

Abstract
Single-step electron tunnelling reactions can transport charges over distances of 15–20 Åin proteins. Longer-range transfer requires multi-step tunnelling processes along redox chains, often referred to as hopping. Long-range hopping via oxidized radicals of tryptophan and tyrosine, which has been identified in several natural enzymes, has been demonstrated in artificial constructs of the blue copper protein azurin. Tryptophan and tyrosine serve as hopping way stations in high-potential charge transport processes. It may be no coincidence that these two residues occur with greater-than-average frequency in O2- and H2O2-reactive enzymes. We suggest that appropriately placed tyrosine and/or tryptophan residues prevent damage from high-potential reactive intermediates by reduction followed by transfer of the oxidizing equivalent to less harmful sites or out of the protein altogether.
Keywords: electron transfer, hopping, protein radical, azurin, cytochrome P450
1. Background
It is well established that multi-step tunnelling, called hopping, is required for functional charge transport in many redox enzymes (examples include ribonucleotide reductase [1–9], photosystem II [10–12], DNA photolyase [13–21], MauG [22–25] and cytochrome c peroxidase [26,27]). Here, we advance the hypothesis that many such enzymes, most especially those that generate high-potential intermediates during turnover, could be irreversibly damaged if the intermediates are not inactivated in some way. We suggest that appropriately placed tyrosine (Tyr) and/or tryptophan (Trp) residues can prevent such damage by rapid reduction of the intermediates followed by transfer of the oxidizing equivalent to less harmful sites or out of the protein altogether [28]. A protective role of this sort will not be apparent in catalytic rates and binding constants; the enzyme survival time is more likely to be an indicator of this function.
The blue copper protein Pseudomonas aeruginosa azurin has been a test-bed for mechanistic investigations of Trp and Tyr radical formation in biological electron transfer (ET) reactions [29–33]. Our initial investigations revealed that CuI oxidation by a photoexcited ReI-diimine (ReI(CO)3(dmp), dmp=4,7-dimethyl-1,10-phenanthroline) covalently bound at His124 on a His124Gly123Trp122Met121 β-strand (ReHis124Trp122CuI-azurin) occurs in a few nanoseconds, fully two orders of magnitude faster than documented for single-step electron tunnelling at a 19 Ådonor–acceptor distance [29]. We attributed the accelerated ET to a two-step hopping mechanism involving a Trp•+ radical intermediate. In recent work, we examined ET in ReHis126Trp122CuI-azurin, which has three redox sites at well-defined distances in the protein fold (Re–Trp122(indole)=13.1 Å, dmp–Trp122(indole)=10.0 Å, Re–Cu=25.6 Å) [32]. Near-UV excitation of the Re chromophore leads to prompt CuI oxidation (less than 50 ns), followed by slow back-ET (more than 0.2 μs) to regenerate CuI and ground-state ReI. Spectroscopic measurements performed with varying protein concentrations suggest that the photoinduced ET reactions occur in protein dimers, (ReHis126Trp122CuI)2, and that forward ET is accelerated by intermolecular electron hopping through the interfacial tryptophan: 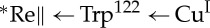 (∥ denotes a protein–protein interface). Solution mass spectrometry confirms a broad oligomer distribution with prevalent monomers and dimers, and the crystal structure of the CuII form reveals two ReHis126Trp122CuII molecules oriented such that redox cofactors Re(dmp) and Trp122-indole on different protein molecules are located in the interface at much shorter intermolecular distances (Re–Trp122(indole)=6.9 Å, dmp–Trp122(indole)=3.5 Åand Re–Cu=14.0 Å) than within single protein folds. Whereas forward ET is accelerated by hopping through Trp122, ReI(dmp•−)∥→CuII ET is sluggish, probably due to poor electronic coupling across the protein–protein interface as well as the absence of an energetically accessible radical intermediate. These findings provided new insights into the factors that regulate Trp•+ radical formation in protein ET reactions involving high-potential oxidants.
