

Abstract
Myelinated axons are patterned into discrete and often repeating domains responsible for the efficient and rapid transmission of electrical signals. These domains include nodes of Ranvier and axon initial segments. Disruption of axonal patterning leads to nervous system dysfunction. In this review we introduce the concept of subcellular patterning as applied to axons and discuss how these patterning events depend on both intrinsic, cytoskeletal mechanisms, and extrinsic, myelinating-glia dependent mechanisms.
Introduction
Patterning is how organisms become segregated into spatially and functionally distinct structures or domains. Patterning is a common process among multicellular organisms as diverse as plants, invertebrates, and vertebrates. Early in development, patterning sets up broad domain distinctions. One of the earliest patterning events determines animal versus vegetal poles, which form the organism or support tissues like the yolk sac, respectively. Soon after, in the gastrulation stage, endoderm, ectoderm, and mesoderm germ layers become differentiated and the body axes are established. As development proceeds, patterning events establish finer and finer degrees of specialization. For example, within the nervous system, different brain regions have distinct neuronal subtypes, cytoarchitectures, and extend long-range axonal projections to highly specific and often unique target regions. An example of this is the patterning of the neocortex into discrete layers, each of which contains a characteristic array of neurons that receive inputs from and project to distinct cell types, have unique developmental origins, and express unique sets of genes and proteins. Overall, these features are established during development by an exquisite interplay of cell-intrinsic and cell-extrinsic signals.
Here, we extend the concept of patterning from regional specification and cell fate induction to patterning of subcellular domains. Traditional patterning emphasizes the differentiation and specialization of cells and groups of cells. In contrast, subcellular patterning is the segregation of spatially distinct, highly organized, and functionally specialized protein complexes within a cell; these processes have also been referred to as compartmentalization and/or subcellular polarization. Exquisite control over the expression, trafficking, and interactions of these protein complexes is required to target and maintain them in their correct subcellular locations at the right time.
In this review, we focus on subcellular patterning events in axons. We emphasize myelinated axons since they are highly polarized with many spatially, structurally, and functionally distinct protein complexes whose formation and positioning are critical for proper nervous system function. As with more traditional patterning mechanisms that depend on interactions between and among cells, patterning of myelinated axons depends on interactions between neurons and glia.
Axons are patterned into repeating excitable and non-excitable domains for the purpose of efficient and rapid transmission of electrical signals. These excitable domains include the axon initial segment (AIS) and nodes of Ranvier, and their disruption by disease or injury severely impairs nervous system function. The AIS, located at the interface between the neuronal cell body and axon (Fig. 1A), integrates synaptic inputs to generate an action potential. Its position and proximity to the cell body is determined by both activity and cell-type. The axon transmits the action potential over very long distances without diminution of speed or amplitude, while minimizing the energy used to propagate the action potential. To do this, axons are wrapped by myelin, which is made by oligodendrocytes and Schwann cells in the CNS and PNS, respectively. Myelin decreases membrane capacitance and increases membrane resistance to minimize the dissipation of ionic current as the action potential propagates along the axon. Gaps in the myelin sheath, called nodes of Ranvier, are located at regular intervals to regenerate the action potential (Fig. 1B–D). The spacing, or patterning, of nodes along the axon influences the speed of action potential propagation (Court et al., 2004). AIS and nodes of Ranvier consist of a common set of ion channels, cell adhesion molecules, and cytoskeletal scaffolds, yet there are differences in the way these molecules are organized along the axon.
Figure 1. Examples of axonal patterning.

A. A cultured hippocampal neuron immunostained to mark the somatodendritic domain (magenta, MAP2), the axon initial segment (green, AnkG), and the axon (red, tubulin). Scale bar, 25 μm.
B. Low magnification view of optic nerve axons immunostained using antibodies against juxtaparanodal Kv1.2 (red), paranodal Caspr (green), nodal NF186 (Magenta). Scale bar, 50 μm.
C. High magnification view of optic nerve axons immunostained using antibodies against juxtaparanodal Kv1.2 (red), paranodal Caspr (green), nodal NF186 (magenta). Scale bar, 20 μm.
D. Triple immunostaining of PNS node of Ranvier immunostained for nodal Na+ channels (green), paranodal Caspr (red), and juxtaparanodal K+ channels (blue). A phase contrast image of the myelin sheath was merged with the fluorescence image to show the outline of the myelin sheath. Scale bar, 5 μm.
In this review, we will use nodes of Ranvier and AIS as representative examples of subcellular patterning. We will emphasize the interplay of cell-intrinsic and cell-extrinsic mechanisms that work together to pattern the axon. Finally, we will discuss some examples of diseases or injuries that disrupt nodes and AIS, and altered axonal physiology.
Axonal patterning: specialized domains for efficient brain function
Nodes of Ranvier and their associated domains
Nodes of Ranvier are ~1 μm-long axonal Na+ channel clusters that occur at gaps in the myelin sheath and regenerate action potentials (Figs. 1B–D, and 2, ➂). Nodes of Ranvier and AIS share a common core protein composition that includes voltage-gated ion channels, the cell adhesion molecules (CAMs) neurofascin 186 (NF186) and NrCAM, and the scaffolding proteins AnkyrinG (AnkG) and βIVspectrin (for a more detailed review of the protein components of nodes and their associated domains, see (Chang and Rasband, 2013)). Nodal clustering of ion channels along the axon confines transmembrane currents to these sites. Together with the reduced membrane capacitance afforded by myelin, nodes greatly increase the speed and efficiency of action potential propagation.
Figure 2.
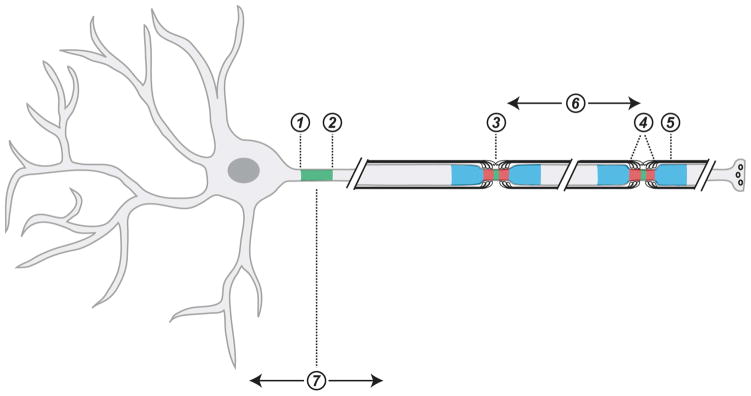
Axons are patterned into many distinct domains. Axons have distinct axonal patterns and boundaries including the proximal axon initial segment (AIS; ➀), the distal AIS and intra-axonal boundary (➁), the node of Ranvier (➂), the paranodal junctions (➃), the juxtaparanodes (➄), internodal length (➅), and axon/dendrite polarity (➆). The patterns ➀, ➁ and ➆ are determined by intrinsic mechanisms while ➂–➆ are determined by extrinsic neuron-glia interactions.
Classic studies in several vertebrate species, including humans, show that the internodal length (the distance between nodes of Ranvier, Fig. 2, ➅, corresponding to the non-excitable region of the axon) and conduction velocity increase with axon diameter. Thus, the periodic spacing, or patterning, of nodes helps to determine conduction velocity (Court et al., 2004). What dictates nodal spacing? In the adult human PNS internodal lengths for a 10 μm diameter axon can vary from ~1 mm in ulnar nerve to half that in facial nerve (Vizoso, 1950), indicating that simple fiber diameter does not predict the spacing of nodes along the axon. Instead, nodal spacing increases as an animal grows. For example, nodal spacing in the lower leg of humans can increase 4-fold from birth to young adulthood as height increases (Vizoso, 1950). However, studies in animal models show that there is a limit to the benefits conferred by increasing internodal length, and that there is a plateau above which increasing internodal length has no benefit to conduction velocity (Simpson et al., 2013). Thus, nodal spacing in the PNS is determined by two properties: the diameter of the axon, and the growth of the part of the animal in which the nerve lies.
