

Abstract
Objective
AMP-activated protein kinase (AMPK) inhibits chondrocyte procatabolic responses to inflammation and biomechanical injury. This study was undertaken to test the hypothesis that peroxisome proliferator–activated receptor γ coactivator 1α (PGC-1α) and FoxO3A, 2 major AMPK downstream targets, mediate the chondroprotective effect of AMPK activation.
Methods
We assessed the activity of AMPKα (threonine 172 phosphorylation) and the expression of PGC-1α and FoxO3A in human chondrocytes and AMPKα1- or AMPKα2-knockout mouse chondrocytes by Western blotting, and in mouse knee cartilage by immunohistochemistry. We also knocked down or overexpressed PGC-1α and FoxO3A by small interfering RNA or plasmid DNA transfection, respectively. We assessed mitochondrial superoxide generation using MitoSOX Red.
Results
Expression of PGC-1α and FoxO3A was enhanced by pharmacologic AMPK activator A-769662 but impaired in AMPKα1−/− or AMPKα2−/− mouse chondrocytes. Reduced expression of PGC-1α and FoxO3A was observed in mouse knee instability-induced osteoarthritis (OA) cartilage and in aged C57BL/6 mouse knee cartilage. Knockdown of PGC-1α and FoxO3A enhanced, but limited the ability of A-769662 to inhibit, phosphorylation of p65 NF-κB (Ser536) and procatabolic responses induced by inflammatory cytokines. Forced expression of PGC-1α and FoxO3A induced increased expression of superoxide dismutase 2 (SOD2) and catalase, but A-769662 failed to increase the expression of SOD2 and catalase in either PGC-1α– or FoxO3A-knockdown chondrocytes. Last, menadione-induced superoxide generation was inhibited by AMPK pharmacologic activators and by overexpression of PGC-1α or FoxO3A.
Conclusion
PGC-1α and FoxO3A limit oxidative stress and at least partially mediate the capacity of AMPK activity to block procatabolic responses in chondrocytes, and therefore have the potential to inhibit the progression of cartilage damage in OA.
In osteoarthritis (OA), abnormalities of chondrocyte differentiation and function lead to disordered cartilage extracellular matrix homeostasis (1–4). Oxidative stress, aging, biomechanical injury, and inflammatory mediators all contribute to the development and progression of OA (1–4), and abnormalities of chondrocyte bioenergetics, including altered glycolysis and mitochondrial function, are increasingly implicated (5–8).
The serine/threonine protein kinase AMP-activated protein kinase (AMPK) is a master regulator of energy homeostasis (9,10). AMPK is a heterotrimeric complex of catalytic a-subunit with regulatory β- and γ-subunits (9,10). Phosphorylation of a conserved threonine within the α-subunit catalytic domain is critical for AMPK activity (9,10). AMPK activity has anti-inflammatory effects, which are partly mediated by inhibition of NF-κB signaling (9,10). AMPK activity is constitutively present in normal articular chondrocytes, but is decreased in human knee OA chondrocytes (11), in normal chondrocytes stimulated with interleukin-1β (IL-1β) and tumor necrosis factor α (TNFa) (11), in chondrocytes after mechanical injury (12), and in aged mouse knee cartilage (12). Several pharmacologic AMPK activators, including 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) and the highly selective AMPK allosteric activator A-769662, attenuate NF-κB activation and chondrocyte procatabolic responses to IL-1β and TNFα and biomechanical injury (11,12). Moreover, exercise, calorie restriction, and some drugs already used for arthritis and other conditions activate AMPK. The AMPK-activating drugs include metformin, methotrexate (which increases AICAR levels), and sodium salicylate and high-dose aspirin (by allosteric effects on AMPK) (13). Hence, understanding how AMPK is chondroprotective is of translational relevance.
AMPK has multiple downstream targets (14,15). Here, we focused on the peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) transcription factor and the FoxO family transcription factor FoxO3A. Both PGC-1α and FoxO3A inhibit NF-κB signaling (14,15). PGC-1α also promotes mitochondrial biogenesis (14). This is pertinent because dysregulated mitochondrial function generates increased reactive oxygen species (ROS), and associated oxidative stress is linked to several age-related tissue degenerative diseases (16–18). These include OA, in which increased ROS promotes cartilage degradation by cleaving collagen and aggrecan, activating matrix metalloproteinases (MMPs) (19–21), and by modulating redox-sensitive signaling pathways, including NF-κB signaling (17,18).
Both PGC-1α and FoxO3A limit cellular oxidative stress by up-regulating antioxidant enzymes, including manganese superoxide dismutase (MnSOD; or SOD2) and catalase (22,23). In this light, reduction of SOD2 expression has been linked with OA progression (24). Alterations in PGC-1α level or activity occur in several disorders associated with oxidative stress, including diabetes, heart disease, and neurodegenerative disease (25–27). FoxO3A deficiency in mice promotes certain tissue inflammatory responses and lymphoid proliferation (28), and is associated with increased ROS accumulation in some cell types (29). Moreover, reduced FoxO3A is seen in mouse heart aging, contributing to cardiomyocyte dysfunction (30).
In this study, we tested the hypothesis that altered PGC-1α and FoxO3A expression and function are intimately linked with decreased AMPK activity in articular chondrocytes, including in mouse OA or aging knee cartilage. Our results link PGC-1α and FoxO3A with AMPK activity as core regulators of catabolic responses to IL-1/β and TNFα and of oxidative stress in articular chondrocytes.
MATERIALS AND METHODS
Reagents
All chemical reagents were from Sigma-Aldrich, unless otherwise indicated. AMPK pharmacologic activators AICAR and A-769662, recombinant human IL-1/β and TNFα, and the human MMP-3 and MMP-13 Quantikine enzyme-linked immunosorbent assay (ELISA) kits were purchased from R&D Systems. Antibodies to phospho-AMPKα (Thr172), AMPKα1, AMPKα2, and phospho-p65 (Ser536) were from Cell Signaling Technology. Antibodies to PGC-1α, FoxO3A, and histone H2B were from Abcam. Human small interfering RNAs (siRNAs) for PGC-1α and FoxO3A and the control siRNA were from Invitrogen.
