

Abstract
Acting as a bridge between the heterogeneous and homogeneous realms, the use of discrete, well-defined, solid-state organometallic complexes for synthesis and catalysis is a remarkably undeveloped field. Here, we present a review of this topic, focusing on describing the key transformations that can be observed at a transition-metal centre, as well as the use of well-defined organometallic complexes in the solid state as catalysts. There is a particular focus upon gas–solid reactivity/catalysis and single-crystal-to-single-crystal transformations.
Keywords: solid state, organometallic, reactivity, catalysis
1. Introduction
Organometallic chemistry, rigorously defined by the chemical synthesis and reactivity of molecules with metal–carbon bonds, is a vibrant area of research with a large variety of practical applications [1]. For example, organometallic complexes are commonly used as catalysts for the production of commodity chemicals, materials such as polymers, and in fine chemical synthesis and medicinal chemistry discovery [1,2]. The majority of discoveries in the area have been performed in the solution phase, with studies in the solid state generally often reserved only for structural analysis; for example, single-crystal X-ray crystallography and, to a significantly lesser extent, solid-state nuclear magnetic resonance spectroscopy. By contrast to the solution phase, studies on the synthesis of, and catalysis using, organometallic complexes in the solid phase have attracted significantly less attention, even though there are potential benefits to this approach, such as: improved selectivities in synthesis that comes from spatially confined environments, improved isolated yields of products and the attenuation of decomposition pathways allowing for products that might be kinetically unstable in solution to be observed in the solid state.
Heterogeneous catalysis using the surfaces of metals or ionic platform materials is a well-researched area of chemistry with many industrial uses [3–5]. However, the mechanism of the binding, catalytic steps and product release may be multifarious or difficult to resolve when using such an extended material. There may also be a limited number of ‘active sites’ for catalysis. By contrast, homogeneous catalysis tends to be significantly more well defined, allowing for the mechanistic pathways to be readily probed using a wide variety of analytical and kinetic techniques, and often uses a single active metal centre that can be tuned by modifying the supporting ligand set, often with exquisite control with regard to reactivity and selectivity [1,6]. This greater degree of molecular control using a single-site catalyst can thus allow for stereoselective or regiospecific reactivity to be more readily tuned [7,8]. Moreover, the solvent (or sometimes ligand, for example M⋯H−C agostic interactions [9]) can play a role in stabilizing the active site(s) on the metal often necessary for reactivity, by forming weak interactions that are readily replaced by incoming substrates, so-called ‘virtual’ [10] or operationally unsaturated vacant sites [11].
Acting as a bridge between the heterogeneous and homogeneous realms, the use of discrete, well-defined, solid-state organometallic complexes for synthesis and catalysis is a remarkably undeveloped field (figure 1) [12–14]. In principle, if using reagents in the gas phase that can penetrate the lattice (i.e. a solid–gas reaction), or when in contact with a solvent that will not dissolve the organometallic species but solvates substrates and products, the active organometallic species can partake in the same processes observed in the solution phase, such as ligand substitution, oxidative addition, reductive elimination and insertion reactions. These resulting single-site catalysts thus bring together the benefits of heterogeneous catalysis (i.e. recyclability and easy removal from the reaction mixture) with the potential for intimate control over the transformations that the metal–ligand environment promotes in a homogeneous system [15,16]; for example, the high degrees of selectivity and mechanistic control associated with a well-defined metal–ligand environment, while harnessing the particular benefits of a solid-state environment. Further advantages of such an approach in catalysis include the simple separation of products from the catalyst [17,18], while avoiding the use of solvents in such solid–gas processes has potential economic and environmental benefits. Finally, and perhaps most excitingly, such a methodology potentially allows for the study of organometallic species without the complications of unwanted reactivity with the solvent. Such species, if being both low coordinate and of low electron count, could well be intermediates in catalytic processes that are often implicated but not observed in solution-phase chemistry. Solid-state reactivity using well-defined single-site organometallic complexes thus potentially allows for the isolation of otherwise unstable complexes, kinetically trapped in the solid state.
Figure 1.
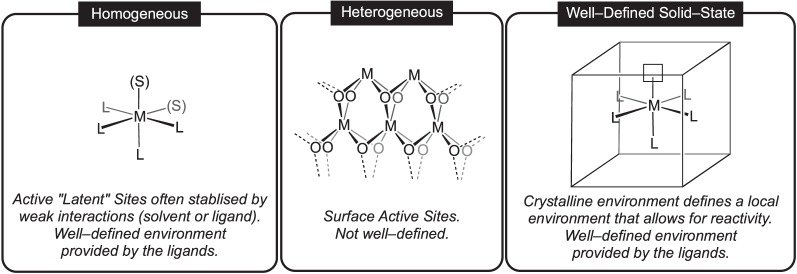
Molecular, heterogeneous and well-defined solid-state organometallic motifs.
2. Scope of this review
In this review, we discuss selected examples of solid-state organometallic synthetic chemistry and catalysis, focusing on describing the key transformations that can be observed, as well as the use of well-defined organometallic complexes in the solid state as catalysts. In particular, we focus on gas–solid reactivity, as this presents the ideal opportunity to explore transformations and catalysis while retaining the crystalline integrity (i.e. single-crystal-to-single-crystal transformations)—which is important for both structural studies (i.e. molecular structures by single-crystal X-ray diffraction) and reactivity (well-defined voids and channels in the lattice to allow for reactivity). We do not attempt to comprehensively review the solid-state organometallic chemistry associated with simple ligand substitution reactions where crystallinity is lost [12], isomerization reactions [19] or mechanochemical transformations [12,19–23]. In addition, the vibrant field of metal–organic framework materials and the encapsulation of active species inside the cavities of these materials is only discussed in passing where appropriate [24,25]. Likewise, we do not cover photocrystallographic techniques that allow for the determination of molecular structures of metastable species generated in the crystal photochemically. This technique has been used, for example, to study linkage isomerization in transition-metal complexes with nitrosyl, dinitrogen, sulfur dioxide and nitrite ligands, when photoactivated by light of an appropriate wavelength [26]; or photoinduced spin-crossover transitions, the structural consequences of which can be measured by photocrystallography [27]. Related to this is the development of ‘crystalline molecular flasks’ [28,29] in which self-assembled cages (e.g. from Pd2+ ions and triazene ligands) can be used to encapsulate and stabilize highly reactive complexes formed by photo-irradiation, such as the photodissociated complex (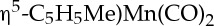 [30].
[30].
This is not the first time the general area of solid-state organometallic chemistry has been reviewed. An excellent in-depth account by Coville & Cheng [12] presented the area of solid-state organometallic chemistry in 1998 with updates with regard to isomerization reactions [19] and organometallic reactions that occur in the melt [31]. A recent (2011), short, highlight article by van der Boom [32] describes molecular single-crystal-to-single-crystal transformations in coordination chemistry. Our intention is that this review builds on these contributions, in particular bringing together both recent and older publications on solid–gas reactivity.
3. General considerations
For reactivity to occur in the solid state, the reagents must be able to penetrate the extended structure and access the metal sites; and this suggests that porosity within the extended solid structure will aid reaction [13,33]. The idea of a ‘reaction cavity’ was pioneered by Cohen & Schmidt [34], who proposed that reactions in the solid state occur with the least amount of molecular movement possible. There is, however, good evidence of limited molecular movement within the solid state from X-ray diffraction studies, in particular the rotation of CH3, CF3 and C5H5 groups [35]. The nevertheless constrained environments within a solid structure open up the possibilities of added reaction selectivity, potentially different from that observed in solution. For example, if a reaction cavity could be designed to be chiral then asymmetric reactivity may also be possible [36]. As solid-state reactions proceed, the products will replace the reagents. This generally occurs from the surface downwards, and Kaupp [37] has proposed the idea of ‘phase rebuilding’ where reaction direction is determined by crystallographic faces, and lattice reconstruction occurs over thousands of angstroms at a time. The kinetics of solid-state reactions have proved difficult to measure owing to their inherent complexity, as the reaction may take place at different rates upon the surface and within the interior of a crystalline material. For example, Caulton and co-workers [38] proposed that changes in molecular shape on reaction induce strain in a crystalline material, which in turn promotes micro-cracking. Such cracks will expose more of the interior of the crystal to the gaseous reagent and increase reaction rate. When little change in shape occurs the crystals may become ‘passivated’ by a surface layer of the product, slowing further reactivity.
4. Stoichiometric solid–gas reactions
A very early example of solid-state organometallic synthesis using solid–gas techniques was reported in the 1960s when the oxidative addition of various HX gases (HF, HCl, HBr, HI and H2S) to Vaska-type complexes IrX′(CO)(PPh3)2 (X′=Cl, Br, I, SCN) was reported to form trans-Ir(PPh3)2(X′)(CO)(H)(X) [39]. Similarly, addition of I2 to Pt(acac)2 (acac=acetylacetonate) forms the oxidative addition product trans-PtI2(acac)2 [40]. More recently, Brammer and co-workers [41–43] have shown that the reaction of trans-[CuCl2(n-X-C5H4N)2] (n=3,4; X=Cl, Br) with HCl gas to form [n-X-C5H4NH]2[CuCl4] requires breaking of two Cu−N bonds to form Cu−Cl bonds and N−H bonds. These reactions have been monitored by powder diffraction, including in situ powder synchrotron diffraction [41], showing that microcrystalline products are formed.
Organometallic stoichiometric reactions with gaseous reagents were extensively studied by Bianchini in the 1990s. For example, the displacement of labile N2 from 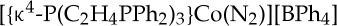 by various gases was studied in a solid–gas reaction (scheme 1) [44]. It was proposed that small gaseous reagents could penetrate the crystal lattice of [BPh4]− anions by dissolving in the hydrophobic regions formed by the tetraphos ligand backbone and anion phenyl groups. As is shown later (§5), displacement of a labile N2 also occurs in single-crystal-to-single-crystal transformations.
by various gases was studied in a solid–gas reaction (scheme 1) [44]. It was proposed that small gaseous reagents could penetrate the crystal lattice of [BPh4]− anions by dissolving in the hydrophobic regions formed by the tetraphos ligand backbone and anion phenyl groups. As is shown later (§5), displacement of a labile N2 also occurs in single-crystal-to-single-crystal transformations.
Scheme 1.
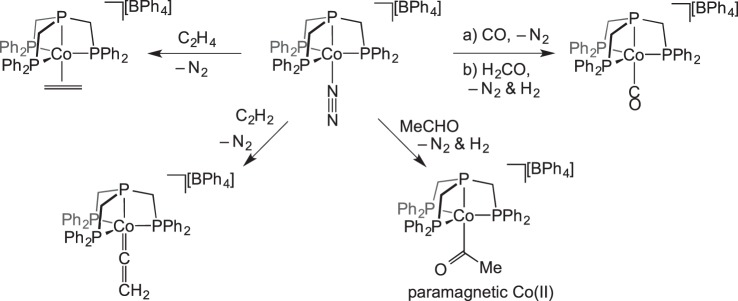
Solid-state gas exchange reactions facilitated by labile N2 ligands.
An interesting case of ligand displacement, in as much as that cleavage of a dimer is occurring in the solid state, comes from the reaction shown in scheme 2 in which Werner and co-workers [45] reported that addition of CO or ethene to a chloride-bridged Rh(I) dimer resulted in the generation of monomeric species, where two dative Rh–Cl bonds had been cleaved by CO or ethene. Other examples of ligand of displacement in the solid state have been reported. Werner and co-workers reported that addition of CO to a Rh-complex that contains a hemilabile [46,47] phosphine–ether ligand results in displacement of one Rh–ether linkage, whereas a dicarbonyl will form in the analogous solution-phase reaction (scheme 3) [45]. Addition of CO to Rh(PPh3)3(OAr) (e.g. Ar=C6Cl5) in the solid state results in an intermediate, tentatively described as five-coordinate Rh(PPh3)3(OAr)(CO), from which washing of the solid with ether removes PPh3 to afford trans-Rh(PPh3)2(OAr)(CO) [48]. Related five-coordinate species can be isolated from addition of CO in the solid state to square planar complexes such as [Ir(COD)(PPh3)(PhCN)][ClO4] to give, for example, [Ir(COD)(PPh3)(CO)2][ClO4] via loss of PhCN [49]. Milstein and co-workers [50] have shown that CO can bind reversibly to a 16-electron Rh(I) nitrosyl pincer complex, in which an equilibrium is established between a five-coordinate, CO-bound, and four-coordinate, CO-free, complex. Concomitant with this addition of CO is the change in NO binding mode from linear to bent, as measured by infrared (IR) spectroscopy.