In contrast, the mechanisms controlling nodal spacing in the CNS are more complicated. While both axon diameter and growth likely influence node spacing, additional activity dependent mechanisms must also dictate nodal spacing to optimize conduction velocity and tune the properties of CNS circuits. One particularly dramatic example of this is seen in the brainstem auditory circuits that calculate interaural time differences for sound localization. Here, neurons in the cochlear nucleus have a single bifurcating axon that innervates ipsilateral and contralateral coincidence detector neurons in the brainstem. These two branches of the same axon, with very different lengths, independently adjust their conduction velocities to achieve the precise temporal integration necessary for sound localization. Remarkably, the conduction velocities of these axon branches correlate with axon diameter and node spacing (Seidl et al., 2014; Seidl et al., 2010). How can two branches from the same axon have different diameters and internodal lengths to control conduction velocity? Although the answer to this question remains unknown, the mechanisms likely depend on local activity-dependent signaling between axons and their closely associated myelinating glial cell, the oligodendrocyte. For example, oligodendrocyte- and Schwann-cell axon signaling regulates neurofilament phosphorylation and spacing (Perrot et al., 2007), which in turn dictates axon diameter (Sanchez et al., 2000). Furthermore, since node of Ranvier formation in the CNS requires extrinsic, oligodendrocyte-derived interactions (see below), node locations must also depend on axon-oligodendrocyte signaling. Together, the data suggest that the CNS nodes of Ranvier are not simply static structures that regenerate action potentials, but that axons are precisely patterned to optimize conduction velocity and circuit functions. Future studies of axon-oligodendrocyte signaling will be needed to clarify the mechanisms regulating this fascinating form of axonal plasticity.
In addition to nodes and internodes, axons are patterned with two other highly organized axonal domains: the paranode (Fig. 1F and 3, ➃) and the juxtaparanode (Fig. 1F and 3, ➄). Paranodes flank each side of the node and are specialized cell-cell junctions formed between the axonal membrane and the paranodal loops of the myelinating glial cell; juxtaparanodes are located adjacent to paranodes and beneath the myelin sheath. The paranodal loops correspond to the end of each individual layer of myelin membrane and spiral around the axon to form the largest known vertebrate intercellular junction. The paranodal junction attaches the myelin membrane to the axon, isolates nodal currents from internodes, functions as a barrier to limit the diffusion of membrane proteins in the axolemma, and helps to cluster Na+ channels at nodes (Feinberg et al., 2010; Rosenbluth, 2009; Susuki et al., 2013; Zonta et al., 2008).
Figure 3. Intrinsic and extrinsic mechanisms of axonal patterning.

A. Two glia-dependent mechanisms cluster NF186 in the axonal membrane. In the PNS, the primary (➀) mechanism depends on interactions with gliomedin and NrCAM at the Schwann cell microvilli, and the secondary (➁) mechanism depends on the barrier function of the paranodal junctions. In the CNS, the primary (➀) mechanism depends on the barrier function of the paranodal junctions, while the secondary (➁) mechanism depends on interactions with secreted NrCAM and chondroitin sulfate proteoglycans found at the nodal extracellular matrix. NF186 functions to recruit nodal AnkG (➂) which then clusters Na+ channels and links the nodal protein complex to the βIV spectrin-based cytoskeleton (➃).
B. AnkB (blue)-dependent gene expression or a temporal delay in AnkG (red) expression may contribute to the intra-axonal boundary.
C. Repulsion or structural exclusion of Ankyrin/Spectrin protein complexes may help assemble distinct AnkB and AnkG-containing axonal domains.
D. Differential transport rates may help establish differences in Ankyrin protein localization along the axon.
E. Differential stability of Ankyrin/Spectrin protein complexes may help establish intra-axonal cytoskeletal boundaries
Juxtaparanodes are characterized by high densities of Kv1 K+ channels (Fig. 1F) that are thought to stabilize membrane potential, especially during myelination, after injury, and during remyelination. Juxtaparanodal Kv1 K+ channel clustering requires neuron-glia interactions between axonal Caspr2 (a homologue of the paranodal Caspr), and glial TAG-1 (a homologue of contactin) (Poliak et al., 2003; Savvaki et al., 2010). Thus, clustering of these K+ channels is another example of neuron-glia interactions that pattern the molecular organization of the axonal membrane.
Assembly of the paranode depends on interactions between the axonal CAMs Caspr and contactin, and the glial 155 kD isoform of neurofascin (NF155) (Bhat et al., 2001; Boyle et al., 2001; Charles et al., 2002; Pillai et al., 2009). Interestingly, like nodes, paranodes are also enriched with ankyrins and spectrins. In the peripheral nervous system, AnkyrinB, βII spectrin, and αII spectrin can be found at paranodes (Ogawa et al., 2006). In the CNS, AnkG is found at paranodes (Rasband et al., 1999). We recently showed that paranodal ankyrins are found in the myelinating glia where they interact with NF155. Genetic ablation of AnkG from oligdendrocytes causes a profound delay in formation and maturation of paranodal junctions (Chang et al., 2014). In axons, assembly of the spectrin-based paranodal cytoskeleton is thought to depend on the adaptor protein 4.1B, which links Caspr to spectrins. Axons lacking protein 4.1B or βII-spectrin have disrupted ion channel clustering and similar phenotypes: proteins normally restricted to juxtaparanodes invade neighboring paranodal domains (Horresh et al., 2010; Zhang et al., 2013). Thus, spectrin-based paranodal submembranous cytoskeletons may be viewed as cytoskeletal boundaries that organize the axon into repeating units of excitable and non-excitable membrane domains. The assembly of these paranodal boundaries depends on axon-glia interactions; loss of Caspr, contactin, or NF155 impairs paranodal junction formation, assembly of the paranodal cytoskeleton, and proteins normally restricted to juxtaparanodes enter paranodal domains (Bhat et al., 2001; Boyle et al., 2001; Ogawa et al., 2006; Pillai et al., 2009).
Although the paranodes, juxtaparanodes, and internodes are excellent examples of axonal patterning, the clustering of Na+ channels at the nodes of Ranvier remains the prototypical, and arguably most interesting and complex example of subcellular axonal patterning. What molecular and cellular mechanisms are responsible for nodal Na+ channel clustering? Remarkably, for this important process, two glial mechanisms have been identified that work together to assemble nodes of Ranvier. These mechanisms include 1) a glia-derived extracellular matrix and cell adhesion molecule protein complex that interacts with and clusters axonal transmembrane NF186, and 2) a paranodal junction-dependent membrane barrier that limits the lateral diffusion of axonal membrane proteins like NF186. These mechanisms converge on AnkG and βIV spectrin since they bind, stabilize, and link Na+ channels and NF186 to the underlying cytoskeleton (Feinberg et al., 2010; Susuki et al., 2013; Zonta et al., 2008).
Ankyrin/spectrin protein complexes function as key structural and scaffolding proteins to pattern the axon. Traditionally, one α- and one β-spectrin subunit combine to form a heterodimer. Two of these heterodimers interact in an anti-parallel arrangement to form a functional tetramer. Spectrin tetramers bind simultaneously to filamentous actin at each end, and to ankyrins at the central region of the tetramer through the β-spectrin subunits. The actin/spectrin/ankyrin complex has been extensively studied in erythrocytes where it anchors membrane proteins and provides structural stability and elasticity to the cell’s membrane (Bennett and Lorenzo, 2013). Interestingly, although PNS and CNS nodes both have clustered axonal Na+ channels, NF186, AnkG, and βIV spectrin, their glia-driven mechanisms of assembly follow different sequences of events and even use different molecules.
