

Abstract
Three receptor tyrosine kinases, Tyro3, Axl, and Mertk (TAM) and their ligands Gas6 and Protein S, have emerged as potent negative regulators of innate immune responses. A number of studies using genetic ablation of TAM loci in mice have elucidated the mechanism of TAM engagement and function during the immune response and removal of apoptotic cells. Following phagocytosis of apoptotic cells or the induction of T-cell dependent adaptive immune responses, ligand-induced TAM signaling dampens proinflammatory cytokine production and thus prevents exaggerated or prolonged inflammation. It is believed that the TAM pathway may play an important role in the pathogenesis of inflammatory bowel disease. Suppression of inflammation and removal of apoptotic cells followed by tissue repair are essential processes for disease remission and the successful management of inflammatory bowel disease. In light of the key role of TAMs in controlling inflammatory responses, here, we review the recent advances on TAM research vis-à-vis the resolution of intestinal inflammation. Targeted activation of TAM receptor tyrosine kinases may represent a potent therapeutic opportunity in inflammatory bowel disease.
Inflammatory bowel disease (IBD) refers to a group of chronic inflammatory disorders that affect mainly the gastrointestinal tract. The 2 main types of IBD are ulcerative colitis (UC) and Crohn’s disease (CD). These diseases seem to be more prevalent in the developed world including North America and Europe. Current estimates suggest that approximately 1.4 million people in the United States have CD or UC (www.cdc.gov/ibd/).
Aminosalicylates have traditionally been considered the first line therapy for IBD, although this concept is evolving, particularly in CD, because of their limited effectiveness in altering the natural history of the disease. Immunomodulator therapy (i.e., Azathioprine) and/or biologic therapy (i.e., infliximab) have been shown to impact health outcomes to a much greater degree, especially in patients with CD.1 However, even with the use of these medications, a significant fraction of patients are nonresponders or have an incomplete response. Newer therapies that target T-cell homing to the intestine such as vedolizumab2,3 will add to the armamentarium but still, newer therapeutic approaches are needed.
Therapeutic efforts have been hampered by the lack of a clear understanding about the pathogenesis of IBD and a realization that the causes are multifactorial. For example, genetic predispositions can be associated with IBD and unbiased approaches such as genome-wide association studies have identified certain IBD susceptibility loci.4 Similarly, environmental influences, including diet and commensal microbiota in the gut have been linked to IBD.5–9 Notwithstanding, the central theme in IBD is the loss of immune homeostasis resulting in chronic inflammation. Some of the IBD susceptibility genes identified by genome-wide association studies, e.g., IL-10, have important immunoregulatory roles.10,11 Commensal microbiota can also clearly shape the immune response (for review see Ref. 6). Therefore, identifying immunoregulatory pathways that maintain physiological mucosal immunity might provide a better understanding of the exact etiology of IBD.
In this review, we discuss the current understanding of an important group of immunoregulatory molecules—the receptor tyrosine kinases (RTKs) Axl and Mertk and their ligands growth-arrest-specific 6 (Gas6) and Protein S (Pros1). The primary mechanism of action of current, frontline IBD therapy centers on dampening the inflammatory immune response.12 These approaches are limited to either neutralization of individual colitogenic cytokines, such as anti-tumor necrosis factor alpha (TNFα) therapy or broad immunosuppression. In contrast, the Axl and Mertk signaling pathway plays a prominent role in the resolution of inflammation through the negative regulation of the innate immune response and the phagocytosis of apoptotic neutrophils. Therefore, an improved understanding of the multifunctional roles of Axl and Mertk in mucosal immunity may prove critical for designing more effective therapies for IBD.
TYRO3, AXL, AND MERTK RECEPTORS AND LIGANDS—STRUCTURAL FEATURES
Three receptors with tyrosine kinase activity form the TAM subgroup—Tyro3, Axl, and Mertk. Lai and Lemke13 initially identified these receptors by cloning fragments encoding their intracellular domains based on homology with tyrosine kinase domains and named them Tyro3, 7, and 12. Subsequently, full-length cDNA of these receptors were cloned in many laboratories. Full-length Axl was independently cloned by 3 groups in 1991. O’Bryan et al14 named the gene Axl—anexelekto, greek for unchecked. Janssen et al15 termed the gene UFO in allusion to the unidentified function of the gene at that time. Rescigno et al16 called it Ark for adhesion-related kinase. The viral and the cellular version of avian Mertk were cloned in 1992 and 1994, respectively, and named v-ryk and c-eyk.17,18 The human ortholog was cloned by Graham et al19 in 1994 and named for its presence in monocytes and in epithelial and reproductive tissues. Lai and Lemke classified these RTKs as a unique subgroup because of sequence identity. The original classification of these RTKs, performed by nothing more than sequence gazing, remarkably withstood the test of bioinformatics-based assembly of the kinome.20 To date, TAM receptors are most closely related to each other and have more distant homology to the macrophage-stimulating protein receptor RON (recepteur d’origine nantais)21 and the hepatocyte growth factor receptor MET (the 3 letter abbreviation suggested by the discoverers22).
The extracellular domain of these single-pass membrane-spanning receptors is composed of 2 immunoglobulin-like domains and 2 fibronectin type III-like domains (Fig. 1). The identity of the ligands that activate the TAM RTKs remained unknown till 1995. Through biochemical and cell-based assays, 2 closely related proteins—Gas6 (growth-arrest-specific 6) and Pros1 (Protein S, named after the city where it was discovered, Seattle23)—were identified as TAM agonists.24 Like the TAM receptors, their ligands also share structural homology. From N- to C-termini, Gas6 and Pros1 feature Gla domains followed by 4 Epidermal Growth Factor-like repeats and 2 tandem laminin G domains that are related to those of the sex hormone binding globulin. The Gas6 and Pros1 Gla domains are approximately 60 amino acid sequences rich in glutamic acid residues that are post-translationally γ–carboxylated in a vitamin K-dependent reaction, enabling these domains to bind the phospholipid phosphatidylserine (PtdSer).25–29 The sex hormone binding globulin–like module is both necessary and sufficient for binding and activating TAM receptors in vitro.30,31 Overall, the 2 TAM ligands share approximately 42% amino acid identity (Fig. 1).
FIGURE 1.
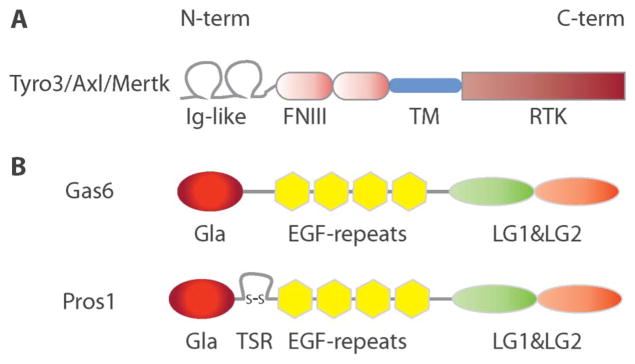
Schematic representation of TAM receptors and ligands protein structure. A, TAM receptors carry 2 immunoglobulin-like domains in their N-terminus, followed by 2 fibronectin type III repeats, a transmembrane region and a tyrosine kinase domain in the C-terminal intracellular region. Overall, TAM receptors share >70% identity in their tyrosine kinase domain. B, TAM agonists, Gas6 and Pros1, carry a GLA domain in their N-terminus, followed by 4 EGF repeats and 2 laminin G domains in their C-terminus. γ-carboxylation of glutamic acid residues in the GLA domain, enable Gas6 and Pros1 to bind to PtdSer. The 2 laminin G domains form a sex hormone binding globulin-like domain, i.e., sufficient to bind and trigger the activation of TAM receptors. Pros1 also carries a thrombin sensitive region. Overall, Gas6 and Pros1, share approximately 42% amino acid identity. EGF, epidermal growth factor.
