

Abstract
Platinum compounds are a mainstay of cancer chemotherapy, with over 50% of patients receiving platinum. But there is a great need for improvement. Major features of the cisplatin mechanism of action involve cancer cell entry, formation mainly of intrastrand cross-links that bend and unwind nuclear DNA, transcription inhibition and induction of cell-death programmes while evading repair. Recently, we discovered that platinum cross-link formation is not essential for activity. Monofunctional Pt compounds such as phenanthriplatin, which make only a single bond to DNA nucleobases, can be far more active and effective against a range of tumour types. Without a cross-link-induced bend, monofunctional complexes can be accommodated in the major groove of DNA. Their biological mechanism of action is similar to that of cisplatin. These discoveries opened the door to a large family of heavy metal-based drug candidates, including those of Os and Re, as will be described.
Keywords: platinum, transition metal, anti-cancer, monofunctional, osmium
1. Introduction
Platinum compounds constitute one of the most widely used classes of anti-cancer drugs. Approximately half of all cancer patients receiving chemotherapy are treated with cisplatin, carboplatin or oxaliplatin (figure 1). Testicular cancer responds particularly well to cisplatin treatment and, following introduction of this drug into clinical use, cure rates have exceeded 95% [1]. Platinum drugs are also used in the treatment of ovarian, bladder, non-small-cell and small-cell lung cancer, melanoma, lymphomas, myelomas, head and neck cancer, and colon cancer [2]. All three of these drugs are square-planar platinum(II) complexes, bearing two inert ‘non-leaving group’ ligands and two labile ‘leaving group’ ligands. Despite the success achieved by these drugs, issues of resistance and toxicity continue to prompt the development of new and improved therapies. Extensive studies in our laboratory and elsewhere have revealed many of the details of the mechanism by which these platinum complexes exert their anti-cancer effect. We currently are applying the experience and mechanistic insights accrued over the past decades to develop novel metal-based therapeutics. Although our work has focused on the use of platinum metal centres, recent findings have convinced us of the validity of pursuing complexes of other transition metals as potential drug candidates. We begin this article with a summary of the mechanism of action of the classical bifunctional platinum(II) drugs and then describe our work on non-classical monofunctional platinum(II) complexes. Finally, we discuss more recent studies to investigate octahedral complexes of earlier third row transition metal ions.
Figure 1.

Compounds discussed in this review.
2. Mechanism of action of classical platinum anti-cancer agents
The mechanism of action of the three platinum drugs approved by the United States Food and Drug Administration (FDA) and the European Medicines Agency (EMA), as well as structurally related platinum(II) complexes, can be divided into four distinct sequential processes: (i) cellular uptake, (ii) aquation/activation, (iii) DNA platination, and (iv) cellular processing leading to apoptosis (figure 2). Each of these processes will be treated in turn.
Figure 2.

Mechanism of action of cisplatin comprising (i) cellular uptake, (ii) aquation/activation, (iii) DNA platination, and (iv) cellular processing leading to apoptosis. (Online version in colour.)
(a). Cellular uptake
Platinum chemotherapy is administered intravenously as a saline solution of the metal complex. The dissolved complex must pass through the cell membrane in order to interact with the target, nuclear DNA. Early reports of the uptake of cisplatin presented conflicting data regarding whether uptake was mediated by passive diffusion or active transport. It is currently accepted that some combination of the two most likely occurs [3]. Recently, much work has centred on the importance of copper transporters, such as CTR1, which can mediate uptake of cisplatin by yeast [4,5]. Although some evidence suggests that in human cells CTR1 can take up cisplatin and deliver it in a form that is capable of binding DNA and triggering apoptosis [6], iconoclastic data continue to emerge [7]. Other membrane proteins may also play a role in the uptake of platinum drugs and, in collaboration with the Giacomini laboratory, we showed that cells overexpressing the organic cation transporters OCT1 and OCT2 were significantly more sensitive to oxaliplatin than cisplatin [8]. This work with organic cation transporters led to our rediscovery and extension of the anti-cancer activity of cation monofunctional compounds, as will be described below [9].
(b). Aquation/activation
Once inside the cell, the platinum complex undergoes ligand substitution. For cisplatin, this chemistry is triggered by the significantly smaller cytosolic chloride ion concentration (approx. 4 mM), compared with that of the extracellular matrix (approx. 100 mM). The lower intracellular chloride ion concentration facilitates transformation to cationic aquated products such as cis-[Pt(NH3)2Cl(OH2)]+ and cis-[Pt(NH3)2(OH2)2]2+. The first-order rate constants of the two aquation steps in pure water are 2.5×10−5 and 3.3×10−5 s−1, respectively, at 25°C [10]. The aquation of cisplatin in the presence of DNA at 37°C was measured by 195Pt NMR spectroscopy [11]. Monofunctional adducts form with a rate constant of 10.2×10−5 s−1, and the rate constant for the closure of the monofunctional adducts to form bifunctional adducts is 9.2×10−5 s−1. The half-life for aquation and DNA binding is therefore approximately 2 h. The terms monofunctional and bifunctional describe the number of covalent bonds formed between the platinum centre and DNA. Carboplatin and oxaliplatin are significantly more stable to aquation, as expected because of the chelating nature of the leaving group ligand. Carboplatin is stable for up to 60 days in water [12], but direct substitution of one of the carboxylates by nucleobases is possible without the need for an aqua intermediate [13]. It has been proposed that carbonate can activate carboplatin [14], but this mechanism is not operative with cisplatin [15]. The aquation of oxaliplatin is also much slower than that of cisplatin, with a rate constant of 1.2×10−6 s−1 at 37°C in 10 mM HEPES buffer (pH 7.4) [16].