The first event leading to Na+ channel clustering in axons is the glia-dependent clustering of NF186 within the axonal membrane (Lambert et al., 1997; Schafer et al., 2006; Susuki et al., 2013). In the PNS, NF186 is initially clustered through interactions with gliomedin and NrCAM, which are found on microvilli at each end of the elongating Schwann cell (Fig. 3A, peripheral nervous system ➀). Gliomedin and NrCAM interact with other extracellular matrix (ECM) molecules to assemble a multimolecular protein complex that functions as a high avidity clustering complex for NF186 (Eshed et al., 2007; Feinberg et al., 2010). In contrast, gliomedin is not found at nodes in the CNS. Instead, paranodal junctions form before the clustering of NF186. As myelin elongates and the paranodal junction migrates along the axon, it accumulates NF186 adjacent to the paranodal junction (Fig. 3A, central nervous system ➀). In both the PNS and CNS, clustered NF186 then acts as an attachment site to recruit its direct binding partner AnkG (Fig. 3A, ➂).
Although the primary mechanism of Na+ channel clustering in the PNS is through gliomedin/NrCAM-dependent clustering of NF186, a secondary, paranodal junction-dependent mechanism can also support Na+ channel clustering (Fig. 3A, peripheral nervous system ➁). For example, gliomedin- and NrCAM-null mice still cluster NF186, AnkG, and Na+ channels, but mice lacking both gliomedin and paranodal junctions do not form Na+ channel clusters (Feinberg et al., 2010). Similarly, although paranodal junction barriers function as the primary mechanism of Na+ channel clustering in the CNS, loss of paranodal junctions does not block Na+ channel clustering. Instead, a second mechanism that depends on interactions between NF186 and a complex set of glia-derived ECM proteins (analogous to the gliomedin/NrCAM-NF186 interactions in the PNS) can substitute for paranodal junctions to assemble CNS nodes (Fig. 3A, central nervous system ➁). In support of this idea, mice lacking both paranodal junctions and CNS nodal ECM proteins die in the first 3 weeks of age and have severely impaired CNS nodal Na+ channel clustering (Susuki et al., 2013).
As described above, the two glia-dependent node assembly mechanisms converge on AnkG to initiate clustering of Na+ channels (Fig. 3A, ➃). Na+ channels have an intracellular ankyrin-binding motif (Garrido et al., 2003; Lemaillet et al., 2003) that is both necessary and sufficient for Na+ channel clustering at nodes (Gasser et al., 2012). The nodal AnkG/Na+ channel complex is stabilized through its association with the βIV spectrin-based submembranous cytoskeleton. Remarkably, during early development or in the absence of AnkG or βIV spectrin, a second ankyrin/spectrin cytoskeletal complex consisting of AnkyrinR and βI spectrin can cluster nodal Na+ channels (Ho et al., 2014). AnkR/βI spectrin complexes, normally thought to functional mainly in erythrocytes, are present in adult myelinated axons but not clustered at nodes. AnkR/βI spectrin has a lower affinity for NF186 and Na+ channels as compared to AnkG/βIV spectrin. This difference in affinities results in a hierarchy of clustering activities with AnkG/βIV spectrin as primary and AnkR/βI spectrin as secondary clustering mechanisms. Loss of both AnkR and AnkG completely blocks the clustering of nodal Na+ channels (Ho et al., 2014).
The importance of the glial-driven clustering mechanisms and the axonal cytoskeleton for CNS node formation was further shown in mice that lacked nodal βIV spectrin and either the paranodal mechanism or the ECM mechanism. These animals showed perinatal lethality, reduced optic nerve conduction velocities, and dramatically impaired clustering of Na+ channels at nodes of Ranvier (Susuki et al., 2013).
Myelination and assembly of nodes of Ranver is a rapid developmental process. How are nodes of Ranvier maintained throughout the life of an organism? Conditional ablation or knockout of the glia-derived factors that assemble nodes including gliomedin and NrCAM in the PNS causes nodes to become destabilized and eventually degenerate (Amor et al., 2014). Loss of axonal NF186 causes destabilization of both PNS and CNS nodes, although in the CNS, paranodal junctions appear to help maintain the nodal Na+ channel clusters (Desmazieres et al., 2014). Despite their primary role in developmental assembly of CNS nodes, loss of paranodal junctions in mature axons only results in a widening of nodal Na+ channel clusters rather than frank dissolution (Pillai et al., 2009). Similarly, loss of AnkG from mature nodes of Ranvier does not cause loss of Na+ channel clustering since AnkR substitutes for AnkG in these axons (Ho et al., 2014). Whether loss of both AnkG and AnkR from mature nodes results in degeneration of nodes remains unknown. Thus, in myelinated axons, glia organize, pattern, and maintain nodes of Ranvier through cytoskeletal scaffolding proteins.
Axon initial segments
The AIS is characterized by a 30–60 μm long stretch of densely packed voltage-gated Na+ and K+ channels located close to the neuronal cell body (Fig. 1A); as mentioned, the position and length of the AIS reflects the physiology and function of each neuron type. For example, the location of Na+ channels along the axons of rabbit retinal ganglion cells and chick auditory neurons in the nucleus laminaris corresponds to the types of stimuli they respond to (Fried et al., 2009; Kuba et al., 2006). The high concentration of ion channels causes the AIS to have a low threshold of activation in response to membrane depolarization (Kole and Stuart, 2008). As at nodes of Ranvier, AIS ion channels are retained by interactions among NF186, ECM molecules, and AnkG/βIV spectrin proteins that link the membrane and AIS-associated proteins to the underlying actin cytoskeleton (for a more detailed review of the molecular composition of the AIS see (Rasband, 2010) and (Leterrier and Dargent, 2013)). A dense network of microtubule bundles is found at the core of the AIS. Although microtubule bundles exist along the entirety of the axon, electron microscopy (EM) shows AIS microtubules coated by associated proteins (Jones et al., 2014). Both short, stable actin filaments as well as long, dynamic actin filaments exist at the AIS, but filamentous actin staining is not dense enough in this region to account for the microtubule coating. Instead, immuno-EM reveals that the coating consists of a variety of AIS-associated proteins (Jones et al., 2014).
Recent experiments using super-resolution microscopy revealed that actin, spectrins, and ankyrins form a periodic and patterned submembranous axonal cytoskeleton (Xu et al., 2013). When axons were viewed in cross-section, these components of the submembranous cytoskeleton formed alternating ring-like structures. The rings of actin were spaced with a periodicity of about 180–190 nm, which corresponds well with the length of a spectrin tetramer. Thus, the subcellular patterning of the actin/spectrin/ankyrin submembranous cytoskeleton is dictated simply by the size and orientation of its constituent parts. This patterned axonal cytoskeleton may function like a latticework of beams and trusses to dissipate the mechanical forces and stresses experienced by the axon, creating a flexible and strong axon (Rasband, 2013). At the AIS, βIV-spectrin subunits interact with AnkG, while the distal axon has a lattice of αII-spectrin, βII-spectrin, and AnkB. Ankyrins have both a spectrin-binding domain and a stretch of ankyrin repeats at their N-termini that allow simultaneous binding of several membrane-associated proteins including voltage-gated Na+ and K+ channels, and cell adhesion molecules (Davis et al., 1993; Garrido et al., 2003; Pan et al., 2006). Because of this ability, AnkG is the master organizer of the AIS (Zhou et al., 1998). Loss of AIS AnkG blocks clustering of all other known AIS proteins (Hedstrom et al., 2007). Thus, AnkG acts as a versatile, yet stable, hub for AIS proteins to be securely anchored to the underlying lattice of axonal actin cytoskeleton.
Besides its central role as the site of AP initiation, the AIS cytoskeleton also regulates the distinction between input and output domains (Fig. 2, ➆). For example, removal of AIS AnkG dismantles the AIS and permits dendritic proteins to enter the axon (Hedstrom et al., 2008). Remarkably, neurons lacking an AIS can even develop spine-like protrusions on their axons (Sobotzik et al., 2009). How does the AIS maintain axon identity? The AIS functions as the site where organelles with dendritic cargoes are refused entry into the axon, while those containing axonal proteins pass through unimpeded. However, the mechanisms that control this property of the AIS remain poorly understood. It has been proposed that a dense actin meshwork within the AIS acts as a cytoplasmic filter to slow protein trafficking and even block entry of dendritic cargoes while allowing axonal cargoes to proceed into the distal axon (Song et al., 2009; Watanabe et al., 2012). This view is supported by experiments where actin-depolymerizing agents dissolve the ‘filter’ and allow mixing of axonal and dendritic proteins (Song et al., 2009; Winckler et al., 1999). However, live-imaging of fluorescently-tagged dendritic and/or axonal proteins showed that organelles containing axonal proteins do not change velocity as they pass through the AIS (Petersen et al., 2014). In contrast, organelles with dendritic cargoes come to an abrupt stop upon entering the AIS. This argues against a simple cytoplasmic filter mechanism that would be expected to gradually slow all organelles. Instead, these data support an active sorting mechanism.