GENETIC DISSECTION OF TAM FUNCTION
TAM RTKs were originally identified using a Schwann cell cDNA library, and their discovery was speculated to support a functional role of their tyrosine kinase activity in neural development.13 Surprisingly, even the simultaneous genetic deletion of all 3 TAM receptors resulted in viable, apparently normal mice.32 Although TAMs do not seem to have a major impact on embryonic development, adult TAM triple knockout mice develop a panoply of degenerative symptoms in their nervous and reproductive systems.32 For example, in the Royal College of Surgeons rat, a classical model of recessively inherited retinal degeneration, the retinal dystrophy locus was mapped to Mertk by positional cloning.33,34 Cultured Royal College of Surgeons retinal pigmental epithelial cells failed to phagocytose rod outer segments.35 Screening the MERTK locus in patients with retinopathies revealed mutations resulting in predicted loss or reduction in MERTK function.36 Additionally, the TAMs in Sertoli cells mediate the phagocytosis of apoptotic germ cells in the testis.37 Consistent with this observation, male TAM triple knockout mice exhibited defective spermatogenesis and were sterile.32 Recently, a role for Mertk in the phagocytosis and elimination of synapses by astrocytes was identified.38 This process leads to synaptic pruning and circuit refinement both during development and in adulthood. A similar TAM function is observed in the immune system. Glenn Matsushima’s laboratory identified the functional role of Mertk in the phagocytosis of apoptotic cells by macrophages—a professional phagocyte in the immune system.39 Shelton Earp’s laboratory went on to show that the failure to clear apoptotic cells associates with a lupus-like disease in Mertk knockout mice.40 Taken together, these results indicate that TAMs are necessary for the removal of apoptotic cells and membranes, and that the lack of TAMs can lead to degeneration of organ function. Hence, the TAMs have been termed homeostatic regulators—their function is mostly dispensable during development but essential in maintaining physiological organ function.
A major insight into the role of TAMs in autoimmune diseases came from the generation of the TAM triple knockout mice.32,41 At birth, the peripheral lymphoid organs of the TAM triple knockout mice are of normal size and weight. However, beginning at approximately 4 weeks after birth, these mice start to display dramatic splenomegaly and lymphadenopathy.41 Both B cells and T cells greatly increase in number and are activated. Furthermore, TAM triple knockouts are characterized by high circulating amounts of autoantibodies against dsDNA and phospholipids, and display clinical features of systemic autoimmunity.41
EVIDENCE FOR A DIRECT ROLE OF TAMs IN THE INHIBITION OF TOLL-LIKE RECEPTOR AND CYTOKINE RECEPTOR SIGNALING
Is autoimmunity in the absence of TAM function a consequence of the failure to clear apoptotic cells, or do TAMs mediate a more direct suppression of the immune response? Lymphocyte activation in TAM triple knockout mice was shown to be non-cell autonomous and due to the hyperactivation of antigen presenting cells (APCs).41 The TAM receptors are expressed in APCs including macrophages and dendritic cells (DC).42 Direct evidence of TAM function in the negative regulation of the innate immune response came from in vitro studies. TAM knockout DC hyper respond to a variety of Toll-like receptor (TLR) agonists producing high amounts of proinflammatory cytokines.43 This is in agreement with a previous observation made by Todd Camenisch et al.44 These authors demonstrated excessive TNFα production and septic shock in Mertk knockout mice after lipopolysaccharide (LPS) administration. Additionally, recombinant Gas6 and Pros1 potently suppressed the activation of DCs and consequent cytokine production triggered by engagement of TLR 3, 4, and 9.43
TAM function as a direct negative regulator of the innate immune response is supported by the following observations in vitro. First, Axl mRNA and protein expression was upregulated by type I interferons produced downstream of TLR activation (Fig. 2A). Type 1 interferons are potent inducers of DC maturation. Therefore, TAM signaling is engaged in APCs as a consequence of immune activation. Second, TAM engagement leads to the upregulation of pleiotropic inhibitors of innate immunity—suppressor of cytokine signaling 1 (Socs1) and Socs3 (Fig. 2B). Socs1 and Socs3 are E3 ubiquitin ligases that lead to the turnover of Toll-interleukin 1 domain-containing adaptor protein (TIRAP) and TNF receptor associated factor 6 (TRAF6), adaptor molecules that function in TLR and NF-κB signaling. Socs1 and Socs3 are also well known inhibitors of JAK-STAT signaling pathway. Importantly, the TAM-dependent upregulation of Socs genes required type I interferon receptor and also STAT1, the very same transcription factor that drives the initial proinflammatory response. This result suggests that components of type I interferon receptor-STAT signaling pathway are hijacked by TAMs to drive Socs upregulation. Third, the upregulation of the Socs genes downstream of type I interferons was contingent on TAMs. In TAM triple knockout DCs, Socs1 induction by interferon alpha was significantly reduced.43
FIGURE 2.

TAM receptors are potent inhibitors of the innate immune response. A, TAM receptors are induced downstream of cytokine receptor signal (i.e., type I interferon receptors) in a STAT dependent manner. B, Subsequently, activation of TAM receptors in conjunction with cytokine receptors (i.e., type I interferon receptors) leads to the induction of the SOCS genes and the suppression of both TLR and cytokine receptor signaling. In a similar fashion, (C) phagocytosis of apoptotic cells in a TAM receptor dependent manner potently inhibits the TLR signaling cascade.
Interestingly, the removal of apoptotic cells has also been associated with the suppression of inflammation during the resolution of the immune response. Roland Tisch’s laboratory identified Mertk as the mediator of the suppression of TLR signaling in DC in the presence of apoptotic cells.45 Incubation of DCs with apoptotic thymocytes inhibited the activation of NF-κB downstream of TLR and the production of TNFα (Fig. 2C). The precise contributions of the 2 aspects of TAM function—phagocytosis and inhibition of TLR/cytokine receptor signaling—in preventing autoimmunity remain to be fully understood in vivo. Raymond Birge’s laboratory has identified distinct tyrosine residues in Mertk that mediate phagocytosis versus TLR inhibition.46 Therefore, Mertk may actually integrate these 2 distinct biological functions for the maintenance of immune homeostasis.