(c). DNA binding
As described above, cisplatin rapidly binds DNA following aquation if the biopolymer is present. The most nucleophilic site on DNA is the N7 position of guanine, which is exposed in the major groove, and these sites are preferentially platinated. An important development in the field of platinum anti-cancer drug research came with the realization, guided by experiments with restriction endonucleases and measurements of DNA buoyant densities, that stretches of guanine residues are preferentially platinated over isolated guanine residues [17]. These experiments, inter alia, led to the recognition of the importance of the ability of cisplatin to cross-link adjacent DNA bases. We and others have worked to elucidate the structures of these cross-links. Crystallographic characterization of the dinucleotide adduct cis-[Pt(NH3)2{d(pGpG)}] revealed that platination induces significant destacking of the guanine rings [18]. Subsequent crystallographic characterization of a site-specifically cisplatin-platinated dodecamer duplex DNA, bearing a 1,2-cis-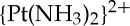 -d(GpG) intrastrand adduct, revealed the significant kink that is introduced into DNA upon platination [19]. Many subsequent studies by us and others have led to the characterization of the structures of the 1,3-cis-
-d(GpG) intrastrand adduct, revealed the significant kink that is introduced into DNA upon platination [19]. Many subsequent studies by us and others have led to the characterization of the structures of the 1,3-cis-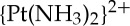 -d(GpTpG) intrastrand adduct formed by cisplatin and carboplatin, the 1,2-cis-{Pt(R,R-DACH)}2+-d(GpG) intrastrand adduct formed by oxaliplatin, the 1,2-cis-{Pt(NH3)(NH2C6H11)}2+-d(GpG) intrastrand adducts formed by satraplatin, and the cis-
-d(GpTpG) intrastrand adduct formed by cisplatin and carboplatin, the 1,2-cis-{Pt(R,R-DACH)}2+-d(GpG) intrastrand adduct formed by oxaliplatin, the 1,2-cis-{Pt(NH3)(NH2C6H11)}2+-d(GpG) intrastrand adducts formed by satraplatin, and the cis-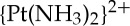 -d(GG) interstrand cross-link formed by cisplatin [20]. In addition, the structure of DNA bearing a 1,2-cis-
-d(GG) interstrand cross-link formed by cisplatin [20]. In addition, the structure of DNA bearing a 1,2-cis-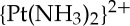 -d(GpG) intrastrand cisplatin adduct complexed with the high mobility group box protein HMGB1 was solved [21] as was the structure of a platinated nucleosome core particle prepared from histone octamer proteins and double-stranded 146-mer DNA bearing the site-specific 1,3-cis-
-d(GpG) intrastrand cisplatin adduct complexed with the high mobility group box protein HMGB1 was solved [21] as was the structure of a platinated nucleosome core particle prepared from histone octamer proteins and double-stranded 146-mer DNA bearing the site-specific 1,3-cis-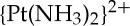 -d(GpTpG) cross-link [22].
-d(GpTpG) cross-link [22].
(d). Cellular processing
When cells experience DNA damage, a common response is to halt progression of the cell cycle so as to allow the damage to be repaired before it passes on to daughter cells, preventing the transmission of dangerous mutation-inducing lesions. The DNA damage that results when cells are treated with cisplatin induces cell cycle arrest at the G2 stage [3]. This arrest is mediated by ATM and ATR via CHK1 and CHK2. Studies suggest that G2 arrest is essential for inducing apoptosis following cisplatin treatment [23]. The main form of DNA repair that is used to remove bifunctional cisplatin lesions is nucleotide excision repair (NER) [3]. This repair scheme uses a multitude of proteins to remove a 30-mer oligonucleotide containing the platinated site and uses the complementary, now single-stranded, DNA to template the synthesis of a new oligonucleotide to replace the excised piece. If the damage cannot be repaired, the cells initiate a process of programmed cell death known as apoptosis. The p53-regulated expression of pro-apoptotic genes including BAX increases following DNA damage, leading to release of cytochrome c from mitochondria, subsequent cleavage of procaspase 9, and activation of caspases 3, 6 and 7 [24]. These caspases degrade components of the cell that are essential for viability.