Scheme 2.
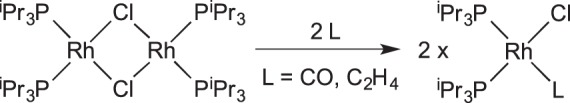
Solid–gas reactivity of a dimeric species to afford monomeric complexes.
Scheme 3.
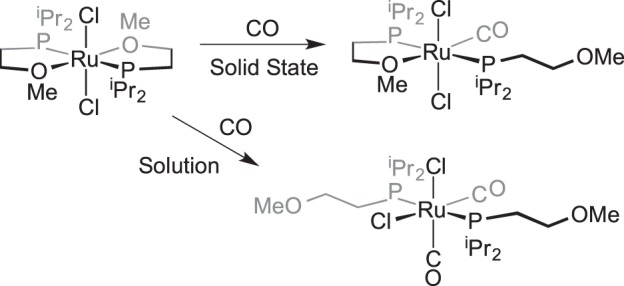
Hemilable ligand displacement in the solid state.
The reversible addition of CO2 to the rhodium and iridium complexes M(CO)(PPh3)2(OH) was reported by Flynn & Vaska [51], although the nature of the M⋯CO2 interaction was not clarified. Very recently, Nolan and co-workers [52] have reported that CO2 rapidly (2 min) inserts into the O−H bond of Ir(COD)(IPr)OH (IPr=1,3-di-isopropyl-imidazolin-2-ylidene) in a solid–gas reaction to form a bimetallic carbonate, 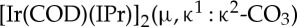 , scheme 4, with the concomitant elimination of water [IPr=1,3-bis(isopropyl)imidazol-2-ylidene)].
, scheme 4, with the concomitant elimination of water [IPr=1,3-bis(isopropyl)imidazol-2-ylidene)].
Scheme 4.
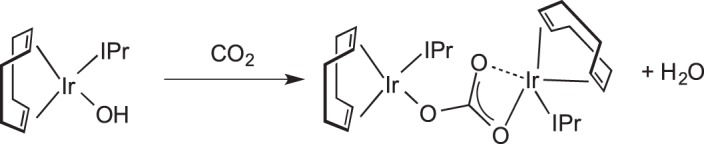
CO2 fixation using an Ir-hydroxide.
A related carbonate complex that comes from the aerobic oxidation of (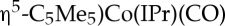 to form a carbonato complex
to form a carbonato complex 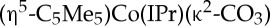 was reported by Radius and co-workers [53]. This process occurs rapidly in air, both in solution and in the solid phase (scheme 5).
was reported by Radius and co-workers [53]. This process occurs rapidly in air, both in solution and in the solid phase (scheme 5).
Scheme 5.
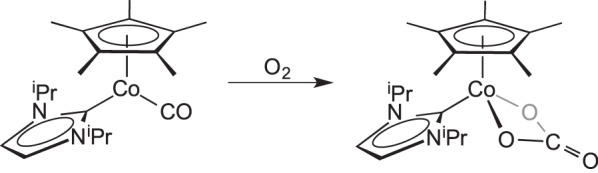
Aerobic oxidation to a carbonyl to form carbonate complex.
Addition of alkynes and alkenes to a metal complex may result in olefin oligomerization through C−H and C−C coupling processes, common transformations in solution-phase organometallic chemistry [1]. Bianchini et al. [54,55] demonstrated that addition of C2H2 to [Ir(triphos)(C2H4)(H)2][BPh4] [triphos=(Ph2PCH2)3CCH3] in the solid state leads to various products, including benzene and butadiene complexes (scheme 6a). Siedle & Newmark [56] have demonstrated trimerization of C2H2 with [Ir(H)2(PPh3)2]3[PW12O40] to form a benzene complex, whereas C−H activation of propene with the same organometallic starting material forms an allyl complex (scheme 6b). Related transformations in which bound ethene in a Rh(I) tripyrazolylhydroborate complex undergoes C−H and C−C bond-forming processes to form allylic species have been reported by Carmona and co-workers [57]. C−Br activation in an η2-azobenzene ligand coordinated to a {Pt(PEt3)2} fragment occurs in the solid state to give the corresponding Pt-aryl-halide [58]. The oxidative addition of a C−Cl bond is reported in the solid-state transformation of the zwitterionic Rh(I) η6-arene complex 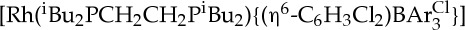
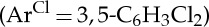 into the dimeric Rh(III) complex
into the dimeric Rh(III) complex 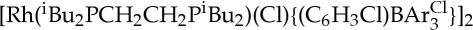 [59]. Interestingly, this process generates two isomers in the solid state, whereas the same process in solution only accesses the thermodynamic isomer. Bond isomerizations in the solid state, involving C−C cleavage, between Ru-alkynylketones and Ru-vinylidenes have been followed using IR spectroscopy, and kinetic data were obtained for this transformation. This is a very rare example of such an analysis of reactivity in solid-state organometallic chemistry, and, from these data, the authors propose a mechanism controlling the reaction that invokes nucleation and nuclei growth rather than diffusion, chemical reaction or phase boundary-controlled steps [60].
[59]. Interestingly, this process generates two isomers in the solid state, whereas the same process in solution only accesses the thermodynamic isomer. Bond isomerizations in the solid state, involving C−C cleavage, between Ru-alkynylketones and Ru-vinylidenes have been followed using IR spectroscopy, and kinetic data were obtained for this transformation. This is a very rare example of such an analysis of reactivity in solid-state organometallic chemistry, and, from these data, the authors propose a mechanism controlling the reaction that invokes nucleation and nuclei growth rather than diffusion, chemical reaction or phase boundary-controlled steps [60].
Scheme 6.

(a,b) Reaction of cationic iridium complexes with alkenes and alkynes in the solid state resulting in C–C couplings and/or C–H activation.
Reversible hydrogen addition to metal complexes is of considerable interest with regard to processes such as hydrogenation, hydrodesulfurization and hydrogen storage applications [1]. The reversible addition of three molecules of hydrogen to 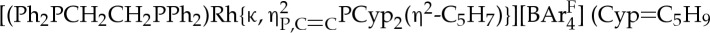 ,
, 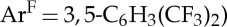 to form [(Ph2PCH2CH2PPh2)Rh(PCyp3)(H)2(H2)][BArF4] has been reported, in which a dihydrogen ligand is coordinated, and the phosphine–alkene ligand has been hydrogenated [61]. In the solid phase, the product loses two molecules of hydrogen under application of a vacuum to form a red intermediate proposed to be the Rh(I) complex
to form [(Ph2PCH2CH2PPh2)Rh(PCyp3)(H)2(H2)][BArF4] has been reported, in which a dihydrogen ligand is coordinated, and the phosphine–alkene ligand has been hydrogenated [61]. In the solid phase, the product loses two molecules of hydrogen under application of a vacuum to form a red intermediate proposed to be the Rh(I) complex 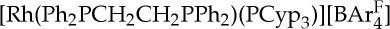 , in which an agostic interaction from the phosphine is proposed. This red intermediate then slowly loses a further equivalent of H2 via an alkyl dehydrogenation (C−H activation and β-elimination) to reform
, in which an agostic interaction from the phosphine is proposed. This red intermediate then slowly loses a further equivalent of H2 via an alkyl dehydrogenation (C−H activation and β-elimination) to reform 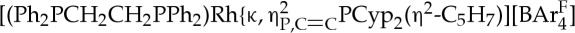 . In solution, no red Rh(I) intermediate is observed, and removal of the dihydrogen ligand under vacuum results, instead, in a Rh(III) dihydride (scheme 7).
. In solution, no red Rh(I) intermediate is observed, and removal of the dihydrogen ligand under vacuum results, instead, in a Rh(III) dihydride (scheme 7).
Scheme 7.
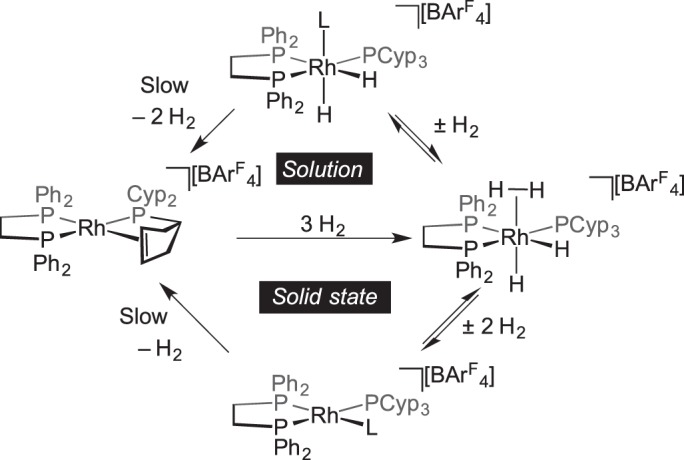
Reversible addition of hydrogen and C–H activation in solution and the solid state. L=agostic or solvent interactions.
The simple reversible addition of dihydrogen to monometallic [62] and multi-metallic cluster [63–66] species in the solid state has been reported by a number of groups. In these instances, the H2-free complexes are often electronically unsaturated and stabilized by bulky ligands around the metal centre(s). An example shown in scheme 8 is the reversible addition of two molecules of H2 to the cluster 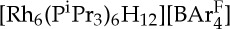 to give
to give 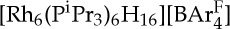 [67]. Related coordinate unsaturation in clusters enabled via loss of a weakly bound ligand in the solid state (such as NCMe) allows for solid–gas reactivity of Os3(CO)11L (L=NCMe) with CO, NH3 and H2 [68].
[67]. Related coordinate unsaturation in clusters enabled via loss of a weakly bound ligand in the solid state (such as NCMe) allows for solid–gas reactivity of Os3(CO)11L (L=NCMe) with CO, NH3 and H2 [68].
Scheme 8.
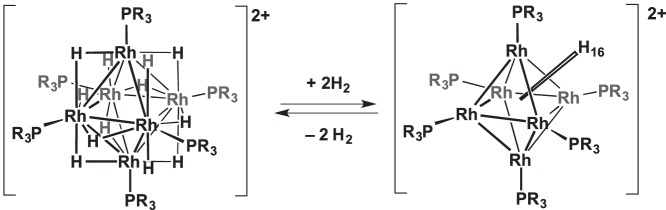
Reversible H2 addition to 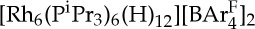 . R=iPr.
. R=iPr.
Deliberate installation of coordinate and electronic unsaturation at a metal centre means that solid–gas reactions can be made particularly facile. Caulton and co-workers have reported that 16-electron Ru(CO)2(PtBu2Me)2 adds H2, Cl2 or O2 to give the corresponding 18-electron complexes cis,cis,trans-Ru(H)2(CO)2(PtBu2Me)2, cis,cis,trans-Ru(Cl)2(CO)2(PtBu2Me)2 and Ru(η2-O2)(CO)2(PtBu2Me)2, respectively. Interestingly, reaction with HSiMe3 gave the dihydride Ru(H)2(CO)2(PtBu2Me)2 dissolved in liquid Me3SiSiMe3, formed by a dehydrocoupling process. Reaction with CO is particularly slow, and this is postulated to be due to the fact that the product, Ru(CO)3(PtBu2Me)2, is a similar size to the organometallic reactant, and this results in surface passivation rather than the fracturing of the crystal (and concomitant faster ingress of gas) that is suggested to occur when product and starting material geometries are different [38]. The electronically and coordinatively unsaturated complex 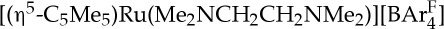 adds H2, N2, O2, CO and ethylene in the solid state, with the latter shown to be reversible. These new complexes (except the CO adduct) were unstable in solution and were characterized by elemental analysis [69].
adds H2, N2, O2, CO and ethylene in the solid state, with the latter shown to be reversible. These new complexes (except the CO adduct) were unstable in solution and were characterized by elemental analysis [69].
5. Single-crystal-to-single-crystal transformations
A single-crystal-to-single-crystal (SC−SC) transition is one in which crystallinity is retained throughout a reaction, allowing the product to be characterized directly by single-crystal X-ray crystallography without recourse to recrystallization from solution [32]. For crystallinity to be retained, only a very small structural reorganization can be tolerated to avoid the break-up of the lattice. Crystal size also probably affects reaction time in SC−SC transitions because of surface area to volume ratio implications. Molecular designs that enable such transitions to take place involve use of bulky ligands or anions which dominate the packing and thus can create a rigid, porous, structure in which smaller movements around the metal centre are made possible [13,70]. SC−SC reactions present excellent possibilities for selectivity to be controlled within a solid-state environment, as the reaction cavity necessarily must remain well defined throughout to preserve crystallinity.