(∥ denotes a protein–protein interface). Solution mass spectrometry confirms a broad oligomer distribution with prevalent monomers and dimers, and the crystal structure of the CuII form reveals two ReHis126Trp122CuII molecules oriented such that redox cofactors Re(dmp) and Trp122-indole on different protein molecules are located in the interface at much shorter intermolecular distances (Re–Trp122(indole)=6.9 Å, dmp–Trp122(indole)=3.5 Åand Re–Cu=14.0 Å) than within single protein folds. Whereas forward ET is accelerated by hopping through Trp122, ReI(dmp•−)∥→CuII ET is sluggish, probably due to poor electronic coupling across the protein–protein interface as well as the absence of an energetically accessible radical intermediate. These findings provided new insights into the factors that regulate Trp•+ radical formation in protein ET reactions involving high-potential oxidants.
Work by theorists has shed much light on the main factors controlling biological ET reactions [34,35]: and, in our experimental programme, we have found semiclassical ET theory [36] to be particularly useful in analyses of results. Notably, given a particular spatial arrangement of redox cofactors, we can predict driving force dependences of the relative time constants for single-step (τss=1/kss) and multi-step (τhop) electron transport [31]. Alternatively, given the redox and reorganization energetics, we can predict the hopping propensity for different cofactor arrangements [33]. We considered azurins labelled with Ru(bpy)2(im)(HisX)2+ (bpy=2,2′-bipyridine; im=imidazole; HisX=surface histidine) labelled at three surface sites (RuHis107, RuHis124 and RuHis126) and examined the hopping advantage (τss/τhop) for a protein with a generalized intermediate (Int) situated between a diimine-RuIII oxidant and CuI [33]. In all cases, the greatest hopping advantage occurs in systems where the Int–RuIII distance (r1) is up to 5 Å shorter than the Int–CuI distance (r2). The hopping advantage increases as systems orient nearer a linear donor–Int–acceptor configuration, owing to minimized intermediate tunnelling distances. The smallest predicted hopping advantage is in RuHis124-azurin, which has the shortest Ru–Cu distance of the three proteins. The hopping advantage is nearly lost as ΔG° for the first step (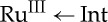 ) rises above +0.15 eV. Isoergic initial steps provide a wide distribution of arrangements, where advantages as great as 104 are possible (for a fixed donor–acceptor distance of 23.7 or 25.4 Å). A slightly exergonic Int→RuIII step provides an even larger distribution of arrangements for productive hopping, which will be the case as long as the driving force for the first step is not more favourable than that for overall transfer.
) rises above +0.15 eV. Isoergic initial steps provide a wide distribution of arrangements, where advantages as great as 104 are possible (for a fixed donor–acceptor distance of 23.7 or 25.4 Å). A slightly exergonic Int→RuIII step provides an even larger distribution of arrangements for productive hopping, which will be the case as long as the driving force for the first step is not more favourable than that for overall transfer.
We tested these predictions experimentally in three Ru–His-labelled azurins using nitrotyrosinate (NO2TyrO−) as a redox intermediate (RuHis107(NO2TyrOH)109; RuHis124(NO2TyrOH)122; and RuHis126(NO2TyrOH)122; E°′(NO2TyrO•/−)≈1.02 V versus NHE) [33]. The first two systems have cofactor placements that are close to the predicted optimum; the last system has a larger first-step distance, which is predicted to decrease the hopping advantage. The phenol pKa of 3-nitrotyrosine (7.2) permitted us to work at near-neutral pH, rather than the high pH (more than 10) required for hopping with tyrosinate. ET via nitrotyrosinate avoids the complexities associated with the proton-coupled redox reactions of tyrosine [37–39]. We found specific rates of CuI oxidation more than 10 times greater than those of single-step ET in the corresponding azurins lacking NO2TyrOH, confirming that NO2TyrO− accelerates long-range ET. In this case, the proposed reaction sequence is [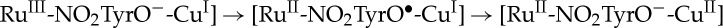 , although the nitrotyrosyl radical intermediate was not detected by transient spectroscopy in any of the proteins investigated. The results are in excellent agreement with hopping maps developed using semiclassical ET theory and parameters derived from our body of protein ET measurements [31–42].