3. Monofunctional platinum anti-cancer agents
(a). Pyriplatin: rediscovery of the potential of monofunctional platinum(II) complexes
Much of the anti-cancer research conducted in our laboratory has centred on uncovering aspects of the mechanism of bifunctional platinum compounds of the kind clinically used [3,25,26]. As mentioned above, we investigated the role of the organic cation transporters in the mechanism of action of oxaliplatin [8]. Greater expression of this protein in cancer cells correlated with cytotoxicity. Following this study, we prepared a variety of platinum complexes with organic ligands chosen such that each complex bore an overall positive charge. The hypothesis was that such constructs should act as more efficient substrates for the OCTs. The complex that gave the best results, far better than those of oxaliplatin, was cationic cis-diamminepyridinechloroplatinum(II), also referred to as cDPCP or pyriplatin (figure 1) [27]. The anti-cancer activity of this compound had been investigated previously in an animal model [28], but only a small number of follow-up studies on related compounds were performed [29]. In addition to significant organic character and cationic charge, pyriplatin is of interest because only one labile chloride ligand is present in the coordination sphere. Consequently, pyriplatin is expected to form only one covalent bond with DNA, precluding the bending induced by bifunctional platinum complexes such as cisplatin. Monofunctional platinum complexes had long been considered inactive [30]. The possibility that a significantly different type of DNA adduct would be formed by pyriplatin, however, suggested that it might have a novel spectrum of activity. Investigation of the cytotoxicity of pyriplatin in cultured mammalian cancer cell lines revealed a distinct spectrum of activity, although the overall potency of the compound was more than an order of magnitude less than that of cisplatin [27,31].
The potential mechanistic differences between pyriplatin and conventional platinum-based anti-cancer agents prompted further study. In the course of this investigation, we determined the crystal structure of a DNA duplex site-specifically platinated with pyriplatin [27]. The crystallographic results revealed that, unlike bifunctional cross-links, the monofunctional adduct induced very little change in the overall structure of the DNA double helix (figure 3).
Figure 3.
The crystal structures of duplex DNA oligonucleotides platinated with either (a) cisplatin, PDB: 1AIO or (b) pyriplatin, PDB: 3CO3. (Online version in colour.)
The adduct was, however, capable of stalling RNA polymerase II (RNA pol II), and transcription inhibition was proposed to be the origin of its cell-killing activity. A structural analysis of RNA pol II stalled at a site-specific pyriplatin lesion suggested that, by increasing the steric bulk of the N-heterocyclic ligand, it might be possible to increase transcription inhibition and consequential cytotoxicity [32]. From a panel of different N-heterocyclic ligands, Am, a series of complexes having the formula cis-[Pt(NH3)2Cl(Am)]+ was prepared [33]. The compound for which Am is phenanthridine showed the most significant results and was termed phenanthriplatin (figure 1).
(b). Phenanthriplatin: a potent monofunctional complex
Phenanthriplatin, like pyriplatin, exhibits a distinct spectrum of activity in cultured cancer cells [33]. When the data from the panel of 60 cell lines maintained by the National Cancer Institute (NCI60) were investigated using the COMPARE algorithm [34,35], the spectrum of activity of phenanthriplatin was distinct, not only from that of cisplatin but also from that of any other platinum agent that had previously been analysed by this assay. Unlike pyriplatin, the overall potency of phenanthriplatin is significantly greater than that of cisplatin. Phenanthriplatin DNA adducts are also able to inhibit RNA pol II [33]. A detailed study of the kinetics of RNA pol II transcription of site-specifically phenanthriplatin-platinated DNA probes revealed that CTP typically inserts in an error-free manner opposite to the platinated dG, and that further elongation is arrested [36].
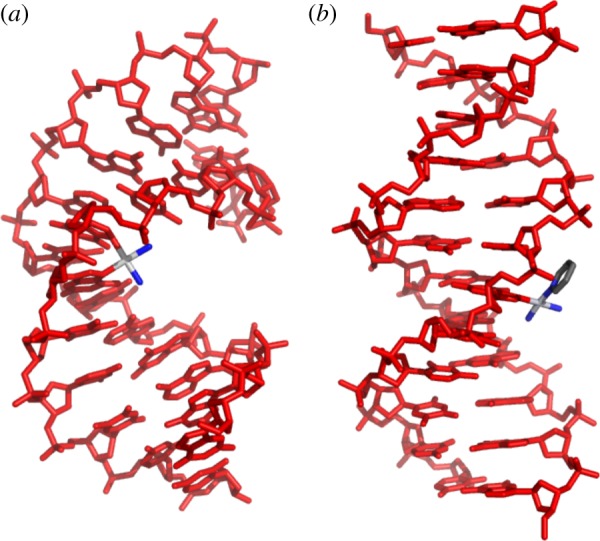
A number of the features of the molecular structure of the phenanthriplatin complex cation merit discussion. Phenanthridine coordinates to platinum such that the plane of the aromatic ligand is perpendicular to the metal coordination plane. The asymmetry of the phenanthridine ligand about the metal coordination plane results in one face of the complex being partially occluded by a C−H group and has been proposed to act as a steric block (figure 4) [33], reducing the rate of deactivation by biological thiols in a manner similar to that proposed for picoplatin, cis-ammine(α-picoline)dichloroplatinum(II) [37]. The asymmetry of the phenanthridine ligand also causes the complex to be chiral [38]. The conformational dynamics of diastereomeric analogues of phenanthriplatin confirm that the two enantiomers of phenanthriplatin rapidly interconvert, and, therefore, no advantage is gained by using enantiomerically resolved material in anti-cancer activity assays [39].
