end-point of allosteric transduction in the trypsin fold. In the case of small cofactors like Na+, perturbation of S195 is sufficient to abrogate the allosteric effect. In the case of macromolecular cofactors like thrombomodulin and factor Va, the residual enhancement of catalytic activity (Figure 4C and Table 2) is accounted for by the additional effect that these cofactors exert on the substrates protein C (27, 57) and prothrombin (58), respectively.
Figure 4.
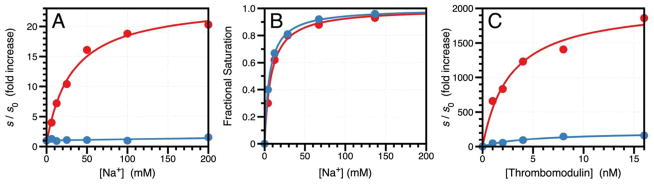
(A) Effect of Na+ on the value of s=kcat/Km for the hydrolysis of FPR by thrombin wild-type (red circles) and S195T (blue circles). Under the effect of a cofactor, the value of s increases according to the linkage expression s=(s0Kd+s1x)/(Kd+x) (28), where s0 and s1 are the asymptotic values of s at x=0 and x=∞, respectively, and Kd is the apparent equilibrium dissociation constant for the cofactor. The values of s are expressed in units of s0 to facilitate comparison. In the case of wild-type, the value of s increases from s0=3.8±0.1 μM−1s−1 to s1=92±2 μM−1s−1. In the case of S195T, the activity drops drastically and the Na+ effect is practically abrogated (s0=1.8±0.1 mM−1s−1 and s1=2.3±0.1 mM−1s−1). Experimental conditions are: 0.1% PEG8000, 5 mM Tris, pH 8.0 at 25 °C. (B) Na+ binding curve for wild-type (red circles) and S195T (blue circles) obtained by titration of the intrinsic fluorescence of the protein. The total change in fluorescence was 11% for wild-type and 13% for S195T. Mutation of S195 has little effect on the value of the equilibrium dissociation constant Kd that changes from 8.2±0.8 mM in the wild-type to 6.2±0.2 mM in the mutant. Experimental conditions are: 0.1% PEG8000, 50 mM Tris, pH 8.0 at 15 °C. (C) Effect of thrombomodulin on the value of s=kcat/Km for the hydrolysis of protein C by thrombin wild-type (red circles) and S195T (blue circles). The values of s are expressed in units of s0 as in panel (A) to facilitate comparison. In the case of wild-type (red circles), the value of s increases 2,100-fold from s0=0.15±0.01 mM−1s−1 to s1=310±10 mM−1s−1. In the case of S195T (blue circles), the activity drops drastically and the effect of thrombomodulin is significantly reduced (s0=0.0020±0.0001 mM−1s−1 and s1=0.36±0.02 mM−1s−1). The value of Kd=2.7±0.1 nM for thrombomodulin binding to wild-type is practically identical to the value of Kd=2.6±0.1 nM for the S195T mutant. Experimental conditions are: 0.1% PEG8000, 145 mM NaCl, 5 mM CaCl2, 5 mM Tris, pH 7.4 at 37 °C.
Figure 5.
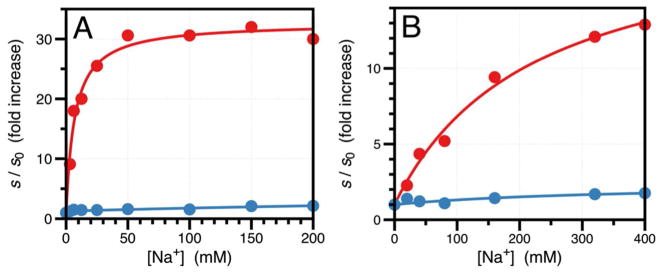
Effect of Na+ on the value of s=kcat/Km for the hydrolysis of a chromogenic substrate by wild-type (red) and mutant S195T (blue) of activated protein C (A) and clotting factor Xa (B). The values of s are expressed in units of s0 as in Figure 4A to facilitate comparison. As observed for thrombin (Figure 4A), the value of s in the wild-type increases from s0=2.1±0.1 mM−1s−1 to s1=69±2 mM−1s−1 for activated protein C (A) and from s0=1.4±0.1 μM−1s−1 to s1=28±1 μM−1s−1 for factor Xa (B). In the case of the S195T mutant, activity drops significantly and the Na+ effect is practically abrogated (s0=0.11±0.01 mM−1s−1 and s1=0.20±0.01 mM−1s−1 for activated protein C; s0=18±1 mM−1s−1, s1=38±1 mM−1s−1 for factor Xa). Experimental conditions are: (A) 0.1% PEG8000, 5 mM EDTA, 5 mM Tris, pH 8.0 at 25 °C; (B) 0.1% PEG8000, 5 mM EDTA, 50 mM HEPES, pH 8.0 at 25°C.
