

Significance
The somatic hypermutation of immunoglobulin (Ig) variable (V) regions is required to produce high-affinity protective antibodies. Activation-induced deaminase (AID) initiates the accumulation of mutations in the V regions at frequencies that are a million times higher than the normal mutation rates in non-Ig genes. On the basis of the in vivo pattern of mutations in the highly used IGHV3-23*01 human V region and the manipulation of DNA sequence motifs in that V region in a mutating B-cell line and biochemically, we have concluded that particular DNA sequence motifs focus and influence AID activity on those parts of the V region that affect antigen binding and should be considered in vaccine strategies.
Keywords: AID, cytosine deamination, complementarity-determining regions, somatic hypermutation, IGHV3-23
Abstract
Activation-induced deaminase (AID) mediates the somatic hypermutation (SHM) of Ig variable (V) regions that is required for the affinity maturation of the antibody response. An intensive analysis of a published database of somatic hypermutations that arose in the IGHV3-23*01 human V region expressed in vivo by human memory B cells revealed that the focus of mutations in complementary determining region (CDR)1 and CDR2 coincided with a combination of overlapping AGCT hotspots, the absence of AID cold spots, and an abundance of polymerase eta hotspots. If the overlapping hotspots in the CDR1 or CDR2 did not undergo mutation, the frequency of mutations throughout the V region was reduced. To model this result, we examined the mutation of the human IGHV3-23*01 biochemically and in the endogenous heavy chain locus of Ramos B cells. Deep sequencing revealed that IGHV3-23*01 in Ramos cells accumulates AID-induced mutations primarily in the AGCT in CDR2, which was also the most frequent site of mutation in vivo. Replacing the overlapping hotspots in CDR1 and CDR2 with neutral or cold motifs resulted in a reduction in mutations within the modified motifs and, to some degree, throughout the V region. In addition, some of the overlapping hotspots in the CDRs were at sites in which replacement mutations could change the structure of the CDR loops. Our analysis suggests that the local sequence environment of the V region, and especially of the CDR1 and CDR2, is highly evolved to recruit mutations to key residues in the CDRs of the IgV region.
After an encounter with antigen and subsequent migration into the germinal centers of the secondary lymphoid organs, B cells undergo a regulated cascade of mutational events that occur at a very high frequency and are largely restricted to the variable (V) and switch (S) regions of the Ig heavy chain locus and the V region of the light chain locus. These mutagenic events are responsible for the somatic hypermutation (SHM) of the V regions and the class switch recombination of the constant (C) regions that are required for protective antibodies (1, 2). Both SHM and class switch recombination are initiated by activation-induced deaminase (AID) that preferentially deaminates the dC residues in WRC (W = A/T, R = A/G) hotspot motifs at frequencies 2–10-fold higher than SYC (S = G/C; Y = C/T) cold spots (3–7). During V region SHM, the resulting dU:G mismatch can then be replicated during S-phase to produce transition mutations, be processed by uracil-DNA glycosylase 2 and apurinic/apyrimidinic endonucleases through the base excision repair pathway to produce both transitions and transversions (8–10), or be recognized by MutS homolog (MSH)2/MSH6 of the mismatch repair (MMR) complex that recruits the low-fidelity polymerase eta (Polη) to generate additional mutations at neighboring A:T residues (11).
The specificity of AID targeting to the Ig gene has been under intense investigation. Studies have shown that AID deamination and mutagenesis targets single-stranded DNA substrates generated during transcription (12, 13). Transcription-associated proteins and RNA processing factors also participate in the AID mutational process and, in some cases, physically interact with AID (14–17). In addition, other transacting proteins (18, 19), including chaperones (20), chromatin modifiers and remodelers (21–23), cell cycle regulators (24), developmental factors (25), and cis-acting sequences (26, 27), appear to affect mutations. However, all of these factors also have pleiotropic effects on non-Ig genes in B cells, so they do not appear to be solely responsible for the targeting of AID-induced mutations to the Ig V. Because many non-Ig genes are also highly transcribed in activated B cells and AID appears to occupy many sites in such cells (28, 29) and can also cause mutations in non-B cells, it is important to understand why the very high rate of mutation in the SHM is not seen in other highly expressed genes in B cells. In addition, we are still learning about how the V regions can undergo such high rates of mutation and still assemble their heavy and light chain V regions to produce a stable antigen-binding site that can undergo affinity maturation and changes in fine specificity. Although it is clear that mutations in the complementary determining regions (CDRs) play a critical role in antigen binding, recent studies reveal that some of these highly mutated sites in the CDRs do not directly interact with antigen but have an important role in generating protective broadly neutralizing antibodies to influenza and HIV (1, 30, 31).