Figure 4.
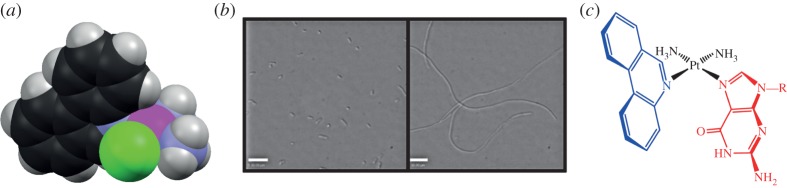
(a) Space-filling representation of the structure of the phenanthriplatin complex cation demonstrating the steric hindrance provided by phenanthridine ligand. (b) Light micrographs of E. coli that were (right) or were not (left) treated with 15 μM phenanthriplatin for 2 h. (c) The preferred diastereomer of cis-[Pt(NH3)2(phenanthridine)(9-alkylguanine)]2+ observed in the solid state and solution when R is methyl or ethyl. (Online version in colour.)
The large, planar aromatic phenanthridine ligand might implicate intercalation as a DNA-binding mode for phenanthriplatin, but analysis of competition Scatchard plots obtained by probing the affinity of ethidium bromide for DNA in the presence of this novel monofunctional platinum compound confirmed that it interacts with DNA in a purely covalent manner [33]. The interaction of phenanthriplatin with DNA in Escherichia coli provided further evidence to support the hypothesis that DNA is the ultimate biological target of this anti-cancer agent (figure 4). Unlike monofunctional platinum(II) complexes with little or no anti-cancer activity, phenanthriplatin was able to replicate the filamentous growth morphology that cisplatin induces in E. coli [40]. This phenotype arises from induction of the bacterial SOS response as a result of DNA damage.
Small molecule models of the phenanthriplatin–DNA lesion were prepared by substituting the chloride ligand of phenanthriplatin for 9-alkylguanine [39]. The guanine derivatives coordinate via the nucleophilic N7 position and are oriented perpendicular to the coordination plane. In the same fashion as phenanthridine, guanine is asymmetric about the platinum coordination plane and consequently serves as a source of chirality. As a result of the chirality about both phenanthridine– and guanine–platinum bonds, diastereomers arise. Diastereomerism therefore arises upon platination of DNA irrespective of the chirality of the double helix or the chiral carbon atoms of the deoxyribose rings. Surprisingly, the model complexes exhibited a preference for one diastereomeric form both in solution and in the solid state (figure 4). The origin of this preference is hydrogen bonding between the O6 carbonyl of the guanine ring and the cis-coordinated ammine ligand.
Studies of translesion DNA synthesis past phenanthriplatin adducts demonstrated that this complex can block not only RNA polymerases but also DNA polymerases. DNA pol η is a particularly promiscuous translesion synthase that is capable of bypassing naturally occurring cyclobutane pyrimidine dimers as well as the cisplatin 1,2-cis-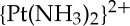 -d(GpG) cross-link [41]. A detailed investigation of the kinetics of lesion bypass by DNA pol η indicates that the incoming dCTP is able to properly insert opposite a platinated dG, but that further elongation is stalled [42]. These biochemical results were corroborated by an X-ray crystallographic study of pol η stalled at different stages of translesion synthesis. The structural data also confirmed that the orientational preference, observed in the small molecule studies described above [39], was preserved in the macromolecular structures and that the preferred diastereomer is the one responsible for arresting the polymerase [42].
-d(GpG) cross-link [41]. A detailed investigation of the kinetics of lesion bypass by DNA pol η indicates that the incoming dCTP is able to properly insert opposite a platinated dG, but that further elongation is stalled [42]. These biochemical results were corroborated by an X-ray crystallographic study of pol η stalled at different stages of translesion synthesis. The structural data also confirmed that the orientational preference, observed in the small molecule studies described above [39], was preserved in the macromolecular structures and that the preferred diastereomer is the one responsible for arresting the polymerase [42].
4. Other third row transition metal complexes
In addition to providing potent non-classical anti-cancer agents that are being actively pursued as possible clinical candidates, our studies of monofunctional Pt(II) complexes revealed that other third row transition metal complexes might be worth consideration as anti-cancer drug candidates. Although other researchers have investigated the anti-cancer potential of complexes of these metals, we long refrained from doing so. One reason was that, based on studies of cisplatin, DNA base cross-linking as encountered with bifunctional Pt(II) complexes appeared to be essential for activity [25,30]. The bending of DNA induced by cross-linking blocks the areas above and below the coordination plane and precludes DNA cross-linking by octahedral complexes. Third row transition metal complexes to the left of platinum in the periodic table tend to be six- rather than four-coordinate. The crystal structure of pyriplatin-platinated DNA [27] highlights the less congested environment about the metal centre (figure 5), suggesting that octahedral compounds may be able to form monofunctional adducts with DNA.
Figure 5.