The spatial, structural, and functional properties of AIS must be precisely specified during development, and regulated throughout life, to produce reliably functioning neurons. During development, axon-intrinsic patterning events assemble and position the AIS. Later, AIS location may change in response to synaptic activity from other neurons (Kuba et al., 2010). AnkG is the first AIS component to cluster in the proximal axon during development and is the master organizer and maintainer of the AIS (Jenkins and Bennett, 2001). Indeed, the AIS is usually defined by AnkG clustering rather than by anatomy. Loss of AnkG from developing neurons blocks AIS assembly (Zhou et al., 1998), and ablation or proteolysis of AnkG in mature neurons dismantles the AIS (Hedstrom et al., 2008; Schafer et al., 2009). Unlike nodes of Ranvier, the initial assembly of the AIS is a neuron-intrinsic event requiring no external factors.
What neuron-intrinsic patterning events restrict AnkG to the proximal axon during development? To begin to address this question, Galiano et al. (Galiano et al., 2012) examined the progression of AnkG clustering at the AIS of developing cortical neurons in vivo. They found that AnkG accumulation only occurs after axon specification. Intriguingly, they observed an apparent intra-axonal boundary limiting AnkG’s location along the axon (Fig. 2, ➁). With further development and beginning at the boundary, AnkG then backfilled the axon towards the soma. These observations suggested that AnkG could be restricted to the AIS by exclusion from the distal axon rather than by active recruitment to the proximal axon. To test this potential mechanism, Galiano et al. (Galiano et al., 2012) searched for cytoskeletal proteins that could function as a boundary. Remarkably, by analogy to the paranodal junction of myelinated axons, they found AnkB and its spectrin binding partners, αII- and βII-spectrin accumulate in the growth cone during axon development, then backfill the axon up to the point where AnkG clustering eventually occurs. These findings suggested that the AnkB/αII-/βII-spectrin-based submembranous cytoskeleton defines the intra-axonal boundary. Additional support for this idea came from experiments where AnkB was overexpressed, resulting in a proximal shift in the intra-axonal boundary. In contrast, when AnkG was overexpressed, the boundary shifted towards the distal axon. Finally, ablation of AnkB, αII-, or βII-spectrin permitted AnkG to bypass the boundary and form clusters in the distal axon. Together, these experiments suggested that AnkB, αII- and βII-spectrin form a submembranous cytoskeletal protein complex that excludes AnkG and its spectrin binding partner βIV-spectrin; this mutual exclusion results in a balance between axonal AnkB and AnkG (and their associated spectrins) that creates an apparent intra-axonal boundary. A similar pattern of complementary AnkG/AnkB localization can be found in rod photoreceptors, where AnkG is localized to rod outer segments while AnkB is localized to inner segments (Kizhatil et al., 2009). Thus, it is possible that the same mechanism of mutual ankyrin exclusion facilitates the subcellular patterning of photoreceptors. Interestingly, a recent study from the Bennet lab (Jenkins et al., 2015) shows that AIS clustering of AnkG requires the alternatively spliced giant exon of AnkG. Furthermore, in contrast to conventional views of ankyrin-spectrin interactions, this exon is also required to recruit βIV spectrin to the AIS (Jenkins et al., 2015). How this giant exon contributes to AnkG clustering at the AIS remains unknown.
Although these experiments provided a significant conceptual advance in our understanding of cytoskeleton-dependent instrinsic axon patterning mechanisms, many questions remain. For example, it remains unclear how intra-axonal boundary location is reproducibly established. In the case of the paranodal junction, where a similar spectrin-based intra-axonal boundary is found, the interaction between axon and myelinating glial cell assembles and defines the boundary location. In the following paragraphs we speculate about the mechanisms that could underlie assembly of a boundary in the absence of instructive signals from other cell types. One possibility that has precedence in regional patterning and cell fate determination is temporal induction of gene expression. If accumulation of a particular level of AnkB within the distal axon triggers activation of ANK3 expression (ANK3 encodes AnkG), or if ANK3 expression is delayed relative to ANK2 expression (ANK2 encodes AnkB), this could contribute to how the AnkB/AnkG boundary is formed (Fig. 3B). Consistent with this idea, immunoblots of developing hippocampal neurons show AnkB protein expression precedes that of AnkG, and simple overexpression of these proteins shifted the position of the boundary (Galiano et al., 2012). However, mechanisms of axonal compartmentalization or patterning in Drosophila argue against timing of protein expression as the sole determinant of intra-axonal boundaries (Katsuki et al., 2009). For example, ROBO and DRL proteins are localized in the distal and proximal axon, respectively, with a boundary between the two located near the midpoint of the axon. Forced expression of ROBO, or ROBO and DRL simultaneously, does not alter the ROBO/DRL boundary. Surprisingly, blocking dynamin-mediated endocytosis shifted the boundary, suggesting that patterning of boundaries also depends on endocytosis and protein trafficking. Thus, patterning by transcriptional regulation may be more common for patterning of boundaries between cells than within cells.
Alternative mechanisms contributing to AIS boundary formation might include direct inhibition between AnkB and AnkG (Fig. 3C). Although these scaffolds share structural homology and can even bind many of the same membrane proteins, it is unclear why they do not occupy the same domains. Direct inhibition may not depend solely on the ankyrins, but rather the ankyrin/spectrin complex consisting of AnkB/βII-spectrin and AnkG/βIV-spectrin. In this view, the structural features of specific ankyrin/spectrin networks may preclude mixing of AnkB and AnkG. In addition, differential axonal trafficking efficiencies of AnkB compared to AnkG could contribute to higher levels of AnkB/αII-/βII-spectrin in the distal axon (Fig. 3D). Consistent with this idea, efficient trafficking of AnkB/αII-/βII-spectrin is mediated by interactions with the microtubule motor Kif3 through the adaptor Kap3 (Galiano et al., 2012).
Another potential mechanism for the specification of a precise intra-axonal boundary draws inspiration from traditional patterning, where gradients of diffusible morphogens induce transcription factor activity in a concentration dependent manner to pattern cells and groups of cells at reproducible locations in the body. Applying this concept to the AIS, specification of the intra-axonal boundary may depend on a gradient of a protein or post-translational protein modifications along the axon, with high levels near the soma, and low levels towards the distal axon. If high ‘concentrations’ of the protein or modification decrease stability or prevent incorporation of AnkB/αII-/βII-spectrin protein complexes into the submembranous cytoskeleton, then a reproducible proximal boundary would form at the threshold for inhibition. However, in the distal axon, below the threshold for inhibition, AnkB/αII-/βII-spectrin could be efficiently incorporated into the submembranous cytoskeleton. Thus, block of AnkB accumulation near the soma would leave the proximal axon free for AnkG and βIV spectrin to accumulate, resulting in a sharp and reproducible intra-axonal AnkB/AnkG boundary (Fig. 3D). This model is consistent with previous studies that suggest the AIS may be a ‘hot-spot’ for protein phosphorylation (Buffington et al., 2012; Yoshimura and Rasband, 2014), and studies showing phosphorylation of microtubules contributes to AIS barrier function (Li et al., 2011). Thus, we propose the intra-axonal boundary for AnkG clustering is more likely to depend on a combination of protein-protein interactions, protein modifications, and trafficking rather than simply timing of ankyrin expression (Fig. 3A–D).