Taken together, these findings suggest that the DNA sequence of Ig V regions has evolved so that AID will mutate certain subregions while protecting those parts of the V region that are required to maintain the overall structure and function of the protein (32). This evolutionary process is reflected in the fact that codons used in the CDRs are more likely to create AID hotspots and to result in replacement mutations than the codons used in the frameworks (FWs) (7, 32, 33). There is the potential for an even more intricate process (32), as hotspots for AID can assume a variety of sequences, each undergoing mutations at different frequencies (6, 7, 34). Particularly interesting is the WGCW motif, which provides two overlapping WRC motifs on opposing strands of the DNA (3, 5, 7). These overlapping AID hotspots can have very high rates of mutation in the V and S regions (3, 7, 35–39), but it is unclear whether they influence the global mutation pattern of the whole V region.
To examine this, we have studied the frequency and distribution of mutations in IGVH3-23*01. The IGVH3 family represents about half of the functional genomic human V regions (40), and IGVH3-23 is particularly interesting because it is the most commonly used V region during normal immune responses (41–43), as well as in some B-cell malignancies (44). Fortunately, there is a large database available for bioinformatic analysis with independently mutated productive and nonproductive human IGHV3-23*01 sequences (45, 46). By analyzing the patterns of mutation in the nonproductive genes in this memory B-cell database, we found that the overlapping AID hotspot (OHS), WGCW, especially AGCT, has a marked propensity to undergo AID deamination, especially in CDR2 of human IGHV3-23*01, and that if the OHS in CDRs do not undergo mutation, there is a decrease in the mutation frequency throughout the V region. We used a biochemical assay and the Ramos cell line where we could modify some of the AGCTs in the CDR1 and CDR2 by site-directed mutagenesis to analyze their relative susceptibility to mutation and found that there is a significant decrease not only in the frequency of mutation in the overlapping hotspots but also throughout the V region, when this motif was removed from the CDRs. These findings suggest that additional levels of complexity exist in the sequence of the V region that facilitate the generation of highly protective antibodies.
Results
Overlapping Hotspots in Human IGHV3-23*01 Accumulate High Frequencies of Mutation in Vivo.
To assess the role of the local sequence context in recruiting AID-induced mutations in human heavy chain V regions, we first reexamined the relative frequency and distribution of 2,657 independent mutations in IGHV3-23*01 V regions from circulating memory B cells of 24 normal individuals reported by Ohm-Laursen and colleagues (45, 46). These mutations were from 438 V regions that had premature stop codons or frame shifts that likely arose during the creation of the CDR3 and were extracted by Ohm-Laursen and colleagues (45, 46) from a larger database of IGHV3-23*01 V region sequences. Because the mutations were at sites of variable, diversity, and joining (VDJ) region rearrangement, the V regions are presumed to have been nonproductive throughout the differentiation of the B-cell clone that contained them, so the subsequent mutations that arose in those V regions were not subjected to antigen selection (45, 46). Because none of the V regions shared the same CDR3, the observed mutations arose from different B-cell clones, and each of these nonproductive V regions reflects sets of independent mutational events (45, 46). The upper panel of Fig. 1 shows the frequency of mutations that accumulated at each position. The black vertical lines are mutations in dC on either strand and are presumed to have been initiated by the direct action of AID, whereas the gray vertical lines are mutations in A:T residues and are presumed be a result of error-prone MMR (47). After recent precedents (1, 30, 48), the CDRs indicated by the horizontal gray bars at the top of the figure are based on the Kabat criteria (49) combined with analysis of the crystal structure of the IGHV3-23*01 V region, rather than the IMGT definition (48). It is obvious that the frequency of these independent and unique somatic mutations in AID hotspots, including the overlapping hotspots, differed greatly at different sites throughout the V region, with some of the higher frequencies occurring in CDR1 and CDR2 (Fig. 1).