Table 2.
Kinetic parameters for activation of prothrombin by factor Xa wild-type and mutant S195T
| − Cofactor Va
|
+ Cofactor Va
|
|||||
|---|---|---|---|---|---|---|
| kcat/Km (μM−1s−1) | kcat (s−1) | Km (μM) | kcat/Km (μM−1s−1) | kcat (s−1) | Km (μM) | |
| wta | 0.12±0.01 | 0.022±0.002 | 0.19±0.01 | 380±10 | 42±2 | 0.11±0.01 |
| S195T | 0.31±0.03 10−3 | 0.049±0.003 10−3 | 0.16±0.01 | 62±3 10−3 | 21±1 10−3 | 0.34±0.02 |
From (32). Experimental conditions are: 25 μM phospholipids, 150 mM NaCl, 5 mM CaCl2, 0.1% PEG8000, 20 mM Tris, pH 7.4 at 25 °C, with or without 40 nM cofactor Va. Mutant S195T is 1 nM (− cofactor Va) or 40 pM (+cofactor Va).
Discussion
Previous studies have addressed the important question as to why Ser and not Thr brokers catalysis in trypsin-like proteases (24). In addition to being an intermediate between the alternative AGY and TCN codons for Ser in the genetic code – two choices significantly represented in the trypsin family (15, 16) – Thr constitutes an obvious functional analog for Ser. In fact, Thr retains the nucleophilicity of Ser, its H-bonding capabilities and a side chain of comparable volume that should be compatible with the constraints of the active site region. The expectation is that the S195T substitution of a trypsin-like protease should translate into perturbation of activity still compatible with biological function. On the other hand, the S195T substitution in trypsin causes a complete loss of activity whose molecular origin was attributed to the methyl group of Thr preventing H-bonding interaction between the Oγ atom of T195 and the Nε2 atom of the catalytic H57 required for efficient catalysis (24). Subsequent modeling calculations (22) and observational analysis (23) have supported this conclusion with the contention that a H-bonding interaction between T195 and H57 would require a rotation of the side chain impeded by the methyl group coming into direct collision with the neighbor C42-C58 disulfide bond or H57 itself. Altogether, previous functional and computational studies have rationalized the evolutionary preference of Ser over Thr in terms of an irremediable steric clash caused by Thr within the active site of the protease.
The results reported here extend and correct the conclusions from previous functional (24) and computational (22, 23) studies. The role of S195 has been analyzed extensively with site-directed substitutions that completely abolish activity (13, 24). Mutants of S195 provide invaluable reagents for structural biology and have assisted in the elucidation of the critical need of S195 for catalysis. However, the S195T substitution in thrombin, activated protein C and factor Xa reduces but does not abrogate activity and affords an opportunity to explore the functional role of S195 in ways not before possible. The presence of an additional methyl group on the side chain of Thr relative to Ser biases the mobility of the nucleophile within the active site and stabilizes rotamers incompatible with substrate binding (Figure 2B). The dihedral angle of T195 documented by the crystal structure of the thrombin mutant S195T in the free form is not documented in any of the structures with S195 currently in the PDB and points the methyl group toward an incoming substrate. However, the structure bound to PPACK demonstrates that productive substrate binding and hydrolysis are possible in the S195T mutant without causing steric clash with the catalytic H57 or the C42-C58 disulfide bond. Rapid kinetics prove that the substrate selects a reactive rotamer of the nucleophile from a pre-existing ensemble of reactive and unreactive rotamers and that the relative distribution of these rotamers defines the level of activity of the protease. The distribution is shifted toward reactive rotamers when Ser is present as a nucleophile (Figure 2B) and toward unreactive rotamers in the presence of Thr (Figure 1A), thereby explaining the significant loss of catalytic activity in the S195T substitution.