Human and mouse articular chondrocytes
All human and mouse experiments were performed in compliance with VA institutionally reviewed and approved human subject and animal research protocols. The human knee chondrocytes were isolated from cadaver donors at autopsy and were graded macroscopically according to a modified Outerbridge scale (31). Only the normal chondrocytes (grade I; intact cartilage surface) or mild OA chondrocytes (grade II; minimal fibrillation) were used. Human chondrocytes were cultured in high-glucose Dulbecco’s modified Eagle’s medium with 10% fetal calf serum, 100 μg/ml streptomycin, and 100 IU/ml penicillin at 37°C, and no later than first-passage chondrocytes were used for all experiments. The mouse chondrocytes were isolated from the femoral head cartilage of 6–8-week-old mice. Unless otherwise indicated, chondrocytes were plated at 2.5 × 105 cells per well in 250 μl of medium in 12-well plates for each treatment.
Experimental OA models in mice
Joint instability-induced OA was induced in 2-month-old C57BL/6 mice by medial meniscectomy, and animals were killed 8 weeks later. Aging C57BL/6 mice were kept under normal conditions, and knee joints were compared at 6, 12, and 24 months of age. Knee joints from mice with surgical OA and from aging mice were resected, fixed in 10% zinc-buffered formalin (Z-Fix; Anatech) for 2 days, decalcified in TBD-2 (Shandon) for 72 hours, and paraffin embedded using standard protocols.
Immunohistochemistry
Mouse knee cartilage sections were pretreated with trypsin (0.05%) for 10 minutes before being treated with 3% (volume/volume) H2O2 for 15 minutes. The sections were then blocked with 10% goat serum for 2 hours at room temperature. After washing with Tris buffered saline (TBS), rabbit antibodies to phospho-AMPKα (Thr172) (1:50 dilution), PGC-1α (1:50 dilution), and FoxO3A (1:50 dilution), and the negative control rabbit IgG (1 μg/ml) were applied to the sections and incubated overnight at 4°C Next, sections were washed with TBS, incubated with biotinylated goat anti-rabbit IgG secondary antibody for 30 minutes, and then incubated for 30 minutes using a Histostain Plus kit (Invitrogen). Finally, sections were washed and incubated with 3,3′-diaminobenzidine substrate for 2–5 minutes.
Quantification of positive-staining chondrocytes
Positive-staining cells in each mouse knee section were counted from noncalcified areas of the femoral condyle and tibia. The cellularity was quantified by counting the number of cells stained with hematoxylin on corresponding, adjacent sections. The number of positive cells for each antibody was expressed as the percentage of positive-staining cells (by immunohistochemistry) relative to the number of cells stained with hematoxylin in corresponding sections.
Knockdown or overexpression of PGC-1α and FoxO3A in human knee articular chondrocytes
Normal cultured human knee articular chondrocytes (passage 1) were transfected with either siRNAs for PGC-1α and FoxO3A and the nontarget control using the X-tremeGene siRNA transfection reagent (Roche Applied Science) or plasmids of pcDNA4-PGC-1α and pcDNA3-FoxO3A (Addgene) and the pcDNA3 vector control using the GenJet Plus transfection reagent (SignaGen), according to the recommendations of the manufacturer. Levels of expression of PGC-1α and FoxO3A were examined by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot analysis. Densitometry was performed using the ImageJ program (National Institutes of Health).
Western blotting
Cells were lysed in cell lysis buffer (20 mM Tris HCl [pH 7.5], 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, and 1 mM Na3VO4) with protease inhibitor cocktail (Roche). Cytosolic and nuclear proteins were prepared using a nuclear/cytosol fractionation kit according to the recommendations of the manufacturer (BioVision). Cell lysates, cytosolic proteins, and nuclear proteins were separated by SDS-PAGE using 4–20% gradient gels and transferred onto nitrocellulose membranes (Bio-Rad), probed with antibodies, exposed to SuperSignal West Pico chemiluminescent substrate (Thermo Scientific), and visualized by radiography.
Measurement of the release of nitric oxide (NO), MMP-3, MMP-13, and superoxide
Levels of NO and MMP-3 and MMP-13 in conditioned media were assayed using the Griess reaction method (11) and ELISA, respectively. To study the chondrocyte response to oxidative stress, chondrocytes were treated with menadione (100 μM) for 1 hour, and production of superoxide by mitochondria was visualized by fluorescence microscopy using MitoSOX Red reagent. Results are presented as the percentage of cells that stained positively for MitoSOX Red.
Statistical analysis
All data were uniformly expressed as the mean ± SD. Statistical analysis was performed by two-way analysis of variance with Bonferroni post hoc test using GraphPad Prism 6. P values less than 0.05 were considered significant.
RESULTS
Regulation of the expression of PGC-1α and FoxO3A by AMPK in human articular chondrocytes
AMPK activation of human chondrocytes using A-769662 resulted in increased phosphorylation of AMPKα and was associated with increased expression of PGC-1α and FoxO3A (Figure 1A), with an average of 2.7-, 3.1-, and 2.9-fold induction, respectively, as determined by densitometry in 3 different experiments. Conversely, decreased expression of PGC-1α and FoxO3A were observed in AMPKα1-knockout mouse chondrocytes and AMPKα2-knockout mouse chondrocytes (Figures 1B and C). Hence, AMPK was linked with the regulation of chondrocyte expression of PGC-1α and FoxO3A.
Figure 1.
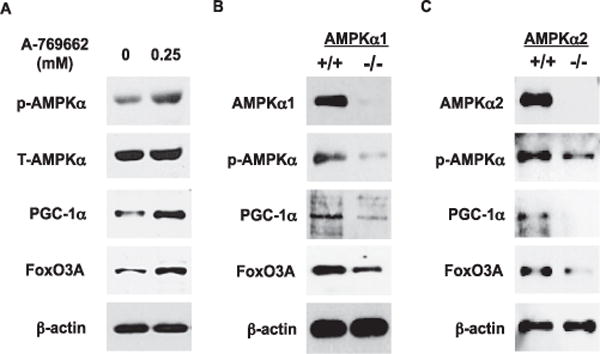
Regulation of the expression of peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) and FoxO3A by AMP-activated protein kinase (AMPK) in articular chondrocytes. A, Phosphorylation of AMPKα and expression of PGC-1α and FoxO3A in cultured human knee chondrocytes (passage 1) stimulated with A-769662 (0.25 mM) for 2 hours, analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting. B and C, Phosphorylation of AMPKα and expression of PGC-1α and FoxO3A in chondrocytes isolated from the femoral head articular cartilage of AMPKα1-knockout mice (B) and AMPKα2-knockout mice (C), analyzed by SDS-PAGE and Western blotting. Results are representative of 3 individual experiments. T-AMPKα = total AMPKα.