Extrinsic influences in the form of neuronal activity can also influence organization of the axonal membrane by inducing changes in AIS structure and location. Using the chick auditory system, Kuba et al. (Kuba et al., 2010) deprived auditory neurons of sensory input by removing the cochlea. In response, AIS length increased to make the synaptically deprived neurons more excitable. In a complementary study, Grubb and Burrone (Grubb and Burrone, 2010) increased the activity of cultured hippocampal neurons using either high extracellular K+ to induce chronic depolarization, or channelrhodopsin to drive neuronal firing (Grubb and Burrone, 2010). In both paradigms the length of the AIS did not change, but the entire AIS translocated to a more distal position along the axon resulting in decreased neuronal excitability. In a more recent study, Kuba et al. (Kuba et al., 2014) showed that under normal conditions the development of the proximal end of the AIS is activity-dependent (Fig. 2, ➀), while the position of the distal end of the AIS is activity-independent (Fig. 2, ➁), a conclusion that fits well with the concept of an ankyrin/spectrin-dependent intra-axonal boundary as described above. It remains to be determined what activity-dependent molecular mechanisms regulate where the AIS begins in relation to the neuronal cell body. Together, these observations show the AIS is not static, but can respond to changes in neuronal activity. Such changes likely represent an elegant homeostatic mechanism to regulate overall neuronal activity in the face of Hebbian plasticity and to protect neuronal networks from runaway potentiation or depression (Turrigiano and Nelson, 2000).
Axonal patterning and disease
Given the important role of axonal patterning for neuronal function, we propose that some neurological disorders may include or result from defects in axonal patterning. Disrupted patterning could result from mutations in key proteins responsible for assembly of excitable domains, or be secondary to other pathologies or compensatory changes, such as demyelination or altered neuronal excitability. Genetic testing, along with increasing knowledge of the components and functions of the AIS and nodes of Ranvier have provided a backdrop for an expanding body of literature consistent with this idea. For example, although mutations in AnkG have been linked (primarily by genome-wide association studies) to various complex cognitive disorders including bipolar disease and schizophrenia (Ferreira et al., 2008; Schulze et al., 2009), evidence for a causative role of these variants or disrupted axonal patterning remains to be found. More recently, mutations in ANK3 were identified that cause intellectual disability (ID), attention deficit hyperactivity disorder (ADHD), and autism spectrum disorder (ASD) (Iqbal et al., 2013). In one patient with ADHD, ASD, sleep disturbance, delayed language, and ID, genetic testing revealed a mutation that disrupts all AnkG isoforms. The mutation occurred between the ankyrin repeat domain (membrane protein-binding domain) and the spectrin binding domain, suggesting that any protein made from the mutant allele would not be able to link membrane proteins at the AIS to the underlying actin cytoskeleton. In a separate case, a familial recessive ANK3 mutation was found in three affected siblings, all presenting with moderate ID, hypotonia, spasticity, sleep disturbances, and behavioral problems including aggression and hyperactivity. Genetic testing revealed a single base pair deletion in ANK3, leading to a frame-shift mutation and premature stop codon in exon 42 (C-terminus of AnkG). In addition to these cases, whole exome sequencing and copy number variation analysis on patients with autism have identified five additional de novo changes within ANK3 (four missense mutations and one duplication on chromosome 10 including the ANK3 locus (Sanders et al., 2012; Sebat et al., 2007)). Although these studies clearly implicate mutant AnkG in the pathology of the disease, future studies will be required to determine if the mutations cause altered axonal patterning.
While AnkG is an obvious genetic risk locus, other cytoskeletal proteins involved in axonal patterning and function have also been implicated in neurological disorders. For example, mutations in SPTAN1 (αII-spectrin) cause early-onset West Syndrome with drug resistant seizures, cerebral hypomyelination, severe developmental delay, spastic quadriplegia, and pontocerebellar atrophy (Saitsu et al., 2010; Writzl et al., 2012). Studies of these mutations revealed aggregates of spectrins within neurons, and disrupted AnkG and Na+ channel clustering at the AIS (Saitsu et al., 2010). These observations are consistent with the reported role of the αII-/βII-spectrin-based cytoskeleton for proper assembly of the AIS (Galiano et al., 2012), and may include altered node of Ranvier formation given the importance of the paranodal cytoskeleton. Other axonal proteins necessary for axonal patterning that are associated with human neurological disorders include Caspr2, which is found both at AIS and juxtaparanodes (Ogawa et al., 2008; Poliak et al., 1999). Mutations and variants of CNTNAP2 (Caspr2) are very strong risk factors for ASD, although it remains unknown if axonal patterning is perturbed in these patients. Intriguingly, autoantibodies against Caspr2 have also been shown to be associated with limbic encephalitis, cerebellar ataxia, and epilepsy (Balint et al., 2013; Lilleker et al., 2013). Recently, a pathogenic homozygous frameshift mutation in Caspr was identified in four unrelated human families. The mutation caused arthrogryposis multiple congenita, characterized by congenital contractures and reduced mobility. Patients showed marked reduction in nerve conduction velocity and disruptions in axoglial contact flanking nodes (Laquerriere et al., 2014).
Not only can mutations in AIS- and node-associated proteins contribute to neurological disorders, but other genetic diseases can also indirectly alter ion channel clustering along axons. For example, Angelman’s syndrome is a developmental disorder caused by mutations in the UBE3A gene that encodes a ubiquitin ligase, and characterized by severe ID, ASD, and epilepsy. Although UBE3A is not at the AIS and does not interact with AnkG, mice lacking UBE3A have increased expression of AnkG, increased AIS length, increased Nav1.6 Na+ channels at the AIS, and altered intrinsic membrane properties in hippocampal neurons (Kaphzan et al., 2011). Remarkably, genetic reduction of the Na+/K+-ATPase restored membrane properties and AIS structure (Kaphzan et al., 2013). This observation is consistent with the concept that neuronal excitability can modulate axonal patterning.
Axon organization can also be disrupted by injury or non-genetic diseases. For example, optic nerve crush or ischemic injury, as occurs during stroke, activate calpains causing proteolysis of the AIS cytoskeletal proteins AnkG and βIV spectrin (Schafer et al., 2009). Loss of AnkG from the AIS cytoskeleton is accompanied by loss of clustered Na+ channels and neuronal polarity (Hedstrom et al., 2008), resulting in dysfunctional neurons. Autoimmune diseases, including multiple sclerosis and Guillain-Barre syndrome (GBS), disrupt axonal patterning. Since myelinating glia are required for both the assembly and maintenance of nodes of Ranvier, demyelination results in loss of the regularly spaced Na+ and K+ channel clusters normally found at nodes and juxtaparanodes (Craner et al., 2004; Dugandzija-Novakovic et al., 1995; Rasband et al., 1998). Recent studies of the pathomechanisms of GBS have even uncovered autoantibodies against nodal antigens like gliomedin and NF186 that directly attack and disrupt nodes of Ranvier (Susuki et al., 2007; Devaux et al., 2012; Ng et al., 2012). These observations have now led to the proposal of a new category of peripheral autoimmune neuropathies termed ‘nodopathies’ (Susuki, 2013; Uncini et al., 2013). Taken together, these observations suggest that disrupted axonal patterning is a common sequela of many nervous system injuries and diseases. We propose that therapeutic strategies aimed at axon maintenance and regeneration will require a detailed understanding of the developmental mechanisms responsible for axonal patterning.
In conclusion, the studies described here reveal both axon-intrinsic and glia-dependent extrinsic mechanisms that together pattern the axon into excitable and non-excitable domains. While many of the essential mechanisms of developmental assembly and protein components have been described, much remains unknown. We propose that future studies of axonal patterning should focus on mechanisms regulating plasticity of these domains, and diseases or injuries that have as a core pathology the disruption of excitable domains.