There is an additional benefit in using Ser and not Thr as nucleophile in the trypsin fold. Several proteases carrying S195 such as tissue-type plasminogen activator (59), clotting factors VIIa (60) and Xa (56) feature extremely low activity toward their physiological targets in the absence of cofactors, namely fibrin, tissue factor and cofactor Va, respectively. Thrombin itself has minuscule activity toward the anticoagulant protein C in the absence of thrombomodulin (61) and significantly reduced activity toward synthetic and procoagulant substrates in the absence of Na+ (28, 62). The observation that the S195T mutation abrogates the allosteric effect of Na+ in thrombin, activated protein C and factor Xa (Figures 4A and 5) and significantly reduces the allosteric effect of thrombomodulin in thrombin (Figure 4C) and of cofactor Va in factor Xa (Table 2) proves that the absolute selection of Ser over Thr also ensures optimal allosteric regulation in the trypsin fold.
The wide range of rotamers accessible to S195 (Figure 2B) suggests that the level of activity of the protease may be fine tuned by small rearrangements of the nucleophile within the active site. This energetically favorable landscape is not accessible to Thr and compromises allosteric transduction in the trypsin fold. A paradigm shift emerges where a rich landscape of catalytic regimens is generated by little perturbation of structure, possibly through changes in protein entropy (63) and dynamics (64, 65). Consequently, the action of allosteric effectors must be precisely exercised through structural changes that culminate in optimization of the rotamer of S195. The subtlety underscoring the role of S195 as the end-point of allosteric transduction in the trypsin fold explains why the architecture of the active site of thrombin changes little upon binding of Na+ (28) or thrombomodulin (57, 66), and why it is difficult to engineer Na+-dependent allostery in proteases devoid of Na+ binding (67, 68), or to reproduce the allosteric effect of thrombomodulin on thrombin (69) and of cofactor Va in the prothrombinase complex (32).
Acknowledgments
We are grateful to Ms. Tracey Baird for help with illustrations.
Footnotes
This work was supported in part by the National Institutes for Health Research Grants HL049413, HL073813, and HL112303.
References
- 1.Page MJ, Di Cera E. Serine peptidases: classification, structure and function. Cell Mol Life Sci. 2008;65:1220–1236. doi: 10.1007/s00018-008-7565-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rawlings ND, Morton FR, Kok CY, Kong J, Barrett AJ. MEROPS: the peptidase database. Nucleic Acids Res. 2008;36:D320–D325. doi: 10.1093/nar/gkm954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Puente XS, Sanchez LM, Gutierrez-Fernandez A, Velasco G, Lopez-Otin C. A genomic view of the complexity of mammalian proteolytic systems. Biochem Soc Trans. 2005;33:331–334. doi: 10.1042/BST0330331. [DOI] [PubMed] [Google Scholar]
- 4.Perona JJ, Craik CS. Structural basis of substrate specificity in the serine proteases. Protein Sci. 1995;4:337–360. doi: 10.1002/pro.5560040301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hedstrom L. Serine protease mechanism and specificity. Chem Rev. 2002;102:4501–4524. doi: 10.1021/cr000033x. [DOI] [PubMed] [Google Scholar]
- 6.Blow DM, Birktoft JJ, Hartley BS. Role of a buried acid group in the mechanism of action of chymotrypsin. Nature. 1969;221:337–340. doi: 10.1038/221337a0. [DOI] [PubMed] [Google Scholar]
- 7.Craik CS, Roczniak S, Largman C, Rutter WJ. The catalytic role of the active site aspartic acid in serine proteases. Science. 1987;237:909–913. doi: 10.1126/science.3303334. [DOI] [PubMed] [Google Scholar]
- 8.Sprang S, Standing T, Fletterick RJ, Stroud RM, Finer-Moore J, Xuong NH, Hamlin R, Rutter WJ, Craik CS. The three-dimensional structure of Asn102 mutant of trypsin: role of Asp102 in serine protease catalysis. Science. 1987;237:905–909. doi: 10.1126/science.3112942. [DOI] [PubMed] [Google Scholar]
- 9.Blow DM. The tortuous story of Asp ... His ..Ser: structural analysis of alpha-chymotrypsin. Trends Biochem Sci. 1997;22:405–408. doi: 10.1016/s0968-0004(97)01115-8. [DOI] [PubMed] [Google Scholar]