ACTIVATION OF TAM SIGNALING AT THE INTERFACE OF THE INNATE AND ADAPTIVE IMMUNE RESPONSE
Activation of TAM receptors by their ligands Gas6 and Pros1 is a 2-step mechanism. Gas6/Pros1 needs to bind PtdSer, which is exposed on the outer leaflet of the plasma membrane during apoptosis.26–28 This binding is believed to induce a conformational change in Gas6 and Pros1 that enables its bioactivity necessary for activating the TAM receptors.47 Nevertheless, the engagement of TAM signaling is not limited to the removal of apoptotic cells. TAM engagement and its anti-inflammatory effect can also occur independent of apoptotic cells. We have recently discovered that TAM activation occurs at the interface of the innate and adaptive immune response.48 Activated APCs present antigen to T cells and provide the cytokine milieu appropriate for the activation and lineage-specific differentiation of T cells.49 Once, this adaptive immune arm is engaged, an antigen-specific response ensues. In contrast, the initial inflammatory response is broad and if persistent or exaggerated, can cause collateral damage.50,51 Therefore, a priori, the adaptive immune response, once activated, should be able to temper the innate system. Experimental evidence demonstrated that the TAM ligand Pros1 was expressed in activated, but not resting, T cells.48,52 Additionally, generation of mice in which Pros1 was specifically ablated in T cells revealed that T-cell derived Pros1 was able to suppress APC activation and cytokine production in an antigen-specific, TAM-dependent manner. T-cell–specific Pros1 knockout mice showed a general increased immune response on immunization.48
Remarkably, the requirement for PtdSer was conserved during T cell-derived Pros1 mediated engagement of TAMs on APCs. Activated T cells transiently express intermediate levels of PtdSer on their cell surface, in comparison with apoptotic cells.48,53 Blocking available PtdSer with excess Annexin V inhibited T-cell–mediated suppression of APC activation. In summary, activation of APCs lead to increased expression of TAM receptors. After APC-dependent engagement of the adaptive immune response, activated T cells produce Pros1 to engage these receptors on the APC. This mechanism leads to the inhibition of the innate immune response and the maintenance of immune homeostasis (Fig. 3).
FIGURE 3.

TAM signaling is activated at the interface of the innate and adaptive immune response. (1) TLR signaling triggers the activation of DCs and the induction of TAM receptors. (2) Activated DCs present antigen to T cells and induce the exposure of PtdSer and the expression of the TAM agonist Pros1 on activated T cells. (3) T cell-derived Pros1 activates the TAM receptors on DCs to limit the magnitude of the DC response.
IMMUNE HOMEOSTASIS AND IBD
The gut microenvironment provides a particularly challenging context for the maintenance of immune homeostasis. The gut is a home to more microorganisms than cells in our own body. An extensive mucosal immune system has evolved to protect against invading pathogens, yet coexist with commensal microbiota. Resident F4/80hiCX3CR1hi macrophages in the lamina propria are highly phagocytic and produce vast amounts of IL-10, contributing to the maintenance of intestinal homeostasis.54–56
Exposure to infectious agents, microabrasions, and localized disruption of the epithelial barrier allow microorganisms to come in contact with the mucosal immune system. The immune system has the task to efficiently control the invading micro-organisms. Neutrophils are the first responders to invading bacteria and are avid phagocytes.57 Following phagocytosis and killing of bacteria, neutrophils themselves die by apoptosis. Ly6Chi monocytes are also recruited to the site of injury and differentiate into CX3CR1int macrophages that secrete cytokines and contribute to the initial inflammatory response.58–60
After dealing with the threat of the pathogen, the immune system initiates the resolution of inflammation. Macrophages are endowed with the task of removal of apoptotic cells, including neutrophils. After clearance of apoptotic debris, a switch toward tissue repair occurs. This switch coincides with a transition from the production of proinflammatory to proresolution mediators. For example, phagocytosis of apoptotic neutrophils leads to the production of PGE2,61 and PGE2 induces the expression of 15-lipooxygenase and the production of lipoxins.62 Phagocytosis of apoptotic cells also induces the production of a panoply of tissue repair mediators including cytokines, such as TGFβ and IL-10, growth factors, such as vascular endothelial growth factor and enzymes that favor tissue remodeling, such as Arginase.61,63–65 This type of macrophage state has been termed “alternative activation.”66 Therefore, a state of “controlled inflammation” is essential for conferring protection in the gut but avoiding tissue damage.
In IBD, not only is there an excessive and prolonged inflammation characterized by over-production of proinflammatory cytokines, tissue repair is also compromised. CD, characterized by transmural inflammation, is often associated with ulcers and/or fistulas, as well as, intestinal fibrosis leading to stricture formation.67 Similarly, mouse models of colitis are characterized by enhanced production of inflammatory cytokines along with an increased neutrophil infiltration, accumulation of apoptotic neutrophils and excessive tissue damage. It is in this setting, that TAM signaling in IBD is of utmost importance.68 The function of the TAM pathway, including negative regulation of inflammation, removal of apoptotic cells and potential induction of the tissue repair response, suggests that it is an ideal candidate to mediate the resolution of the inflammatory response (Fig. 4).
FIGURE 4.

TAM signaling in the intestinal mucosa. A, Damage to the intestinal mucosa (e.g., in the context of bacterial infection) leads to neutrophil infiltration. Once the threat has been controlled, a tissue repair response ensues. Apoptotic neutrophils are cleared by intestinal macrophages in a TAM-dependent manner. This process associates with the switch of TAM expressing macrophages from a proinflammatory M1 (INOS and tumor necrosis factor alpha) profile to a tissue repair, alternative M2 state (IL-10, Relm-α and TGFβ). B, Damage to the intestinal mucosa, in the absence of TAM signaling, leads to an accumulation of apoptotic neutrophils and failure of intestinal macrophages to acquire an alternative activation state that associates with severe injury.
TAM SIGNALING IN IBD
Axl and Mertk expression have not been reported in mouse intestinal mucosa under physiological conditions. However, Axl and Mertk are readily detected in murine intestinal lamina propria macrophages on Dextran sodium sulfate (DSS)-induced inflammation.68 The expression of Mertk in myeloid cells is consistent with the original report describing the identification of Mertk, and its expression in human peripheral blood derived mononuclear cells, bone marrow mononuclear cells, and monocytes.19 Mertk expression has been reported in alveolar macrophages, where this RTK is functionally important for phagocytosis of apoptotic cells.69 More recently, large scale gene expression profiling of tissue resident macrophages including peritoneal macrophages, red pulp splenic macrophages, lung macrophages, and microglia identified Mertk as an universal marker of mature tissue resident macrophages.70
Axl is more broadly expressed in both hematopoietic and in nonhematopoietic cells. Murine and human DCs express significant levels of Axl.43,71 Human and murine wound macrophages respond to PGE2 by Axl phosphorylation and downstream induction of oncostatin M, a potent cytokine that mediates wound closure during the initial phase of wound healing and tissue repair.72 Nonhematopoietic cells such as endothelial and smooth muscle cells also express Axl.73,74 Axl expression is induced during neointima formation following carotid artery injury, a process important for tissue repair after vessel damage.74
Whether Axl has a similar repair function in the context of colitis-associated intestinal injury is not well understood. Notwithstanding, Axl−/−Mertk−/− mice exhibit an exaggerated response to DSS characterized by a more severe loss of body weight and signs of colitis in comparison with wild-type mice.68 Colonoscopy in Axl−/−Mertk−/− mice revealed increased granularity, loss of vasculature and translucency, and looser stool consistency. The increased severity of colitis was also confirmed by histopathological features such as ulcerations, crypt hyperplasia, crypt loss, leukocyte infiltration, and edema. Consistent with the dual function of TAM RTKs in phagocytosis of apoptotic cells and inhibition of innate immune signaling, Axl−/−Mertk−/− mice had increased load of apoptotic Ly6G+ neutrophils and increased interferon gamma and TNFα production.68 Axl−/−Mertk−/− lamina propria macrophages responded to the inflammatory trigger by producing increased amounts of proinflammatory mediators such as iNOS and TNFα, whereas they failed to produce adequate amounts of tissue repair factors such as Resistin-like molecule alpha (RELMα), IL-10, and TGFβ.68
It is interesting to note that MERTK is highly expressed in response to IL-10 in a subtype of alternatively activated human macrophages (M2c macrophages) and functions in the clearance of apoptotic cells.75 Nonetheless, unlike mice knockout for genes coding for molecules important in intestinal barrier function, such as the Muc2−/− mice that lack the goblet-cell-derived secretory mucine Muc2,76 Axl−/−Mertk−/− mice do not develop spontaneous colitis. This is consistent with the activation of the TAM pathway as a consequence of induced inflammation.