Decreased expression of PGC-1α and FoxO3A in knee cartilage samples from aging mice and from mice with OA
First, we examined cellularity by hematoxylin staining and analyzed the expression of PGC-1α and FoxO3A by immunohistochemistry in knee cartilage sections from 6-month-old (young), 12-month-old (middle-aged), and 24-month-old (old) mice (with Osteoarthritis Research Society International [OARSI] [32] scores of 0, 0, and 1 respectively). As seen in Figure 2, aging-associated reduction in cellularity was observed. However, the numbers of chondrocytes in the noncalcified region of both femoral and tibial cartilage that stained positively for PGC-1α and FoxO3A appeared to be directly proportional to the numbers of chondrocytes that stained positively for phosphorylated AMPKα in all age groups (Figure 2A). Notably, the numbers of chondrocytes that stained positively for phosphorylated AMPKα, PGC-1α, and FoxO3A decreased only slightly in 12-month-old animals, but significantly in 24-month-old animals, compared with those in 6-month-old animals (Figure 2B).
Figure 2.
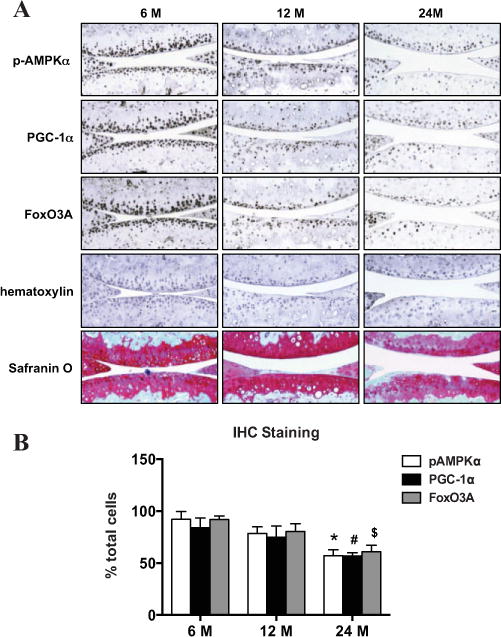
Concomitant reduction of the phosphorylation of AMPKα and expression of PGC-1α and FoxO3A in aging mouse knee cartilage. A, Phosphorylation of AMPKα and expression of PGC-1α and FoxO3A in knee sections from 6-month-old, 12-month-old, and 24-month-old mice. Mouse knee sections were analyzed by immunohistochemistry (IHC), as described in Materials and Methods. Sections were stained with hematoxylin and Safranin O for assessment of proteoglycan content and cellularity in cartilage, respectively. Original magnification × 10. B, Percentage of cells staining positively for AMPKα, PGC-1α, and FoxO3A, relative to the total number of cells staining for hematoxylin. Bars show the mean ± SD (n = 4 mice per age group). Data are representative of 2 individual experiments. * = P < 0.0001; # = P < 0.0007; $ = P < 0.0002, versus 6-month-old mice. See Figure 1 for other definitions.
We previously observed decreased phosphorylation of AMPKα in mouse OA knee cartilage (11). In this study, we examined the expression of PGC-1α and FoxO3A by immunohistochemistry in knee sections from mice with surgical instability–induced OA and from sham-operated control mice (with OARSI scores of 5 and 0, respectively). Although cellularity was significantly reduced, reductions of >50% in the percentages of chondrocytes in the noncalcified region of both the femoral and tibial cartilage that stained positively for PGC-1α and FoxO3A were seen in mouse OA knee cartilage compared to sham-operated mouse knee cartilage (Figure 3). Notably, in the boxed regions in the panels showing mouse OA knee cartilage in Figure 3, very few cells showed positive staining for PGC-1α and FoxO3A. The boxed areas in the OA panels exhibited relatively good cellularity, as indicated by positive hematoxylin staining, showing that decreased expression of PGC-1α and FoxO3A was not due to loss of chondrocytes in that region. These data were consistent with a concomitant reduction in the phosphorylation of AMPKα and the expression of PGC-1α and FoxO3A in mouse knee cartilage in association with aging and OA.
Figure 3.
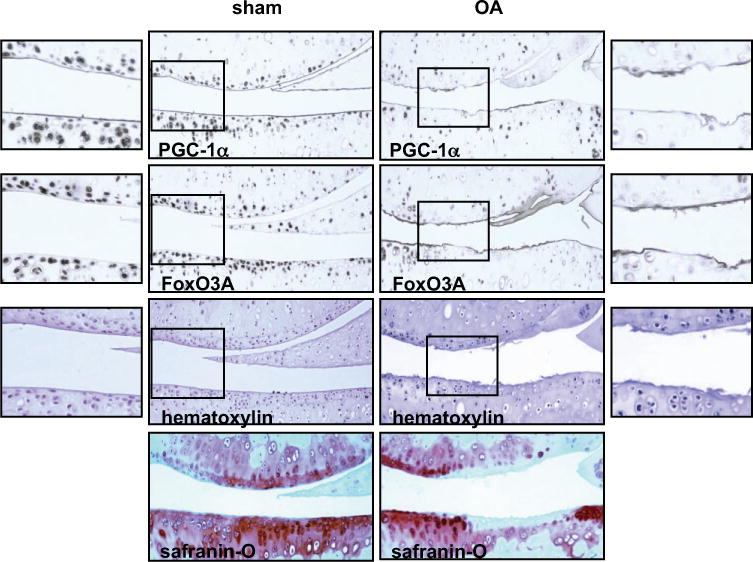
Decreased expression of peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) and FoxO3A in knee cartilage from mice with osteoarthritis (OA). Knee sections from mice with surgical instability-induced knee OA (verified by Safranin O staining) and sham-operated control mice were analyzed for expression of PGC-1α and FoxO3A by immunohistochemistry as described in Materials and Methods. The cellularity of each section was assessed by hematoxylin staining. Panels on the far left and far right show higher-magnification views of the boxed areas in the sham and OA panels, respectively. Boxed areas show the noncalcified region of both femoral and tibial cartilage. Original magnification × 10.