Acknowledgments
This work was supported by NIH grants NS044916 and NS069688, and a grant from the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation. We apologize to colleagues whose relevant work was not discussed due to space limitations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amor V, Feinberg K, Eshed-Eisenbach Y, Vainshtein A, Frechter S, Grumet M, Rosenbluth J, Peles E. Long-Term Maintenance of Na+ Channels at Nodes of Ranvier Depends on Glial Contact Mediated by Gliomedin and NrCAM. J Neurosci. 2014;34:5089–5098. doi: 10.1523/JNEUROSCI.4752-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balint B, Regula JU, Jarius S, Wildemann B. Caspr2 antibodies in limbic encephalitis with cerebellar ataxia, dyskinesias and myoclonus. J Neurol Sci. 2013;327:73–74. doi: 10.1016/j.jns.2013.01.040. [DOI] [PubMed] [Google Scholar]
- Bennett V, Lorenzo DN. Spectrin- and ankyrin-based membrane domains and the evolution of vertebrates. Current topics in membranes. 2013;72:1–37. doi: 10.1016/B978-0-12-417027-8.00001-5. [DOI] [PubMed] [Google Scholar]
- Bhat MA, Rios JC, Lu Y, Garcia-Fresco GP, Ching W, St Martin M, Li J, Boyle ME, Berglund EO, Murai KK, Weber L, Peles E, Ranscht B. Contactin orchestrates assembly of the septate-like junctions at the paranode in myelinated peripheral nerve. Neuron. 2001;30:385–397. doi: 10.1016/s0896-6273(01)00296-3. [DOI] [PubMed] [Google Scholar]
- Buffington SA, Sobotzik JM, Schultz C, Rasband MN. IκBα is not required for axon initial segment assembly. Mol Cell Neurosci. 2012;50:1–9. doi: 10.1016/j.mcn.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KJ, Rasband MN. Excitable domains of myelinated nerves: axon initial segments and nodes of Ranvier. Current topics in membranes. 2013;72:159–192. doi: 10.1016/B978-0-12-417027-8.00005-2. [DOI] [PubMed] [Google Scholar]
- Chang KJ, Zollinger DR, Susuki K, Sherman DL, Makara MA, Brophy PJ, Cooper EC, Bennett V, Mohler PJ, Rasband MN. Glial ankyrins facilitate paranodal axoglial junction assembly. Nat Neurosci. 2014;17:1673–1681. doi: 10.1038/nn.3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles P, Tait S, Faivre-Sarrailh C, Barbin G, Gunn-Moore F, Denisenko-Nehrbass N, Guennoc AM, Girault JA, Brophy PJ, Lubetzki C. Neurofascin is a glial receptor for the paranodin/Caspr-contactin axonal complex at the axoglial junction. Curr Biol. 2002;12:217–220. doi: 10.1016/s0960-9822(01)00680-7. [DOI] [PubMed] [Google Scholar]
- Court FA, Sherman DL, Pratt T, Garry EM, Ribchester RR, Cottrell DF, Fleetwood-Walker SM, Brophy PJ. Restricted growth of Schwann cells lacking Cajal bands slows conduction in myelinated nerves. Nature. 2004;431:191–195. doi: 10.1038/nature02841. [DOI] [PubMed] [Google Scholar]
- Craner MJ, Newcombe J, Black JA, Hartle C, Cuzner ML, Waxman SG. Molecular changes in neurons in multiple sclerosis: altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proc Natl Acad Sci U S A. 2004;101:8168–8173. doi: 10.1073/pnas.0402765101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JQ, McLaughlin T, Bennett V. Ankyrin-binding proteins related to nervous system cell adhesion molecules: candidates to provide transmembrane and intercellular connections in adult brain. J Cell Biol. 1993;121:121–133. doi: 10.1083/jcb.121.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmazieres A, Zonta B, Zhang A, Wu LM, Sherman DL, Brophy PJ. Differential stability of PNS and CNS nodal complexes when neuronal neurofascin is lost. J Neurosci. 2014;34:5083–5088. doi: 10.1523/JNEUROSCI.4662-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaux JJ, Odaka M, Yuki N. Nodal proteins are target antigens in Guillain-Barre syndrome. Journal of the peripheral nervous system : JPNS. 2012;17:62–71. doi: 10.1111/j.1529-8027.2012.00372.x. [DOI] [PubMed] [Google Scholar]
- Dugandzija-Novakovic S, Koszowski AG, Levinson SR, Shrager P. Clustering of Na+ channels and node of Ranvier formation in remyelinating axons. J Neurosci. 1995;15:492–503. doi: 10.1523/JNEUROSCI.15-01-00492.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einheber S, Chesler M, Rosenbluth J, et al. Axon-glia interactions and the domain organization of myelinated axons requires neurexin IV/Caspr/Paranodin. Neuron. 2001;30:369–383. doi: 10.1016/s0896-6273(01)00294-x. [DOI] [PubMed] [Google Scholar]
- Eshed Y, Feinberg K, Carey DJ, Peles E. Secreted gliomedin is a perinodal matrix component of peripheral nerves. J Cell Biol. 2007;177:551–562. doi: 10.1083/jcb.200612139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg K, Eshed-Eisenbach Y, Frechter S, Amor V, Salomon D, Sabanay H, Dupree JL, Grumet M, Brophy PJ, Shrager P, et al. A glial signal consisting of gliomedin and NrCAM clusters axonal Na+ channels during the formation of nodes of Ranvier. Neuron. 2010;65:490–502. doi: 10.1016/j.neuron.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira MA, O’Donovan MC, Meng YA, Jones IR, Ruderfer DM, Jones L, Fan J, Kirov G, Perlis RH, Green EK, et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet. 2008;40:1056–81058. doi: 10.1038/ng.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried SI, Lasker AC, Desai NJ, Eddington DK, Rizzo JF., 3rd Axonal sodium-channel bands shape the response to electric stimulation in retinal ganglion cells. J Neurophysiol. 2009;101:1972–1987. doi: 10.1152/jn.91081.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galiano MR, Jha S, Ho TS, Zhang C, Ogawa Y, Chang KJ, Stankewich MC, Mohler PJ, Rasband MN. A distal axonal cytoskeleton forms an intra-axonal boundary that controls axon initial segment assembly. Cell. 2012;149:1125–1139. doi: 10.1016/j.cell.2012.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido JJ, Giraud P, Carlier E, Fernandes F, Moussif A, Fache MP, Debanne D, Dargent B. A targeting motif involved in sodium channel clustering at the axonal initial segment. Science. 2003;300:2091–2094. doi: 10.1126/science.1085167. [DOI] [PubMed] [Google Scholar]
- Gasser A, Ho TSY, Cheng X, Chang KJ, Waxman SG, Rasband MN, Dib-Hajj S. An ankyrinG-binding motif is necessary and sufficient for targeting Nav1.6 Na+ channels to axon initial segments and nodes of Ranvier. J Neurosci. 2012;32:7232–7243. doi: 10.1523/JNEUROSCI.5434-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb MS, Burrone J. Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature. 2010;465:107–1074. doi: 10.1038/nature09160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedstrom KL, Ogawa Y, Rasband MN. AnkyrinG is required for maintenance of the axon initial segment and neuronal polarity. J Cell Biol. 2008;183:635–640. doi: 10.1083/jcb.200806112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedstrom KL, Xu X, Ogawa Y, Frischknecht R, Seidenbecher CI, Shrager P, Rasband MN. Neurofascin assembles a specialized extracellular matrix at the axon initial segment. J Cell Biol. 2007;178:875–886. doi: 10.1083/jcb.200705119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TS, Zollinger DR, Chang KJ, Xu M, Cooper EC, Stankewich MC, Bennett V, Rasband MN. A hierarchy of ankyrin-spectrin complexes clusters sodium channels at nodes of Ranvier. Nat Neurosci. 2014;17:1664–1672. doi: 10.1038/nn.3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horresh I, Bar V, Kissil JL, Peles E. Organization of myelinated axons by Caspr and Caspr2 requires the cytoskeletal adapter protein 4.1B. J Neurosci. 2010;30:2480–2489. doi: 10.1523/JNEUROSCI.5225-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal Z, Vandeweyer G, van der Voet M, Waryah AM, Zahoor MY, Besseling JA, Roca LT, Vulto-van Silfhout AT, Nijhof B, Kramer JM, et al. Homozygous and heterozygous disruptions of ANK3: at the crossroads of neurodevelopmental and psychiatric disorders. Hum Mol Genet. 2013;22:1960–1970. doi: 10.1093/hmg/ddt043. [DOI] [PubMed] [Google Scholar]
