

Abstract
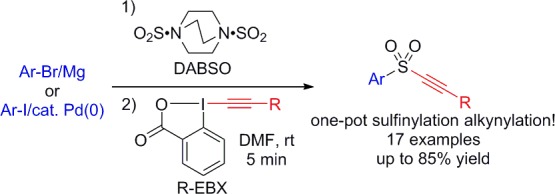
A one-pot three-component protocol for the preparation of arylsulfonyl alkynes through the reaction of ethynyl-benziodoxolone (EBX) reagents, DABSO (DABCO·SO2), and either organomagnesium reagents or aryl iodides with a palladium catalyst is reported. A broad range of aryl and heteroarylalkynyl sulfones were obtained in 46–85% overall yield.
Aryl sulfone-containing compounds display a variety of biological activities. Several of them are marketed drugs for treatment of human diseases. Examples include the antimigraine Vioxx (1),1a the antibacterial Dapsone (2)1b and the antiandrogen Casodex (3)1c (Figure 1).
Figure 1.

Biologically active aryl sulfones.
Among all sulfones, (aryl)alkynyl sulfones constitute an important class of building blocks due to their versatile reactivity. The sulfonyl unit has a strong electron-withdrawing character that enhances the reactivity of the triple bond.2 Therefore, sulfonyl acetylenes are widely utilized in cycloadditions and conjugate additions reactions. They were also found to react in certain cases with organometallic reagents or radicals through 1,3-addition to generate anions or radical intermediates, followed by an elimination of the sulfonyl group to give disubstituted alkynes.3
Up to now, the preparation of sulfones could be achieved by numerous approaches, the most frequent one being the oxidation of the corresponding thiols.4 Another convenient method is based on the reaction of sulfinate salts with electrophiles.5 If the introduction of an aryl or an alkynyl group is desired on the sulfinate, hypervalent iodine reagents have emerged as reagents of choice, due to their exceptional reactivity.6 For example, Manolikakes and co-workers reported that diaryl sulfones could be synthesized from arylsulfinate and diaryliodonium salts (Scheme 1A, eq 1).7 Electrophilic alkynyl iodonium salts are also widely used in alkynylation reactions,8 and their utilization for the preparation of alkynyl sulfones from sulfinate salts were reported by the groups of Stang9 and Moran (Scheme 1A, eq 2).10 However, the scope of arylsulfinate salts reported was limited (only PhSO2Na and p-TolSO2Na), probably due to the low number of commercially available sulfinate salts. Recently, Willis and co-workers showed that DABSO (DABCO·SO2, the combination of DABCO and sulfur dioxide) can serve as a surrogate of SO2 for the in situ formation of sulfinate salts in the synthesis of sulfonamides.11 More importantly, Willis and co-workers12 and Rocke and co-workers13 independently demonstrated a one-pot synthesis of sulfones from organolithium/magnesium and organozinc reagents, DABSO, and alkyl halides (Scheme 1B, eq 3). In addition, transition metal-catalyzed, e.g. palladium14 (eq 4) and gold,15 as well as metal-free16 sulfinate salt formation with DABSO were also reported for sulfone synthesis. These recent breakthroughs have greatly simplified the synthesis of sulfones. However, a one-pot protocol for the preparation of arylalkynyl sulfones has still not been reported. The synthesis of this class of compounds is especially challenging, as the products themselves are also highly reactive.
Scheme 1. Recent Reports on Sulfone Synthesis.

Our group is interested in alkyne synthesis using electrophilic alkynylation reagents, in particular ethynyl-benziodoxolones (EBX, 4).17,18 These cyclic hypervalent iodine reagents display high reactivity, but are more stable than the traditionally used alkynyliodonium salts. We therefore thought they would be well-suited to develop the first one-pot three-component synthesis of alkynyl sulfones. Herein we would like to present the successful implementation of this approach using EBX reagents 4, DABSO, and organomagnesium/lithium reagents or aryl iodides with a palladium catalyst (Scheme 2).
Scheme 2. Our Approach toward the Synthesis of Alkynyl Sulfones.

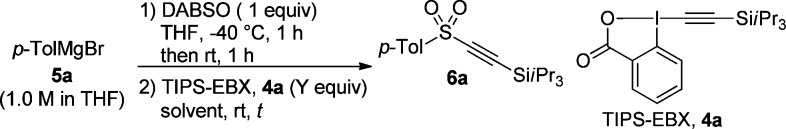
The preparation of sulfonyl alkynes starting from tolyl magnesium bromide (5a) with TIPS-EBX (4a) and DABSO was examined first. Initially, the protocol developed by Willis for preparation of sulfonamides11a was utilized. Unfortunately, the desired product was not obtained when using THF only as solvent. To our delight, 16% yield of 6a was observed when THF was removed and replaced by DMF (Table 1, entry 1).