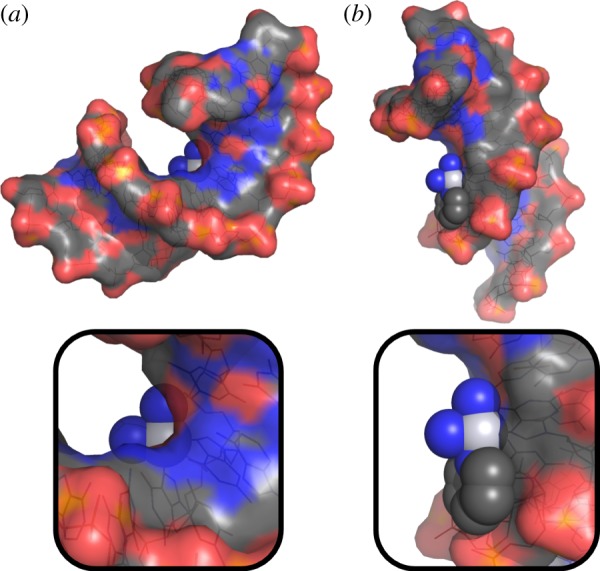
Top: surface representations of the cisplatin (a) and pyriplatin (b) adducts on duplex DNA with the atoms of the platinum lesion shown as spheres. The structures are oriented such that the platinum coordination plane lies in the plane of the page. Bottom: magnification of the platinum lesion illustrating the occlusion of the space above the platinum atom in the cisplatin lesion. (Online version in colour.)
(a). Osmium(VI) complexes operate on widely disparate cellular targets
Our first investigation centred on a series of octahedral Os(VI) nitrido complexes, mer-[OsN(L)Cl3], where L is 2,2′-bipyridine, 1,10-phenanthroline, 3,4,7,8-tetramethyl-1,10-phenanthroline, or 4,7-diphenyl-1,10-phenanthroline [43]. One of the most striking findings of this study was that peripheral ligand modification dramatically altered the cellular mechanism of action. Complexes having 1,10-phenanthroline and 4,7-diphenyl-1,10-phenanthroline ligands, Os-1 and Os-2, respectively (figure 1), are the most potent and the mechanism by which they induce cell death was investigated in detail. The initial hypothesis, based on previous reports of cytotoxic osmium complexes, was that these compounds would damage DNA. Compound Os-1 is able to cleave the sugar–phosphate backbone of DNA as revealed by gel electrophoresis experiments. Compound Os-2, however, could not. Moreover, unlike the analogue lacking phenyl substituents, it did not appreciably bind to DNA, as determined by osmium atomic absorption spectroscopy. Immunoblotting analyses confirmed that the unsubstituted species Os-1 induces phosphorylation of the histone protein H2AX, as well as CHK1, CHK2 and p53, which is indicative of DNA damage. The upregulation of p53 and phosphorylation of CHK1 arrest the cell cycle at the G2/M phase as measured by DNA flow cytometry (figure 6) [43].
Figure 6.

Mechanism of action of the osmium-based anti-cancer agents Os-1 and Os-2. (Adapted with permission from Suntharalingam et al. [43]. Copyright XXXX 2013 American Chemical Society.) (Online version in colour.)
The high accumulation of osmium in the cytoplasm of cells treated with Os-2 suggested that it may be able to induce endoplasmic reticulum (ER) stress. Co-treatment of cells with known inhibitors of ER stress reduced cytotoxicity, and immunoblotting analyses indicated expression of proteins related to the unfolded protein response, a major cellular reaction to ER stress. Microscopic examination of cells treated with both Os-2 and a fluorescent ER-staining dye revealed the organelle to be significantly engorged by comparison with those of untreated cells [43].
(b). An osmium(VI) compound that targets cancer stem cells
Cancer stem cells, or CSCs, are a small sub-population of tumour cells that have been linked to cancer relapse [44]. The association arises from the ability of these cells to self-renew, differentiate and remain untouched by conventional chemotherapy and radiotherapy [44,45]. The realization that CSCs may contribute to cancer recurrence prompted an intense search for small molecules that specifically target CSCs in the expectation that these compounds can be combined with conventional chemotherapeutic agents to provide more sustained responses [46]. The Os(VI) nitrido complex Os-1 is specifically toxic to CSCs. [47]. The breast cancer cell line HMLER can be enriched in CSCs and this enriched population can be used to assess the selectivity of anti-cancer agents for CSCs [48]. Os-1 depleted CSCs from an enriched population of breast cancer cells. Furthermore, the compound inhibits the formation of breast CSC mammospheres to a similar extent to salinomycin, the most selective breast CSC-targeting compound identified to date (figure 7) [46]. Mammospheres are discrete spheroid structures formed by breast cancer progenitor cell lines [49]. These results demonstrate the ability of transition metal complexes to specifically target CSCs and suggest their utility in chemotherapy aimed at targeting both tumour and tumour progenitor cells.
Figure 7.

Efficacy of a variety of chemical agents, including Os-1, in preventing mammosphere formation by CSC-enhanced HMLER breast cancer cells. Scale bar, 0.3 mm. (Online version in colour.)