If gaseous reagents require access to the interior of the crystal then empty or partially filled channels throughout the lattice may be necessary [33]. This was suggested by Brookhart and co-workers [13], who noted the channels of disordered toluene throughout the crystalline lattice of (POCOP)IrL (POCOP=1,3-[OP{2,4,6-C6H2(CF3)3}2]2C6H3, L=N2, CO, NH3, C2H4, O2). They reported a series of SC−SC gas transfer reactions using this system (scheme 9). Interestingly, the precursor complex studied (L=N2) is stable under vacuum, but readily reacts with more strongly binding gases, which suggests that an associative mechanism for gas exchange is operating within the interior of the crystal. These SC−SC transformations presumably occur as the packing in the lattice is dominated by the large tris-(CF3) substituted aryl groups on the pincer ligand, meaning that changes in the ancillary ligands around Ir (e.g. CO for N2) result in minimal structural reorganization.
Scheme 9.
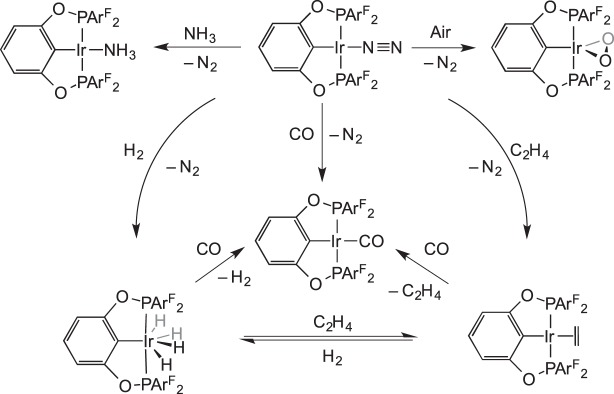
SC–SC transitions using Ir–pincer complexes. ArF=2,4,6-C6H2(CF3)3.
Such transitions can also be reversible. van Koten and co-workers [71,72] reported the reversible addition of SO2 to (NCN)PtCl (NCN=C6H2-5-(OH)-1,3-(CH2NMe2)2), which induced significant changes in the geometry of the metal centre, from square planar to pseudo-square pyramidal, and remarkably such a large structural change does not result in the loss of crystallinity, albeit these processes occur in the microcrystalline powder state and the transformations are monitored by powder diffraction techniques and IR spectroscopy rather than by single-crystal X-ray diffraction (crystals suitable for such analysis were grown independently). The authors speculate that these processes are likely to involve local solutions of the reactants that recrystallize at a comparable rate to solute formation. They proposed that this material could be used as a gas-triggered switch, with SO2 uptake signalled either by colour change or by crystal expansion. SC−SC transitions may also occur sequentially. In an elegant example, Crudden and co-workers [70] presented a double SC−SC gas exchange reaction using the (SIPr)2RhCl(L) system 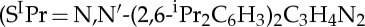 , L=N2, O2 or CO; scheme 10). First, N2 is replaced with O2 to give a dioxygen adduct, and then O2 is replaced with CO, without crystal degradation at either stage.
, L=N2, O2 or CO; scheme 10). First, N2 is replaced with O2 to give a dioxygen adduct, and then O2 is replaced with CO, without crystal degradation at either stage.
Scheme 10.
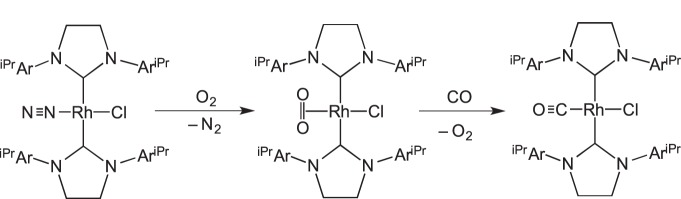
Sequential SC–SC gas transfer transitions. AriPr=2,6- .
.
Alcohol uptake (i.e. MeOH, EtOH and iPrOH) by the non-porous coordination polymer [Ag4(O2C(CF2)2CF3)4(TMP)3]n (A) is reversible in a SC−SC transformation by a solid–vapour process (TMP=2,3,5,6-tetramethylpyrazine). In an elegant sequence of substitution reactions, consecutive alcohols are introduced into the lattice: A-EtOH  A-MeOH
A-MeOH  A-iPrOH
A-iPrOH  A-EtOH without loss of single crystallinity. These ligand substitution reactions are accommodated by changes in coordination geometry at specific Ag(I) centres, and specifically alcohol insertion occurs into one-quarter of the Ag−O carboxylate bonds [73,74]. As this material does not have significant porosity, a mechanism is proposed in which concerted motion of the disordered fluoroalkyl chains allows for the transport of the alcohol molecules within the crystals. Powder X-ray crystallography techniques were also used to follow these transformations.
A-EtOH without loss of single crystallinity. These ligand substitution reactions are accommodated by changes in coordination geometry at specific Ag(I) centres, and specifically alcohol insertion occurs into one-quarter of the Ag−O carboxylate bonds [73,74]. As this material does not have significant porosity, a mechanism is proposed in which concerted motion of the disordered fluoroalkyl chains allows for the transport of the alcohol molecules within the crystals. Powder X-ray crystallography techniques were also used to follow these transformations.
SC−SC transitions can also take place in a suspension of non-solvating liquid, for example in polymeric platform materials. McKeown and co-workers [33] reported such SC−SC transitions using Fe(MeOH)2(phthalocyanine). Large interconnected voids (8 nm3) run through these structures that are defined by a cubic assembly of six of the phthalocyanine groups. These voids allow liquid penetration, and axial ligands can be reversibly, and rapidly, displaced by a variety of exogeneous ligands (scheme 11). Interestingly, monodentate ligands bind preferentially to an axial binding site within this cubic assembly, whereas bidentate ligands selectively bind to link neighbouring cubic assemblies together. Selective exchange between water and methanol has been observed as a SC−SC transformation in the trinuclear iron complexes [Fe3(μ3-O)(μ2-CH3COO)6(C5H5NO)2(L)][ClO4] (L=H2O, MeOH) [75], whereas heterolytic dissociation of water at a bis(μ-oxo)divanadiumpolyoxometallate, which models the interactions of water with metal oxide surfaces, was found to occur as a SC−SC transformation.
Scheme 11.

(a) Porous phthalocyanine–derivative complex (PNC[vL–Fe–cL]) which can undergo SC–SC ligand (L) exchanges when immersed in organic solvents. AriPr=2,6-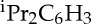 . (b) Example of two sequential ligand exchanges, displaying the linking of two metal centres by a bidentate ligand (phthalocyanine derivative simplified to a flat square).
. (b) Example of two sequential ligand exchanges, displaying the linking of two metal centres by a bidentate ligand (phthalocyanine derivative simplified to a flat square).
In the absence of added reagents, SC−SC transitions can occur in the form of a simple phase change within the crystal lattice. Balch and co-workers [77] reported a reversible phase change when acetone or dichloromethane vapour is passed over a crystal of β-Au2(μ-Ph2PCH2CH2PPh2)2I2⋅(OCMe)2, even though no additional solvent incorporation is observed. Two equivalents of acetone are present in both phases. The reverse reaction occurs if the crystal is left in air (scheme 12).
Scheme 12.
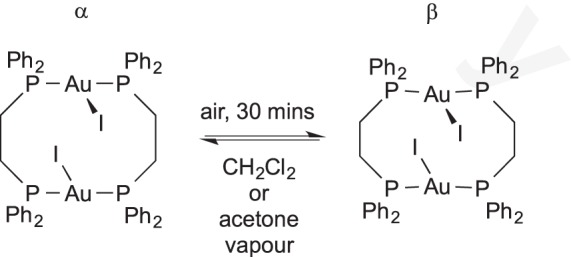
Phase change SC–SC transition driven by drying in air and reversible by exposure to CH2Cl2 or acetone vapour.
Brill, Rheingold and co-workers [78] of 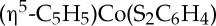 in the single-crystal phase at room temperature could be reversed upon warming to 423 K while still maintaining crystallinity (scheme 13). The dimeric species is thermodynamically favoured in the crystalline lattice at room temperature, but the monomer is favoured thermodynamically in solution, presumably owing to a dominant entropy term in solution.
in the single-crystal phase at room temperature could be reversed upon warming to 423 K while still maintaining crystallinity (scheme 13). The dimeric species is thermodynamically favoured in the crystalline lattice at room temperature, but the monomer is favoured thermodynamically in solution, presumably owing to a dominant entropy term in solution.
Scheme 13.
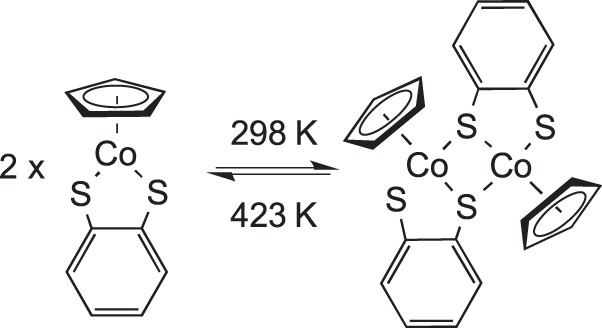
Reversible dimerization in a single crystal.
Another SC−SC transformation that occurs without additional reagent is the reversible C−C activation in 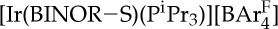 , which contains a rare example of an agostic M ⋯ C −C bond (BINOR-S=1,2,4,5,6,8-dimetheno-s-indacene). In this complex, reversible C−C cleavage occurs to form an equilibrium mixture, in the crystalline phase, of dynamically disordered C−C activated [Ir(V)] and C−C agostic [Ir(III)] complexes—the ratio of which changes with temperature. Interestingly, the
, which contains a rare example of an agostic M ⋯ C −C bond (BINOR-S=1,2,4,5,6,8-dimetheno-s-indacene). In this complex, reversible C−C cleavage occurs to form an equilibrium mixture, in the crystalline phase, of dynamically disordered C−C activated [Ir(V)] and C−C agostic [Ir(III)] complexes—the ratio of which changes with temperature. Interestingly, the 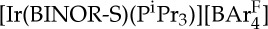 complex is itself made from a solid-state organometallic reaction, where cyclodimerization of two norbornadiene ligands occurs to yield the BINOR-S ligand (scheme 14) [79].
complex is itself made from a solid-state organometallic reaction, where cyclodimerization of two norbornadiene ligands occurs to yield the BINOR-S ligand (scheme 14) [79].
Scheme 14.
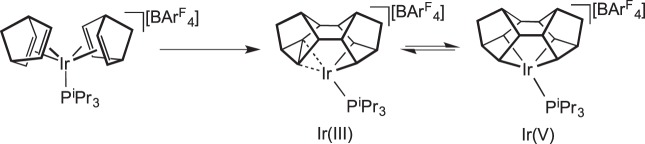
Formation of a BINOR-S complex and reversible SC–SC C–C cleavage.
SC−SC reactions present possibilities for directly forming reactive complexes, which may not be accessible cleanly using solution routes. C−N oxidative cleavage in the PNP pincer complex 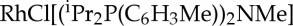 to give
to give 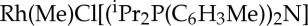 occurs in the solid state in a SC−SC transformation [80]. Saliently, when performed in the bulk, this preparative route is cleaner than that in solution [81]. Richter-Addo and co-workers [82] were able to crystallographically characterize biologically important porphyrin complexes which bind NO. Examples include (TPP)Fe(NO)(OC(=O)CF3), [(TPP)Fe(NO)(H2O)][(TPP)Fe(H2O)][OC(=O)CF3]2 [83] (TPP = tetraphenylporphyrin) and [(oep)Fe(NO)(S-2,6-(CF3CONH)2C6H3)] [84] (oep = octaethylporphyrinato dianion). These complexes were formed by addition of NO gas to the unsaturated precursors in the solid state. This transformation was not possible using solution crystallization methods, as in solution a mixture of by-products form instead (scheme 15) [82–84].
occurs in the solid state in a SC−SC transformation [80]. Saliently, when performed in the bulk, this preparative route is cleaner than that in solution [81]. Richter-Addo and co-workers [82] were able to crystallographically characterize biologically important porphyrin complexes which bind NO. Examples include (TPP)Fe(NO)(OC(=O)CF3), [(TPP)Fe(NO)(H2O)][(TPP)Fe(H2O)][OC(=O)CF3]2 [83] (TPP = tetraphenylporphyrin) and [(oep)Fe(NO)(S-2,6-(CF3CONH)2C6H3)] [84] (oep = octaethylporphyrinato dianion). These complexes were formed by addition of NO gas to the unsaturated precursors in the solid state. This transformation was not possible using solution crystallization methods, as in solution a mixture of by-products form instead (scheme 15) [82–84].