- 10.Bobofchak KM, Pineda AO, Mathews FS, Di Cera E. Energetic and structural consequences of perturbing Gly-193 in the oxyanion hole of serine proteases. J Biol Chem. 2005;280:25644–25650. doi: 10.1074/jbc.M503499200. [DOI] [PubMed] [Google Scholar]
- 11.Hartley BS, Kilby BA. The reaction of p-nitrophenyl esters with chymotrypsin and insulin. Biochem J. 1954;56:288–297. doi: 10.1042/bj0560288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carter P, Wells JA. Engineering enzyme specificity by “substrate-assisted catalysis”. Science. 1987;237:394–399. doi: 10.1126/science.3299704. [DOI] [PubMed] [Google Scholar]
- 13.Carter P, Wells JA. Dissecting the catalytic triad of a serine protease. Nature. 1988;332:564–568. doi: 10.1038/332564a0. [DOI] [PubMed] [Google Scholar]
- 14.Di Cera E. Site-specific analysis of mutational effects in proteins. Adv Protein Chem. 1998;51:59–119. doi: 10.1016/s0065-3233(08)60651-8. [DOI] [PubMed] [Google Scholar]
- 15.Brenner S. The molecular evolution of genes and proteins: a tale of two serines. Nature. 1988;334:528–530. doi: 10.1038/334528a0. [DOI] [PubMed] [Google Scholar]
- 16.Krem MM, Di Cera E. Molecular markers of serine protease evolution. Embo J. 2001;20:3036–3045. doi: 10.1093/emboj/20.12.3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Polgar L. The catalytic triad of serine peptidases. Cell Mol Life Sci. 2005;62:2161–2172. doi: 10.1007/s00018-005-5160-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCaldon P, Argos P. Oligopeptide biases in protein sequences and their use in predicting protein coding regions in nucleotide sequences. Proteins. 1988;4:99–122. doi: 10.1002/prot.340040204. [DOI] [PubMed] [Google Scholar]
- 19.Seemuller E, Lupas A, Stock D, Lowe J, Huber R, Baumeister W. Proteasome from Thermoplasma acidophilum: a threonine protease. Science. 1995;268:579–582. doi: 10.1126/science.7725107. [DOI] [PubMed] [Google Scholar]
- 20.Buller AR, Freeman MF, Wright NT, Schildbach JF, Townsend CA. Insights into cis-autoproteolysis reveal a reactive state formed through conformational rearrangement. Proc Natl Acad Sci U S A. 2012;109:2308–2313. doi: 10.1073/pnas.1113633109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marc F, Weigel P, Legrain C, Glansdorff N, Sakanyan V. An invariant threonine is involved in self-catalyzed cleavage of the precursor protein for ornithine acetyltransferase. J Biol Chem. 2001;276:25404–25410. doi: 10.1074/jbc.M100392200. [DOI] [PubMed] [Google Scholar]
- 22.Gokey T, Baird TT, Jr, Guliaev AB. Conformational dynamics of threonine 195 and the S1 subsite in functional trypsin variants. J Mol Model. 2012;18:4941–4954. doi: 10.1007/s00894-012-1541-x. [DOI] [PubMed] [Google Scholar]
- 23.Buller AR, Townsend CA. Intrinsic evolutionary constraints on protease structure, enzyme acylation, and the identity of the catalytic triad. Proc Natl Acad Sci U S A. 2013;110:653–661. doi: 10.1073/pnas.1221050110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baird TT, Jr, Wright WD, Craik CS. Conversion of trypsin to a functional threonine protease. Protein Sci. 2006;15:1229–1238. doi: 10.1110/ps.062179006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pozzi N, Chen Z, Zapata F, Pelc LA, Barranco-Medina S, Di Cera E. Crystal structures of prethrombin-2 reveal alternative conformations under identical solution conditions and the mechanism of zymogen activation. Biochemistry. 2011;50:10195–10202. doi: 10.1021/bi2015019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rezaie AR, Esmon CT. The function of calcium in protein C activation by thrombin and the thrombin-thrombomodulin complex can be distinguished by mutational analysis of protein C derivatives. J Biol Chem. 1992;267:26104–26109. [PubMed] [Google Scholar]
- 27.Pozzi N, Barranco-Medina S, Chen Z, Di Cera E. Exposure of R169 controls protein C activation and autoactivation. Blood. 2012;120:664–670. doi: 10.1182/blood-2012-03-415323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pineda AO, Carrell CJ, Bush LA, Prasad S, Caccia S, Chen ZW, Mathews FS, Di Cera E. Molecular dissection of Na+ binding to thrombin. J Biol Chem. 2004;279:31842–31853. doi: 10.1074/jbc.M401756200. [DOI] [PubMed] [Google Scholar]