Fig. 1.
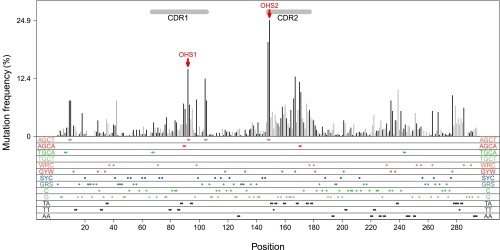
Demonstration of the mutation distribution of IGHV3-23*01 in vivo. The database includes V regions that are mutated (264) and not mutated (174). Each CDR3 was unique; they are not shown here. The x axis shows the mutation sites within the V region. The y axis is the mutation frequency for each site. Black lines show the mutation frequencies that occurred at G:C sites. Gray lines demonstrate the mutation frequencies at A:T sites. Each colored dot in the bottom panels represents an AID hot/cold/neutral spot or Polη hotspot, as labeled at both edges. Double dots are AID overlapping hotspots (WGCW) or two adjacent Polη hotspots. The red arrows point to the OHS1 and OHS2 overlapping hotspots, as labeled. Horizontal gray bars on top are CDRs as labeled.
Characteristics of AID Hotspot Motifs and Broader Sequence Requirements.
At least some of the reasons for these different frequencies of mutation became obvious when we examined the motifs that were associated with the mutations. These are annotated in the lower horizontal panels of Fig. 1, where the different motifs displayed at the edges of each panel are represented by colored dots to indicate where they are located within the V region sequence. For example, the first horizontal bar in the lower panel shows a red dot at residue 149, representing the C in the AGCT hotspot motif on the upper strand and an adjacent red dot representing the C at residue 148 in the 3′-TCGA-5′ AID hotspot on the lower strand. This overlapping AGCT hotspot in CDR2 (OHS2 marked by the arrow) has accumulated approximately two times as many mutations as each of the two AGCT overlapping hotspots in CDR1 (both P values are less than 10−6 in χ2 test), and almost three times as many mutations as there are in the AGCT at residues 9 and 10 in FW1 (P < 10−6). The AGCTs in CDR1 and CDR2 have more mutations than the AGCA in each of those CDRs. The AGCT in the middle of CDR1 (OHS1 marked by an arrow in Fig. 1) is also part of an adjoining AGCA overlapping hotspot (AGCAGCT) on the same strand, resulting in a tight cluster of four hotspots on the two strands (Fig. 1 and Fig. S1).
The relatively high frequency of mutations in OHS2 and in other G:C and A:T bases in CDR2 and of G:Cs in other sites such as around residue 280 in FW3 suggest that some neighboring sequences could be contributing to the high frequency of mutation of certain parts of the V regions. It is particularly interesting that CDR1 and CDR2 are relatively devoid of AID cold spots (SYC and GRS, where R = A/G, S = G/C and Y = C/T) and are rich in AID hotspots (Fig. 1). Overall, there is a statistically significant enrichment of hotspots in CDRs versus FWs (Fig. S2A; Fisher test, P = 7 × 10−4). Approximately 50% of the G:C bases in CDR1 and CDR2 are in AID hotspots (Fig. S2A), and CDR1 has 15% of the remaining G:C bases in cold spots; there are no cold spots in CDR2. In contrast, less than 22% of the G:C bases in the FWs are in AID hotspots. In addition, there are more WA Polη hotspots in CDR1 and CDR2 than in FW1 and FW2. Approximately 45% of the A:T residues in CDR1 and CDR2 are in Polη hotspots, and most of these are in TA motifs, which are more susceptible to errors (50–52), whereas in FW1 and FW2, the percentage of A:T residues that are in TA motifs is much lower (Fig. S2B). Again, this difference is significant (P = 6.5 × 10−3). The clusters of TA Polη hotspots in CDR2 and, to a lesser extent, in CDR1 are associated with many Polη mutations. FW3 has as many Polη mutations as CDR1, but most of them are in AA motifs, which is very infrequent in the other parts of the V region (Fig. 1 and Fig. S2B). This nonrandom distribution of motifs and the frequency of associated mutations suggest that the overall sequence context, as well as specific motifs, is important in targeting AID and Polη mutations.