, although the nitrotyrosyl radical intermediate was not detected by transient spectroscopy in any of the proteins investigated. The results are in excellent agreement with hopping maps developed using semiclassical ET theory and parameters derived from our body of protein ET measurements [31–42].
2. Protecting P450s?
The cytochromes P450 are members of a superfamily of haem oxygenases that perform two broad functional roles: xenobiotic metabolism and biosynthesis [43,44]. In mammals, these functions include drug metabolism, conversion of lipophilic molecules to more polar products for enhanced elimination, steroid biosynthesis and conversion of polyunsaturated fatty acids to biologically active products [43,45]. P450s are monooxygenases that incorporate one oxygen atom from O2 into the substrate while the second is reduced to H2O [43,44]. Substrate binding triggers reduction of the ferric enzyme to the ferrous state by a reductase with reducing equivalents originating in NAD(P)H. Oxygen binding, followed by delivery of a second electron, induces O−O bond cleavage, producing H2O and a ferryl-porphyrin cation radical (Cmpd-1). Cmpd-1 transfers an O atom to substrate, regenerating the ferric haem [43]. The UniProtKB/Swiss-Prot database indicates that P450s are rich in aromatic amino acids: 90% of the sequences in the P450 family (956 sequences) have above-average occurrence of Phe residues; and 68% of the sequences have more Trp residues than average. Phe residues are unlikely to participate as real intermediates in ET reactions, but their role in enhanced superexchange coupling for long-range ET remains unresolved [46–48]. Cytochrome P450 BM3 (CYP102A1) is frequently used as a soluble surrogate for human microsomal P450 s [44]. A Trp96 residue is found within 5 Åof the haem in the structure of CYP102A1 [49] (figure 1), and sequence alignment (UniProtKB) in the P450 family suggests that Trp is conserved at this position in more than 75% of the members of this group. The most common alternative residue at this position is His. Interestingly, of the 698 sequences with Trp at this position, all but five derive from eukaryotic sources, whereas about half of the proteins with His at this position derive from bacterial or archaeal sources. The strong conservation of the Trp96 residue has been noted previously [50]. An ET mediator role for this residue had been suggested, but replacement of Trp96 with Ala, Phe or Tyr has little impact on catalytic turnover kinetics [50]. To the best of our knowledge, no role other than structural has been reported for this highly conserved Trp residue in P450 [44].
Figure 1.
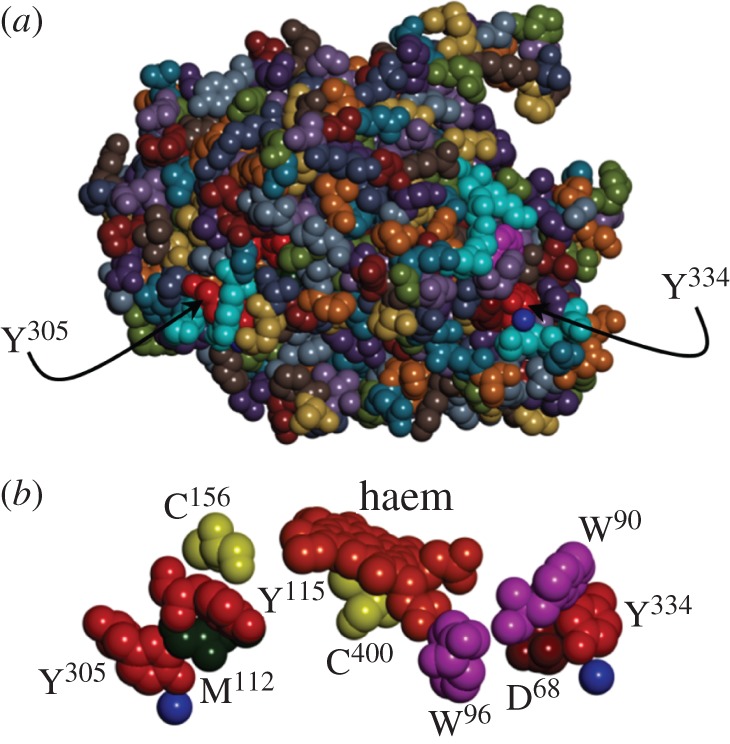
(a) Space-filling structural model of the haem domain of CYP102A1 (PDB #2IJ2) highlighting the surface locations of terminal residues in two potential radical transfer pathways (Tyr334 and Tyr305). (b) Space-filling model of the residues comprising CYP102A1 putative radical transfer pathways. Blue spheres represent structurally resolved water molecules. (Online version in colour.)