Increased phosphorylation of p65 NF-κB and procatabolic responses to IL-1β and TNFα after knockdown of PGC-1α and FoxO3A expression in human chondrocytes
Since decreased AMPK activity in human chondrocytes leads to induction of chondrocyte procatabolic responses to IL-1β and TNFα via activation of NF-κB (11), and PGC-1α and FoxO3A are effective inhibitors of NF-κB signaling (14,15), we tested whether PGC-1α and FoxO3A mediate the anticatabolic effect of AMPK. To do so, we used a loss-of-function approach to knock down expression of PGC-1α and FoxO3A via siRNA transfection in normal human knee chondrocytes (Figure 4A). Densitometric analysis indicated that PGC-1α expression and FoxO3A expression were reduced by 80% and 60%, respectively. We found that phosphorylation of p65 NF-κB was increased at baseline and was further enhanced in response to IL-1β and TNFα in either PGC-1α–knockdown or FoxO3A-knockdown chondrocytes (Figure 4B), suggesting increased NF-κB activation.
Figure 4.
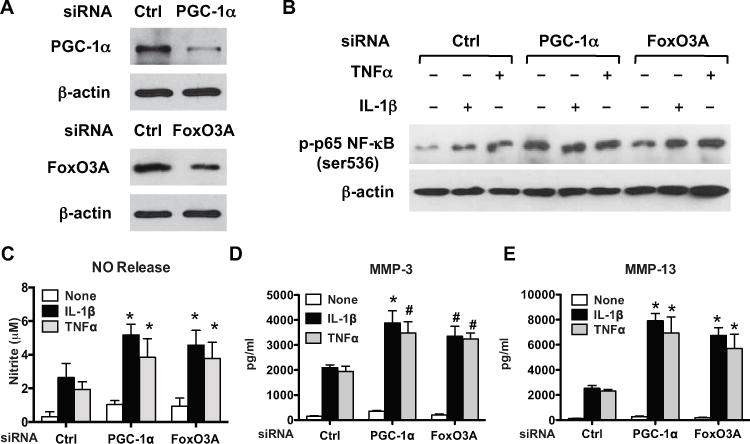
Enhanced phosphorylation of p65 NF-κB and procatabolic responses to interleukin-1β (IL-1β) and tumor necrosis factor α (TNFα) after knockdown of peroxisome proliferator–-activated receptor γ coactivator 1α (PGC-1α) and FoxO3A in chondrocytes. Cultured human knee chondrocytes (passage 1) were transfected with PGC-1α small interfering RNA (siRNA) or FoxO3A siRNA and the nontarget control for 48 hours as described in Materials and Methods. A, Western blot of the expression of PGC-1α and FoxO3A B, Western blot of the phosphorylation of the p65 subunit of NF-κB in transfected cells stimulated with IL-1β (10 ng/ml) or TNFα (20 ng/ml) for 6 hours. C–E, Release of nitric oxide (NO) (C), matrix metalloproteinase 3 (MMP-3) (D), and MMP-13 (E) in conditioned media from transfected cells stimulated with IL-1β (10 ng/ml) or TNFα (20 ng/ml) for 18 hours. NO was analyzed by Griess reaction, and MMP-3 and MMP-13 were analyzed by enzyme-linked immunosorbent assay. Bars show the mean ± SD. Data are representative of 3 individual experiments. * = P < 0.001; # = P < 0.01, versus control.
To further confirm this result, we examined the expression of cytosolic IκBα and nuclear p65 NF-κB in either PGC-1α–knockdown or FoxO3A-knockdown chondrocytes, as well as in chondrocytes overexpressing PGC-1α or FoxO3A in response to IL-1β (results are available from the author upon request). Stimulation of the control cells (either for siRNA or plasmid vector alone) with IL-1β resulted in decreased cytosolic IκBα expression (indicating degradation of IκBα) and increased nuclear p65 NF-κB expression. In chondrocytes with knockdown of PGC-1α or FoxO3A, there was a further reduction in cytosolic IκBα expression and increase in nuclear p65 NF-κB subunit expression. In contrast, in chondrocytes overexpressing PGC-1α and FoxO3A, there was less degradation of cytosolic IκBα and decreased nuclear expression of p65 NF-κB subunit. We also noticed that the basal level of cytosolic IκBα was slightly decreased in chondrocytes with knockdown of PGC-1α and FoxO3A, and was increased in chondrocytes overexpressing PGC-1α and FoxO3A. Increased NF-κB activation was associated with increased NO production and release of MMP-3 and MMP-13 (Figures 4C–E). These results indicated that decreased expression of PGC-1α and FoxO3A promoted chondrocyte procatabolic responses.
PGC-1α and FoxO3A at least partially mediate the capacity of AMPK to inhibit phosphorylation of p65 NF-κB and procatabolic responses induced by IL-1β in human chondrocytes
To further define whether PGC-1α and FoxO3A mediate inhibition of inflammatory cytokine–induced procatabolic effects by AMPK activation in human chondrocytes, we stimulated PGC-1α–knockdown and FoxO3A-knockdown chondrocytes, which had a knockdown efficiency of 82% and 86%, respectively (Figure 5A), with IL-1β in the presence or absence of A-769662 (0.25 mM). A-769662 significantly increased phosphorylation of AMPKα at the basal level (Figure 5B), inhibited dephosphorylation of AMPKα by IL-1β (Figure 5B), and attenuated IL-1β–induced phosphorylation of p65 NF-κB (Figure 5B) in the control cells. However, A-769662 only partially inhibited IL-1β–induced phosphorylation of p65 NF-κB in either PGC-1α–knockdown or FoxO3A-knockdown chondrocytes (Figure 5B).
Figure 5.