Scheme 15.
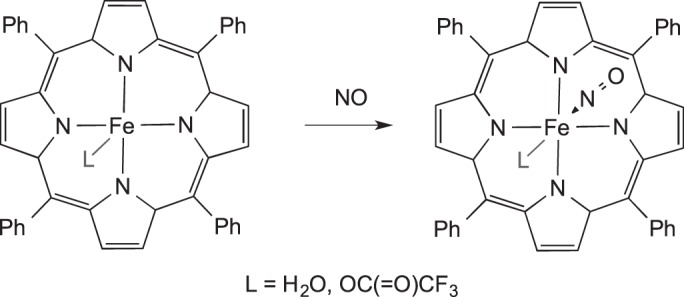
Formation (NO)–Fe–porphyrin complexes by SC–SC transitions.
Perhaps the most dramatic exploitation of solid-state SC−SC transformations in stabilizing highly reactive complexes comes from the addition of H2 to the complex [Rh(NBD)(iBu2PCH2CH2PiBu2)][BArF4] (NBD=C7H8, ArF=3,5-C6H3(CF3)2) that results in the isolation and structural characterization of a transition-metal sigma–alkane complex in which the hydrogenated organic fragment (NBA=C7H12) binds to the metal through two Rh⋯ H−C sigma interactions [Rh(η2,η2-NBA)(iBu Bu2)][BArF4] (scheme 16) [85]. Such alkane complexes are exceedingly rare, with only two examples having been previously characterized in the solid state by single-crystal X-ray diffraction, in which there is a close approach of the alkane H−C bond to a metal centre [86,87]. Ultimately, this alkane complex undergoes a further transformation, in the solid state, to form a complex in which the alkane ligand has been lost and the [BArF4]− anion coordinates. An intermediate, mono-hydrogenated, species was proposed
Bu2)][BArF4] (scheme 16) [85]. Such alkane complexes are exceedingly rare, with only two examples having been previously characterized in the solid state by single-crystal X-ray diffraction, in which there is a close approach of the alkane H−C bond to a metal centre [86,87]. Ultimately, this alkane complex undergoes a further transformation, in the solid state, to form a complex in which the alkane ligand has been lost and the [BArF4]− anion coordinates. An intermediate, mono-hydrogenated, species was proposed 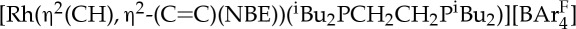 (NBE=C7H10), which also adds H2 in the solid state to give the alkane complex, as shown by solid state NMR spectroscopy.
(NBE=C7H10), which also adds H2 in the solid state to give the alkane complex, as shown by solid state NMR spectroscopy.
Scheme 16.
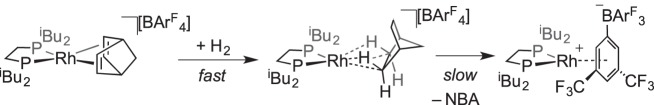
Formation of an alkane complex in the solid state.
(a). Overview of the structural changes associated with single-crystal-to-single-crystal transformations
Selected examples of SC−SC transitions are displayed in table 1 with their respective crystallographic volume change (z=1 equivalent). Minimal structural changes are apparently necessary to allow for SC−SC transitions, and most examples reported show less than 4% volume change in the lattice volume. The outlier of van Koten and co-workers’ example of SO2 uptake is exceptional because it involves a very large percentage volume change (15.7%), with expansion predominantly along one axis. However, this occurs in the microcrystalline phase without loss of crystallinity as measured by powder diffraction, and SC−SC experiments were not successful. Noteworthy is the 10% change in volume associated with reversible alcohol addition in the Ag-carboxylates, this perhaps being associated with the fact that this material is a coordination polymer in the solid state, which might allow for increased flexibility without loss of crystallinity.
Table 1.
Examples of SC–SC transitions and the crystallographic volume changes involved. POCOP=1,3-[OP{C6H2(CF3)3-2,4,6}2]2C6H3; NCN=C6H2-5-(OH)-1,3(CH2NMe2)2; SiPr=N,N′-(2,6-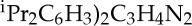 ; TPP= tetraphenylporphyrin; oep=octaethylporphyrinato dianion; NBD=C7H8; NBA= C7H12; ArF=3,5-C6H3(CF3)2; TMP=2,3,5,6-tetramethylpyrazine.
; TPP= tetraphenylporphyrin; oep=octaethylporphyrinato dianion; NBD=C7H8; NBA= C7H12; ArF=3,5-C6H3(CF3)2; TMP=2,3,5,6-tetramethylpyrazine.
| entry | starting complex space group, volume (Å3), z | product space group, volume (Å3), (z) | [change in volume (Å3)] and percentage change. For z=1 equivalent | |
|---|---|---|---|---|
| 1 | Brookhart and co-workers [13] | (POCOP)Ir(N2);  , 3002.11(14), z=2 , 3002.11(14), z=2 |
(POCOP)Ir(O2);  , 3046.3(10), z=2 , 3046.3(10), z=2 |
[+22] +1.4% |
(POCOP)Ir(CO);  , 3000.1(2), z=2 , 3000.1(2), z=2 |
[−1] −0.06% | |||
(POCOP)Ir(C2H4);  , 3020.6(2), z=2 , 3020.6(2), z=2 |
[+9] +0.6% | |||
(POCOP)Ir(H)2(H2);  , 2986.6(4), z=2 , 2986.6(4), z=2 |
[−8] −0.6% | |||
(POCOP)Ir(NH3);  , 3003.56(19), z=2 , 3003.56(19), z=2 |
[+1] +0.04% | |||
| 2 | van Koten and co-workers [71] | (NCN)PtCl; Pna21, 1346.0(4), z=4 | (NCN)PtCl(SO2); Pna21, 1557.5(4), z=4 | [+53] +15.7% |
| 3 | Crudden and co-workers [70] | (SIPr)RhCl(N2); P21212, 2855.15(8), z=2 | (SIPr)RhCl(O2); P21212, 2852.13(12), z=2 | [−3] −0.1% |
| (SIPr)RhCl(O2); P21212, 2852.13(12), z=2 | (SIPr)RhCl(CO); P21212, 2861.5(3), z=2 | [+9] +0.4% | ||
| 4 | McKeown and co-workers [33] | PNC[ctBuNC-Fe-vtBuNC]; Pn–3n, 52348.5(7), z=12 | PNC[ctBuNC-Fe-v(py)0.7(tBuNC)0.3]; Pn–3n, 52736(2), z=12 | [+32] +0.7% |
| PNC[cMeOH-Fe-vMe(C3N2H3)]; Pn–3n, 52819.6(12), z=12 | [+39] +0.8% | |||
| PNC[vtBuNC-Fe-c(4,4-bipy)-Fe-vtBuNC)]; Pn–3n, 54263.4(5), z=12 | [+160] +3.7% | |||
| PNC[vN2-Fe-cCN(C6H4)NC-Fe-vN2)]; Pn–3n, 53204(5), z=12 | [+71] +1.6% | |||
| 5 | Balch and co-workers [77] | α-Au2(μtPh2PCH2CH2-PPh2)2I2.(OCMe)2; P21/c, 2824.28(13) z=2 | βAu2(μ–Ph2PCH2CH2PPh2)2I2.(OCMe)2;  , 1411.99(7), z=1 , 1411.99(7), z=1 |
[−0.2] ∼0% |
| 6 | Brill, Rheingold and co-workers [78] | (η5-C5H5)Co(S2C6H4); P21/c, 2159.1(6), z=8 | [(η5-C5H5)Co(S2C6H4)]2;P21/c, 1031.2(3), z=2 (dimer) | [−12] −4.5%a |
| 7 | Richter-Addo and co-workers [82–84] | (TPP)Fe{OC(=O)CF3};  , 2093.1(3), z=2 , 2093.1(3), z=2 |
(TPP)Fe(NO){OC(=O)CF3};  , 2144(3), z=2 , 2144(3), z=2 |
[+25] +2.4% |
[(TPP)Fe(H2O)][OC(=O)CF3];  , 1833.4(3), z=2 , 1833.4(3), z=2 |
[(TPP)Fe(NO)(H2O)][(TPP)Fe(H2O)] [OC(=O)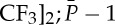 , 3818.2(16), z=2 , 3818.2(16), z=2 |
[+38] +4.1%b | ||
[(oep)Fe{S-2,6-(CF3CONH)2C6H3}];  , 2266.3(6), z=2 , 2266.3(6), z=2 |
[(oep)Fe(NO){S-2,6-(CF3CONH)2C6H3}];  , 2360(2), z=2 , 2360(2), z=2 |
[+47] +4.1% | ||
| 8 | Weller and co-workers [85] | [Rh(iBu2P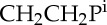 Bu2)(η2η2-NBD)][BArF4]; C 2/c, 5957.62(18), z=4 Bu2)(η2η2-NBD)][BArF4]; C 2/c, 5957.62(18), z=4 |
[Rh(iBu2P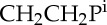 Bu2)(η2η2-NBA)][BArF4]; C 2/c, 6044.1(3), z=4 Bu2)(η2η2-NBA)][BArF4]; C 2/c, 6044.1(3), z=4 |
[+22] +1.5% |
| 9 | Ozerov and co-workers [80,81] | RhCl[(iPr2P(C6H3Me))2NMe]; P212121, 5955(4), z=8 | Rh(Me)Cl[(iPr2P(C6H3Me))2N]; P212121, 5976(4), z=8 | [+21] +0.4% |
| 10 | Brammer and co-workers [74] | [Ag4(O2C(CF2)2CF3)4(TMP)3]n;  , 1405.34(6), z=1 , 1405.34(6), z=1 |
[Ag4(O2C(CF2)2CF3)4(TMP)3(iPrOH)]n;  , 1545.9(2), z=1 , 1545.9(2), z=1 |
[+140.6] +10.0% |
6. Catalysis in the solid state
(a). Heterogeneous organometallic catalysts
The heterogenization of single-site catalysts brings together the benefits of heterogeneous catalysis (i.e. recyclability and ease of removal from the reaction mixture) with the potential for intimate control over transformations that occur at the metal centre that is provided by the local ligand environment in a homogeneous system [15,16]. There are a number of strategies that can be used to facilitate the heterogenization of well-defined organometallic catalyst systems, including surface-supported organometallic chemistry in which a platform material such as silica, zeolites or metal oxides support directly, or indirectly via linker groups, the organometallic complex [88–98]. Metal organic frameworks (MOFs) are also particularly attractive as one-, two- and three-dimensional assemblies can be created in which the metal atoms often act as the geometry-enforcing linkage points, but can also be envisaged as potential active sites for catalysis [24]. In addition, a MOF may simply act as a reaction cavity, with the organometallic species as a host/guest material [99]. Within the context of this review that concentrates on the reactivity of well-defined organometallic complexes in the solid state these materials, as they are often not well defined at the metal centre of interest, are not included here.
(b). Self-supported organometallic catalysis
Self-supported catalysts invoke an active metal centre in which the ligand environment also acts as the platform microporous material [100]. The advantage in many of these systems, compared with heterogenized organometallic catalysts, is that they are well defined at the metal–centre and thus more amenable to structural and spectroscopic investigation. Early reports of self-supported catalysts include the linkage polymers of [RhCl(CO)(1,4-(CN)2C6H4)]n which can hydrogenate and isomerize 1-hexene, with no leaching of complexes into solution [101]. The active rhodium site was created by photolytic dissociation of the CO ligand. A similar system was formed with two bridging ligands per metal, enabling a well-defined three-dimensional-stacked layer structure to form. The surface and corner positions of this structure are likely to contain unsaturated metal centres which are catalytically active; however, the interior sites are proposed to be totally inactive [102,103]. Multi-dentate oxime, thiourea, phosphine and NHC ligands have been used to form frameworks with Pd-centres for use in Suzuki–Miyaura C−C coupling reactions [104–107]. For example, Karimi & Akhavan [107] reported a coordination polymer of palladium with a linking bidentate NHC ligand, which is insoluble in water, resulting in C−C coupling catalysis that could be performed using water as the substrate and product solvent (scheme 17). Although the authors used the mercury test, which probes for nanoparticle formation, which showed no loss in activity, it is difficult to unequivocally prove that nanoparticles are in no way involved for such systems.