Table 1. Optimization of the Arylalkynyl Sulfone Synthesis.
| entrya | equiv of 4a | solvent | t | yield (%)d |
|---|---|---|---|---|
| 1 | 1.2 | DMFb | 2 h | 16 |
| 2 | 1.2 | DMFb | 14 h | 0 |
| 3 | 1.2 | DMF/H2Ob,c | 2 h | 15 |
| 4 | 1.2 | DMF/H2Ob,c | 14 h | 0 |
| 5 | 1.2 | DMF | 2 h | 65 |
| 6 | 1.2 | DMSO | 2 h | 0 |
| 7 | 1.5 | DMF | 2 h | 50 |
| 8 | 1.5 | DMF | 1 h | 50 |
| 9 | 1.5 | DMF | 30 min | 75 |
| 10 | 1.5 | DMF | 5 min | 78 |
| 11 | 1.2 | DMF | 5 min | 80 |
| 12 | 1.1 | DMF | 5 min | 75 |
0.06 mmol p-tolylmagnesium bromide (5a) was used in 0.2 mL of THF. 0.2 mL of solvent was added for the second step (final concentration: 0.13 M).
THF was removed before adding 0.2 mL of solvent (final concentration: 0.3 M).
Ratio (v/v) = 5/1.
The yield was obtained based on 1H NMR analysis using 1,3,5-trimethoxybenzene as internal reference.
Longer reaction time (14 h), and DMF/H2O (v/v = 5/1) as a solvent system for the alkynylation did not give better results (Table 1, entries 2–4). On the other hand, we were pleased to observe an increased yield of 6a (65%) when THF was not removed before adding DMF for the alkynylation step (Table 1, entry 5). In contrast, the addition of DMSO was not successful (Table 1, entry 6). A fast examination of TIPS-EBX 4a loading and reaction time (Table 1, entries 6–12) showed that 1.2 equiv of 4a and 5 min reaction time resulted in the production of 6a in 80% yield (Table 1, entry 11). The lower yields observed with longer reaction times are probably due to the high reactivity of the formed alkynyl sulfone 6a.
With optimized conditions in hand we examined the scope of the one-pot sulfinylation alkynylation from organometallic reagents with TIPS-EBX 4a (Scheme 3). Acetylene 6a was obtained in good yield on preparative scale (85%). We further expanded the utility of this protocol by preparing the organomagnesium/lithium reagents immediately before use. Phenylalkynyl sulfone 6b could be synthesized in 85% overall yield starting from bromobenzene 5b. Other electron-rich and electron-poor functional groups were well-tolerated. For example, p-tert-butyl, p-methoxy, and p-dimethylaminosulfonyl alkynes 6c, 6d, and 6e were obtained in good yields (79, 79, and 85% respectively). p-Chloro and p-fluorophenylsulfonyl alkynes 6f, and 6g were also synthesized in 70 and 85% yield, respectively.
Scheme 3. Scope of the One-Pot Sulfinylation Alkynylation.

Reaction conditions: 0.20 mmol of 5 (1.0 equiv), 0.20 mmol of Mg (1.0 equiv), 0.20 mmol of DABSO (1.0 equiv), and THF (0.65 mL) were used for the first step. 0.24 mmol of TIPS-EBX 4a (1.2 equiv) and DMF (0.65 mL) were added for the second step. Isolated yield after purification on column chromatography is given.
Commercial Grignard reagent was used.
0.5 M organometallic reagent in THF.
R-Li was generated from the corresponding C–H bond with n-BuLi.
Commercial n-BuLi was used.
o-Tolyl sulfinate magnesium salt gave a lower yield of product 6h (52%). In addition, alkyne 6i was also obtained from 1-bromonaphthalene in 83% yield. The heteroaryl bromides 5-bromo-N-methylindole (5j) and 2-bromo-thiophene (5k) gave products 6j and 6k in 72 and 79% yield, respectively. Sulfone 6l was synthesized in 70% yield starting from furan 5l using a selective lithiation at C2. However, 2-pyridinyl and 2-pyrimidinyl alkynyl sulfones 6m and 6n could not be synthesized using this one-pot protocol. Allylsulfonyl alkyne 6o was obtained in 46% yield. Sulfones 6p and 6q could not be obtained when starting from the corresponding organolithium reagents.