The oxygenation chemistry catalysed by some P450s is tightly coupled to substrate hydroxylation: one mole of product is produced for each mole of O2 consumed. In many enzymes, particularly the eukaryotic proteins with broad substrate specificities, hydroxylation is much less efficiently coupled to O2 reduction (frequently less than 10%) [51–53]. When the enzyme does not transfer an O atom to substrate, it can produce reactive oxygen species (ROS: 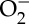 , H2O2) or a second H2O molecule [54]. The production of ROS can lead to rapid degradation of the enzyme. In the case of oxidase chemistry (formation of 2H2O from O2), two reducing equivalents must be delivered by sources other than the substrate. Uncoupled P450 catalysis leads to oxidation of bilirubin and uroporphyrinogen [55,56]. The physiological consequences of this uncoupling could involve uroporphyria and lowered plasma bilirubin concentrations [56]. Halogenated P450 substrates, including environmental toxins such as polyhalogenated biphenyls, are associated with inhibition of monooxygenase activity and uncoupled O2 consumption [55,56]. We suggest that redox-active Tyr and/or Trp residues in P450, quite possibly analogues of Trp96, protect the enzyme from oxidative degradation during uncoupled catalysis.
, H2O2) or a second H2O molecule [54]. The production of ROS can lead to rapid degradation of the enzyme. In the case of oxidase chemistry (formation of 2H2O from O2), two reducing equivalents must be delivered by sources other than the substrate. Uncoupled P450 catalysis leads to oxidation of bilirubin and uroporphyrinogen [55,56]. The physiological consequences of this uncoupling could involve uroporphyria and lowered plasma bilirubin concentrations [56]. Halogenated P450 substrates, including environmental toxins such as polyhalogenated biphenyls, are associated with inhibition of monooxygenase activity and uncoupled O2 consumption [55,56]. We suggest that redox-active Tyr and/or Trp residues in P450, quite possibly analogues of Trp96, protect the enzyme from oxidative degradation during uncoupled catalysis.
3. Parting shots
Across the six enzyme classes (oxidoreductases, transferases, hydrolases, lyases, isomerases and ligases) defined by the Enzyme Data Bank of the Swiss Institute of Bioinformatics, greater than half of the oxidoreductases have Tyr and Trp frequencies above the database average (figure 2). For the remaining five classes, most proteins have Trp frequencies below the database average, and only among the ligases does a majority of proteins have Tyr frequencies substantially above the database average. Refinement of this analysis reveals that, of the O2-reactive oxidoreductases, 66% have Tyr frequencies above the database average and 81% have above-average Trp frequencies. Could the direct participation of Tyr and Trp residues in enzymatic redox chemistry be far more common than currently recognized? We think so!
Figure 2.

Occurrence frequencies relative to the UniProtKB/Swiss-Prot database average of aromatic residues (Phe, Trp and Tyr) in different enzyme classes. (Online version in colour.)
Funding statement
Our work on biological electron transfer processes is supported by the National Institutes of Health (DK-019038) and the Arnold and Mabel Beckman Foundation.