The TAM ligands have also been implicated in limiting colonic inflammation. Gas6−/− mice are more susceptible to DSS.77 DSS-treated Gas6−/− mice display reduced Socs1/3 gene expression and increased NF-κB activation in colon tissue. The cellular compartment producing Gas6 to engage the TAMs within the intestinal mucosa is not well defined. However, bone marrow-transplant approaches have suggested that both radioresistant and radiosensitive cells can be the source of Gas6 during induced-inflammatory responses in the gut.77
The other known TAM ligand, Pros1, also has an important function in the context of IBD. The T-cell specific ablation of Pros1 in mice caused enhanced colitis in a T-cell transfer model.48 IBD in humans is characterized by an abundance of colitogenic T cells.67 When T cells sans T regs are transferred into Rag−/− mice, these animals develop colitis.78,79 This is dependent on the gut microbiota and is triggered by antigen-specific DCs.80 When Pros1 deleted, naive T cells were transferred into Rag−/−, the mice showed increased numbers of colitogenic interferon gamma and IL-17A expressing T cells.48 These features were associated with an acceleration of colitis onset as determined by colonoscopy. These findings are in agreement with the function of T cell-derived Pros1 in tempering DC response by activating DC TAM receptors and inhibiting TLR signaling.
In humans, PROS1 deficiencies have been reported in both UC and CD patients. Three independent association studies reported the reduced amounts of circulating PROS1 in patients with either CD or UC.81–83 Furthermore, multiple case reports support this association.84–86 Additionally, PROS1 deficiencies have been reported in autoimmune diseases such as systemic lupus erythematosus.87,88 The most well-known function of PROS1 is as an anticoagulant.89–91 PROS1 is a cofactor of activated Protein C in the degradation of Factor Va and VIIIa in the clotting cascade. PROS1 in human circulates free or bound to C4BP. Mutations in PROS1 that leads to reduced levels of expression and/or function, increased levels of C4BP or the presence of circulating antibodies against PROS1 can compromise its function leading to PROS1 deficiencies.92–94 Intriguingly, the TAM-independent function of PROS1 as an anticoagulant versus the TAM-dependent function as an anti-inflammatory, has not been experimentally dissociated. It is likely that the loss of either or both of these functions may be important in the context of IBD. In fact, IBD has been associated with an increased risk of thrombosis since as early as 1936. Bargen and Barker95 reported extensive arterial and venous thrombosis in patients with IBD. To date, whether the hypercoagulable state in PROS1-deficient patients directly contributes to IBD or merely increases the risk of thrombosis in patients with IBD remains unknown. Furthermore, direct experimental evidence to indicate that T cells in patients with IBD with reduced levels of plasma PROS1 are also impaired in their capacity to engage TAM receptors is lacking.
TAM SIGNALING IN COLITIS-ASSOCIATED CANCER
Full-length human AXL was originally cloned from primary human myeloid leukemia cells.14,15 Similarly, MERTK was cloned from a B-lymphoblastoid expression library19 and TYRO3 from teratocarcinoma and hepatocarcinoma cells.96,97 Axl and/or Mertk are overexpressed in a variety of cancers including but not limited to leukemias, glioblastoma, melanoma, pancreatic cancer, breast cancer, and lung cancer. Tyro3 is overexpressed in multiple myeloma and acute myeloid leukemia (for review see Ref. 98). Interestingly, overexpression of TAM components, rather than activating mutations, seems to be the common theme in oncogenic TAM function.
The oncogenic function of TAMs was anticipated based on the transforming capacity of v-ryk. v-ryk is a viral oncogene from the avian retrovirus RPL30.18 The cellular homolog of this viral oncogene was identified as Mertk.17 Multiple aspects of cancer biology including cell proliferation, migration, and invasion, apoptosis resistance and cell survival, and angiogenesis have been linked to TAM signaling (for review see Ref. 98). Apart from the autocrine or cell autonomous role of TAM signaling in cancer cells, Loges et al99 demonstrated an interesting TAM signaling axis between tumor cells and tumor-associated macrophages. Tumor-infiltrating macrophages express higher levels of Gas6 than their splenic counterparts, suggesting that tumor microenvironment-derived factors such as IL-10 and macrophage colony stimulating factor (M-CSF) lead to Gas6 upregulation. This Gas6, in turn, acts on TAM receptors in tumor cells to promote tumor cell proliferation. Cancer progression in various model systems have been inhibited by interfering with TAM signaling through the use of dominant-negative constructs, silencing, soluble ectodo-main, antibodies, and small molecule inhibitors.100–106 Recently, BergenBio announced a phase I clinical trial of its Axl kinase inhibitor.107
Inhibiting an oncogene has obvious therapeutic potential. However, the role of TAM signaling as a critical negative regulator of inflammation presents an interesting paradox. Chronic inflammation and failure of tissue repair has long been associated with cancer. Rudolf Virchow interpreted his 1863 discovery of “lymphoreticular infiltrate” in cancer tissue as suggestive of a chronic inflammatory origin of cancers.108 In 1986, Dvorak109 described cancer as a wound that never heals. Chronic inflammation as at least a permissive, if not instructive, factor in cancer has been experimentally established through pioneering efforts in a number of laboratories.110–114
The increased risk of colorectal cancer (CRC) is not only linked to inherited mutations in genes such as APC (familial adenomatous polyposis), MHL1/MSH2,6/PMS2 (hereditary non-polyposis colon cancer/lynch syndrome), LKB1 or PTEN (hamar-tomatous polyps) but also to inflammation. Two important factors that increase the risk of CRC include the extent of inflammatory disease and its duration. For example, in patients with left-sided UC or pancolitis, the approximate cumulative incidence of CRC is 8% after 20 years and 18% after 30 years of persistent disease.115 The median duration of disease before diagnosis of CRC is 15 years in CD and 18 years in UC. For this reason, surveillance strategies are recommended after 8 to 10 years of disease.
Specifically, IBD has been associated with the development of dysplasia and colitis-associated cancer (CAC), a subtype of CRC. Both familial and sporadic forms of CRC exhibit a characteristic sequence of gene mutations along the adenoma-carcinoma axis, first described by Fearon and Vogelstein116 (commonly called Vogelgram). CAC shares many of the gene mutations associated with CRC such as TP53, APC, and K-RAS, although the sequence of these mutations along the adenoma–carcinoma axis is different.117 Using mouse models, Michael Karin’s laboratory has established a critical function of NF-κB and inflammation in CAC.118 Sergei Grivennikov et al119 demonstrated that the cytokine IL-6 produced by lamina propria myeloid cells stimulate the proliferation of tumor-initiating cells and the development of CAC. Is TAM signaling oncogenic in cancer or does it help to reduce inflammation and prevent cancer?