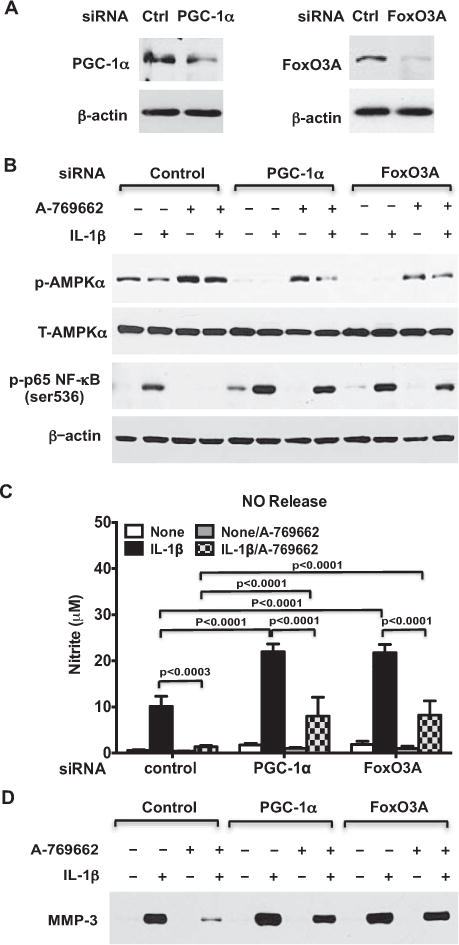
Pharmacologic AMP-activated protein kinase (AMPK) activation inhibits IL-1β–induced procatabolic responses via PGC-1α and FoxO3A. Cultured human knee chondrocytes (passage 1) were trans-fected with PGC-1α siRNA or FoxO3A siRNA and the nontarget control for 48 hours as described in Materials and Methods. A, Western blot of the expression of PGC-1α and FoxO3A B, Western blot of phosphory-lated and total AMPKα (T-AMPKα) and the phosphorylated p65 subunit of NF-κB in transfected cells stimulated with IL-1β (10 ng/ml) in the presence or absence of A-769662 (0.25 mM) for 18 hours. C and D, Release of NO (C) and MMP-3 (D) in conditioned media from transfected cells stimulated with IL-1β (10 ng/ml) in the presence or absence of A-769662 (0.25 mM) for 18 hours, analyzed by Griess reaction (for NO) and Western blotting (for MMP-3). Bars in C show the mean ± SD. Data are representative of 3 individual experiments. See Figure 4 for other definitions.
Similar to the results shown in Figure 4C, IL-1β–induced NO release was significantly enhanced in both PGC-1α–knockdown and FoxO3A-knockdown chondrocytes compared with control cells. A-769662 nearly completely attenuated IL-1β–induced NO release in the control cells. In comparison, A-769662 still significantly inhibited IL-1β–induced NO release in both PGC-1α–knockdown and FoxO3A-knockdown chondrocytes, but the inhibition of IL-1β–induced NO release by A-769662 was only 2.7-fold in PGC-1α–knockdown and 2.6-fold in FoxO3A-knockdown chondrocytes, compared with 8-fold in the control cells. In parallel, inhibition of IL-1β–induced MMP-3 release by A-769662 was much less in either PGC-1α– knockdown or FoxO3A-knockdown chondrocytes, as compared with the control cells. These results suggested that PGC-1α and FoxO3A at least partially transduced the capacity of AMPK to inhibit chondrocyte matrix catabolism.
PGC-1α and FoxO3A mediate the capacity of AMPK to limit oxidative stress in human chondrocytes
AMPK regulates redox balance (14,15). Since PGC-1α and FoxO3A up-regulate antioxidants, which include SOD2 and catalase (22,23), we tested whether PGC-1α and FoxO3A mediate the capacity of AMPK to up-regulate SOD2 and catalase expression. As shown in Figures 6A and B, expression of SOD2 and catalase was partially reduced in both PGC-1α–knockdown chondrocytes and FoxO3A-knockdown chondrocytes, but was increased by A-769662 in the control cells. Similarly, increased expression of SOD2 and catalase was observed in chondrocytes overexpressing PGC-1α or FoxO3A achieved via transfection (Figure 6C). However, A-769662 did not increase the expression of SOD2 and catalase in either PGC-1α–knockdown or FoxO3A-knockdown chondrocytes (Figures 6A and B). These data suggest that PGC-1a and FoxO3A, at least in part, mediate A-769662 to up-regulate SOD2 and catalase.
Figure 6.
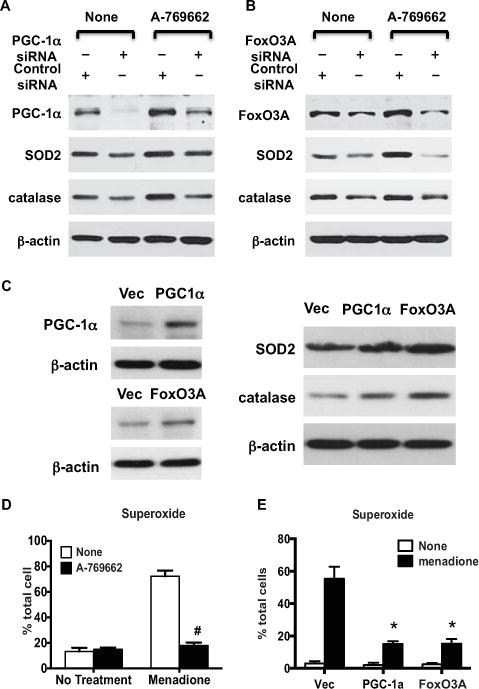
Pharmacologic AMP-activated protein kinase (AMPK) activation protects against excessive oxidative stress in chondrocytes via PGC-1α and FoxO3A. A and B, Western blots of the expression of PGC-1α, FoxO3A, superoxide dismutase 2 (SOD2), and catalase in cultured human knee chondrocytes (passage 1) transfected with PGC-1α siRNA (A) or FoxO3A siRNA (B) and the nontarget control and left untreated or treated with A-769662. C, Western blot of the expression of PGC-1α, FoxO3A, SOD2, and catalase in cultured human knee chondrocytes transfected with PGC-1α and FoxO3A cDNA plasmids and their vector (Vec) controls for 48 hours, as described in Materials and Methods. D, Superoxide generation, measured by MitoSOX staining, in cultured human knee chondrocytes (passage 1) treated with A-769662 (0.25 mM) for 1 hour, followed by menadione (100 μM) for 1 hour. Bars show the mean ± SD percent of the total number of cells (n = 3 replicates per condition). Data are representative of 3 individual experiments. # = P < 0.001 versus untreated control. E, Superoxide generation, measured by MitoSOX staining, in chondrocytes transfected with PGC-1α and FoxO3A cDNA plasmids and the vector control. Bars show the mean ± SD percent of the total number of cells (n = 3 replicates per condition). Data are representative of 3 individual experiments. * = P < 0.0001 versus vector control. See Figure 4 for other definitions.