Further extension of the scope of the one-pot sulfonyl alkynylation was focused on p-tolyl Grignard (5a) with R-EBX reagents (Scheme 4). Alkyne 7a was made in 79% yield using t-Bu-EBX 4b.18f Unfortunately, Me-EBX 4c,18k Ph-EBX 4d,18f and n-Hex-EBX 4e(18f) were not able to react with the in situ formed p-tolyl sulfinate salt to deliver the desired alkynes 7b, 7c, and 7d respectively. Interestingly, dihydrofuran 8 was obtained in 93% yield when the EBX reagent 4f was utilized in the alkynylation.
Scheme 4. Iodine Reagent Scope of the One-Pot Sulfinylation Alkynylation.

Reaction conditions: 0.20 mmol of p-tolylMgBr (5a) (1.0 equiv), 0.20 mmol of DABSO (1.0 equiv) and THF (0.65 mL) were used for the first step. 0.24 mmol of R-EBX 4 (1.2 equiv) and DMF (0.65 mL) were added for the second step. Isolated yield after purification on column chromatography is given.
At least three different mechanisms could be considered for the alkynylation step in this one-pot protocol. A first possibility frequently occurring with hypervalent iodine reagents is direct nucleophilic attack of sulfinate onto the iodine atom of the benziodoxolone, followed by a C–S bond formation via a reductive elimination step.6 Our group has recently discovered by computation an alternative mechanism for the alkynylation of thiols involving a concerted three-atom transition state including the iodine, the sulfur and the α-carbon atom of the alkyne.18k However, these two mechanisms are less likely to be involved in the alkynylation of sulfinates because (i) dihydrofuran 8 was obtained and this product most probably results from 1,5-C–H insertion of a carbene intermediate, and (ii) EBX reagents 4c–f did not give the desired alkyne products, in contrast to the high yields observed with thiols.18k These facts suggest that the reaction mechanism is different for sulfinates and most probably involves a third alternative: a conjugate addition of the sulfinate onto the β-alkynyl carbon of ethynyl-benziodoxolone, followed by an α-elimination of the aryl iodide to give a carbene intermediate, and finally a 1,2-shift to form the alkyne.10,19
One disadvantage of the developed sulfonylation-alkynylation protocol involving organo-magnesium or -lithium reagents is that it cannot be applied to substrates sensitive to strong bases or nucleophiles. In order to further enhance the generality of the one-pot approach for the synthesis of alkynyl sulfones, we then examined a Pd-catalyzed ammonium sulfinate salt formation starting from aryl iodides which can proceed under much milder conditions (Scheme 1B, eq 4).14 Alkynes 6a (82%) and 6d (68%) could be synthesized in comparable yields using the Pd-catalyzed sulfinylation in the first step (Scheme 5). Gratifyingly, alkynes 6r, 6s and 6t bearing potentially base- and nucleophile sensitive hydroxy, methyl ester, and methyl ketone groups were synthesized successfully in 39–60% yield.
Scheme 5. Scope of the Pd-Catalyzed One-Pot Sulfinylation Alkynylation.

Reaction conditions: 0.20 mmol of ArI 9 (1.0 equiv), 10 μmol of Pd(OAc)2 (5 mol %), 15 μmol of PAd2Bu (7.5 mol %), 0.20 mmol DABSO (1.0 equiv), Et3N (3.0 equiv) and iPrOH (1.7 mL) were used for the first step. 0.24 mmol of TIPS-EBX 4a (1.2 equiv) and DMF (0.65 mL) were used for the second step. Isolated yield after purification on column chromatography is given.
In conclusion, we report a simple one-pot protocol for sulfonylation-alkynylation starting either from organomagnesium/lithium reagents or from aryl iodides with a palladium catalyst, DABSO, and EBX reagents, in up to 85% overall yield. The method from organomagnesium/lithium reagents gives an unprecedented efficient access to aryl-alkynyl sulfones bearing benzene rings with electron-poor or electron-rich substituents, as well as heterocycles. The complementary Pd-catalyzed protocol can be used for substrates sensitive to the use of strongly basic or nucleophilic organometallic reagents. Extension of the scope and investigations on the mechanism of the sulfonyl alkynylation are currently underway and will be reported in due course.
Acknowledgments
EPFL, F. Hoffmann-La Roche, Ltd., and ERC (starting Grant iTools4MC, number 334840) are acknowledged for financial support.