5. Future directions
The foregoing results have inspired a more extensive exploration of complexes of the earlier third row transition metals. We have already uncovered the ability of certain high-valent rhenium species to trigger a relatively unexplored cell-killing pathway. We have also recently obtained promising results with organometallic tantalum complexes. Although platinum complexes have proved very important in the clinical management of cancer, and will continue to be a subject of much research in our laboratory, the diversity offered by other metal species should provide access to compounds capable of exerting biological activities not commonly exhibited by drugs in the clinic today. In particular, chemotherapeutic regimens containing a compound capable of selectively targeting cancer stem cells, such as the Os(VI) compound described above, will allow cancers to be driven into remission and eliminate recurrence.
Acknowledgements
This work was supported by grant no. CA034992 from the National Cancer Institute.
Glossary
| Am | N-heterocyclic amine |
| ATM | ataxia telangiectasia mutated protein |
| ATR | ataxia telangiectasia and Rad3 related protein |
| BAX | BCL2-associated X protein |
| cDPCP | cis-diamminepyridinechloroplatinum(II) |
| CHK1 | checkpoint kinase 1 |
| CHK2 | checkpoint kinase 2 |
| CSCs | cancer stem cells |
| CTP | cytidine triphosphate |
| CTR1 | copper transporter 1 |
| dCTP | 2′-deoxycytidine triphosphate |
| dG | 2′-deoxyguanosine |
| DNA | deoxyribonucleic acid |
| DNA pol η | DNA polymerase η |
| EMA | European Medicines Agency |
| ER | endoplasmic reticulum |
| FDA | US Food and Drug Administration |
| G2 phase | gap 2 (between DNA synthesis and mitosis) phase |
| H2AX | H2A histone family, member X |
| HMLER | tumorigenic human mammary epithelial cell line |
| HMGB1 | high mobility group box protein 1 |
| NER | nucleotide excision repair |
| NMR | nuclear magnetic resonance |
| OCT1/2 | organic cation transporter 1/2 |
| R,R-DACH | R,R-1,2-diaminocyclohexane |
| RNA pol II | RNA polymerase II |
References
- 1.Howlader N, et al. 2012. SEER cancer statistics review, 1975–2009. Bethesda, MD: National Cancer Institute. [Google Scholar]
- 2.Wheate NJ, Walker S, Craig GE, Oun R. 2010. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans. 39, 8113–8127. ( 10.1039/C0dt00292e) [DOI] [PubMed] [Google Scholar]
- 3.Wang D, Lippard SJ. 2005. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 4, 307–320. ( 10.1038/Nrd1691) [DOI] [PubMed] [Google Scholar]
- 4.Lin XJ, Okuda T, Holzer A, Howell SB. 2002. The copper transporter CTR1 regulates cisplatin uptake in?. Saccharomyces cerevisiae. 62, 1154–1159. ( 10.1124/mol.62.5.1154) [DOI] [PubMed] [Google Scholar]
- 5.Ishida S, Lee J, Thiele DJ, Herskowitz I. 2002. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc. Natl Acad. Sci. USA 99, 14298–14302. ( 10.1073/pnas.162491399) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Howell SB, Safaei R, Larson CA, Sailor MJ. 2010. Copper transporters and the cellular pharmacology of the platinum-containing cancer drugs. Mol. Pharmacol. 77, 887–894. ( 10.1124/mol.109.063172) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ivy KD, Kaplan JH. 2013. A re-evaluation of the role of hCTR1, the human high-affinity copper transporter, in platinum-drug entry into human cells. Mol. Pharmacol. 83, 1237–1246. ( 10.1124/mol.113.085068) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang S, et al. 2006. Organic cation transporters are determinants of oxaliplatin cytotoxicity. Cancer Res. 66, 8847–8857. ( 10.1158/0008-5472.CAN-06-0769) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnstone TC, Park GY, Lippard SJ. 2014. Understanding and improving platinum anticancer drugs—phenanthriplatin. Anticancer Res. 34, 471–476. [PMC free article] [PubMed] [Google Scholar]
- 10.Reishus JW, Martin DS., Jr 1961. cis-Dichlorodiammineplatinum(II). Acid hydrolysis and isotopic exchange of the chloride ligands. J. Am. Chem. Soc. 83, 2457–2462. ( 10.1021/ja01472a009) [DOI] [Google Scholar]
- 11.Bancroft DP, Lepre CA, Lippard SJ. 1990. 195Pt NMR kinetic and mechanistic studies of cis- and trans-diamminedichloroplatinum(II) binding to DNA. J. Am. Chem. Soc. 112, 6860–6871. ( 10.1021/ja00175a020) [DOI] [Google Scholar]
- 12.Canovese L, Cattalini L, Chessa G, Tobe ML. 1988. Kinetics of the displacement of cyclobutane-1,1-dicarboxylate from diammine(cyclobutane-1,1-dicarboxylato)platinum(II) in aqueous solution. J. Chem. Soc. Dalton Trans. 2135–2140. ( 10.1039/dt9880002135) [DOI] [Google Scholar]