References
- 1.Sjöberg BM. 1997. Ribonucleotide reductases—a group of enzymes with different metallosites and a similar mechanism. Struct. Bonding 88, 139–173. ( 10.1007/3-540-62870-3_5) [DOI] [Google Scholar]
- 2.Stubbe J, Nocera DG, Yee CS, Chang MCY. 2003. Radical initiation in the class I ribonucleotide reductase: long-range proton-coupled electron transfer? Chem. Rev. 103, 2167–2201. ( 10.1021/cr020421u) [DOI] [PubMed] [Google Scholar]
- 3.Stubbe J, van der Donk WA. 1998. Protein radicals in enzyme catalysis. Chem. Rev. 98, 705–762. ( 10.1021/cr9400875) [DOI] [PubMed] [Google Scholar]
- 4.Argirevic T, Riplinger C, Stubbe J, Neese F, Bennati M. 2012. ENDOR spectroscopy and DFT calculations: evidence for the hydrogen-bond network within α2 in the PCET of E. coli ribonucleotide reductase. J. Am. Chem. Soc. 134, 17661–17670. ( 10.1021/ja3071682) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holder PG, Pizano AA, Anderson BL, Stubbe J, Nocera DG. 2012. Deciphering radical transport in the large subunit of class I ribonucleotide reductase. J. Am. Chem. Soc. 134, 1172–1180. ( 10.1021/ja209016j) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Offenbacher AR, Minnihan EC, Stubbe J, Barry BA. 2013. Redox-linked changes to the hydrogen-bonding network of ribonucleotide reductase β2. J. Am. Chem. Soc. 135, 6380–6383. ( 10.1021/ja3032949) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Worsdorfer B, et al. 2013. Function of the diiron cluster of Escherichia coli class Ia ribonucleotide reductase in proton-coupled electron transfer. J. Am. Chem. Soc. 135, 8585–8593. ( 10.1021/ja401342s) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yokoyama K, Smith AA, Corzilius B, Griffin RG, Stubbe J. 2011. Equilibration of tyrosyl radicals (Y356•, Y731•, Y730•) in the radical propagation pathway of the Escherichia coli class Ia ribonucleotide reductase. J. Am. Chem. Soc. 133, 18420–18432. ( 10.1021/ja207455k) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Offenbacher AR, Burns LA, Sherrill CD, Barry BA. 2013. Redox-linked conformational control of proton-coupled electron transfer: Y122 in the ribonucleotide reductase β2 subunit. J. Phys. Chem. B 117, 8457–8468. ( 10.1021/jp404757r) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boussac A, Rappaport F, Brettel K, Sugiura M. 2013. Charge recombination in Sn TyrZ• QA−• radical pairs in D1 protein variants of photosystem II: long range electron transfer in the Marcus inverted region. J. Phys. Chem. B 117, 3308–3314. ( 10.1021/jp400337j) [DOI] [PubMed] [Google Scholar]
- 11.Keough JM, Zuniga AN, Jenson DL, Barry BA. 2013. Redox control and hydrogen bonding networks: proton-coupled electron transfer reactions and tyrosine Z in the photosynthetic oxygen-evolving complex. J. Phys. Chem. B 117, 1296–1307. ( 10.1021/jp3118314) [DOI] [PubMed] [Google Scholar]
- 12.Sjoholm J, Styring S, Havelius KGV, Ho FM. 2012. Visible light induction of an electron paramagnetic resonance split signal in photosystem II in the S2 state reveals the importance of charges in the oxygen-evolving center during catalysis: a unifying model. Biochemistry 51, 2054–2064. ( 10.1021/bi2015794) [DOI] [PubMed] [Google Scholar]
- 13.Sancar A. 2003. Structure and function of DNA photolyase and cryptochrome blue-light photoreceptors. Chem. Rev. 103, 2203–2238. ( 10.1021/cr0204348) [DOI] [PubMed] [Google Scholar]