The direct dissection of prooncogenic and antioncogenic role of TAM function in CAC remains unaddressed. A couple of studies have investigated the role of TAM signaling in colon cancer although these studies were not dedicated to CAC in particular. In early studies investigating RTK in colon cancer cells, TAM expression was reported to be similar in cancer versus matched control tissue except in a case of liver metastasis and a peritoneal metastasis of colon cancer.120 However, recent studies have reported that high AXL expression correlates with poor survival in this disease.121,122 Several lines of in vitro evidence also suggest that AXL may function as an oncogene in human colon cancer.121,122
In contrast, in vivo studies in mouse models support an antioncogenic role. Gas6−/− mice were more susceptible to azoxy-methane–dextran sodium sulfate (AOM-DSS)-induced CAC.77 Gas6−/− mice developed a significantly greater number of Proliferating cell nuclear antigen (PCNA)- and c-Myc- positive polyps, produced higher levels of TNFα, CXCL1, and CCL2 and increased NF-κB activation after AOM-DSS treatment, in comparison with wild-type mice. Gas6−/− mice also had reduced survival after AOM-DSS treatment. Similarly, Axl−/−Mertk−/− mice had more numerous and larger polyps after AOM-DSS treatment, accounting for an increased colonoscopic tumor score in comparison with wild-type mice.68 Additionally, in a model of mouse colon cancer driven by mutations in the Apc loci (ApcMin), the loss of Gas6 rendered the animal more susceptible.77 ApcMinGas6−/− mice had increased tumor load and reduced survival in comparison to ApcMinGas6+/+ mice. Therefore, the role of TAM signaling in the gut in a mouse model of CAC and CRC is consistent with its anti-inflammatory function, but contrary to its prooncogenic role. In light of recent developments in systemic targeting of TAM RTKs in cancer with small molecules and biologics, we believe that this is an outstanding issue that needs additional investigation.
CONCLUSIONS
Discovered in the early 1990s and without a known ligand for about half a decade thereafter, the TAM RTKs have now been established as critical negative regulators of the innate immune response. After the engagement of the adaptive immune response, TAM ligands are produced. These act on TAM receptors in APCs to inhibit TLR and type I interferon receptor signaling. TAMs are also important for the removal of apoptotic neutrophils. Given the particular challenges of immune homeostasis in the intestine, maintaining a fine balance between an adequate inflammatory response to invading pathogens and swift resolution so as to prevent overzealous reactions, TAM function may play a crucial role in this organ. Therefore, altered TAM function may contribute to the etiology or pathogenesis of IBD. Although the investigation of TAM function during intestinal inflammation and its resolution are revealing important mechanisms of intestinal homeostasis, important questions remain unresolved. For example, the source of TAM ligands and the precise identity of effector cell types in which TAM signaling functions during resolution of intestinal inflammation need to be defined. The signaling pathways engaged during the removal of apoptotic cells versus TLR inhibition remain to be contrasted. Additionally, TAM function as an oncogene and its role as a negative regulator of inflammation present an apparent contradiction in the context of IBD and CAC. It will be important to dissect the individual versus the combinatorial role of TAM RTKs in CRC to shed more light on the tumor-promoting versus antitumor effects of these RTKs. Combining in vivo pharmacological approaches of targeted TAM activation and inhibition, along with the development of improved genetic tools such as cell type–specific knockouts, will not only increase our understanding of the basic biology of IBD but also reveal therapeutic opportunities to target this signaling pathway for the restitution of organ function in patients with IBD.
Acknowledgments
Supported by grants from the National Institutes of Health (R01 AI089824 to C.V.R. and S.G., Arizona Cancer Center GI Spore CA95060 subaward to S.G.), by the Crohn’s and Colitis Foundation (C.V.R. and S.G.), and a Pilot Grant from the Yale Comprehensive Cancer Center C.V.R.).
Footnotes
The authors have no conflicts of interest to disclose.
References
- 1.Colombel JF, Sandborn WJ, Reinisch W, et al. Infliximab, azathioprine, or combination therapy for Crohn’s disease. N Engl J Med. 2010;362:1383–1395. doi: 10.1056/NEJMoa0904492. [DOI] [PubMed] [Google Scholar]
- 2.Feagan BG, Rutgeerts P, Sands BE, et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2013;369:699–710. doi: 10.1056/NEJMoa1215734. [DOI] [PubMed] [Google Scholar]
- 3.Sandborn WJ, Feagan BG, Rutgeerts P, et al. Vedolizumab as induction and maintenance therapy for Crohn’s disease. N Engl J Med. 2013;369:711–721. doi: 10.1056/NEJMoa1215739. [DOI] [PubMed] [Google Scholar]
- 4.Graham DB, Xavier RJ. From genetics of inflammatory bowel disease towards mechanistic insights. Trends Immunol. 2013;34:371–378. doi: 10.1016/j.it.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Zoete MR, Flavell RA. Interactions between nod-like receptors and intestinal bacteria. Front Immunol. 2013;4:462. doi: 10.3389/fimmu.2013.00462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Honda K, Littman DR. The microbiome in infectious disease and inflammation. Annu Rev Immunol. 2012;30:759–795. doi: 10.1146/annurev-immunol-020711-074937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spor A, Koren O, Ley R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat Rev Microbiol. 2011;9:279–290. doi: 10.1038/nrmicro2540. [DOI] [PubMed] [Google Scholar]
- 8.Chinen T, Rudensky AY. The effects of commensal microbiota on immune cell subsets and inflammatory responses. Immunol Rev. 2012;245:45–55. doi: 10.1111/j.1600-065X.2011.01083.x. [DOI] [PubMed] [Google Scholar]
- 9.Leone V, Chang EB, Devkota S. Diet, microbes, and host genetics: the perfect storm in inflammatory bowel diseases. J Gastroenterol. 2013;48:315–321. doi: 10.1007/s00535-013-0777-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Franke A, McGovern DP, Barrett JC, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mowat C, Cole A, Windsor A, et al. Guidelines for the management of inflammatory bowel disease in adults. Gut. 2011;60:571–607. doi: 10.1136/gut.2010.224154. [DOI] [PubMed] [Google Scholar]
- 13.Lai C, Lemke G. An extended family of protein-tyrosine kinase genes differentially expressed in the vertebrate nervous system. Neuron. 1991;6:691–704. doi: 10.1016/0896-6273(91)90167-x. [DOI] [PubMed] [Google Scholar]
- 14.O’Bryan JP, Frye RA, Cogswell PC, et al. axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol Cell Biol. 1991;11:5016–5031. doi: 10.1128/mcb.11.10.5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Janssen JW, Schulz AS, Steenvoorden AC, et al. A novel putative tyrosine kinase receptor with oncogenic potential. Oncogene. 1991;6:2113–2120. [PubMed] [Google Scholar]
- 16.Rescigno J, Mansukhani A, Basilico C. A putative receptor tyrosine kinase with unique structural topology. Oncogene. 1991;6:1909–1913. [PubMed] [Google Scholar]