Supporting Information Available
Experimental procedures and analytical data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- a Prasit P.; Wang Z.; Brideau C.; Chan C. C.; Charleson S.; Gauthier J. Y.; Gordon R.; Guay J.; Gresser M.; Kargman S.; Kennedy B.; Leblanc Y.; Leger S.; Mancini J.; O’Neill G. P.; Ouellet M.; Percival M. D.; Perrier H.; Riendeau D.; Rodger I.; Tagari P.; Therien M.; Vickers P.; Wong E.; Xu L. J.; Young R. N.; Zamboni R. Bioorg. Med. Chem. Lett. 1999, 9, 1773. [DOI] [PubMed] [Google Scholar]; b Artico M.; Silvestri R.; Pagnozzi E.; Bruno B.; Novellino E.; Greco G.; Massa S.; Ettorre A.; Giulia Loi A.; Scintu F.; La Colla P. J. Med. Chem. 2000, 43, 1886. [DOI] [PubMed] [Google Scholar]; c Lopez de Compadre R. L.; Pearlstein R. A.; Hopfinger A. J.; Seyde J. K. J. Med. Chem. 1987, 30, 900. [DOI] [PubMed] [Google Scholar]
- For selected reviews, see:; a Back T. G.; Clary K. N.; Gao D. Chem. Rev. 2010, 110, 4498. [DOI] [PubMed] [Google Scholar]; b Garcia Ruano J. L.; Aleman J.; Parra J.; Marzo L. Eur. J. Org. Chem. 2014, 1577. [Google Scholar]
- Selected examples for the anionic pathway, see:; a Smorada R. L.; Truce W. E. J. Org. Chem. 1979, 44, 3444. [Google Scholar]; b de Lucchi O.; Licini G.; Pasquato L.; Senta M. J. Chem. Soc., Chem. Commun. 1985, 1597. [Google Scholar]; c Garcia Ruano J. L.; Aleman J.; Marzo L.; Alvarado C.; Tortosa M. Angew. Chem., Int. Ed. 2012, 51, 2712. [DOI] [PubMed] [Google Scholar]; d Garcia Ruano J. L.; Aleman J.; Marzo L.; Alvarado C.; Tortosa M.; Diaz-Tendero S.; Fraile A. Chem.—Eur. J. 2012, 18, 8414. [DOI] [PubMed] [Google Scholar]; e Marzo L.; Perez I.; Yuste F.; Aleman J.; Garcia Ruano J. L. Chem. Commun. 2015, 51, 346. [DOI] [PubMed] [Google Scholar]; Selected examples for the radical pathway, see:; f Schaffner A. P.; Darmency V.; Renaud P. Angew. Chem., Int. Ed. 2006, 45, 5847. [DOI] [PubMed] [Google Scholar]; g Hoshikawa T.; Kamijo S.; Inoue M. Org. Biomol. Chem. 2013, 11, 164. [DOI] [PubMed] [Google Scholar]; h Todoroki H.; Iwatsu M.; Urabe D.; Inoue M. J. Org. Chem. 2014, 79, 8835. [DOI] [PubMed] [Google Scholar]
- a Simpkins N. S.Sulphones in Organic Synthesis; Pergamon Press: Oxford, U.K., 1993. [Google Scholar]; b Back T. G. Tetrahedron 2001, 57, 5263. [Google Scholar]
- Aziz J.; Messaoudi S.; Alami M.; Hamze A. Org. Biomol. Chem. 2014, 12, 9743. [DOI] [PubMed] [Google Scholar]
- Zhdankin V. V.Hypervalent Iodine Chemistry: Preparation, Structure, and Synthetic Applications of Polyvalent Iodine Compounds; Wiley: Chichester, U.K., 2014. [Google Scholar]
- a Manolikakes G.; Umierski N. Org. Lett. 2013, 15, 188. [DOI] [PubMed] [Google Scholar]; b Manolikakes G.; Umierski N. Org. Lett. 2013, 15, 4972. [DOI] [PubMed] [Google Scholar]
- a Zhdankin V. V.; Stang P. J. Tetrahedron 1998, 54, 10927. [Google Scholar]; b Brand J. P.; Waser J. Chem. Soc. Rev. 2012, 41, 4165. [DOI] [PubMed] [Google Scholar]
- Tykwinski R. R.; Williamson B. L.; Fischer D. R.; Stang P. J.; Arif A. M. J. Org. Chem. 1993, 58, 5235. [Google Scholar]
- Hamnett D. J.; Moran W. J. Org. Biomol. Chem. 2014, 12, 4156. [DOI] [PubMed] [Google Scholar]