- 14.Taylor JS. 1994. Unraveling the molecular pathway from sunlight to skin cancer. Acc. Chem. Res. 27, 76–82. ( 10.1021/ar00039a003) [DOI] [Google Scholar]
- 15.Li YF, Heelis PF, Sancar A. 1991. Active site of DNA photolyase: tryptophan-306 is the intrinsic hydrogen atom donor essential for flavin radical photoreduction and DNA repair in vitro. Biochemistry 30, 6322–6329. ( 10.1021/bi00239a034) [DOI] [PubMed] [Google Scholar]
- 16.Aubert C, Mathis P, Eker APM, Brettel K. 1999. Intraprotein electron transfer between tyrosine and tryptophan in DNA photolysase from Anacystis nidulans. Proc. Natl Acad. Sci. USA 96, 5423–5427. ( 10.1073/pnas.96.10.5423) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Byrdin M, Eker APM, Vos MH, Brettel K. 2003. Dissection of the triple tryptophan electron transfer chaIn In Escherichia coli DNA photolyase: Trp382 is the primary donor in photoactivation. Proc. Natl Acad. Sci. USA 100, 8676–8681. ( 10.1073/pnas.1531645100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kodali G, Siddiqui SU, Stanley RJ. 2009. Charge redistribution in oxidized and semiquinone E. coli DNA photolyase upon photoexcitation: Stark spectroscopy reveals a rationale for the position of Trp382. J. Am. Chem. Soc. 131, 4795–4807. ( 10.1021/ja809214r) [DOI] [PubMed] [Google Scholar]
- 19.Lukacs A, Eker APM, Byrdin M, Villette S, Pan J, Brettel K, Vos MH. 2006. Role of the middle residue in the triple tryptophan electron transfer chain of DNA photolyase: ultrafast spectroscopy of a Trp→Phe mutant. J. Phys. Chem. B 110, 15654–15658. ( 10.1021/jp063686b) [DOI] [PubMed] [Google Scholar]
- 20.Woiczikowski PB, Steinbrecher T, KubaŖ T, Elstner M. 2011. Nonadiabatic QM/MM simulations of fast charge transfer in Escherichia coli DNA photolyase. J. Phys. Chem. B 115, 9846–9863. ( 10.1021/jp204696t) [DOI] [PubMed] [Google Scholar]
- 21.Aubert C, Vos MH, Mathis P, Eker APM, Brettel K. 2000. Intraprotein radical transfer during photoactivation of DNA photolyase. Nature 405, 586–590. ( 10.1038/35014644) [DOI] [PubMed] [Google Scholar]
- 22.Davidson VL, Liu AM. 2012. Tryptophan tryptophylquinone biosynthesis: a radical approach to posttranslational modification. Biochim. Biophys. Acta 1824, 1299–1305. ( 10.1016/j.bbapap.2012.01.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geng JF, Dornevil K, Davidson VL, Liu AM. 2013. Tryptophan-mediated charge-resonance stabilization in the bis-Fe(IV) redox state of MauG. Proc. Natl Acad. Sci. USA 110, 9639–9644. ( 10.1073/pnas.1301544110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yukl ET, Liu FG, Krzystek J, Shin S, Jensen LMR, Davidson VL, Wilmot CM, Liu A. 2013. Diradical intermediate within the context of tryptophan tryptophylquinone biosynthesis. Proc. Natl Acad. Sci. USA 110, 4569–4573. ( 10.1073/pnas.1215011110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davidson VL, Wilmot CM. 2013. Posttranslational biosynthesis of the protein-derived cofactor tryptophan tryptophylquinone. Annu. Rev. Biochem. 