- 17.Jia R, Hanafusa H. The proto-oncogene of v-eyk (v-ryk) is a novel receptor-type protein tyrosine kinase with extracellular Ig/GN-III domains. J Biol Chem. 1994;269:1839–1844. [PubMed] [Google Scholar]
- 18.Jia R, Mayer BJ, Hanafusa T, et al. A novel oncogene, v-ryk, encoding a truncated receptor tyrosine kinase is transduced into the RPL30 virus without loss of viral sequences. J Virol. 1992;66:5975–5987. doi: 10.1128/jvi.66.10.5975-5987.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graham DK, Dawson TL, Mullaney DL, et al. Cloning and mRNA expression analysis of a novel human protooncogene, c-mer. Cell Growth Differ. 1994;5:647–657. [PubMed] [Google Scholar]
- 20.Manning G, Whyte DB, Martinez R, et al. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 21.Ronsin C, Muscatelli F, Mattei MG, et al. A novel putative receptor protein tyrosine kinase of the met family. Oncogene. 1993;8:1195–1202. [PubMed] [Google Scholar]
- 22.Cooper CS, Park M, Blair DG, et al. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature. 1984;311:29–33. doi: 10.1038/311029a0. [DOI] [PubMed] [Google Scholar]
- 23.Di Scipio RG, Hermodson MA, Yates SG, et al. A comparison of human prothrombin, factor IX (Christmas factor), factor X (Stuart factor), and protein S. Biochemistry. 1977;16:698–706. doi: 10.1021/bi00623a022. [DOI] [PubMed] [Google Scholar]
- 24.Stitt TN, Conn G, Gore M, et al. The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell. 1995;80:661–670. doi: 10.1016/0092-8674(95)90520-0. [DOI] [PubMed] [Google Scholar]
- 25.Huang M, Rigby AC, Morelli X, et al. Structural basis of membrane binding by Gla domains of vitamin K-dependent proteins. Nat Struct Biol. 2003;10:751–756. doi: 10.1038/nsb971. [DOI] [PubMed] [Google Scholar]
- 26.Nakano T, Kawamoto K, Kishino J, et al. Requirement of gamma-carboxyglutamic acid residues for the biological activity of Gas6: contribution of endogenous Gas6 to the proliferation of vascular smooth muscle cells. Biochem J. 1997;323(pt 2):387–392. doi: 10.1042/bj3230387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hasanbasic I, Rajotte I, Blostein M. The role of gamma-carboxylation in the anti-apoptotic function of gas6. J Thromb Haemost. 2005;3:2790–2797. doi: 10.1111/j.1538-7836.2005.01662.x. [DOI] [PubMed] [Google Scholar]
- 28.Anderson HA, Maylock CA, Williams JA, et al. Serum-derived protein S binds to phosphatidylserine and stimulates the phagocytosis of apoptotic cells. Nat Immunol. 2003;4:87–91. doi: 10.1038/ni871. [DOI] [PubMed] [Google Scholar]
- 29.Nyberg P, He X, Hardig Y, et al. Stimulation of sky tyrosine phosphorylation by bovine protein S: domains involved in the receptor-ligand interaction. Eur J Biochem. 1997;246:147–154. doi: 10.1111/j.1432-1033.1997.t01-2-00147.x. [DOI] [PubMed] [Google Scholar]
- 30.Sasaki T, Knyazev PG, Cheburkin Y, et al. Crystal structure of a C-terminal fragment of growth arrest-specific protein Gas6. Receptor tyrosine kinase activation by laminin G-like domains. J Biol Chem. 2002;277:44164–44170. doi: 10.1074/jbc.M207340200. [DOI] [PubMed] [Google Scholar]
- 31.Sasaki T, Knyazev PG, Clout NJ, et al. Structural basis for Gas6-Axl signalling. EMBO J. 2006;25:80–87. doi: 10.1038/sj.emboj.7600912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu Q, Gore M, Zhang Q, et al. Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature. 1999;398:723–728. doi: 10.1038/19554. [DOI] [PubMed] [Google Scholar]
- 33.Nandrot E, Dufour EM, Provost AC, et al. Homozygous deletion in the coding sequence of the c-mer gene in RCS rats unravels general mechanisms of physiological cell adhesion and apoptosis. Neurobiol Dis. 2000;7:586–599. doi: 10.1006/nbdi.2000.0328. [DOI] [PubMed] [Google Scholar]
- 34.D’Cruz PM, Yasumura D, Weir J, et al. Mutation of the receptor tyrosine kinase gene Mertk in the retinal dystrophic RCS rat. Hum Mol Genet. 2000;9:645–651. doi: 10.1093/hmg/9.4.645. [DOI] [PubMed] [Google Scholar]
- 35.Feng W, Yasumura D, Matthes MT, et al. Mertk triggers uptake of photoreceptor outer segments during phagocytosis by cultured retinal pigment epithelial cells. J Biol Chem. 2002;277:17016–17022. doi: 10.1074/jbc.M107876200. [DOI] [PubMed] [Google Scholar]
- 36.Gal A, Li Y, Thompson DA, et al. Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat Genet. 2000;26:270–271. doi: 10.1038/81555. [DOI] [PubMed] [Google Scholar]
- 37.Xiong W, Chen Y, Wang H, et al. Gas6 and the Tyro 3 receptor tyrosine kinase subfamily regulate the phagocytic function of sertoli cells. Reproduction. 2008;135:77–87. doi: 10.1530/REP-07-0287. [DOI] [PubMed] [Google Scholar]
- 38.Chung WS, Clarke LE, Wang GX, et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature. 2013;504:394–400. doi: 10.1038/nature12776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scott RS, McMahon EJ, Pop SM, et al. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature. 2001;411:207–211. doi: 10.1038/35075603. [DOI] [PubMed] [Google Scholar]
- 40.Cohen PL, Caricchio R, Abraham V, et al. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J Exp Med. 2002;196:135–140. doi: 10.1084/jem.20012094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu Q, Lemke G. Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science. 2001;293:306–311. doi: 10.1126/science.1061663. [DOI] [PubMed] [Google Scholar]
- 42.Lemke G, Rothlin CV. Immunobiology of the TAM receptors. Nat Rev Immunol. 2008;8:327–336. doi: 10.1038/nri2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rothlin CV, Ghosh S, Zuniga EI, et al. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. 2007;131:1124–1136. doi: 10.1016/j.cell.2007.10.034. [DOI] [PubMed] [Google Scholar]
- 44.Camenisch TD, Koller BH, Earp HS, et al. A novel receptor tyrosine kinase, Mer, inhibits TNF-alpha production and lipopolysaccharide-induced endotoxic shock. J Immunol. 1999;162:3498–3503. [PubMed] [Google Scholar]
- 45.Sen P, Wallet MA, Yi Z, et al. Apoptotic cells induce Mer tyrosine kinase-dependent blockade of NF-kappaB activation in dendritic cells. Blood. 2007;109:653–660. doi: 10.1182/blood-2006-04-017368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tibrewal N, Wu Y, D’mello V, et al. Autophosphorylation docking site Tyr-867 in Mer receptor tyrosine kinase allows for dissociation of multiple signaling pathways for phagocytosis of apoptotic cells and down-modulation of lipopolysaccharide-inducible NF-kappaB transcriptional activation. J Biol Chem. 2008;283:3618–3627. doi: 10.1074/jbc.M706906200. [DOI] [PubMed] [Google Scholar]
- 47.Tanabe K, Nagata K, Ohashi K, et al. Roles of gamma-carboxylation and a sex hormone-binding globulin-like domain in receptor-binding and in biological activities of Gas6. FEBS Lett. 1997;408:306–310. doi: 10.1016/s0014-5793(97)00448-1. [DOI] [PubMed] [Google Scholar]