Next, we tested whether A-769662 limits oxidative stress in chondrocytes. To do so, we pretreated human chondrocytes with A-769662 (0.5 mM) for 1 hour before stimulation with menadione (100 μM), which is an inducer of ROS, for 1 hour. Superoxide generation was then assessed using MitoSOX Red. As seen in Figure 6D, A-769662 almost completely abolished superoxide generation in response to menadione. Chondrocytes with overexpression of PGC-1α or FoxO3A exhibited decreased capacity to induce superoxide generation by menadione (Figure 6E), as demonstrated by a reduced number of cells showing positive MitoSOX Red staining (15% and 15.25%, respectively, compared with 55.3% in the vector control).
DISCUSSION
Impairments in AMPK, PGC-1α, and FoxO3A can be linked, and contribute to mitochondrial dysfunction, one of the hallmarks of aging (33). In this study, we discovered that the AMPK heterotrimer, bearing either the AMPKα1 or AMPKα2 subunit, supports constitutive expression of PGC-1α and FoxO3A in chondrocytes. Moreover, we observed that expression levels of PGC-1α and FoxO3A were significantly reduced in aging mouse knee cartilage, as well as in mouse OA cartilage, which was correlated with reduced phosphorylation of AMPKα. Our loss-of-function studies demonstrated increased phosphorylation of p65 NF-κB and procatabolic responses to IL-1β and TNFα in either PGC-1α–knockdown or FoxO3A-knockdown chondrocytes. PGC-1α is known to bind to the p65 subunit of NF-κB in human cardiac cells, and activation of NF-κB signaling increased the interaction between p65 and PGC-1α, which consequently reduces the expression of PGC-1α (34). FoxO3A is shown to inhibit NF-κB activity in T cells, and the lack of FoxO3A generated an autoinflammatory condition in mice (28). Hence, NF-κB signaling is likely one of the major chondroprotective targets of both PGC-1α and FoxO3A.
This study reinforced evidence that AMPK activation and related signaling decline during aging (15), and suggests that impaired AMPK signaling could contribute to aging as a major risk factor for the development of OA (3,4). Low-grade inflammatory processes present in aging tissues may be among the events suppressing AMPK signaling (15). Importantly, our results suggest that reduced capacity for AMPK activation and signaling in aging chondrocytes could lead to decreased expression of PGC-1α and FoxO3A, with consequently increased matrix catabolism, therefore contributing to OA development and progression.
The results of this study may help explain why an age-related decrease in expression levels of antioxidants such as catalase (35) and SOD2 (7) is seen in chondrocytes and why SOD2 expression is decreased in OA chondrocytes (24). In this study, we determined that activation of AMPK limits chondrocyte oxidative stress via PGC-1α and FoxO3A (14,15). Specifically, we observed that the highly selective allosteric AMPK activator A-769662 inhibited superoxide generation in chondrocytes in response to menadione. In addition, we demonstrated increased levels of expression of SOD2 and catalase, which correlated with decreased superoxide generation in chondrocytes overexpressing PGC-1α and FoxO3A. Our data suggest that pharmacologic activation of AMPK merits investigation as a potential approach to limit chondrocyte oxidative stress in OA.
PGC-1α and FoxO3A are closely related, since FoxO3A is a direct transcription regulator of PGC-1α, and PGC-1α itself can augment the transcription activity of FoxO3A (22). However, the molecular mechanisms by which AMPK regulates PGC-1α and FoxO3A in chondrocytes were not determined in this study. Significantly, in cells other than chondrocytes, AMPK has been demonstrated to induce PGC-1α expression by stimulating the cAMP response element binding protein (36) and possibly GATA-4 in cultured cells (37,38). AMPK can also phosphorylate PGC-1α at threonine 177 and serine 538 to directly enhance activity (39). Similarly, AMPK directly phosphorylates FoxO3A at 6 serine/threonine residues, and AMPK induces activation of FoxO3A-mediated transcription (40).
One limitation of the analyses in this study is that PGC-1α and FoxO3A have the potential to be chondroprotective by mechanisms beyond antioxidant regulatory genes. For example, target genes of the AMPK/FoxO3A pathway, associated with longevity, are involved in defense against not only oxidative stress, but also DNA damage, e.g., growth arrest and DNA damage–inducible protein 45 (41). Additionally, AMPK activation induces autophagy in chondrocytes (42,43), and autophagy is a protective or homeostatic mechanism in normal cartilage, but is deficient in aging and OA cartilage (44). AMPK/FoxO3A signaling can also promote autophagy partly by stimulating the expression of inducers of autophagy, including Bcl-2/adenovirus E1B 19-kd protein-interacting protein 3 and autophagy protein 12 (45). It would be of interest to ascertain if reduction in AMPK and FoxO3A activity contributes to deceased autophagy in aging and OA cartilage. Last, aging is associated with a lower renewal of mitochondria, which is mediated by deficiency in PGC-1α support of mitochondrial biogenesis (46). However, testing whether decreased AMPK/PGC-1α signaling directly causes decreased mitochondrial biogenesis and mitochondrial dysfunction in aged and OA chondrocytes was beyond the scope of this study, as were direct analyses of OA in young or old AMPK-deficient mice.