- a Woolven H.; Gonzalez-Rodriguez C.; Marco L.; Thompson A. L.; Willis M. C. Org. Lett. 2011, 13, 4876. [DOI] [PubMed] [Google Scholar]; For a recent review on the use of DABSO, see:; b Deeming A. S.; Emmett E. J.; Richards-Taylor S. R.; Willis M. C. Synthesis 2014, 46, 2701. [Google Scholar]
- Deeming A. S.; Russell C. J.; Hennessy A. J.; Willis M. C. Org. Lett. 2014, 16, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocke B. N.; Bahnck K. B.; Herr M.; Lavergne S.; Mascitti V.; Perreault C.; Polivkova J.; Shavnya A. Org. Lett. 2014, 16, 154. [DOI] [PubMed] [Google Scholar]
- Emmett E. J.; Hayter B. R.; Willis M. C. Angew. Chem., Int. Ed. 2014, 53, 10204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Johnson M. W.; Bagley S. W.; Mankad N. P.; Bergman R. G.; Mascitti V.; Toste F. D. Angew. Chem., Int. Ed. 2014, 53, 4404. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ye S.; Wang H.; Xiao Q.; Ding Q.; Wu J. Adv. Synth. Catal. 2014, 356, 3225. [Google Scholar]
- Zhang D.; An Y.; Li Z.; Wu J. Angew. Chem., Int. Ed. 2014, 53, 2451. [DOI] [PubMed] [Google Scholar]; b Zhang D.; Li Y.; An Y.; Wu J. Chem. Commun. 2014, 50, 8886. [DOI] [PubMed] [Google Scholar]
- a Ochiai M.; Masaki Y.; Shiro M. J. Org. Chem. 1991, 56, 5511. [Google Scholar]; b Zhdankin V. V.; Kuehl C. J.; Krasutsky A. P.; Bolz J. T.; Simonsen A. J. J. Org. Chem. 1996, 61, 6547. [DOI] [PubMed] [Google Scholar]
- Selected examples:; a Brand J. P.; Charpentier J.; Waser J. Angew. Chem., Int. Ed. 2009, 48, 9346. [DOI] [PubMed] [Google Scholar]; b Brand J. P.; Waser J. Angew. Chem., Int. Ed. 2010, 49, 7304. [DOI] [PubMed] [Google Scholar]; c Fernandez Gonzalez D.; Brand J. P.; Waser J. Chem.—Eur. J. 2010, 16, 9457. [DOI] [PubMed] [Google Scholar]; d Nicolai S.; Erard S.; Fernandez Gonzalez D.; Waser J. Org. Lett. 2010, 12, 384. [DOI] [PubMed] [Google Scholar]; e Nicolai S.; Piemontesi C.; Waser J. Angew. Chem., Int. Ed. 2011, 50, 4680. [DOI] [PubMed] [Google Scholar]; f Brand J. P.; Chevalley C.; Scopelliti R.; Waser J. Chem.—Eur. J. 2012, 18, 5655. [DOI] [PubMed] [Google Scholar]; g Fernández González D.; Brand J. P.; Mondière R.; Waser J. Adv. Synth. Catal. 2013, 355, 1631. [Google Scholar]; h Frei R.; Waser J. J. Am. Chem. Soc. 2013, 135, 9620. [DOI] [PubMed] [Google Scholar]; i Li Y.; Brand J. P.; Waser J. Angew. Chem., Int. Ed. 2013, 52, 6743. [DOI] [PubMed] [Google Scholar]; j Chen C. C.; Waser J. Chem. Commun. 2014, 50, 12923. [DOI] [PubMed] [Google Scholar]; k Frei R.; Wodrich M. D.; Hari D. P.; Borin P.-A.; Chauvier C.; Waser J. J. Am. Chem. Soc. 2014, 136, 16563. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a review, see:; l Brand J. P.; Fernandez Gonzalez D.; Nicolai S.; Waser J. Chem. Commun. 2011, 47, 102. [DOI] [PubMed] [Google Scholar]
- a Ochiai M.; Kunishima M.; Nagao Y.; Fuji K.; Shiro M.; Fujita E. J. Am. Chem. Soc. 1986, 108, 8281. [Google Scholar]; b Ochiai M.; Ito T.; Takaoka Y.; Masaki Y.; Kunishima M.; Tani S.; Nagao Y. J. Chem. Soc., Chem. Commun. 1990, 118. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.