- 48.Carrera Silva EA, Chan PY, Joannas L, et al. T cell-derived protein S engages TAM receptor signaling in dendritic cells to control the magnitude of the immune response. Immunity. 2013;39:160–170. doi: 10.1016/j.immuni.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science. 2010;327:291–295. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Coombes JL, Powrie F. Dendritic cells in intestinal immune regulation. Nat Rev Immunol. 2008;8:435–446. doi: 10.1038/nri2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lambrecht BN, Hammad H. The role of dendritic and epithelial cells as master regulators of allergic airway inflammation. Lancet. 2010;376:835–843. doi: 10.1016/S0140-6736(10)61226-3. [DOI] [PubMed] [Google Scholar]
- 52.Smiley ST, Boyer SN, Heeb MJ, et al. Protein S is inducible by interleukin 4 in T cells and inhibits lymphoid cell procoagulant activity. Proc Natl Acad Sci U S A. 1997;94:11484–11489. doi: 10.1073/pnas.94.21.11484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fischer K, Voelkl S, Berger J, et al. Antigen recognition induces phosphatidylserine exposure on the cell surface of human CD8+ T cells. Blood. 2006;108:4094–4101. doi: 10.1182/blood-2006-03-011742. [DOI] [PubMed] [Google Scholar]
- 54.Denning TL, Wang YC, Patel SR, et al. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat Immunol. 2007;8:1086–1094. doi: 10.1038/ni1511. [DOI] [PubMed] [Google Scholar]
- 55.Medina-Contreras O, Geem D, Laur O, et al. CX3CR1 regulates intestinal macrophage homeostasis, bacterial translocation, and colitogenic Th17 responses in mice. J Clin Invest. 2011;121:4787–4795. doi: 10.1172/JCI59150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Niess JH, Brand S, Gu X, et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science. 2005;307:254–258. doi: 10.1126/science.1102901. [DOI] [PubMed] [Google Scholar]
- 57.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159–175. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- 58.Yona S, Kim KW, Wolf Y, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38:79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rivollier A, He J, Kole A, et al. Inflammation switches the differentiation program of Ly6Chi monocytes from antiinflammatory macrophages to inflammatory dendritic cells in the colon. J Exp Med. 2012;209:139–155. doi: 10.1084/jem.20101387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bain CC, Scott CL, Uronen-Hansson H, et al. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol. 2013;6:498–510. doi: 10.1038/mi.2012.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fadok VA, Bratton DL, Konowal A, et al. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Levy BD, Clish CB, Schmidt B, et al. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol. 2001;2:612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- 63.Golpon HA, Fadok VA, Taraseviciene-Stewart L, et al. Life after corpse engulfment: phagocytosis of apoptotic cells leads to VEGF secretion and cell growth. FASEB J. 2004;18:1716–1718. doi: 10.1096/fj.04-1853fje. [DOI] [PubMed] [Google Scholar]
- 64.Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest. 2002;109:41–50. doi: 10.1172/JCI11638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Johann AM, Barra V, Kuhn AM, et al. Apoptotic cells induce arginase II in macrophages, thereby attenuating NO production. FASEB J. 2007;21:2704–2712. doi: 10.1096/fj.06-7815com. [DOI] [PubMed] [Google Scholar]
- 66.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 67.Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. 2009;361:2066–2078. doi: 10.1056/NEJMra0804647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bosurgi L, Bernink JH, Delgado Cuevas V, et al. Paradoxical role of the proto-oncogene Axl and Mer receptor tyrosine kinases in colon cancer. Proc Natl Acad Sci U S A. 2013;110:13091–13096. doi: 10.1073/pnas.1302507110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hu B, Jennings JH, Sonstein J, et al. Resident murine alveolar and peritoneal macrophages differ in adhesion of apoptotic thymocytes. Am J Respir Cell Mol Biol. 2004;30:687–693. doi: 10.1165/rcmb.2003-0255OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gautier EL, Shay T, Miller J, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. 2012;13:1118–1128. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Scutera S, Fraone T, Musso T, et al. Survival and migration of human dendritic cells are regulated by an IFN-alpha-inducible Axl/Gas6 pathway. J Immunol. 2009;183:3004–3013. doi: 10.4049/jimmunol.0804384. [DOI] [PubMed] [Google Scholar]
- 72.Ganesh K, Das A, Dickerson R, et al. Prostaglandin E2 induces oncostatin M expression in human chronic wound macrophages through Axl receptor tyrosine kinase pathway. J Immunol. 2012;189:2563–2573. doi: 10.4049/jimmunol.1102762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Korshunov VA, Mohan AM, Georger MA, et al. Axl, a receptor tyrosine kinase, mediates flow-induced vascular remodeling. Circ Res. 2006;98:1446–1452. doi: 10.1161/01.RES.0000223322.16149.9a. [DOI] [PubMed] [Google Scholar]
- 74.Melaragno MG, Wuthrich DA, Poppa V, et al. Increased expression of Axl tyrosine kinase after vascular injury and regulation by G protein-coupled receptor agonists in rats. Circ Res. 1998;83:697–704. doi: 10.1161/01.res.83.7.697. [DOI] [PubMed] [Google Scholar]
- 75.Zizzo G, Hilliard BA, Monestier M, et al. Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J Immunol. 2012;189:3508–3520. doi: 10.4049/jimmunol.1200662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Van der Sluis M, De Koning BA, De Bruijn AC, et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology. 2006;131:117–129. doi: 10.1053/j.gastro.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 77.Akitake-Kawano R, Seno H, Nakatsuji M, et al. Inhibitory role of Gas6 in intestinal tumorigenesis. Carcinogenesis. 2013;34:1567–1574. doi: 10.1093/carcin/bgt069. [DOI] [PubMed] [Google Scholar]
- 78.Powrie F, Correa-Oliveira R, Mauze S, et al. Regulatory interactions between CD45RBhigh and CD45RBlow CD4+ T cells are important for the balance between protective and pathogenic cell-mediated immunity. J Exp Med. 1994;179:589–600. doi: 10.1084/jem.179.2.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Powrie F, Leach MW, Mauze S, et al. Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity. 1994;1:553–562. doi: 10.1016/1074-7613(94)90045-0. [DOI] [PubMed] [Google Scholar]
- 80.Feng T, Wang L, Schoeb TR, et al. Microbiota innate stimulation is a prerequisite for T cell spontaneous proliferation and induction of experimental colitis. J Exp Med. 2010;207:1321–1332. doi: 10.1084/jem.20092253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Koutroubakis IE, Sfiridaki A, Mouzas IA, et al. Resistance to activated protein C and low levels of free protein S in Greek patients with inflammatory bowel disease. Am J Gastroenterol. 2000;95:190–194. doi: 10.1111/j.1572-0241.2000.01683.x. [DOI] [PubMed] [Google Scholar]
- 82.Owczarek D, Cibor D, Salapa K, et al. Anti-inflammatory and anticoagulant properties of the protein C system in inflammatory bowel disease. Pol Arch Med Wewn. 2012;122:209–216. doi: 10.20452/pamw.1261. [DOI] [PubMed] [Google Scholar]
- 83.Cakal B, Gokmen A, Yalinkilic M, et al. Natural anticoagulant protein levels in Turkish patients with inflammatory bowel disease. Blood Coagul Fibrinolysis. 2010;21:118–121. doi: 10.1097/MBC.0b013e328335d025. [DOI] [PubMed] [Google Scholar]