Scheme 17.
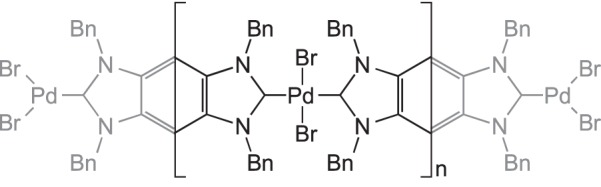
A coordination polymer with Pd capable of catalysing heterogeneous Suzuki–Miyaura cross coupling in water.
More complex microporous structures can also be formed with a mixture of metal sites. For instance, copper, nickel or palladium porphyrin moieties can be combined with rhodium–polycarboxylate linkages in which both metal sites may exhibit cooperative effects for hydrogenation reactions [108,109]. Kaskel and co-workers [110] have recently reported the formation of a microporous organometallic network based upon a rhodium alkene fragment linked to a rigid tetraphenylsilane backbone (scheme 18). While an accurate structural determination has proved difficult, the framework appeared to be air-stable unlike its homogeneous analogue [Rh(NBD)2][BF4]. This material catalysed transfer hydrogenation reactions.
Scheme 18.
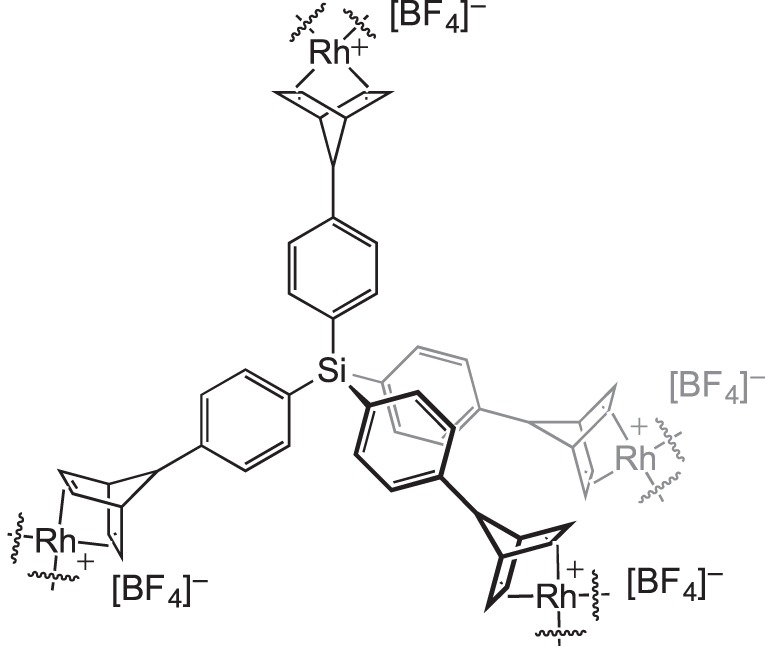
A microporous organometallic framework based upon rhodium alkene coordination.
(c). Solid-state organometallic catalysis without a support
Solid-state catalysis using well-defined organometallic complexes that are not incorporated into a platform material is a relatively undeveloped field. Bianchini et al. [14] introduced the concept with simple ethene hydrogenation reactions using [(triphos)Ir(H)2(C2H4)][BPh4] at 343 K (scheme 19). The catalyst was active in the solid state, in a mechanism proposed to operate via hydride migration to form an Ir−(C2H5) species, which can react with further H2 followed by reductive elimination of ethane. In solution, the same species was not catalytically active, because a coordinatively saturated dimeric bridging hydride species rapidly forms in the presence of H2 which was inactive for further reactions. Although some of the inactive dimeric species is also formed in the solid-state reaction, it appears to form at a slower rate than in solution. This highlights the ability of the solid state to maintain the integrity of the reactive species by playing a role in protecting them from deactivation pathways that require structural reorganization. The [BPh4]− anions are proposed to create a hydrophobic lattice structure ideal for allowing the passage of small hydrocarbon gases. The catalytic trimerization of ethyne to form benzene was also investigated by Bianchini and co-workers, who showed that a η4-benzene complex (formed itself from a solid–gas reaction) is an active pre-catalyst active at 373 K in the solid state (scheme 20) [55].
Scheme 19.

Catalytic ethene hydrogenation in the solid state versus solution.
Scheme 20.
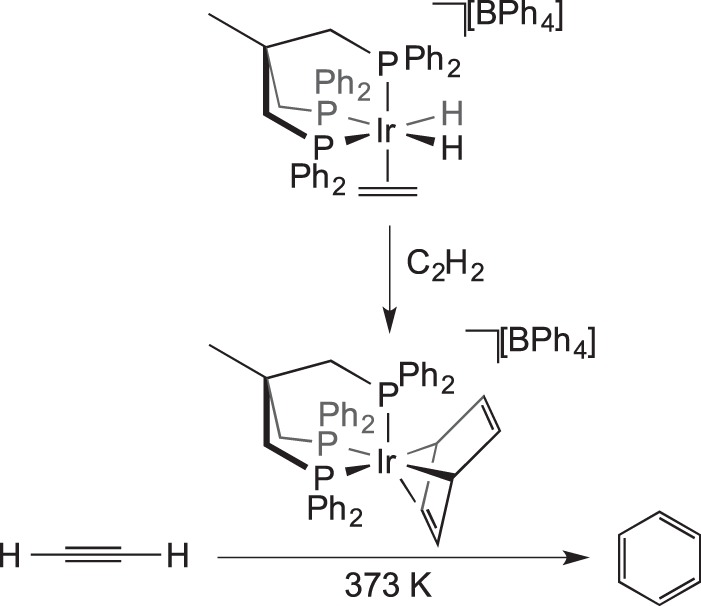
Trimerization of ethyne using a solid-state catalyst.
Siedle & Newmark [56] reported the room temperature catalytic activity of iridium phosphine cations partnered with Keggin-type trianions, [Ir(H)2(PPh3)2]3[PW12O40] (scheme 21), with the hydrogenation of ethene, propene and 1-hexene demonstrated. The isomerization of 1-hexene to a mixture of cis- and trans-2-hexenes and 3-hexenes was also reported, presumably via reversible C−H activation accessing an allyl-iridium–hydride intermediate. The authors do not comment on the rates of reaction. Similar to the findings of Bianchini, addition of excess ethyne forms benzene in catalytic quantities, with an iridium–benzene complex expected to act as the pre-catalyst, although the reaction is reported to be slow. The catalytic dimerization of CF2=CFCl to cis- and trans-1,2-dichlorohexafluorocyclobutane complexes was also reported.
Scheme 21.
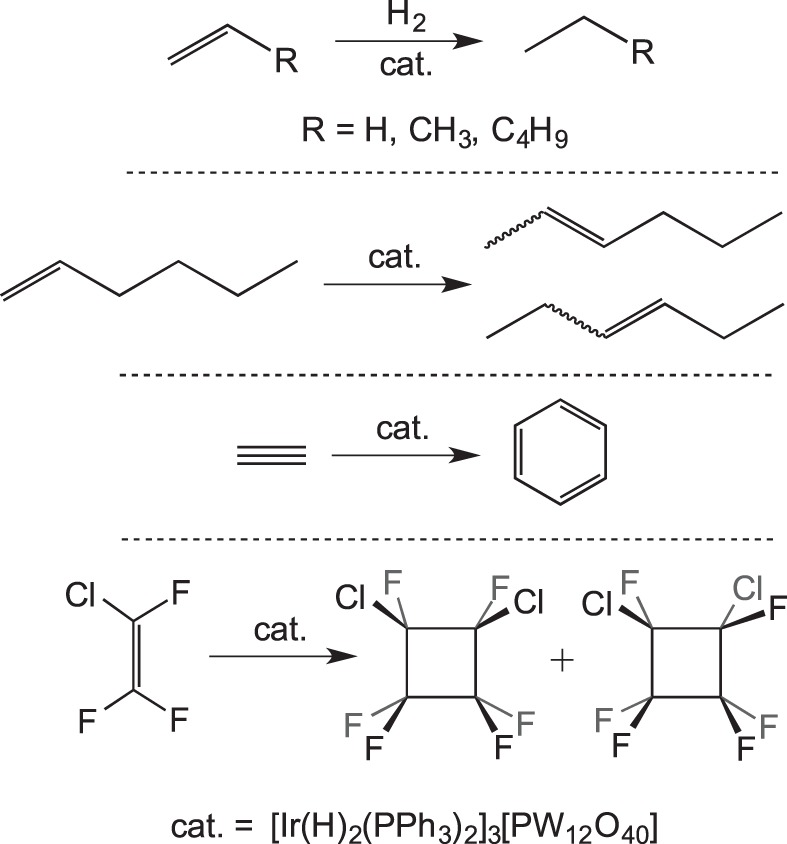
Catalytic reactions using [Ir(H)2(PPh3)2]3[PW12O40].
Limbach and co-workers [111] have reported on the solid-state catalysed hydrogenation of ethene using Vaska’s complex, Ir(CO)Cl(PPh3)2, by following the reaction products by gas-phase 1H NMR spectroscopy. In solution, the product of H2 addition (which presumably related to the active catalyst) is a cis-dihydride/trans-phosphine species. In the solid state, this is not formed, and it was proposed that this was due to ligand reorientation being inhibited. The authors thus suggested a different pathway for hydrogenation in the solid state and solution (scheme 22) that invokes a dihydrogen intermediate as the active species in the solid state.
Scheme 22.
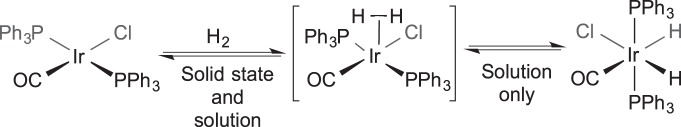
Reaction of Vaska’s complex with H2 in the solid state and solution.
Brookhart et al. [13] have reported the hydrogenation of ethene using single crystals of (POCOP)Ir(N2), as monitored by gas-phase NMR spectroscopy. At 298 K, the reaction requires 5 h to reach 95% conversion, but at 348 K 99% conversion occurs within 30 min. At this higher temperature, lattice-incorporated toluene is lost, and this is proposed to be responsible for the higher activity. (POCOP)Ir(C2H4) is the suggested resting state. Remarkable selective catalytic hydrogenation inside single crystals was also presented. By passivating the surface sites of crystals of (POCOP)Ir(N2) with a layer of (POCOP)Ir(CO), an incomplete crystal-to-crystal transition occurs. The resulting material can selectively hydrogenate ethene in the presence of propene, with a 25:1 preference at 348 K (scheme 23) [13]. It was proposed that the porous crystals allow the smaller ethene and hydrogen molecules access to the active interior metal sites, whereas the larger propene molecules cannot penetrate the surface. In the absence of surface passivation, only a small selectivity is seen in favour of hydrogenation of ethene (1.8:1 ratio of ethane to propane produced at 298 K), consistent with this.
Scheme 23.
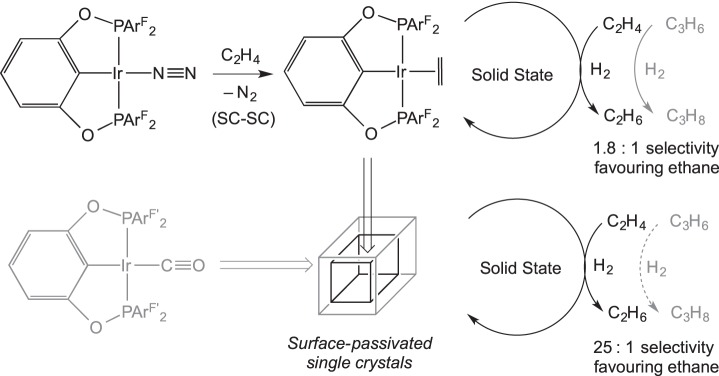
Hydrogenation of ethene using single crystals, and the selective hydrogenation of ethene in the presence of propene using surface-passivated single crystals.
Mul and co-workers [112] have reported upon the palladium-catalysed CO/C2H4 co-polymerization reaction which is operated under gas phase or slurry conditions in industry. The catalyst becomes incorporated into the growing polymer chain, and so its nature as a heterogeneous or homogeneous catalyst is not well defined. Mul et al. chose to investigate the mechanism by using microcrystalline (Ph2PCH2CH2CH2PPh2)Pd(CH3)(OTf) deposited onto a gold surface. They were able to probe the first few turnovers of reaction using polarization modulation reflection–absorption IR spectroscopy (scheme 24). The findings suggest that ethene insertion into the Pd–acyl bond is actually CO-assisted. The incoming CO is able to displace the chelating ketone group more easily than ethene, but can itself then exchange with ethene. This subtlety had not been previously revealed by solution studies.