Association of Mutations in the OHS1 and OHS2 with Mutations Throughout the V Region.
Because the relative frequency of mutation in OHS2 and, to a lesser degree, in the other overlapping hotspots is so high, it is possible that those mutations serve as an activation or entry site for AID mutations of the whole V region. To address whether the presence or absence of mutations in the overlapping hotspots is associated with differences in the characteristics or distribution of mutations elsewhere in the V region, we compared the mutations per V region and the overall mutation frequency in V regions that did and did not have mutations in each of the overlapping AGCT AID hotspots (Fig. 2). To focus on the mutations in the rest of V region, we subtracted the mutations that had occurred in the GC bases of that particular AGCT. As shown in Fig. 2A, in those V regions where OHS2 was mutated on both or either strand (OHS2 mutated), there were many more mutations elsewhere in those V regions than in V regions that did not have any mutations in OHS2 (13 vs. 4). This was associated with a significantly higher overall frequency of mutations throughout the V region in which OHS2 had undergone mutation compared with those V regions in which OHS2 is not mutated (4.5 × 102 vs. 1.5 × 102 mutations/bp; P < 10−6). This decrease is reflected by the mutations at both G:C and A:T bases throughout the whole V region (Fig. 3). This would be expected because most of the mutations in A:T are the result of the recruitment of error-prone mismatch repair by the G:U mismatch created by AID. Even though there were many fewer mutations throughout the V region in those Vs that did not undergo mutation in OHS2 (Fig. 2A), the spatial distribution of mutations, excluding OHS2 itself, is similar (Pearson correlation of site-by-site mutation frequencies, r = 0.70; P < 10−6). In particular, the absence of mutations in OHS2 was associated with fewer mutations in OHS1, as well as in the AGCTs at the other overlapping hotspots in FW1 and at the 3′ end of CDR1 (Fig. 3). The effect of the mutations in overlapping hotspots in OHS1 (arrow in Fig. 1) was similar to that of OHS2 (Fig. 2B), with a statistically significant decrease in the overall frequency of mutation elsewhere in the V region, as well as fewer mutations/V regions.
Fig. 2.

Analysis of the mutations in subsets of the memory B-cell database. (A) Analysis of OHS2 mutated and unmutated V regions. The first subset in the first column is the V regions that have both GC in OHS2 mutated or either G or C mutated (shown in the arrow in Fig.1). The second column is the V regions that have no mutations at either G or C in OHS2 and one or more additional mutations. The mutation frequencies do not include the mutations that occurred at the GC sites in OHS2, so that only the mutations outside of those GC sites are compared. The bolded number represents the frequency of mutation in V regions in which neither G nor C of OHS2 was mutated and represents a significantly decreased frequency of mutation compared with those V regions in which G or C of OHS2 or both GC were mutated [P < 10−6, calculated in SHMTool program in which the χ2 test is implemented (76)]. (B) Analysis of OHS1 mutated and unmutated V regions. (C) Analysis of 104/105 GC in CDR1 mutated and unmutated V regions. (D) Analysis of 9/10 GC sites in FW1 mutated and unmutated V regions. In B, C, and D, the same analysis was performed as in A, and the bolded values are significantly decreased (P < 10−6).
Fig. 3.

Comparison of the two subsets of the memory B-cell database. (Top) V regions that have mutations in both or neither CDR2 overlapping hotspots mutated (OHS2 mutated). (Bottom) V regions in which neither strand of OHS2 is mutated (OHS2 unmutated) and the V regions have mutations at other sites. The red arrows point to OHS1 and OHS2. The gray bars on top are the CDRs as labeled. The vertical black lines show the frequency of mutations at G:C sites, and the vertical gray lines show the frequency of mutations at A:T sites. The x axis shows the position of each of the mutations. The y axis shows the mutation frequency.