In conclusion, AMPK regulates chondrocyte catabolism and antioxidative stress capacity via both PGC-1α and FoxO3A. Reductions in AMPK and PGC-1α and FoxO3A are linked in aging cartilage, and share the potential to contribute to OA development and progression. Because pharmacologic activation of AMPK is achievable by drugs already used for arthritis and other conditions (e.g., sodium salicylate, high-dose aspirin, methotrexate, and metformin), and because AMPK activation is promoted by exercise and dietary factors (47,48), the effects of pharmacologic AMPK activators on articular cartilage degradation in OA merit further translational investigation.
Acknowledgments
We gratefully acknowledge the Sample Acquisition core of the NIH Program Project Grant on Cartilage Aging and Osteoarthritis (AG-007996) for human knee cartilage grading and chondrocyte isolation.
Supported by the Research Service of the Department of Veterans Affairs, the NIH (grants AG-007996 to Drs. Lotz and Terkeltaub, HL-077360 to Dr. Terkeltaub, and AR-106796 to Dr. Liu-Bryan), and the Arthritis Foundation (Innovative Science grant to Dr. Liu-Bryan).
Footnotes
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Liu-Bryan had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Lotz, Terkeltaub, Liu-Bryan.
Acquisition of data. Zhao, Petursson, Viollet, Liu-Bryan.
Analysis and interpretation of data. Liu-Bryan.
References
- 1.Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ [review] Arthritis Rheum. 2012;64:1697–707. doi: 10.1002/art.34453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goldring MB, Goldring SR. Osteoarthritis. J Cell Physiol. 2007;213:626–34. doi: 10.1002/jcp.21258. [DOI] [PubMed] [Google Scholar]
- 3.Lotz M, Loeser RF. Effects of aging on articular cartilage homeostasis. Bone. 2012;51:241–8. doi: 10.1016/j.bone.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loeser RF. Aging and osteoarthritis. Curr Opin Rheumatol. 2011;23:492–6. doi: 10.1097/BOR.0b013e3283494005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wolff KJ, Ramakrishnan PS, Brouillette MJ, Journot BJ, McKinley TO, Buckwalter JA, et al. Mechanical stress and ATP synthesis are coupled by mitochondrial oxidants in articular cartilage. J Orthop Res. 2013;31:191–6. doi: 10.1002/jor.22223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martin JA, Martini A, Molinari A, Morgan W, Ramalingam W, Buckwalter JA, et al. Mitochondrial electron transport and glycolysis are coupled in articular cartilage. Osteoarthritis Cartilage. 2012;20:323–9. doi: 10.1016/j.joca.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruiz-Romero C, Lopez-Armada MJ, Blanco FJ. Mitochondrial proteomic characterization of human normal articular chondrocytes. Osteoarthritis Cartilage. 2006;14:507–18. doi: 10.1016/j.joca.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 8.Ruiz-Romero C, Carreira V, Rego I, Remeseiro S, Lopez-Armada MJ, Blanco FJ. Proteomic analysis of human osteoarthritic chondrocytes reveals protein changes in stress and glycolysis. Proteom-ics. 2008;8:495–507. doi: 10.1002/pmic.200700249. [DOI] [PubMed] [Google Scholar]
- 9.Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev. 2009;89:1025–78. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- 10.Witczak CA, Sharoff CG, Goodyear LJ. AMP-activated protein kinase in skeletal muscle: from structure and localization to its role as a master regulator of cellular metabolism. Cell Mol Life Sci. 2008;65:3737–55. doi: 10.1007/s00018-008-8244-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Terkeltaub R, Yang B, Lotz M, Liu-Bryan R. Chondrocyte AMP-activated protein kinase activity suppresses matrix degradation responses to proinflammatory cytokines interleukin-1β and tumor necrosis α. Arthritis Rheum. 2011;63:1928–37. doi: 10.1002/art.30333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Petursson F, Husa M, June R, Lotz M, Terkeltaub R, Liu-Bryan R. Linked decreases in liver kinase B1 and AMP-activated protein kinase activity modulate matrix catabolic responses to biomechani-cal injury in chondrocytes. Arthritis Res Therapy. 2013;15:R77. doi: 10.1186/ar4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O’Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature. 2013;493:346–55. doi: 10.1038/nature11862. [DOI] [PubMed] [Google Scholar]
- 14.Salminen A, Hyttinen JM, Kaarniranta K. AMP-activated protein kinase inhibits NF-kB signaling and inflammation: impact on health span and lifespan. J Mol Med (Berl) 2011;89:667–76. doi: 10.1007/s00109-011-0748-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salminen A, Kaarniranta K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res Rev. 2012;11:230–41. doi: 10.1016/j.arr.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 16.Blanco FJ, Lopez-Armada MJ, Maneiro E. Mitochondrial dysfunction in osteoarthritis. Mitochondrion. 2004;4:715–28. doi: 10.1016/j.mito.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 17.Loeser RF. Molecular mechanisms of cartilage destruction in osteoarthritis. J Musculoskelet Neuronal Interact. 2008;8:303–6. [PubMed] [Google Scholar]
- 18.Henrotin YE, Bruckner P, Pujol JP. The role of reactive oxygen species in homeostasis and degradation of cartilage. Osteoarthritis Cartilage. 2003;11:747–55. doi: 10.1016/s1063-4584(03)00150-x. [DOI] [PubMed] [Google Scholar]
- 19.Rajagopalan S, Meng XP, Ramasamy S, Harrison DG, Galis ZS. Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro: implications for atherosclerotic plaque stability. J Clin Invest. 1996;98:2572–9. doi: 10.1172/JCI119076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petersen SV, Oury TD, Ostergaard L, Valnickova Z, Wegrzyn J, Thogersen IB, et al. Extracellular superoxide dismutase (EC-SOD) binds to type I collagen and protects against oxidative fragmentation. J Biol Chem. 2004;279:13705–10. doi: 10.1074/jbc.M310217200. [DOI] [PubMed] [Google Scholar]