Scheme 24.
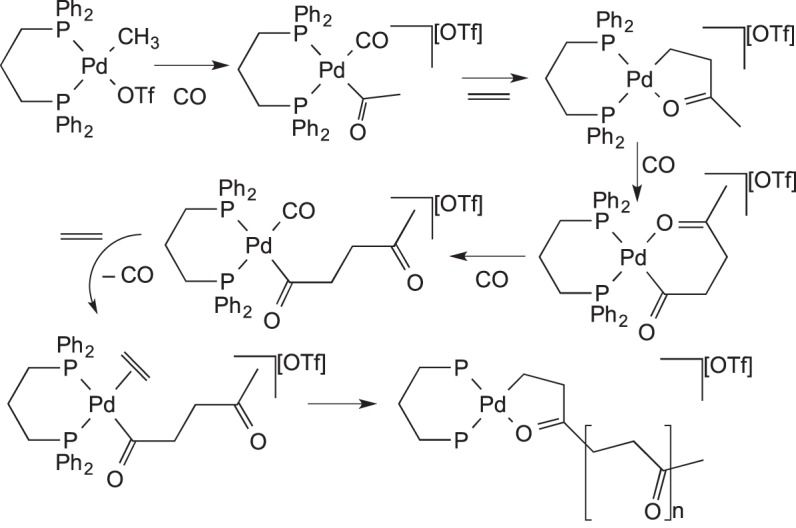
The proposed mechanism for the copolymerization of CO and ethene using a palladium catalyst in the solid state.
Dorta et al. [113] formed a variety of chiral crystalline organometallic complexes with either chiral phosphine ligands or chiral counterions, with the idea to investigate asymmetric catalysis. Unfortunately, this was not achieved, in this instance, and this interesting area has yet to be successfully developed.
7. Conclusion and future outlook
We hope that this review has shown that solid-state organometallic chemistry can offer significant advantages over the solution phase, especially when kinetic stability, or overall reaction selectivity, are different in the solid state compared with solution. Particularly exciting is the opportunity presented for SC−SC transformations in the solid state, as this methodology not only provides synthetic routes to new complexes but also enables direct structural analysis by single-crystal X-ray diffraction. However, for such transformations to proceed there is a requirement for minimal structural reorganization, and the appropriate design of systems that allow for this (e.g. large anions or bulky ligand groups) thus needs to be considered. An exciting prospect exists, which has not been developed to a significant extent, for such transformations to also result in catalytic processes. One example we suggest here is the selective transformation of hydrocarbons (alkane upgrading), where both solid–gas reactivity and selectivity from the local spatially well-defined environment will be important in producing effective and efficient catalysis. It will be fascinating to see how the field evolves as more examples of solid-state organometallic transformations are reported.
Acknowledgements
The EPSRC for funding SDP (EP/L505031/1).
References
- 1.Hartwig JF. 2010. Organotransition metal chemistry. Sausalito, CA: University Science Books [Google Scholar]
- 2.Crabtree RH. 2005. The organometallic chemistry of the transition metals Hoboken, NJ: John Wiley & Sons Inc; 4th edn [Google Scholar]
- 3.Bowker M. 1998. Basis and applications of heterogeneous catalysis. New York, NY: OUP [Google Scholar]
- 4.Smart LE, Moore EA. 2005. Solid state chemistry: an introduction New York, NY: CRC Press; 3rd edn [Google Scholar]
- 5.Thomas JM, Thomas WJ. 2005. Principles and practice of heterogeneous catalysis Weinheim, Germany: VCH Publishers; 3rd edn [Google Scholar]
- 6.Karunadasa HI, Montalvo E, Sun YJ, Majda M, Long JR, Chang CJ. 2012. A molecular MoS$_2$ edge site mimic for catalytic hydrogen generation. Science 335, 698–702. ( 10.1126/science.1215868) [DOI] [PubMed] [Google Scholar]
- 7.Halpern J. 1982. Mechansim and stereoselectivity of asymmetric hydrogenation. Science 217, 401–407. ( 10.1126/science.217.4558.401) [DOI] [PubMed] [Google Scholar]
- 8.Gonzalez-Rodriguez C, Pawley RJ, Chaplin AB, Thompson AL, Weller AS, Willis MC. 2011. Rhodium-catalyzed branched-selective alkyne hydroacylation: a ligand-controlled regioselectivity switch. Angew. Chem. Int. Ed. 50, 5134–5138. ( 10.1002/anie.201100956) [DOI] [PubMed] [Google Scholar]
- 9.Brookhart M, Green MLH, Parkin G. 2007. Agostic interactions in transition metal compounds. Proc. Natl Acad. Sci. USA 104, 6908–6914. ( 10.1073/pnas.0610747104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strauss SH. 1993. The search for larger and more weakly coordinating anions. Chem. Rev. 93, 927–942. ( 10.1021/cr00019a005) [DOI] [Google Scholar]
- 11.Verat AY, Pink M, Fan H, Tomaszewski J, Caulton KG. 2008. [(tBu2PCH2SiMe2)2N]Rh(I)?: Rapidly reversible H-C(sp3) and H-C(sp2) bond cleavage by rhodium(I). Organometallics 27, 166–168. ( 10.1021/om701165n) [DOI] [Google Scholar]
- 12.Coville NJ, Cheng L. 1998. Organometallic chemistry in the solid state. J. Organometal. Chem. 571, 149–169. ( 10.1016/s0022-328x(98)00914-0) [DOI] [Google Scholar]
- 13.Huang Z, White PS, Brookhart M. 2010. Ligand exchanges and selective catalytic hydrogenation in molecular single crystals. Nature 465, 598–601. ( 10.1038/nature09085) [DOI] [PubMed] [Google Scholar]
- 14.Bianchini C, Farnetti E, Graziani M, Kaspar J, Vizza F. 1993. Molecular solid–gas organometallic chemistry. Catalytic and stoichiometric transformations of ethyne at iridium. J. Am. Chem. Soc. 115, 1753–1759. ( 10.1021/ja00058a021) [DOI] [Google Scholar]
- 15.Jones C. 2010. On the stability and recyclability of supported metal–ligand complex catalysts: myths, misconceptions and critical research needs. Top. Catal. 53, 942–952. ( 10.1007/s11244-010-9513-9) [DOI] [Google Scholar]
- 16.Madhavan N, Jones CW, Weck M. 2008. Rational approach to polymer-supported catalysts: synergy between catalytic reaction mechanism and polymer design. Acc. Chem. Res. 41, 1153–1165. ( 10.1021/ar800081y) [DOI] [PubMed] [Google Scholar]
- 17.Tsoukala A, Peeva L, Livingston AG, Bjørsvik H-R. 2011. Separation of reaction product and palladium catalyst after a Heck coupling reaction by means of organic solvent nanofiltration. Chem. Sus. Chem. 5, 188–193. ( 10.1002/cssc.201100355) [DOI] [PubMed] [Google Scholar]
- 18.Hagen J. 1999. Industrial catalysis: a practical approach. Weinheim, Germany: Wiley-VCH [Google Scholar]
- 19.Coville NJ, Levendis DC. 2002. Organometallic chemistry: structural isomerization reactions in confined environments. Eur. J. Inorg. Chem. 2002, 3067–3078. () [DOI] [Google Scholar]
- 20.Hernández JG, Macdonald NAJ, Mottillo C, Frišèiæ T. Butler IS. 2014. A mechanochemical strategy for oxidative addition: remarkable yields and stereoselectivity in the halogenation of organometallic Re(I) complexes. Green Chem. 16, 1087–1092. ( 10.1039/c3gc42104j) [DOI] [Google Scholar]
- 21.Egbert JD, Slawin AMZ, Nolan SP. 2013. Synthesis of N-heterocyclic carbene gold complexes using solution-phase and solid-state protocols. Organometallics 32, 2271–2274. ( 10.1021/om301187a) [DOI] [Google Scholar]
- 22.Lewínski J, Dutkiewicz M, Lesiuk M, Sliwínski W, Zelga K, Justyniak I, Lipkowski J. 2010. Solid-state conversion of the solvated dimer [{tBuZn(μ–OtBu)(thf)}2] into a long overlooked trimeric [{tBuZnOtBu}3] species. Angew. Chem. Int. Ed. 49, 8266–8269. ( 10.1002/anie.201004504) [DOI] [PubMed] [Google Scholar]
- 23.Braga D, Giaffreda SL, Grepioni F, Pettersen A, Maini L, Curzi M, Polito M. 2006. Mechanochemical preparation of molecular and supramolecular organometallic materials and coordination networks. Dalton Trans. 1249–1263. ( 10.1039/B516165G) [DOI] [PubMed] [Google Scholar]
- 24.Furukawa H, Cordova KE, O’Keeffe M, Yaghi OM. 2013. The chemistry and applications of metal-organic frameworks. Science 341, 1230444( 10.1126/science.1230444) [DOI] [PubMed] [Google Scholar]
- 25.Liu J, Chen L, Cui H, Zhang J, Zhang L, Su C-Y. 2014. Applications of metal-organic frameworks in heterogeneous supramolecular catalysis. Chem. Soc. Rev. 43, 6011–6061. ( 10.1039/C4CS00094C) [DOI] [PubMed] [Google Scholar]
- 26.Hatcher LE, Raithby PR. 2013. Solid-state photochemistry of molecular photo-switchable species: the role of photocrystallographic techniques. Acta Crystallogr. C 69, 1448–1456. ( 10.1107/S010827011303223X) [DOI] [PubMed] [Google Scholar]
- 27.Coppens P. 2009. The new photocrystallography. Angew. Chem. Int. Ed. 48, 4280–4281. ( 10.1002/anie.200900910) [DOI] [PubMed] [Google Scholar]
- 28.Inokuma Y, Kawano M, Fujita M. 2011. Crystalline molecular flasks. Nat. Chem. 3, 349–358. ( 10.1038/nchem.1031) [DOI] [PubMed] [Google Scholar]
- 29.Horiuchi S, Murase T, Fujita M. 2012. A remarkable organometallic transformation on a cage-incarcerated dinuclear ruthenium complex. Angew. Chem. Int. Ed. 51, 12029–12031. ( 10.1002/anie.201206325) [DOI] [PubMed] [Google Scholar]
- 30.Kawano M, Kobayashi Y, Ozeki T, Fujita M. 2006. Direct crystallographic observation of a coordinatively unsaturated transition-metal complex in situ generated within a self-assembled cage. J. Am. Chem. Soc. 128, 6558–6559. ( 10.1021/ja0609250) [DOI] [PubMed] [Google Scholar]
- 31.Bala MD, Coville NJ. 2007. Organometallic chemistry in the melt phase. J. Organometal. Chem. 692, 709–730. ( 10.1016/j.jorganchem.2006.10.046) [DOI] [Google Scholar]
- 32.van der Boom ME. 2011. Consecutive molecular crystalline-state reactions with metal complexes. Angew. Chem. Int. Ed. 50, 11846–11848. ( 10.1002/anie.201105940) [DOI] [PubMed] [Google Scholar]
- 33.Bezzu CG, Helliwell M, Warren JE, Allan DR, McKeown NB. 2010. Heme-like coordination chemistry within nanoporous molecular crystals. Science 327, 1627–1630. ( 10.1126/science.1184228) [DOI] [PubMed] [Google Scholar]
- 34.Cohen MD, Schmidt GMJ. 1964. 383. Topochemistry. Part I. A survey. J. Chem. Soc. 1996–2000. ( 10.1039/jr9640001996) [DOI] [Google Scholar]
- 35.Braga D. 1992. Dynamical processes in crystalline organometallic complexes. Chem. Rev. 92, 633–665. ( 10.1021/cr00012a007) [DOI] [Google Scholar]
- 36.Sakamoto M. 1997. Absolute asymmetric synthesis from achiral molecules in the chiral crystalline environment. Chem. Eur. J. 3, 684–689. ( 10.1002/chem.19970030506) [DOI] [Google Scholar]
- 37.Kaupp G. 1996. Comprehensive supramolecular chemistry (eds Atwood JL, Davies JED, MacNicol DD, Votgle F.). New York, NY: Pergamon. [Google Scholar]
- 38.Oliván M, Marchenko AV, Coalter JN, Caulton KG. 1997. Gas/solid reactivity of unsaturated ruthenium-containing molecular solids. J. Am. Chem. Soc. 119, 8389–8390. ( 10.1021/ja971608j) [DOI] [Google Scholar]