The decrease in mutations throughout the IGHV3-23*01 V region associated with the lack of mutation in OHS1 and in OHS2 suggested that OHS1 and OHS2 might serve as activation or entry sites for AID to initiate the process of SHM in a particular V region. However, these were purely correlations, and it is difficult to imagine how an AID-induced mutation at one dC in the V region would influence the frequency of mutations at distant dCs, as it appears that most of the time there is only one AID-induced mutation during each cell cycle (53–55). This raised the alternative possibility that the results described in Figs. 2 A and B and 3 are the result of AID mutation being a stochastic process, in which the frequency at each site reflects the susceptibility of that site to AID deamination. To examine these two different hypotheses further, we determined whether mutations in the other AGCTs in IGHV3-23*01 were associated with changes in the frequency of mutation elsewhere in those V regions. The GC sites in the AGCT motif at residues 104 and 105 in the 3′ end of CDR1 (Fig. 1 and Fig. S1) were as highly mutated as OHS1. As with OHS1 and OHS2, the lack of mutations of the C on either strand in the AGCT 104/105 was associated with a decrease in mutation throughout the V region (Fig. 2C). We also examined the AGCT at residues 9 and 10 in FW1 (Fig. 1 and Fig. S1). This site is preceded by a TGCA overlapping hotspot, but there are very few mutations in either G or C of TGCAs in IGHV3-23*01 (Fig. 1). As can be seen in Fig. 2D, the lack of mutations in the AGCT in residues 9 and 10 in FW1 was, as with OHS1 and OHS2, associated with a significantly lower frequency of mutations throughout the rest of the V region.
The finding that the lack of mutation at each of the AGCTs was correlated with a similar decrease in the frequency of mutation throughout the V regions made it less likely that the AGCTs in CDR1 and CDR2 were routinely acting as the activation or entry site for the whole V region. Rather, these findings suggested that the frequency of mutations at each of the AGCTs was largely the result of a stochastic process that reflected their inherent susceptibility to AID and the overall mutational state of that particular B cell at the time that mutation was occurring. Nevertheless, it was quite possible that on some occasions, the initial mutation, for example, at OHS2 did act as an entry or activation site for subsequent mutations in that V region. We therefore set out to further evaluate the role of the overlapping AGCT hotspot in the V region mutagenesis process experimentally.
OHS Targeting in Vitro.
Because the substrate for AID in vivo is ssDNA (12), we used a biochemical system to compare the ability of purified AID to mutate the germ line IGHV3-23*01 WT gene (3-23WT) and a variant IGHV3-23*01 modified gene (3-23Mod) in which OHS1 and OHS2 and their immediate surrounding sequences had been modified to replace the overlapping hotspot motifs with neutral and cold spots (Fig. 4A). We focused on OHS2 because it was the most mutated AGCT in IGHV3-23*01 in unselected memory B cells, and the V regions that did not undergo mutation at that site had the fewest mutations elsewhere in the V region (Figs. 2 and 3). We also modified the AGCT in OHS1 because it and OHS2 were embedded in the same CAGCTAT motif that occurs only at these two sites in IGHV3-23*01, and the lack of mutation in OHS1 was also associated with a decrease in mutation elsewhere in the V region (Fig. 2B). The 6 bp that were changed in CDR1 and the six different changes that spanned a 10-bp stretch in CDR2 preserved most of the amino acid sequence. These changes were made because the same modified V region was going to be used in Ramos cell (see following), and we wanted the IgM antibody to continue to be expressed on the Ramos cell surface.
Fig. 4.