- 21.Klamfeldt A, Marklund S. Enhanced breakdown in vitro of bovine articular cartilage proteoglycans by conditional synovial medium: the effect of superoxide dismutase and catalase. Scand J Rheumatol. 1987;16:41–5. [PubMed] [Google Scholar]
- 22.Olmos Y, Valle I, Borniquel S, Tierrez A, Soria E, Lamas S, et al. Mutual dependence of Foxo3a and PGC-1a in the induction of oxidative stress genes. J Biol Chem. 2009;284:14476–84. doi: 10.1074/jbc.M807397200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang C, Ji LL. Role of PGC-1α signaling in skeletal muscle health and disease. Ann N Y Acad Sci. 2012;1271:110–7. doi: 10.1111/j.1749-6632.2012.06738.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scott JL, Gabrielides C, Davidson RK, Swingler TE, Clark IM, Wallis GA, et al. Superoxide dismutase downregulation in osteoarthritis progression and end-stage disease. Ann Rheum Dis. 2010;69:1502–10. doi: 10.1136/ard.2009.119966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1a-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–73. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 26.Sano M, Schneider MD. Energizer: PGC-1 α keeps the heart going. Cell Metab. 2005;1:216–8. doi: 10.1016/j.cmet.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 27.Weydt P, Pineda VV, Torrence AE, Libby RT, Satterfield TF, Lazarowski ER, et al. Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1α in Huntington’s disease neurodegeneration. Cell Metab. 2006;4:349–62. doi: 10.1016/j.cmet.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 28.Lin L, Hron JD, Peng SL. Regulation of NF-κB, Th activation, and autoinflammation by the forkhead transcription factor Foxo3a. Immunity. 2004;21:203–13. doi: 10.1016/j.immuni.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 29.Miyamoto K, Araki KY, Naka K, Arai F, Takubo K, Yamazaki S, et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell. 2007;1:101–12. doi: 10.1016/j.stem.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 30.Li Q, Ceylan-Isik AF, Li J, Ren J. Deficiency of insulin-like growth factor 1 reduces sensitivity to aging-associated cardiomyocyte dysfunction. Rejuvenation Res. 2008;11:725–33. doi: 10.1089/rej.2008.0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamada K, Healey R, Amiel D, Lotz M, Coutts R. Subchondral bone of the human knee joint in aging and osteoarthritis. Osteoarthritis Cartilage. 2002;10:360–9. doi: 10.1053/joca.2002.0525. [DOI] [PubMed] [Google Scholar]
- 32.Glasson SS, Chambers MG, Van Den Berg WB, Little CB. The OARSI histopathology initiative—recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthritis Cartilage. 2010;18(Suppl 3):S17–23. doi: 10.1016/j.joca.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 33.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alvarez-Guardia D, Palomer X, Coll T, Davidson MM, Chan TO, Feldman AM, et al. The p65 subunit of NF-kB binds to PGC-1α, linking inflammation and metabolic disturbances in cardiac cells. Cardiovasc Res. 2010;87:449–58. doi: 10.1093/cvr/cvq080. [DOI] [PubMed] [Google Scholar]
- 35.Brandl A, Hartmann A, Bechmann V, Graf B, Nerlich M, Angele P. Oxidative stress induces senescence in chondrocytes. J Orthop Res. 2011;29:1114–20. doi: 10.1002/jor.21348. [DOI] [PubMed] [Google Scholar]
- 36.Thomson DM, Herway ST, Fillmore N, Kim H, Brown JD, Barrow JR, et al. AMP-activated protein kinase phosphorylates transcription factors of the CREB family. J Appl Physiol. 2008;104:429–38. doi: 10.1152/japplphysiol.00900.2007. [DOI] [PubMed] [Google Scholar]
- 37.Handschin C, Rhee J, Lin J, Tarr PT, Spiegelman BM. An autoregulatory loop controls peroxisome proliferator-activated receptor γ coactivator 1α expression in muscle. Proc Natl Acad Sci USA. 2003;100:7111–6. doi: 10.1073/pnas.1232352100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Irrcher I, Ljubicic V, Kirwan AF, Hood DA. AMP-activated protein kinase-regulated activation of the PGC-1α promoter in skeletal muscle cells. PLoS One. 2008;3:e3614. doi: 10.1371/journal.pone.0003614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc Natl Acad Sci U S A. 2007;104:12017–22. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Greer EL, Oskoui PR, Banko MR, Maniar JM, Gygi MP, Gygi SP, et al. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J Biol Chem. 2007;282:30107–19. doi: 10.1074/jbc.M705325200. [DOI] [PubMed] [Google Scholar]
- 41.Tran H, Brunet A, Grenier JM, Datta SR, Fornace AJ, Jr, DiStefano PS, et al. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science. 2002;296:530–4. doi: 10.1126/science.1068712. [DOI] [PubMed] [Google Scholar]
- 42.Srinivas V, Bohensky J, Shapiro IM. Autophagy: a new phase in the maturation of growth plate chondrocytes is regulated by HIF, mTOR and AMP kinase. Cells Tissues Organs. 2009;189:88–92. doi: 10.1159/000151428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bohensky J, Leshinsky S, Srinivas V, Shapiro IM. Chondrocyte autophagy is stimulated by HIF-1 dependent AMPK activation and mTOR suppression. Pediatr Nephrol. 2010;25:633–42. doi: 10.1007/s00467-009-1310-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carames B, Taniguchi N, Otsuki S, Blanco FJ, Lotz M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 2010;62:791–801. doi: 10.1002/art.27305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van der Vos KE, Coffer PJ. The extending network of FOXO transcriptional target genes. Antioxid Redox Signal. 2011;14:579–92. doi: 10.1089/ars.2010.3419. [DOI] [PubMed] [Google Scholar]
- 46.Gomez-Cabrera MC, Sanchis-Gomar F, Garcia-Valles R, Pareja-Galeano H, Gambini J, Borras C, et al. Mitochondria as sources and targets of damage in cellular aging. Clin Chem Lab Med. 2012;50:1287–95. doi: 10.1515/cclm-2011-0795. [DOI] [PubMed] [Google Scholar]
- 47.Hardie DG. Energy sensing by the AMP-activated protein kinase and its effects on muscle metabolism. Proc Nutr Soc. 2011;70:92–9. doi: 10.1017/S0029665110003915. [DOI] [PubMed] [Google Scholar]
- 48.Spindler SR. Caloric restriction: from soup to nuts. Ageing Res Rev. 2010;9:324–53. doi: 10.1016/j.arr.2009.10.003. [DOI] [PubMed] [Google Scholar]