- 13.Frey U, Ranford JD, Sadler PJ. 1993. Ring-opening reactions of the anticancer drug carboplatin: NMR characterization of cis-[Pt(NH3)2(CBDCA-O)(5′-GMP-N7)] in solution. Inorg. Chem. 32, 1333–1340. ( 10.1021/ic00060a005) [DOI] [Google Scholar]
- 14.Di Pasqua AJ, Goodisman J, Kerwood DJ, Toms BB, Dubowy RL, Dabrowiak JC. 2006. Activation of carboplatin by carbonate. Chem. Res. Toxicol. 19, 139–149. ( 10.1021/tx050261s) [DOI] [PubMed] [Google Scholar]
- 15.Todd RC, Lovejoy KS, Lippard SJ. 2007. Understanding the effect of carbonate ion on cisplatin binding to DNA. J. Am. Chem. Soc. 129, 6370–6371. ( 10.1021/ja071143p) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jerremalm E, Videhult P, Alvelius G, Griffiths WJ, Bergman T, Eksborg S, Ehrsson H. 2002. Alkaline hydrolysis of oxaliplatin—isolation and identification of the oxalato monodentate intermediate. J. Pharm. Sci. 91, 2116–2121. ( 10.1002/jps.10201) [DOI] [PubMed] [Google Scholar]
- 17.Sherman SE, Lippard SJ. 1987. Structural aspects of platinum anticancer drug interactions with DNA. Chem. Rev. 87, 1153–1181. ( 10.1021/Cr00081a013) [DOI] [Google Scholar]
- 18.Sherman S, Gibson D, Wang A, Lippard S. 1985. X-ray structure of the major adduct of the anticancer drug cisplatin with DNA: cis-[Pt(NH3)2d(pGpG)]. Science 230, 412–417. ( 10.1126/science.4048939) [DOI] [PubMed] [Google Scholar]
- 19.Takahara PM, Rosenzweig AC, Frederick CA, Lippard SJ. 1995. Crystal structure of double-stranded DNA containing the major adduct of the anticancer drug cisplatin. Nature 377, 649–652. ( 10.1038/377649a0) [DOI] [PubMed] [Google Scholar]
- 20.Todd RC, Lippard SJ. 2009. Inhibition of transcription by platinum antitumor compounds. Metallomics 1, 280–291. ( 10.1039/B907567d) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohndorf U-M, Rould MA, He Q, Pabo CO, Lippard SJ. 1999. Basis for recognition of cisplatin-modified DNA by high-mobility-group proteins. Nature 399, 708–712. ( 10.1038/21460) [DOI] [PubMed] [Google Scholar]
- 22.Todd RC, Lippard SJ. 2010. Consequences of cisplatin binding on nucleosome structure and dynamics. Chem. Biol. 17, 1334–1343. ( 10.1016/j.chembiol.2010.10.018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sorenson CM, Eastman A. 1988. Mechanism of cis-diamminedichloroplatinum(II)-induced cytotoxicity: role of G2 arrest and DNA double-strand breaks. Cancer Res. 48, 4484–4488. [PubMed] [Google Scholar]
- 24.Haupt S, Berger M, Goldberg Z, Haupt Y. 2003. Apoptosis—the p53 network. J. Cell Sci. 116, 4077–4085. ( 10.1242/Jcs.00739) [DOI] [PubMed] [Google Scholar]
- 25.Lippard SJ. 1987. Chemistry and molecular CSC-enhanced HMLER breast cancer cells biology of platinum anticancer drugs. Pure Appl. Chem. 59, 731–742. ( 10.1351/pac198759060731) [DOI] [Google Scholar]
- 26.Jamieson ER, Lippard SJ. 1999. Structure, recognition, and processing of cisplatin-DNA adducts. Chem. Rev. 99, 2467–2498. ( 10.1021/Cr980421n) [DOI] [PubMed] [Google Scholar]
- 27.Lovejoy KS, Todd RC, Zhang S, McCormick MS, D’Aquino JA, Reardon JT, Sancar A, Giacomini KM, Lippard SJ. 2008. cis-Diammine(pyridine)chloroplatinum(II), a monofunctional platinum(II) antitumor agent: uptake, structure, function, and prospects. Proc. Natl Acad. Sci. USA 105, 8902–8907. ( 10.1073/pnas.0803441105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hollis LS, Amundsen AR, Stern EW. 1989. Chemical and biological properties of a new series of cis-diammineplatinum(II) antitumor agents containing three nitrogen donors: cis-[Pt(NH3)2(N-donor)Cl]+. J. Med. Chem. 32, 128–136. ( 10.1021/Jm00121a024) [DOI] [PubMed] [Google Scholar]
- 29.Johnstone TC, Wilson JJ, Lippard SJ. 2013. Monofunctional and higher-valent platinum anticancer agents. Inorg. Chem. 52, 12234–12249. ( 10.1021/ic400538c) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cleare MJ, Hoeschele JD. 1973. Studies on the antitumor activity of group VIII transition metal complexes. Part I. Platinum(II) complexes. Bioinorg. Chem. 2, 187–210. ( 10.1016/S0006-3061(00)80249-5) [DOI] [Google Scholar]