- Jenkins PM, Kim N, Jones SL, Tseng WC, Svitkina TM, Yin HH, Bennett V. Giant ankyrin-G: A critical innovation in vertebrate evolution of fast and integrated neuronal signaling. Proc Natl Acad Sci U S A. 2015 doi: 10.1073/pnas.1416544112. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins SM, Bennett V. Ankyrin-G coordinates assembly of the spectrin-based membrane skeleton, voltage-gated sodium channels, and L1 CAMs at Purkinje neuron initial segments. J Cell Biol. 2001;155:739–746. doi: 10.1083/jcb.200109026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SL, Korobova F, Svitkina T. Axon initial segment cytoskeleton comprises a multiprotein submembranous coat containing sparse actin filaments. J Cell Biol. 2014;205:67–81. doi: 10.1083/jcb.201401045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaphzan H, Buffington SA, Jung JI, Rasband MN, Klann E. Alterations in intrinsic membrane properties and the axon initial segment in a mouse model of angelman syndrome. J Neurosci. 2011;31:17637–17648. doi: 10.1523/JNEUROSCI.4162-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaphzan H, Buffington SA, Ramaraj AB, Lingrel JB, Rasband MN, Santini E, Klann E. Genetic reduction of the alpha1 subunit of Na/K-ATPase corrects multiple hippocampal phenotypes in Angelman syndrome. Cell Rep. 2013;4:405–412. doi: 10.1016/j.celrep.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuki T, Ailani D, Hiramoto M, Hiromi Y. Intra-axonal patterning: intrinsic compartmentalization of the axonal membrane in Drosophila neurons. Neuron. 2009;64:188–199. doi: 10.1016/j.neuron.2009.08.019. [DOI] [PubMed] [Google Scholar]
- Kizhatil K, Baker SA, Arshavsky VY, Bennett V. Ankyrin-G promotes cyclic nucleotide-gated channel transport to rod photoreceptor sensory cilia. Science. 2009;323:1614–1617. doi: 10.1126/science.1169789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole MH, Stuart GJ. Is action potential threshold lowest in the axon? Nat Neurosci. 2008;11:1253–1255. doi: 10.1038/nn.2203. [DOI] [PubMed] [Google Scholar]
- Kuba H, Adachi R, Ohmori H. Activity-dependent and activity-independent development of the axon initial segment. J Neurosci. 2014;34:3443–3453. doi: 10.1523/JNEUROSCI.4357-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuba H, Ishii TM, Ohmori H. Axonal site of spike initiation enhances auditory coincidence detection. Nature. 2006;444:1069–1072. doi: 10.1038/nature05347. [DOI] [PubMed] [Google Scholar]
- Kuba H, Oichi Y, Ohmori H. Presynaptic activity regulates Na(+) channel distribution at the axon initial segment. Nature. 2010;465:1075–1078. doi: 10.1038/nature09087. [DOI] [PubMed] [Google Scholar]
- Lambert S, Davis JQ, Bennett V. Morphogenesis of the node of Ranvier: co-clusters of ankyrin and ankyrin-binding integral proteins define early developmental intermediates. J Neurosci. 1997;17:7025–7036. doi: 10.1523/JNEUROSCI.17-18-07025.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laquerriere A, Maluenda J, Camus A, Fontenas L, Dieterich K, Nolent F, Zhou J, Monnier N, Latour P, Gentil D, et al. Mutations in CNTNAP1 and ADCY6 are responsible for severe arthrogryposis multiplex congenita with axoglial defects. Hum Mol Genet. 2014;23:2279–2289. doi: 10.1093/hmg/ddt618. [DOI] [PubMed] [Google Scholar]
- Lemaillet G, Walker B, Lambert S. Identification of a conserved ankyrin-binding motif in the family of sodium channel alpha subunits. J Biol Chem. 2003;278:27333–27339. doi: 10.1074/jbc.M303327200. [DOI] [PubMed] [Google Scholar]
- Leterrier C, Dargent B. No Pasaran! Role of the axon initial segment in the regulation of protein transport and the maintenance of axonal identity. Semin Cell Dev Biol. 2013;27:44–51. doi: 10.1016/j.semcdb.2013.11.001. [DOI] [PubMed] [Google Scholar]
- Li X, Kumar Y, Zempel H, Mandelkow EM, Biernat J, Mandelkow E. Novel diffusion barrier for axonal retention of Tau in neurons and its failure in neurodegeneration. EMBO J. 2011;30:4825–4837. doi: 10.1038/emboj.2011.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilleker JB, Jones MS, Mohanraj R. VGKC complex antibodies in epilepsy: diagnostic yield and therapeutic implications. Seizure : the journal of the British Epilepsy Association. 2013;22:776–779. doi: 10.1016/j.seizure.2013.06.004. [DOI] [PubMed] [Google Scholar]
- Ng JK, Malotka J, Kawakami N, Derfuss T, Khademi M, Olsson T, Linington C, Odaka M, Tackenberg B, Pruss H, et al. Neurofascin as a target for autoantibodies in peripheral neuropathies. Neurology. 2012;79:2241–2248. doi: 10.1212/WNL.0b013e31827689ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa Y, Horresh I, Trimmer JS, Bredt DS, Peles E, Rasband MN. Postsynaptic density-93 clusters Kv1 channels at axon initial segments independently of Caspr2. J Neurosci. 2008;28:5731–5739. doi: 10.1523/JNEUROSCI.4431-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa Y, Schafer DP, Horresh I, Bar V, Hales K, Yang Y, Susuki K, Peles E, Stankewich MC, Rasband MN. Spectrins and ankyrinB constitute a specialized paranodal cytoskeleton. J Neurosci. 2006;26:5230–5239. doi: 10.1523/JNEUROSCI.0425-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Z, Kao T, Horvath Z, Lemos J, Sul JY, Cranstoun SD, Bennett MV, Scherer SS, Cooper EC. A common ankyrin-G-based mechanism retains KCNQ and Nav channels at electrically active domains of the axon. J Neurosci. 2006;26:2599–2613. doi: 10.1523/JNEUROSCI.4314-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrot R, Lonchampt P, Peterson AC, Eyer J. Axonal neurofilaments control multiple fiber properties but do not influence structure or spacing of nodes of Ranvier. J Neurosci. 2007;27:9573–9584. doi: 10.1523/JNEUROSCI.1224-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen JD, Kaech S, Banker G. Selective microtubule-based transport of dendritic membrane proteins arises in concert with axon specification. J Neurosci. 2014;34:4135–4147. doi: 10.1523/JNEUROSCI.3779-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai AM, Thaxton C, Pribisko AL, Cheng JG, Dupree JL, Bhat MA. Spatiotemporal ablation of myelinating glia-specific neurofascin (Nfasc NF155) in mice reveals gradual loss of paranodal axoglial junctions and concomitant disorganization of axonal domains. J Neurosci Res. 2009;87:1773–1793. doi: 10.1002/jnr.22015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliak S, Gollan L, Martinez R, Custer A, Einheber S, Salzer JL, Trimmer JS, Shrager P, Peles E. Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron. 1999;24:1037–1047. doi: 10.1016/s0896-6273(00)81049-1. [DOI] [PubMed] [Google Scholar]
- Poliak S, Salomon D, Elhanany H, Sabanay H, Kiernan B, Pevny L, Stewart CL, Xu X, Chiu SY, Shrager P, et al. Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J Cell Biol. 2003;162:1149–1160. doi: 10.1083/jcb.200305018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN. The axon initial segment and the maintenance of neuronal polarity. Nat Rev Neurosci. 2010;11:552–562. doi: 10.1038/nrn2852. [DOI] [PubMed] [Google Scholar]
- Rasband MN. Cytoskeleton: axons earn their stripes. Curr Biol. 2013;23:R197–198. doi: 10.1016/j.cub.2013.01.050. [DOI] [PubMed] [Google Scholar]