- 29.Vogt AD, Bah A, Di Cera E. Evidence of the E*-E equilibrium from rapid kinetics of Na(+) binding to activated protein C and factor Xa. J Phys Chem B. 2010;114:16125–16130. doi: 10.1021/jp105502c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu H, Bush LA, Pineda AO, Caccia S, Di Cera E. Thrombomodulin changes the molecular surface of interaction and the rate of complex formation between thrombin and protein C. J Biol Chem. 2005;280:7956–7961. doi: 10.1074/jbc.M412869200. [DOI] [PubMed] [Google Scholar]
- 31.Yang L, Prasad S, Di Cera E, Rezaie AR. The conformation of the activation peptide of protein C is influenced by Ca2+ and Na+ binding. J Biol Chem. 2004;279:38519–38524. doi: 10.1074/jbc.M407304200. [DOI] [PubMed] [Google Scholar]
- 32.Pozzi N, Chen Z, Pelc LA, Shropshire DB, Di Cera E. The linker connecting the two kringles plays a key role in prothrombin activation. Proc Natl Acad Sci U S A. 2014;111:7630–7635. doi: 10.1073/pnas.1403779111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bah A, Garvey LC, Ge J, Di Cera E. Rapid kinetics of Na+ binding to thrombin. J Biol Chem. 2006;281:40049–40056. doi: 10.1074/jbc.M608600200. [DOI] [PubMed] [Google Scholar]
- 34.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected by oscillation methods. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 35.Bailey S. The CCP4 suite. Programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 36.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 37.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Joosten RP, Joosten K, Cohen SX, Vriend G, Perrakis A. Automatic rebuilding and optimization of crystallographic structures in the Protein Data Bank. Bioinformatics. 2011;27:3392–3398. doi: 10.1093/bioinformatics/btr590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morris AL, MacArthur MW, Hutchinson EG, Thornton JM. Stereochemical quality of protein structure coordinates. Proteins. 1992;12:345–364. doi: 10.1002/prot.340120407. [DOI] [PubMed] [Google Scholar]
- 40.Zhu J, Weng Z. FAST: a novel protein structure alignment algorithm. Proteins. 2005;58:618–627. doi: 10.1002/prot.20331. [DOI] [PubMed] [Google Scholar]
- 41.Gohara DW, Di Cera E. Allostery in trypsin-like proteases suggests new therapeutic strategies. Trends Biotechnol. 2011;29:577–585. doi: 10.1016/j.tibtech.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pozzi N, Vogt AD, Gohara DW, Di Cera E. Conformational selection in trypsin-like proteases. Curr Opin Struct Biol. 2012;22:421–431. doi: 10.1016/j.sbi.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bode W, Turk D, Karshikov A. The refined 1.9-A X-ray crystal structure of D-Phe-Pro-Arg chloromethylketone-inhibited human alpha-thrombin: structure analysis, overall structure, electrostatic properties, detailed active-site geometry, and structure-function relationships. Protein Sci. 1992;1:426–471. doi: 10.1002/pro.5560010402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li H, Robertson AD, Jensen JH. Very fast empirical prediction and rationalization of protein pKa values. Proteins. 2005;61:704–721. doi: 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- 45.Warshel A, Naray-Szabo G, Sussman F, Hwang JK. How do serine proteases really work? Biochemistry. 1989;28:3629–3637. doi: 10.1021/bi00435a001. [DOI] [PubMed] [Google Scholar]
- 46.Vogt AD, Di Cera E. Conformational Selection or Induced Fit? A Critical Appraisal of the Kinetic Mechanism. Biochemistry. 2012;51:5894–5902. doi: 10.1021/bi3006913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prasad S, Cantwell AM, Bush LA, Shih P, Xu H, Di Cera E. Residue Asp-189 controls both substrate binding and the monovalent cation specificity of thrombin. J Biol Chem. 2004;279:10103–10108. doi: 10.1074/jbc.M312614200. [DOI] [PubMed] [Google Scholar]
- 48.Dang QD, Di Cera E. Residue 225 determines the Na(+)-induced allosteric regulation of catalytic activity in serine proteases. Proc Natl Acad Sci U S A. 1996;93:10653–10656. doi: 10.1073/pnas.93.20.10653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Di Cera E, Guinto ER, Vindigni A, Dang QD, Ayala YM, Wuyi M, Tulinsky A. The Na+ binding site of thrombin. J Biol Chem. 1995;270:22089–22092. doi: 10.1074/jbc.270.38.22089. [DOI] [PubMed] [Google Scholar]