In vitro analysis. (A) DNA modification schematic in which 3-23WT is the IGHV3-23*01 WT germ line DNA sequence and 3-23Mod shows the modified IGHV3-23*01 DNA sequence. The highlighted nucleotides in red are the AGCT sites in CDR1 and CDR2. The bold nucleotides are the differences between 3-23WT and 3-23Mod DNAs. The 6-bp AGCAGC in CDR1 of 3-23WT are changed to TCGTCT in 3-23Mod, and another 6-bp AGC...AGC in CDR2 of 3-23WT are changed to GAT...TCG in 3-23Mod. The light blue boxes mark the CDRs as labeled but are not drawn to scale. (B) Mutation frequency generated by the in vitro assay. This table shows the untranscribed (Top) strand DNA mutation frequencies after 1- and 5-min incubations with AID, as labeled on the top of the table. The first row shows the number of V regions sequenced, and the second row tells the percentage of the mutated V regions in the number of the first row. “Average mutation number per V” shows the total mutations divided by number of mutated V regions. “Overall frequency” shows the total mutation frequencies in all of the V regions shown in the first row (mutations/bp). “Frequency in unmodified region” refers to the mutation frequency in the V region excluding the modified area. P values are shown after each comparison between 3-23WT and 3-23Mod and calculated in SHMTool, as described in ref. 76. The bold numbers show the significantly decreased values in 3-23Mod compared with 3-23WT.
As shown in Fig. 4, when the top strand of the 3-23WT (Fig. 4B) and modified IGHV3-23*01 genes (3-23Mod) were incubated with purified AID (6) for 1 min, there was a trend toward fewer mutations in Cs in the modified V regions, but this difference was only marginally significant (P = 0.036). However, this difference was more focused on the overlapping hotspots because the other parts of the V showed no difference (“Frequency in unmodified region” in Fig. 4B; P = 0.63). After 5 min of exposure to AID, there were significantly more mutations in the 3-23WT than 3-23Mod (P < 0.001). This difference was seen not only in the frequency of mutation throughout the V region, including the overlapping hotspots, but also when the mutations in OHS1 and OHS2 and their surrounding sequences in the WT and the modified samples were removed from the calculation and only the rest of the V region (unmodified region) was considered (Fig. 4B). There were no significant differences at either 1 or 5 min when ssDNA representing the bottom strand was incubated with AID. Others have also seen differences between biochemical studies of the top (untranscribed) and bottom (transcribed) strands and attributed these to the fact that the in vivo bottom strand is heavily occupied by RNA polymerase and its many associated factors, including exosomes, so it is more difficult to duplicate the process biochemically (14, 51). Because AID is limited and processive through a slide-and-jump mechanism in this biochemical assay (56, 57), these results suggest that even on single-stranded DNA, the AGCT motifs were more likely to undergo AID-induced deamination and, through AID’s processivity in this in vitro system, increase the frequency of mutation throughout the V region.
The Lack of the OHSs Decreases the Mutation Frequency in Ramos Cells.
The analysis of the unselected Ohm-Laursen (46) memory B-cell data (Figs. 1 and 2) revealed that the deamination of the Cs in OHS2 was associated with different frequencies of mutation in other parts of the V region. However, because in the memory B-cell database there are multiple mutations in each V region, and each of the V regions comes from a clonally different memory B cell that has undergone many rounds of mutation, it is difficult to deduce the order of the mutational events. In an attempt to identify the location of the initial deaminations and the effect of those events on the rest of the V region, we examined mutations of the same germ line IGHV3-23*01 in the Ramos cultured human B-cell line that is like the unselected memory B-cell database and does not undergo antigen selection. We used recombinase-mediated cassette exchange (RMCE) (38, 58) to introduce the WT IGHV3-23*01 (3-23WT) and two modified versions of IGHV3-23*01 (3-23Mod and 3-23AATT) as single copies in the right orientation into the endogenous Ig heavy chain V-region locus of the Ramos human Burkitt’s lymphoma cell line (58). The Ramos cells are a germinal center-like B-cell line that constitutively expresses AID at a low level (59) and undergoes SHM