82, 531–550. ( 10.1146/annurev-biochem-051110-133601) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang N, Kuznetsov A, Nocek JM, Hoffman BM, Crane BR, Hu XQ, Beratan DN. 2013. Distance-independent charge recombination kinetics in cytochrome c–cytochrome c peroxidase complexes: compensating changes in the electronic coupling and reorganization energies. J. Phys. Chem. B 117, 9129–9141. ( 10.1021/jp401551t) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wallrapp FH, Voityuk AA, Guallar V. 2013. In-silico assessment of protein–protein electron transfer. A case study: cytochrome c peroxidase–cytochrome c. PLoS Comput. Biol. 9, e1002990 ( 10.1371/journal.pcbi.1002990) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Butchosa C, Simon S, Voityuk AA. 2010. Electron transfer from aromatic amino acids to guanine and adenine radical cations in π stacked and T-shaped complexes. Org. Biomol. Chem. 8, 1870–1875. ( 10.1039/b927134a) [DOI] [PubMed] [Google Scholar]
- 29.Shih C, et al. 2008. Tryptophan-accelerated electron flow through proteins. Science 320, 1760–1762. ( 10.1126/science.1158241) [DOI] [PubMed] [Google Scholar]
- 30.Blanco-Rodriguez AM, et al. 2011. Phototriggering electron flow through ReI-modified Pseudomonas aeruginosa azurins. Chem. Eur. J. 17, 5350–5361. ( 10.1002/chem.201002162) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Warren JJ, Ener ME, ek A, Winkler JR, Gray HB. 2012. Electron hopping through proteins. Coord. Chem. Rev. 256, 2478–2487. ( 10.1016/j.ccr.2012.03.032) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takematsu K, et al. 2013. Tryptophan-accelerated electron flow across a protein–protein interface. J. Am. Chem. Soc. 134, 15515–15525. ( 10.1021/ja406830d) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Warren JJ, Herrera N, Hill MG, Winkler JR, Gray HB. 2013. Electron flow through nitrotyrosinate in Pseudomonas aeruginosa azurin. J. Am. Chem. Soc. 135, 11151–11158. ( 10.1021/ja403734n) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Renaud N, Berlin YA, Lewis FD, Ratner MA. 2013. Between superexchange and hopping: an intermediate charge-transfer mechanism in poly(A)-poly(T) DNA hairpins. J. Am. Chem. Soc. 135, 3953–3963. ( 10.1021/ja3113998) [DOI] [PubMed] [Google Scholar]
- 35.Zhang Y, Liu C, Balaeff A, Skourtis SS, Beratan DN. 2014. Biological charge transfer via flickering resonance. Proc. Natl Acad. Sci. USA 111, 10049–10054. ( 10.1073/pnas.1316519111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marcus RA, Sutin N. 1985. Electron transfers in chemistry and biology. Biochim. Biophys. Acta 811, 265–322. ( 10.1016/0304-4173(85)90014-X) [DOI] [Google Scholar]
- 37.Hammes-Schiffer S, Stuchebrukhov AA. 2010. Theory of coupled electron and proton transfer reactions. Chem. Rev. 110, 6939–6960. ( 10.1021/cr1001436) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Migliore A, Polizzi NF, Therien MJ, Beratan DN. 2014. Biochemistry and theory of proton-coupled electron transfer. Chem. Rev. 114, 3381–3465. ( 10.1021/cr4006654) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weinberg DR, et al. 2012. Proton-coupled electron transfer. Chem. Rev. 112, 4016–4093. ( 10.1021/cr200177j) [DOI] [PubMed] [Google Scholar]
- 40.Gray HB, Winkler JR. 2010. Electron flow through metalloproteins. Biochim. Biophys. Acta 1797, 1563–1572. ( 10.1016/j.bbabio.2010.05.001) [DOI] [PubMed] [Google Scholar]
- 41.Winkler JR, Gray HB. 2014. Electron flow through metalloproteins. Chem. Rev. 114, 3369–3380. ( 10.1021/cr4004715) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Winkler JR, Gray HB. 2014. Long-range electron tunneling. J. Am. Chem. Soc. 136, 2930–2939. ( 10.1021/ja500215j) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Denisov IG, Makris TM, Sligar SG, Schlichting I. 2005. Structure and chemistry of cytochrome P450. Chem. Rev. 105, 2253–2277. ( 10.1021/cr0307143) [DOI] [PubMed] [Google Scholar]