- 39.Vaska L. 1966. Stereospecific addition of hydrogen halides to tetragonal d8 complexes. J. Am. Chem. Soc. 88, 5325–5327. ( 10.1021/ja00974a055) [DOI] [Google Scholar]
- 40.Cook PM, Dahl LF, Hopgood D, Jenkins RA. 1973. Oxidative-addition reaction of platinum acetylacetonate with iodine in solid state and solution. Crystal structure and equilibrium studies of trans-bis(acetylacetonato)di-iodoplatinum(IV). J. Chem. Soc. Dalton Trans. 294–301. ( 10.1039/dt9730000294) [DOI] [Google Scholar]
- 41.Vitorica-Yrezabal IJ, Sullivan RA, Purver SL, Curfs C, Tang CC, Brammer L. 2011. Synthesis and polymorphism of (4-ClpyH)2[CuCl4]: solid–gas and solid–solid reactions. CrystEngComm 13, 3189–3196. ( 10.1039/c0ce00628a) [DOI] [Google Scholar]
- 42.Minguez Espallargas G, Florence AJ, van de Streek J, Brammer L. 2011. Different structural destinations: comparing reactions of [CuBr2(3-Brpy)2] crystals with HBr and HCl gas. CrystEngComm 13, 4400–4404. ( 10.1039/c1ce05222e) [DOI] [Google Scholar]
- 43.Mínguez Espallargas G, van de Streek J, Fernandes P, Florence AJ, Brunelli M, Shankland K, Brammer L. 2010. Mechanistic insights into a gas–solid reaction in molecular crystals: the role of hydrogen bonding. Angew. Chem. Int. Ed. 49, 8892–8896. ( 10.1002/anie.201003265) [DOI] [PubMed] [Google Scholar]
- 44.Bianchini C, Peruzzini M, Zanobini F. 1991. Solid-state organometallic chemistry of tripodal (polyphosphine)metal complexes. Carbon-hydrogen activation reactions at cobalt(I) encapsulated into the tetraphosphine P(CH2CH2PPh2)3. Organometallics 10, 3415–3417. ( 10.1021/om00056a002) [DOI] [Google Scholar]
- 45.Werner H, Rappert T, Baum M, Stark A. 1993. Metallorganische chemie in fester phase: umlagerungs-, eliminierungs-und additionsreaktionen von organometallverbindungen ohne solvens. J. Organometal. Chem. 459, 319–323. ( 10.1016/0022-328X(93)86085-V) [DOI] [Google Scholar]
- 46.Jeffrey JC, Rauchfuss TB. 1979. Metal complexes of hemilabile ligands. Reactivity and structure of dichlorobis(o-(diphenylphosphino)anisole)ruthenium(II). Inorg. Chem. 18, 2658–2666. ( 10.1021/ic50200a004) [DOI] [Google Scholar]
- 47.Braunstein P, Naud F. 2001. Hemilability of hybrid ligands and the coordination chemistry of oxazoline-based systems. Angew. Chem. Int. Ed. 40, 680–699. () [DOI] [PubMed] [Google Scholar]
- 48.Osakada K, Ishii H. 2004. Solid–gas carbonylation of aryloxide rhodium(I) complexes: stepwise reaction forming Vaska-type complexes. Inorg. Chim. Acta 357, 3007–3013. ( 10.1016/j.ica.2004.02.030) [DOI] [Google Scholar]
- 49.Chin CS, Lee B, Kim S. 1993. Solid-state reactions of iridium (I)-1,5-cyclooctadiene compounds with carbon monoxide: synthesis of cationic (1,5-cyclooctadiene) carbonyliridium (I) complexes. Organometallics 12, 1462–1466. ( 10.1021/om00028a077) [DOI] [Google Scholar]
- 50.Gaviglio C, Ben-David Y, Shimon LJW, Doctorovich F, Milstein D. 2009. Synthesis, structure, and reactivity of nitrosyl pincer-type rhodium complexes. Organometallics 28, 1917–1926. ( 10.1021/om8011536) [DOI] [Google Scholar]
- 51.Flynn BR, Vaska L. 1974. Reversible addition of carbon dioxide to rhodium and iridium complexes. J. Chem. Soc. Chem. Commun. 703–704. ( 10.1039/c39740000703) [DOI] [Google Scholar]
- 52.Truscott BJ, Nelson DJ, Slawin AMZ, Nolan SP. 2014. CO2 fixation employing an iridium(I)-hydroxide complex. Chem. Commun. 50, 286–288. ( 10.1039/C3CC46922K) [DOI] [PubMed] [Google Scholar]
- 53.Durr S, Zarzycki B, Ertler D, Ivanovic-Burmazovic I, Radius U. 2012. Aerobic CO oxidation of a metal-bound carbonyl in a NHC-stabilized cobalt half-sandwich complex. Organometallics 31, 1730–1742. ( 10.1021/om201037w) [DOI] [Google Scholar]
- 54.Bianchini C, Frediani P, Graziani M, Kaspar J, Meli A, Peruzzini M, Vizza F. 1993. Molecular solid–gas organometallic chemistry. Catalytic and stoichiometric transformations of ethyne at iridium. Organometallics 12, 2886–2887. ( 10.1021/om00032a006) [DOI] [Google Scholar]
- 55.Bianchini C, Graziani M, Kaspar J, Meli A, Vizza F. 1994. Molecular solid–gas organometallic chemistry. Catalytic and stoichiometric iridium-assisted C–C bond-forming reactions involving ethyne and ethene. Organometallics 13, 1165–1173. ( 10.1021/om00016a020) [DOI] [Google Scholar]
- 56.Siedle AR, Newmark RA. 1989. Solid-state chemistry of molecular metal oxide clusters. Reactions of microporous dihydridobis(triphenylphosphine)iridium-tungsten complex [(Ph3P)2IrH2]3PW12O40 with small organic molecules. Organometallics 8, 1442–1450. ( 10.1021/om00108a012) [DOI] [Google Scholar]
- 57.Nicasio MC, et al. 1999. Substitution and hydrogenation reactions on rhodium(I)-ethylene complexes of the hydrotris(pyrazolyl)borate ligands Tp’ (Tp’=Tp, TpMe2). Inorg. Chem. 39, 180–188. ( 10.1021/ic990419u) [DOI] [PubMed] [Google Scholar]
- 58.Zenkina O, Altman M, Leitus G, Shimon LJW, Cohen R, van der Boom ME. 2007. From azobenzene coordination to aryl-halide bond activation by platinum. Organometallics 26, 4528–4534. ( 10.1021/om700519v) [DOI] [Google Scholar]
- 59.Pike SD, Weller AS. 2013. C-Cl activation of the weakly coordinating anion [B(3,5-Cl2C6H3)4]- at a Rh(I) centre in solution and the solid-state. Dalton Trans. 42, 12832–12835. ( 10.1039/c3dt51617b) [DOI] [PubMed] [Google Scholar]
- 60.de los Rios I, Bustelo E, Puerta MC, Valerga P. 2010. Isomerization of internal alkynones to vinylidenes in tris(pyrazolyl)borate ruthenium complexes. Solution and solid-state kinetics. Organometallics 29, 1740–1749. ( 10.1021/om100084x) [DOI] [Google Scholar]
- 61.Douglas TM, Weller AS. 2008. Acceptorless, intramolecular, alkyl dehydrogenation in the solid-state in a rhodium phosphine complex; reversible uptake of three equivalents of H2 per molecule. N. J. Chem. 32, 966–969. ( 10.1039/B718615K) [DOI] [Google Scholar]
- 62.Mediati M, Tachibana GN, Jensen CM. 1992. Solid-state and solution dynamics of the reversible loss of hydrogen from the iridium nonclassical polyhydride complexes IrClH2(PR3)2(H2) (R= iso-Pr, Cy, tert-Bu). Inorg. Chem. 31, 1827–1832. ( 10.1021/ic00036a020) [DOI] [Google Scholar]
- 63.Arif AM, Heaton DE, Jones RA, Kidd KB, Wright TC, Whittlesey BR, Atwood JL, Hunter WE, Zhang H. 1987. Synthesis and structures of di- and trinuclear di-tert-butylphosphido and di-tert-butylarsenido complexes of iridium. X-ray crystal structures of [Ir(μ-tert-Bu2E)(CO)2]2 (E=P, As), [Ir(tert-Bu2PH)(CO)]2(μ-H)(μ-tert-Bu2P), [Ir(tert-Bu2PH)(CO)(μ-H)]2(H)(μ-tert-Bu2P), and Ir3(μ-tert-Bu2P)3(CO)5. Inorg. Chem. 26, 4065–4073. ( 10.1021/ic00271a020) [DOI] [Google Scholar]
- 64.Morris RH, Foley HC, Targos TS, Geoffroy GL. 1981. Photoinduced elimination of hydrogen from [Pt2H3(dppm)2]PF6 and [Pt2H2Cl(dppm)2]PF6. J. Am. Chem. Soc. 103, 7337–7339. ( 10.1021/ja00414a050) [DOI] [Google Scholar]
- 65.Brayshaw SK, Ingleson MJ, Green JC, McIndoe JS, Raithby PR, Kociok-Köhn G, Weller AS. 2006. High hydride count rhodium octahedra, [Rh6(PR3)6H12][BArF4]2: synthesis, structures, and reversible hydrogen uptake under mild conditions. J. Am. Chem. Soc. 128, 6247–6263. ( 10.1021/ja0604663) [DOI] [PubMed] [Google Scholar]
- 66.Goodfellow RJ, Hamon EM, Howard JA, Spencer JL, Turner DG. 1984. Cationic platinum hydride clusters: X-ray crystal structures of [Pt4H2(PBut3)4][BF4]2 and [Pt4H7(PBut3)4][BPh4]. J. Chem. Soc. Chem. Commun. 1604–1606. ( 10.1039/C39840001604) [DOI] [Google Scholar]
- 67.Brayshaw SK, Ingleson MJ, Green JC, Raithby PR, Kociok-Köhn G, McIndoe JS, Weller AS. 2005. Holding onto lots of hydrogen: a 12-hydride rhodium cluster that reversibly adds two molecules of H2. Angew. Chem. Int. Ed. 44, 6875–6878. ( 10.1002/anie.200502221) [DOI] [PubMed] [Google Scholar]
- 68.Aime S, Dastru W, Gobetto R, Krause J, Sappa E. 1995. 14, 3( 10.1021/om00007a024) [DOI] [Google Scholar]
- 69.Gemel C, Huffman JC, Caulton KG, Mauthner K, Kirchner K. 2000. Solution and solid–gas reactivity of unsaturated [RuCp(tmeda)] + (tmeda = Me2NC2H4NMe2). J. Organometal. Chem. 593, 342–353. ( 10.1016/S0022-328X(99)00545-8) [DOI] [Google Scholar]
- 70.Zenkina OV, Keske EC, Wang R, Crudden CM. 2011. Double single-crystal-to-single-crystal transformation and small-molecule activation in rhodium NHC complexes. Angew. Chem. Int. Ed. 50, 8100–8104. ( 10.1002/anie.201103316) [DOI] [PubMed] [Google Scholar]
- 71.Albrecht M, Lutz M, Spek AL, van Koten G. 2000. Organoplatinum crystals for gas-triggered switches. Nature 406, 970–974. ( 10.1038/35023107) [DOI] [PubMed] [Google Scholar]
- 72.Albrecht M, Lutz M, Schreurs AMM, Lutz ETH, Spek AL, van Koten G. 2000. Self-assembled organoplatinum(II) supermolecules as crystalline, SO2 gas-triggered switches. J. Chem. Soc. Dalton Trans. 3797–3804. ( 10.1039/b006419j) [DOI] [Google Scholar]
- 73.Libri S, et al. 2008. Ligand substitution within nonporous crystals of a coordination polymer: elimination from and insertion into Ag–O bonds by alcohol molecules in a solid–vapor reaction. Angew. Chem. Int. Ed. 47, 1693–1697. ( 10.1002/anie.200703194) [DOI] [PubMed] [Google Scholar]
- 74.Vitórica-Yrezábal IJ, Mínguez Espallargas G, Soleimannejad J, Florence AJ, Fletcher AJ, Brammer L. 2013. Chemical transformations of a crystalline coordination polymer: a multi-stage solid–vapour reaction manifold. Chem. Sci. 4, 696( 10.1039/c2sc21654j) [DOI] [Google Scholar]