- 31.Lovejoy KS, et al. 2011. Spectrum of cellular responses to pyriplatin, a monofunctional cationic antineoplastic platinum(II) compound, in human cancer cells. Mol. Cancer Ther. 10, 10 ( 10.1158/1535-7163.MCT-11-0250) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang D, Zhu G, Huang X, Lippard SJ. 2010. X-ray structure and mechanism of RNA polymerase II stalled at an antineoplastic monofunctional platinum-DNA adduct. Proc. Natl Acad. Sci. USA 107, 9584–9589. ( 10.1073/pnas.1002565107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park GY, Wilson JJ, Song Y, Lippard SJ. 2012. Phenanthriplatin, a monofunctional DNA-binding platinum anticancer drug candidate with unusual potency and cellular activity profile. Proc. Natl Acad. Sci. USA 109, 11987–11992. ( 10.1073/pnas.1207670109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shoemaker RH. 2006. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 6, 813–823. ( 10.1038/nrc1951) [DOI] [PubMed] [Google Scholar]
- 35.Paull KD, Shoemaker RH, Hodes L, Monks A, Scudiero DA, Rubinstein L, Plowman J, Boyd MR. 1989. Display and analysis of patterns of differential activity of drugs against human tumor cell lines: development of mean graph and COMPARE algorithm. J. Natl Cancer Inst. 81, 1088–1092. ( 10.1093/jnci/81.14.1088) [DOI] [PubMed] [Google Scholar]
- 36.Kellinger MW, Park GY, Chong J, Lippard SJ, Wang D. 2013. Effect of a monofunctional phenanthriplatin-DNA adduct on RNA polymerase II transcriptional fidelity and translesion synthesis. J. Am. Chem. Soc. 135, 13054–13061. ( 10.1021/ja405475y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Y, Guo ZJ, Parkinson JA, Sadler PJ. 1998. Kinetic control of reactions of a sterically hindered platinum picoline anticancer complex with guanosine 5′-monophosphate and glutathione. J. Chem. Soc. Dalton Trans. 3577–3585. ( 10.1039/A806544f) [DOI] [Google Scholar]
- 38.Biagini MC, Ferrari M, Lanfranchi M, Marchiò L, Pellinghelli MA. 1999. Chirality in mononuclear square planar complexes. J. Chem. Soc. Dalton Trans. 1575–1580. ( 10.1039/A808940j) [DOI] [Google Scholar]
- 39.Johnstone TC, Lippard SJ. 2014. The chiral potential of phenanthriplatin and its influence on guanine binding. J. Am. Chem. Soc. 136, 2126–2134. ( 10.1021/ja4125115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnstone TC, Alexander SM, Lin W, Lippard SJ. 2014. Effects of monofunctional platinum agents on bacterial growth: a retrospective study. J. Am. Chem. Soc. 136, 116–118. ( 10.1021/ja411742c) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao Y, Biertümpfel C, Gregory MT, Hua Y-J, Hanaoka F, Yang W. 2012. Structural basis of human DNA polymerase η-mediated chemoresistance to cisplatin. Proc. Natl Acad. Sci. USA 109, 7269–7274. ( 10.1073/pnas.1202681109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gregory MT, Park GY, Johnstone TC, Lee Y-S, Yang W, Lippard SJ. 2014. Structural and mechanistic studies of polymerase η bypass of phenanthriplatin DNA damage. Proc. Natl Acad. Sci. USA 111, 9133–9138. ( 10.1073/pnas.1405739111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suntharalingam K, Johnstone TC, Bruno PM, Lin W, Hemann MT, Lippard SJ. 2013. Bidentate ligands on osmium(VI) nitrido complexes control intracellular targeting and cell death pathways. J. Am. Chem. Soc. 135, 14060–14063. ( 10.1021/ja4075375) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gupta PB, Chaffer CL, Weinberg RA. 2009. Cancer stem cells: mirage or reality?. Nat. Med. 15, 1010–1012. ( 10.1038/nm0909-1010) [DOI] [PubMed] [Google Scholar]
- 45.Reya T, Morrison SJ, Clarke MF, Weissman IL. 2001. Stem cells, cancer, and cancer stem cells. Nature 414, 105–111. ( 10.1038/35102167) [DOI] [PubMed] [Google Scholar]
- 46.Gupta PB, Onder TT, Jiang GZ, Tao K, Kuperwasser C, Weinberg RA, Lander ES. 2009. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 138, 645–659. ( 10.1016/j.cell.2009.06.034) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suntharalingam K, Lin W, Johnstone TC, Bruno PM, Zheng YR, Hemann MT, Lippard SJ. 2014. A breast cancer stem cell-selective, mammospheres-potent osmium(VI) nitrido complex. J. Am. Chem. Soc. 136, 14413–14416. ( 10.1021/ja508808v) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Elenbaas B, Spirio L, Koerner F, Fleming MD, Zimonjic DB, Donaher JL, Popescu NC, Hahn WC, Weinberg RA. 2001. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 15, 50–65. ( 10.1101/Gad.828901) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grimshaw MJ, Cooper L, Papazisis K, Coleman JA, Bohnenkamp HR, Chiapero-Stanke L, Taylor-Papadimitriou J, Burchell JM. 2008. Mammosphere culture of metastatic breast cancer cells enriches for tumorigenic breast cancer cells. Breast Cancer Res. 10, 52 ( 10.1186/bcr2106) [DOI] [PMC free article] [PubMed] [Google Scholar]