- 50.Hedstrom L, Szilagyi L, Rutter WJ. Converting trypsin to chymotrypsin: the role of surface loops. Science. 1992;255:1249–1253. doi: 10.1126/science.1546324. [DOI] [PubMed] [Google Scholar]
- 51.Krem MM, Di Cera E. Evolution of enzyme cascades from embryonic development to blood coagulation. Trends Biochem Sci. 2002;27:67–74. doi: 10.1016/s0968-0004(01)02007-2. [DOI] [PubMed] [Google Scholar]
- 52.Niu W, Chen Z, Bush-Pelc LA, Bah A, Gandhi PS, Di Cera E. Mutant N143P reveals how Na+ activates thrombin. J Biol Chem. 2009;284:36175–36185. doi: 10.1074/jbc.M109.069500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gandhi PS, Chen Z, Mathews FS, Di Cera E. Structural identification of the pathway of long-range communication in an allosteric enzyme. Proc Natl Acad Sci USA. 2008;105:1832–1837. doi: 10.1073/pnas.0710894105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vijayalakshmi J, Padmanabhan KP, Mann KG, Tulinsky A. The isomorphous structures of prethrombin2, hirugen-, and PPACK-thrombin: changes accompanying activation and exosite binding to thrombin. Protein Sci. 1994;3:2254–2271. doi: 10.1002/pro.5560031211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Esmon CT. The protein C pathway. Chest. 2003;124:26S–32S. doi: 10.1378/chest.124.3_suppl.26s. [DOI] [PubMed] [Google Scholar]
- 56.Rosing J, Tans G, Govers-Riemslag JW, Zwaal RF, Hemker HC. The role of phospholipids and factor Va in the prothrombinase complex. J Biol Chem. 1980;255:274–283. [PubMed] [Google Scholar]
- 57.Fuentes-Prior P, Iwanaga Y, Huber R, Pagila R, Rumennik G, Seto M, Morser J, Light DR, Bode W. Structural basis for the anticoagulant activity of the thrombin-thrombomodulin complex. Nature. 2000;404:518–525. doi: 10.1038/35006683. [DOI] [PubMed] [Google Scholar]
- 58.Pozzi N, Di Cera E. Prothrombin structure: unanticipated features and opportunities. Expert Rev Proteomics. 2014;11:653–655. doi: 10.1586/14789450.2014.971763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ding L, Coombs GS, Strandberg L, Navre M, Corey DR, Madison EL. Origins of the specificity of tissue-type plasminogen activator. Proc Natl Acad Sci U S A. 1995;92:7627–7631. doi: 10.1073/pnas.92.17.7627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dickinson CD, Kelly CR, Ruf W. Identification of surface residues mediating tissue factor binding and catalytic function of the serine protease factor VIIa. Proc Natl Acad Sci U S A. 1996;93:14379–14384. doi: 10.1073/pnas.93.25.14379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Esmon NL, Owen WG, Esmon CT. Isolation of a membrane-bound cofactor for thrombin-catalyzed activation of protein C. J Biol Chem. 1982;257:859–864. [PubMed] [Google Scholar]
- 62.Di Cera E. Thrombin. Mol Aspects Med. 2008;29:203–254. doi: 10.1016/j.mam.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cooper A, Dryden DT. Allostery without conformational change. A plausible model. Eur Biophys J. 1984;11:103–109. doi: 10.1007/BF00276625. [DOI] [PubMed] [Google Scholar]
- 64.Henzler-Wildman K, Kern D. Dynamic personalities of proteins. Nature. 2007;450:964–972. doi: 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- 65.Motlagh HN, Wrabl JO, Li J, Hilser VJ. The ensemble nature of allostery. Nature. 2014;508:331–339. doi: 10.1038/nature13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Adams TE, Li W, Huntington JA. Molecular basis of thrombomodulin activation of slow thrombin. J Thromb Haemost. 2009;7:1688–1695. doi: 10.1111/j.1538-7836.2009.03563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vindigni A, Di Cera E. Role of P225 and the C136-C201 disulfide bond in tissue plasminogen activator. Protein Sci. 1998;7:1728–1737. doi: 10.1002/pro.5560070807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Page MJ, Bleackley MR, Wong S, MacGillivray RTA, Di Cera E. Conversion of trypsin into a Na+ activated enzyme. Biochemistry. 2006;45:2987–2993. doi: 10.1021/bi052481a. [DOI] [PubMed] [Google Scholar]
- 69.Berg DT, Wiley MR, Grinnell BW. Enhanced protein C activation and inhibition of fibrinogen cleavage by a thrombin modulator. Science. 1996;273:1389–1391. doi: 10.1126/science.273.5280.1389. [DOI] [PubMed] [Google Scholar]