at a rate that is 20–50 times lower than the estimates for the rate of mutation in vivo (4, 58–60). After RMCE replacement, cells were cultured for ∼3 mo. The inserted germ line IGHV3-23*01 was sequenced by paired-end 250 × 250-bp deep sequencing, using the Illumina MiSeq platform. We combined the mutations from three subclones of one Ramos clone transfected with the germ line IGHV3-23*01 (3-23WT) and two subclones from another independently transfected Ramos 3-23WT clone. Although the frequency of mutation in Ramos was much lower than in vivo (see the different scales in Ramos and memory B cell in Fig. 5), the distribution and relative frequency of mutations in both G:C and A:T bases in the various parts of the V region in vivo and the Ramos cell line were quite similar (r = 0.66, Pearson correlation of site by site mutation frequencies) and were even higher if we compare only the mutations at G:C sites (r = 0.72) (Fig. 5). Nevertheless, there are clear differences. The relatively highly mutated OHS1 in vivo has relatively fewer mutations in Ramos, and the GC in the overlapping hotspot at residue 104 and 105 at the 3′ end of CDR1 has relatively more mutations (Fig. 5). In addition, the AGCT at residues 9 and 10 have more mutations than OHS1 and as many as the AGCT at residues 104/105. As has been reported previously for the endogenous Ramos V region (58, 59), 90% of the mutations were in G:C sites, and 10% were in A:T residues, most of which were in Polη hotspots. This relatively lower frequency of mutation in A:T residues in the Ramos cell line has been reported by others (4, 58, 60). Consistent with this lower frequency of mutations at A:T residues in Ramos, there are many fewer mutations in the A:T residues just 3′ to OHS2 and at the 3′ end of the V region (Fig. 5) than are seen in vivo (Fig. 1). Because the OHS2 still had by far the most mutations in the Ramos cell line expressing the WT IGHV3-23*01, and OHS1 was within the same CAGCTAT context, we introduced the same modified IGHV3-23*01 (3-23Mod) that we had used for the biochemical experiments into the endogenous heavy chain locus in Ramos cells (Fig. 6A).
Fig. 5.

Comparison of the mutation pattern of IGHV3-23*01 in Ramos cell lines (Ramos 3-23WT) with that from memory B-cell database (memory B-cell). The y axis shows the mutation frequency. (Upper) Y axis is gapped from 0.012 to 0.098 and is a different scale than in the lower panel because of the lower overall frequency of mutations and the relatively higher frequencies in OHS2 in Ramos. CDRs are showed in the gray horizontal boxes. Black lines and gray vertical bars represent G:C and A:T mutations frequencies, respectively. Red arrows point to OHS1 and OHS2.
Fig. 6.

DNA modification in both CDR1 and CDR2 changed the mutation frequency of V region in Ramos. (A) DNA modification schematic is the same as shown in Fig. 4A. (B) Mutation analysis in 3-23WT Ramos subclones and DNA-modified Ramos subclones (3-23Mod). The first column has the values from a pool of all of the V regions of five 3-23WT subclones, and the second column contains the values from a pool of five 3-23Mod subclones. The first row shows the numbers of the total V regions obtained from the deep sequencing for 3-23WT and 3-23Mod. In the parenthesis are the numbers of mutated V regions. The second row has the percentages of mutated V regions in total V regions shown in the first row. “Overall frequency” represents the mutations in all of the V regions (shown in the first row) that occurred throughout the whole V region, including the mutations that occurred in the modified regions, calculated in SHMTool (76). “Frequency in unmodified region” shows the mutation rates after the mutations in the modified area (12-bp differences totally in CDR1 and CDR2 between 3-23WT and 3-23Mod) are excluded from 3-23WT and 3-23Mod V regions, and they are also calculated in SHMTool. Bold values in 3-23Mod for mutation frequencies are significantly decreased values compared with 3-23WT (P < 10−6, calculated by SHMtool). The bold numbers are percentage of mutated V regions, the P value is <0.001 (χ2 test). (C) Mutation distribution comparison between 3-23WT and 3-23Mod. Upper part is 3-23WT, lower part is 3-23Mod, as labeled. The x axis gives the site for each mutation of V region. The y axis shows the mutation frequency for each site. Each vertical line represents the mutation frequency at the corresponding site, as labeled in x axis. Red arrows point to OHS1 and OHS2.