- 44.Whitehouse CJC, Bell SG, Wong LL. 2012. P450BM3 (CYP102A1): connecting the dots. Chem. Soc. Rev. 41, 1218–1260. ( 10.1039/c1cs15192d) [DOI] [PubMed] [Google Scholar]
- 45.Johnson EF, Stout CD. 2013. Structural diversity of eukaryotic membrane cytochrome P450s. J. Biol. Chem. 288, 17082–17090. ( 10.1074/jbc.R113.452805) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Amdursky N, Molotskii M, Gazit E, Rosenman G. 2010. Elementary building blocks of self-assembled peptide nanotubes. J. Am. Chem. Soc. 132, 15632–15636. ( 10.1021/ja104373e) [DOI] [PubMed] [Google Scholar]
- 47.Hauser CAE, Zhang SG. 2010. Nanotechnology: peptides as biological semiconductors. Nature 468, 516–517. ( 10.1038/468516a) [DOI] [PubMed] [Google Scholar]
- 48.Vargas M, Malvankar NS, Tremblay PL, Leang C, Smith JA, Patel P, Snoeyenbos-West O, Nevin KP, Lovley DR. 2013. Aromatic amino acids required for pili conductivity and long-range extracellular electron transport in Geobacter sulfurreducens. mBio 4, e00270-13 ( 10.1128/mBio.00210-13) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Girvan HM, Seward HE, Toogood HS, Cheesman MR, Leys D, Munro AW. 2007. Structural and spectroscopic characterization of P450 BM3 mutants with unprecedented P450 heme iron ligand sets—new heme ligation states influence conformational equilibria in P450 BM3. J. Biol. Chem. 282, 564–572. ( 10.1074/jbc.M607949200) [DOI] [PubMed] [Google Scholar]
- 50.Munro AW, Malarkey K, McKnight J, Thomson AJ, Kelly SM, Price NC, Lindsay JC, Coggins JR, Miles JS. 1994. The role of tryptophan-of cytochrome P450 BM3 from Bacillus megaterium in catalytic function—evidence against the ‘covalent switching’ hypothesis of P450 electron-transfer.”, Biochem. J. 303, 423–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Denisov IG, Baas BJ, Grinkova YV, Sligar SG. 2007. Cooperativity in cytochrome P450 3A4: linkages in substrate binding, spin state, uncoupling, and product formation. J. Biol. Chem. 282, 7066–7076. ( 10.1074/jbc.M609589200) [DOI] [PubMed] [Google Scholar]
- 52.Grinkova YV, Denisov IG, McLean MA, Sligar SG. 2013. Oxidase uncoupling in heme monooxygenases: human cytochrome P450 CYP3A4 in nanodiscs. Biochem. Biophys. Res. Commun. 430, 1223–1227. ( 10.1016/j.bbrc.2012.12.072) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Staudt H, Lichtenb F, Ullrich V. 1974. Role of NADH in uncoupled microsomal monoxygenations. Eur. J. Biochem. 46, 99–106. ( 10.1111/j.1432-1033.1974.tb03601.x) [DOI] [PubMed] [Google Scholar]
- 54.Puntarulo S, Cederbaum AI. 1998. Production of reactive oxygen species by microsomes enriched in specific human cytochrome P450 enzymes. Free Rad. Biol. Med. 24, 1324–1330. ( 10.1016/S0891-5849(97)00463-2) [DOI] [PubMed] [Google Scholar]
- 55.De Matteis F, Dawson SJ, Pons N, Pipino S. 2002. Bilirubin and uroporphyrinogen oxidation by induced cytochrome P4501A and cytochrome P4502B—role of polyhalogenated biphenyls of different configuration. Biochem. Pharmacol. 63, 615–624. ( 10.1016/S0006-2952(01)00851-6) [DOI] [PubMed] [Google Scholar]
- 56.De Matteis F, Ballou DP, Coon MJ, Estabrook RW, Haines DC. 2012. Peroxidase-like activity of uncoupled cytochrome P450: studies with bilirubin and toxicological implications of uncoupling. Biochem. Pharmacol. 84, 374–382. ( 10.1016/j.bcp.2012.04.016) [DOI] [PubMed] [Google Scholar]