- Rasband MN, Peles E, Trimmer JS, Levinson SR, Lux SE, Shrager P. Dependence of nodal sodium channel clustering on paranodal axoglial contact in the developing CNS. J Neurosci. 1999;19:7516–7528. doi: 10.1523/JNEUROSCI.19-17-07516.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN, Trimmer JS, Schwarz TL, Levinson SR, Ellisman MH, Schachner M, Shrager P. Potassium channel distribution, clustering, and function in remyelinating rat axons. J Neurosci. 1998;18:36–47. doi: 10.1523/JNEUROSCI.18-01-00036.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbluth J. Multiple functions of the paranodal junction of myelinated nerve fibers. J Neurosci Res. 2009;87:3250–3258. doi: 10.1002/jnr.22013. [DOI] [PubMed] [Google Scholar]
- Saitsu H, Tohyama J, Kumada T, Egawa K, Hamada K, Okada I, Mizuguchi T, Osaka H, Miyata R, Furukawa T, et al. Dominant-negative mutations in alpha-II spectrin cause West syndrome with severe cerebral hypomyelination, spastic quadriplegia, and developmental delay. Am J Hum Genet. 2010;86:881–891. doi: 10.1016/j.ajhg.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez I, Hassinger L, Sihag RK, Cleveland DW, Mohan P, Nixon RA. Local control of neurofilament accumulation during radial growth of myelinating axons in vivo. Selective role of site-specific phosphorylation. J Cell Biol. 2000;151:1013–1024. doi: 10.1083/jcb.151.5.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savvaki M, Theodorakis K, Zoupi L, Stamatakis A, Tivodar S, Kyriacou K, Stylianopoulou F, Karagogeos D. The expression of TAG-1 in glial cells is sufficient for the formation of the juxtaparanodal complex and the phenotypic rescue of tag-1 homozygous mutants in the CNS. J Neurosci. 2010;30:13943–13954. doi: 10.1523/JNEUROSCI.2574-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Custer AW, Shrager P, Rasband MN. Early events in node of Ranvier formation during myelination and remyelination in the PNS. Neuron Glia Biol. 2006;2:69–79. doi: 10.1017/S1740925X06000093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Jha S, Liu F, Akella T, McCullough LD, Rasband MN. Disruption of the axon initial segment cytoskeleton is a new mechanism for neuronal injury. J Neurosci. 2009;29:13242–13254. doi: 10.1523/JNEUROSCI.3376-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze TG, Detera-Wadleigh SD, Akula N, Gupta A, Kassem L, Steele J, Pearl J, Strohmaier J, Breuer R, Schwarz M, et al. Two variants in Ankyrin 3 (ANK3) are independent genetic risk factors for bipolar disorder. Molecular psychiatry. 2009;14:487–491. doi: 10.1038/mp.2008.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidl AH, Rubel EW, Barria A. Differential conduction velocity regulation in ipsilateral and contralateral collaterals innervating brainstem coincidence detector neurons. J Neurosci. 2014;34:4914–4919. doi: 10.1523/JNEUROSCI.5460-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidl AH, Rubel EW, Harris DM. Mechanisms for adjusting interaural time differences to achieve binaural coincidence detection. J Neurosci. 2010;30:70–80. doi: 10.1523/JNEUROSCI.3464-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson AH, Gillingwater TH, Anderson H, Cottrell D, Sherman DL, Ribchester RR, Brophy PJ. Effect of limb lengthening on internodal length and conduction velocity of peripheral nerve. J Neurosci. 2013;33:4536–4539. doi: 10.1523/JNEUROSCI.4176-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobotzik JM, Sie JM, Politi C, Del Turco D, Bennett V, Deller T, Schultz C. AnkyrinG is required to maintain axo-dendritic polarity in vivo. Proc Natl Acad Sci U S A. 2009;106:17564–17569. doi: 10.1073/pnas.0909267106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song AH, Wang D, Chen G, Li Y, Luo J, Duan S, Poo MM. A selective filter for cytoplasmic transport at the axon initial segment. Cell. 2009;136:1148–1160. doi: 10.1016/j.cell.2009.01.016. [DOI] [PubMed] [Google Scholar]
- Susuki K. Node of Ranvier disruption as a cause of neurological diseases. ASN neuro. 2013;5:209–219. doi: 10.1042/AN20130025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susuki K, Chang KJ, Zollinger DR, Liu Y, Ogawa Y, Eshed-Eisenbach Y, Dours-Zimmermann MT, Oses-Prieto JA, Burlingame AL, Seidenbecher CI, et al. Three mechanisms assemble central nervous system nodes of Ranvier. Neuron. 2013;78:469–482. doi: 10.1016/j.neuron.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susuki K, Rasband MN, Tohyama K, Koibuchi K, Okamoto S, Funakoshi K, Hirata K, Baba H, Yuki N. Anti-GM1 antibodies cause complement-mediated disruption of sodium channel clusters in peripheral motor nerve fibers. J Neurosci. 2007;27:3956–3967. doi: 10.1523/JNEUROSCI.4401-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Hebb and homeostasis in neuronal plasticity. Curr Opin Neurobiol. 2000;10:358–364. doi: 10.1016/s0959-4388(00)00091-x. [DOI] [PubMed] [Google Scholar]
- Uncini A, Susuki K, Yuki N. Nodo-paranodopathy: beyond the demyelinating and axonal classification in anti-ganglioside antibody-mediated neuropathies. Clinical neurophysiology : official journal of the International Federation of Clinical Neurophysiology. 2013;124:1928–1934. doi: 10.1016/j.clinph.2013.03.025. [DOI] [PubMed] [Google Scholar]
- Vizoso AD. The relationship between internodal length and growth in human nerves. Journal of anatomy. 1950;84:342–353. [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Al-Bassam S, Miyazaki Y, Wandless TJ, Webster P, Arnold DB. Networks of polarized actin filaments in the axon initial segment provide a mechanism for sorting axonal and dendritic proteins. Cell Rep. 2012;2:1546–1553. doi: 10.1016/j.celrep.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winckler B, Forscher P, Mellman I. A diffusion barrier maintains distribution of membrane proteins in polarized neurons. Nature. 1999;397:698–701. doi: 10.1038/17806. [DOI] [PubMed] [Google Scholar]
- Writzl K, Primec ZR, Strazisar BG, Osredkar D, Pecaric-Meglic N, Kranjc BS, Nishiyama K, Matsumoto N, Saitsu H. Early onset West syndrome with severe hypomyelination and coloboma-like optic discs in a girl with SPTAN1 mutation. Epilepsia. 2012;53:e106–110. doi: 10.1111/j.1528-1167.2012.03437.x. [DOI] [PubMed] [Google Scholar]
- Xu K, Zhong G, Zhuang X. Actin, Spectrin, and Associated Proteins Form a Periodic Cytoskeletal Structure in Axons. Science. 2013;339:30495–30501. doi: 10.1126/science.1232251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura T, Rasband MN. Axon initial segments: diverse and dynamic neuronal compartments. Curr Opin Neurobiol. 2014;27C:96–102. doi: 10.1016/j.conb.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Susuki K, Zollinger DR, Dupree JL, Rasband MN. Membrane domain organization of myelinated axons requires betaII spectrin. J Cell Biol. 2013;203:437–443. doi: 10.1083/jcb.201308116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Lambert S, Malen PL, Carpenter S, Boland LM, Bennett V. AnkyrinG is required for clustering of voltage-gated Na channels at axon initial segments and for normal action potential firing. J Cell Biol. 1998;143:1295–1304. doi: 10.1083/jcb.143.5.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zonta B, Tait S, Melrose S, Anderson H, Harroch S, Higginson J, Sherman DL, Brophy PJ. Glial and neuronal isoforms of Neurofascin have distinct roles in the assembly of nodes of Ranvier in the central nervous system. J Cell Biol. 2008;181:1169–1177. doi: 10.1083/jcb.200712154. [DOI] [PMC free article] [PubMed] [Google Scholar]