- 75.Supriya S, Das SK. 2007. Reversible single crystal to single crystal transformation through Fe-O(H)Me/Fe-OH2 bond formation/bond breaking in a gas-solid reaction at an ambient condition. J. Am. Chem. Soc. 129, 3464–3465. ( 10.1021/ja067572p) [DOI] [PubMed] [Google Scholar]
- 76.Uehara K, Mizuno N. 2011. Heterolytic dissociation of water demonstrated by crystal-to-crystal core interconversion from (μ -oxo)divanadium to bis(μ -hydroxo)divanadium substituted polyoxometalates. J. Am. Chem. Soc. 133, 1622–1625. ( 10.1021/ja108245g) [DOI] [PubMed] [Google Scholar]
- 77.Lim SH, Olmstead MM, Balch AL. 2013. Inorganic topochemistry. Vapor-induced solid state transformations of luminescent, three-coordinate gold(I) complexes. Chem. Sci. 4, 311–318. ( 10.1039/c2sc20820b) [DOI] [Google Scholar]
- 78.Miller EJ, Brill TB, Rheingold AL, Fultz WC. 1983. A reversible chemical reaction in a single crystal. The dimerization of η5-cyclopentadienyl(o-dithiobenzene)cobalt [CpCo(S2C6H4-o)]. J. Am. Chem. Soc. 105, 7580–7584. ( 10.1021/ja00364a020) [DOI] [Google Scholar]
- 79.Chaplin AB, Green JC, Weller AS. 2011. C-C activation in the solid state in an organometallic σ -complex. J. Am. Chem. Soc. 133, 13162–13168. ( 10.1021/ja2047599) [DOI] [PubMed] [Google Scholar]
- 80.Ozerov OV, Guo C, Papkov VA, Foxman BM. 2004. Facile oxidative addition of N-C and N-H bonds to monovalent rhodium and iridium. J. Am. Chem. Soc. 126, 4792–4793. ( 10.1021/ja049659l) [DOI] [PubMed] [Google Scholar]
- 81.Weng W, Guo C, Moura C, Yang L, Foxman BM, Ozerov OV. 2005. Competitive activation of N-C and C-H bonds of the PNP framework by monovalent rhodium and iridium. Organometallics 24, 3487–3499. ( 10.1021/om050346o) [DOI] [Google Scholar]
- 82.Xu N, Goodrich LE, Lehnert N, Powell DR, Richter-Addo GB. 2013. Preparation of the elusive [(por)Fe(NO)(O-ligand)] complex by diffusion of nitric oxide into a crystal of the precursor. Angew. Chem. Int. Ed. 52, 3896–3900. ( 10.1002/anie.201208063) [DOI] [PubMed] [Google Scholar]
- 83.Xu N, Powell DR, Richter-Addo GB. 2011. Nitrosylation in a crystal: remarkable movements of iron porphyrins upon binding of nitric oxide. Angew. Chem. Int. Ed. 50, 9694–9696. ( 10.1002/anie.201103329) [DOI] [PubMed] [Google Scholar]
- 84.Xu N, Powell DR, Cheng L, Richter-Addo GB. 2006. The first structurally characterized nitrosyl heme thiolate model complex. Chem. Commun. 2030–2032. ( 10.1039/b602611g) [DOI] [PubMed] [Google Scholar]
- 85.Pike SD, Thompson AL, Algarra ASG, Apperley DC, Macgregor SA, Weller AS. 2012. Synthesis and characterization of a rhodium(I) σ -alkane complex in the solid state. Science 337, 1648–1651. ( 10.1126/science.1225028) [DOI] [PubMed] [Google Scholar]
- 86.Evans DR, Drovetskaya T, Bau R, Reed CA, Boyd PDW. 1997. Heptane coordination to an iron(II) porphyrin. J. Am. Chem. Soc. 119, 3633–3634. ( 10.1021/ja970008h) [DOI] [Google Scholar]
- 87.Castro-Rodriguez I, Nakai H, Gantzel P, Zakharov LN, Rheingold AL, Meyer K. 2003. Evidence for alkane coordination to an electron-rich uranium center. J. Am. Chem. Soc. 125, 15734–15735. ( 10.1021/ja0379316) [DOI] [PubMed] [Google Scholar]
- 88.Basset JM, Lefebvre F, Santini C. 1998. Surface organometallic chemistry: some fundamental features including the coordination effects of the support. Coord. Chem. Rev. 178, 1703–1723. ( 10.1016/s0010-8545(98)00159-3) [DOI] [Google Scholar]
- 89.Copéret C, Basset JM. 2007. Strategies to immobilize well-defined olefin metathesis catalysts: supported homogeneous catalysis vs. surface organometallic chemistry. Adv. Syn. Catal. 349, 78–92. ( 10.1002/adsc.200600443) [DOI] [Google Scholar]
- 90.Staub H, Guillet-Nicolas R, Even N, Kayser L, Kleitz F, Fontaine F-G. 2011. Substantiating the influence of pore surface functionalities on the stability of Grubbs catalyst in mesoporous SBA-15 silica. Chem. Eur. J. 17, 4254–4265. ( 10.1002/chem.201002740) [DOI] [PubMed] [Google Scholar]
- 91.Serna P, Gates BC. 2011. A bifunctional mechanism for ethene dimerization: catalysis by rhodium complexes on zeolite HY in the absence of halides. Angew. Chem. Int. Ed. 50, 5528–5531. ( 10.1002/anie.201008086) [DOI] [PubMed] [Google Scholar]
- 92.Bhirud VA, Ehresmann JO, Kletnieks PW, Haw JF, Gates BC. 2005. Rhodium complex with ethylene ligands supported on highly dehydroxylated MgO: synthesis, characterization, and reactivity. Langmuir 22, 490–496. ( 10.1021/la052268f) [DOI] [PubMed] [Google Scholar]
- 93.Gajan D, Coperet C. 2011. Silica-supported single-site catalysts: to be or not to be? A conjecture on silica surfaces. N. J. Chem. 35, 2403–2408. ( 10.1039/c1nj20506d) [DOI] [Google Scholar]
- 94.Samantaray MK, et al. 2013. Evidence for metal-surface interactions and their role in stabilizing well-defined immobilized Ru-NHC alkene metathesis catalysts. J. Am. Chem. Soc. 135, 3193–3199. ( 10.1021/ja311722k) [DOI] [PubMed] [Google Scholar]
- 95.Thomas JM, Maschmeyer T, Johnson BFG, Shephard DS. 1999. Constrained chiral catalysts. J. Mol. Catal. A, Chem. 141, 139–144. ( 10.1016/S1381-1169(98)00257-X) [DOI] [Google Scholar]
- 96.Thomas JM, Raja R. 2008. Exploiting nanospace for asymmetric catalysis: confinement of immobilized, single-site chiral catalysts enhances enantioselectivity. Acc. Chem. Res. 41, 708–720. ( 10.1021/ar700217y) [DOI] [PubMed] [Google Scholar]
- 97.Hu A, Ngo HL, Lin W. 2003. Chiral, porous, hybrid solids for highly enantioselective heterogeneous asymmetric hydrogenation of β-keto esters. Angew. Chem. Int. Ed. 42, 6000–6003. ( 10.1002/anie.200351264) [DOI] [PubMed] [Google Scholar]
- 98.Hu A, Ngo HL, Lin W. 2003. Chiral porous hybrid solids for practical heterogeneous asymmetric hydrogenation of aromatic ketones. J. Am. Chem. Soc. 125, 11490–11491. ( 10.1021/ja0348344) [DOI] [PubMed] [Google Scholar]
- 99.Genna DT, Wong-Foy AG, Matzger AJ, Sanford MS. 2013. Heterogenization of homogeneous catalysts in metal-organic frameworks via cation exchange. J. Am. Chem. Soc. 135, 10586–10589. ( 10.1021/ja402577s) [DOI] [PubMed] [Google Scholar]
- 100.Wang Z, Chen G, Ding K. 2008. Self-supported catalysts. Chem. Rev. 109, 322–359. ( 10.1021/cr800406u) [DOI] [PubMed] [Google Scholar]
- 101.Efraty A, Feinstein I. 1982. Catalytic hydrogenation and isomerization of 1-hexene with [RhCl(CO)(1,4-(CN)2C6H4)]n in the dark and under irradiation. Inorg. Chem. 21, 3115–3118. ( 10.1021/ic00138a038) [DOI] [Google Scholar]
- 102.Feinstein-Jaffe I, Efraty A. 1987. Heterogeneous catalysis with coordination polymers: hydrogenation and isomerization of 1-hexene in the presence of [Rh(diisocyano-biphenyl)2Cl]n. J. Mol. Catal. 40, 1–7. ( 10.1016/0304-5102(87)80001-9) [DOI] [Google Scholar]
- 103.Lawrence SA, Sermon PA, Feinstein-Jaffe I. 1989. An investigation into the role of cooperative effects in polymeric rhodium(I) bis-4,4′-diisocyanobiphenyl chloride catalysts in olefin hydrogenation reactions. J. Mol. Catal. 51, 117–127. ( 10.1016/0304-5102(89)80092-6) [DOI] [Google Scholar]
- 104.Liu Q-P, Chen Y-C, Wu Y, Zhu J, Deng J-G. 2006. Self-supported heterogeneous star-shaped oxime-palladacycle catalysts for the Suzuki coupling reactions. Synlett 2006, 1503–1506. ( 10.1055/s-2006-941585) [DOI] [Google Scholar]
- 105.Chen W, Li R, Wu Y, Ding L-S, Chen Y-C. 2006. Self-supported thiourea-palladium complexes: highly air-stable and recyclable catalysts for the Suzuki reaction in neat water. Synthesis 2006, 3058–3062. ( 10.1055/s-2006-942531) [DOI] [Google Scholar]
- 106.Yamada YMA, Maeda Y, Uozumi Y. 2006. Novel 3D coordination palladium network complex: a recyclable catalyst for Suzuki–Miyaura reaction. Org. Lett. 8, 4259–4262. ( 10.1021/ol0615026) [DOI] [PubMed] [Google Scholar]
- 107.Karimi B, Akhavan PF. 2009. Main-chain NHC-palladium polymer as a recyclable self-supported catalyst in the Suzuki–Miyaura coupling of aryl chlorides in water. Chem. Commun. 3750–3752. ( 10.1039/b902096a) [DOI] [PubMed] [Google Scholar]
- 108.Mori W, Sato T, Ohmura T, Nozaki Kato C, Takei T. 2005. Functional microporous materials of metal carboxylate: gas-occlusion properties and catalytic activities. J. Solid State Chem. 178, 2555–2573. ( 10.1016/j.jssc.2005.07.009) [DOI] [Google Scholar]
- 109.Sato T, Mori W, Kato CN, Yanaoka E, Kuribayashi T, Ohtera R, Shiraishi Y. 2005. Novel microporous rhodium(II) carboxylate polymer complexes containing metalloporphyrin: syntheses and catalytic performances in hydrogenation of olefins. J. Catal. 232, 186–198. ( 10.1016/j.jcat.2005.02.007) [DOI] [Google Scholar]
- 110.Stoeck U, Nickerl G, Burkhardt U, Senkovska I, Kaskel S. 2012. Modular construction of a porous organometallic network based on rhodium olefin complexation. J. Am. Chem. Soc. 134, 17335–17337. ( 10.1021/ja305482a) [DOI] [PubMed] [Google Scholar]
- 111.Matthes J, Pery T, Gründemann S, Buntkowsky G, Sabo-Etienne S, Chaudret B, Limbach H-H. 2004. Bridging the gap between homogeneous and heterogeneous catalysis: ortho/para H2 conversion, hydrogen isotope scrambling, and hydrogenation of olefins by Ir(CO)Cl(PPh3)2. J. Am. Chem. Soc. 126, 8366–8367. ( 10.1021/ja0475961) [DOI] [PubMed] [Google Scholar]
- 112.Mul WP, Oosterbeek H, Beitel GA, Kramer G-J, Drent E. 2000. In situ monitoring of a heterogeneous palladium-based polyketone catalyst. Angew. Chem. Int. Ed. 39, 1848–1851. () [DOI] [PubMed] [Google Scholar]
- 113.Dorta R, Shimon L, Milstein D. 2004. Rhodium complexes with chiral counterions: achiral catalysts in chiral matrices. J. Organometal. Chem. 689, 751–758. ( 10.1016/j.jorganchem.2003.12.012) [DOI] [Google Scholar]