- 84.Bonanni I, Bellotti P, Brignone M, et al. Protein S deficiency. Description of a case associated with chronic inflammatory bowel disease. Minerva Med. 2002;93:309–313. [PubMed] [Google Scholar]
- 85.Wyshock E, Caldwell M, Crowley JP. Deep venous thrombosis, inflammatory bowel disease, and protein S deficiency. Am J Clin Pathol. 1988;90:633–635. doi: 10.1093/ajcp/90.5.633. [DOI] [PubMed] [Google Scholar]
- 86.Kempton CL, Bagby G, Collins JF. Ulcerative colitis presenting as pur-pura fulminans. Inflamm Bowel Dis. 2001;7:319–322. doi: 10.1097/00054725-200111000-00007. [DOI] [PubMed] [Google Scholar]
- 87.Suh CH, Hilliard BA, Li S, et al. TAM receptor ligands in lupus: protein S but not Gas6 levels reflect disease activity in systemic lupus erythematosus. Arthritis Res Ther. 2010;12:R146. doi: 10.1186/ar3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Recarte-Pelz P, Tàssies D, Espinosa G, et al. Vitamin K-dependent proteins GAS6 and protein S and TAM receptors in patients of systemic lupus erythematosus: correlation with common genetic variants and disease activity. Arthritis Res Ther. 2013;15:R41. doi: 10.1186/ar4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dahlback B. The tale of protein S and C4b-binding protein, a story of affection. Thromb Haemost. 2007;98:90–96. [PubMed] [Google Scholar]
- 90.Burstyn-Cohen T, Heeb MJ, Lemke G. Lack of protein S in mice causes embryonic lethal coagulopathy and vascular dysgenesis. J Clin Invest. 2009;119:2942–2953. doi: 10.1172/JCI39325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Saller F, Brisset AC, Tchaikovski SN, et al. Generation and phenotypic analysis of protein S-deficient mice. Blood. 2009;114:2307–2314. doi: 10.1182/blood-2009-03-209031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gandrille S, Borgel D, Sala N, et al. Protein S deficiency: a database of mutations: summary of the first update. Thromb Haemost. 2000;84:918. [PubMed] [Google Scholar]
- 93.Bertolaccini ML, Sanna G, Ralhan S, et al. Antibodies directed to protein S in patients with systemic lupus erythematosus: prevalence and clinical significance. Thromb Haemost. 2003;90:636–641. doi: 10.1160/TH03-03-0151. [DOI] [PubMed] [Google Scholar]
- 94.Saibeni S, Vecchi M, Valsecchi C, et al. Reduced free protein S levels in patients with inflammatory bowel disease: prevalence, clinical relevance, and role of anti-protein S antibodies. Dig Dis Sci. 2001;46:637–643. doi: 10.1023/a:1005675921664. [DOI] [PubMed] [Google Scholar]
- 95.Bargen JA, Barker NW. Extensive arterial and venous thrombosis complicating chronic ulcerative colitis. Arch Intern Med. 1936;58:17–31. [Google Scholar]
- 96.Polvi A, Armstrong E, Lai C, et al. The human TYRO3 gene and pseudogene are located in chromosome 15q14-q25. Gene. 1993;134:289–293. doi: 10.1016/0378-1119(93)90109-g. [DOI] [PubMed] [Google Scholar]
- 97.Ohashi K, Mizuno K, Kuma K, et al. Cloning of the cDNA for a novel receptor tyrosine kinase, Sky, predominantly expressed in brain. Oncogene. 1994;9:699–705. [PubMed] [Google Scholar]
- 98.Linger RM, Keating AK, Earp HS, et al. TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv Cancer Res. 2008;100:35–83. doi: 10.1016/S0065-230X(08)00002-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Loges S, Schmidt T, Tjwa M, et al. Malignant cells fuel tumor growth by educating infiltrating leukocytes to produce the mitogen Gas6. Blood. 2010;115:2264–2273. doi: 10.1182/blood-2009-06-228684. [DOI] [PubMed] [Google Scholar]
- 100.Meyer AS, Miller MA, Gertler FB, et al. The receptor AXL diversifies EGFR signaling and limits the response to EGFR-targeted inhibitors in triple-negative breast cancer cells. Sci Signal. 2013;6:ra66. doi: 10.1126/scisignal.2004155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang Z, Lee JC, Lin L, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012;44:852–860. doi: 10.1038/ng.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Linger RM, Lee-Sherick AB, DeRyckere D, et al. Mer receptor tyrosine kinase is a therapeutic target in pre-B-cell acute lymphoblastic leukemia. Blood. 2013;122:1599–1609. doi: 10.1182/blood-2013-01-478156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Brandao LN, Winges A, Christoph S, et al. Inhibition of MerTK increases chemosensitivity and decreases oncogenic potential in T-cell acute lymphoblastic leukemia. Blood Cancer J. 2013;3:e101. doi: 10.1038/bcj.2012.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schlegel J, Sambade MJ, Sather S, et al. MERTK receptor tyrosine kinase is a therapeutic target in melanoma. J Clin Invest. 2013;123:2257–2267. doi: 10.1172/JCI67816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Vajkoczy P, Knyazev P, Kunkel A, et al. Dominant-negative inhibition of the Axl receptor tyrosine kinase suppresses brain tumor cell growth and invasion and prolongs survival. Proc Natl Acad Sci U S A. 2006;103:5799–5804. doi: 10.1073/pnas.0510923103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li Y, Ye X, Tan C, et al. Axl as a potential therapeutic target in cancer: role of Axl in tumor growth, metastasis and angiogenesis. Oncogene. 2009;28:3442–3455. doi: 10.1038/onc.2009.212. [DOI] [PubMed] [Google Scholar]
- 107.Sheridan C. First Axl inhibitor enters clinical trials. Nat Biotechnol. 2013;31:775–776. doi: 10.1038/nbt0913-775a. [DOI] [PubMed] [Google Scholar]
- 108.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 109.Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–1659. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 110.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science. 2013;339:286–291. doi: 10.1126/science.1232227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Balkwill FR, Mantovani A. Cancer-related inflammation: common themes and therapeutic opportunities. Semin Cancer Biol. 2012;22:33–40. doi: 10.1016/j.semcancer.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 114.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001;48:526–535. doi: 10.1136/gut.48.4.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 117.Itzkowitz SH, Yio X. Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflammation. Am J Physiol Gastrointest Liver Physiol. 2004;287:G7–G17. doi: 10.1152/ajpgi.00079.2004. [DOI] [PubMed] [Google Scholar]
- 118.Greten FR, Eckmann L, Greten TF, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 119.Grivennikov S, Karin E, Terzic J, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Craven RJ, Xu LH, Weiner TM, et al. Receptor tyrosine kinases expressed in metastatic colon cancer. Int J Cancer. 1995;60:791–797. doi: 10.1002/ijc.2910600611. [DOI] [PubMed] [Google Scholar]
- 121.Yuen HF, McCrudden CM, Huang YH, et al. TAZ expression as a prognostic indicator in colorectal cancer. PLoS One. 2013;8:e54211. doi: 10.1371/journal.pone.0054211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Dunne PD, McArt DG, Blayney JK, et al. AXL is a key regulator of inherent and chemotherapy-induced invasion and predicts a poor clinical outcome in early-stage colon cancer. Clin Cancer Res. 2014;20:164–175. doi: 10.1158/1078-0432.CCR-13-1354. [DOI] [PMC free article] [PubMed] [Google Scholar]