As shown in Fig. 6B, using deep sequencing, we obtained 2,587,368 V region sequences in total and 715,886 mutated V regions from a combination of five subclones of two independently transfected Ramos clones expressing the 3-23WT V region, and 1,260,675 V regions in total and 76,977 mutated V regions from a combination of five Ramos cell clones expressing the modified IGHV3-23*01 (3-23Mod) V region in which OHS1 and OHS2 and their neighboring bases had been replaced (Fig. 6A). In the WT Ramos IGHV3-23*01, 27.7% of the V regions were mutated, whereas in the modified Ramos with the modified IgVH3-23*01, only 16% of the Vs are mutated (Fig. 6B). This indicates that the loss of OHS1 and OHS2 and their neighboring sequences resulted in a 42% decrease in the number of V regions mutated (P < 0.001). Similarly, the loss of OHS1 and OHS2 and their neighboring bases also resulted in a statistically significant (P < 10−6) decrease in the overall frequency of mutation (16.4 × 10−4 vs. 5.8 × 10−4) (Fig. 6B). This decrease is largely a result of the loss of mutations in the modified area of 3-23Mod, especially at the OHS2 site, which is so highly mutated in 3-23WT (Fig. 6C). However, there still is a significant decrease in mutation frequency of 3-23Mod compared with 3-23WT when the mutations in the modified region were subtracted from both the WT and modified frequencies, so we could evaluate only the effect on the other parts of the V region (“Frequency in unmodified region” in Fig. 6B). This suggests that in the Ramos cells, OHS1 and OHS2 and their surrounding sequences have an effect on the mutations on the rest of the V region. It is important to note that although the frequency of mutations outside the overlapping hotspots is relatively low in Ramos, as shown in Fig. 5, there are many mutations distributed throughout the V region. Because we only sequenced the V regions from clones in which the mRNA and protein of AID were similar in the WT and modified clones, this difference was not a result of differences in AID abundance.
To examine the contribution of a single AGCT motif, as we had done in Fig. 2A for OHS2 in vivo, we compared the effect of changing only the GC sites in OHS2 in Ramos cells by creating cells with a V region containing an AATT in place of the AGCT in OHS2 (3-23AATT; Fig. 7A). We combined all of the sequences from four subclones of 3-23AATT and compared this more restricted change to the same 3-23WT data shown in Fig. 6 because the experiments in Fig. 6 and Fig. 7 were all done in parallel. The absence of only the Cs on both strands of OHS2 resulted in the same statistically significant decrease in the percentage of V regions that were mutated (P < 0.001) as when the larger modifications were made in both CDR1 and CDR2 (Fig. 7B vs. Fig. 6B). This is because such a large percentage of the mutations in Ramos cells are in OHS2 (Fig. 7C). There is a ∼2.7-fold decrease in the overall frequency of mutation in 3-23AATT clones when the OHS2 is removed, which is the same as the decrease in mutations when both OHS1 and OHS2 are removed (Fig. 6). When the mutations in the modified sites were subtracted, the mutation frequency in 3-23AATT was lower than that in 3-23WT (“Frequency in unmodified region” in Fig. 7B). We have carried out identical experiments on completely different Ramos subclones using 150 × 150 bp Illumina sequencing and obtained the same results. These differences between 3-23AATT and 3-23WT confirm that OHS2 plays a role in mutating the whole V region.
Fig. 7.

DNA modification in OHS2 changed the mutation frequency in Ramos. (A) DNA modification schematic: 3-23WT represents IGHV3-23*01 WT germ line DNA sequence, and 3-23AATT shows the GC sites of OHS2-modified IGHV3-23*01 DNA sequence. The highlighted nucleotides are the differences between 3-23WT and 3-23Mod. The light blue boxes mark the CDRs as labeled. (B) Mutation analysis in V regions of 3-23WT and DNA modified 3-23AATT subclones. The first column has the values obtained from the same V regions as in Fig. 6B because the experiments in Figs. 6 and 7 were done in parallel. The second column contains the values from the combination of the V regions from four 3-23AATT subclones. The rows showed the numbers obtained by the same analysis as in Fig. 6B for 3-23AATT. Bold values in 3-23AATT for mutation frequencies are significantly decreased values compared with 3-23WT (P < 10−6, calculated by SHMtool). In the bold values for percentage of mutated V regions, the P value is <0.001 (χ2 test). Here the modified area only includes GC sites in OHS2. (C) Mutation distribution comparison between 3-23WT and 3-23AATT. The upper part is 3–23WT, and the lower part is 3–23AATT. The rest is the same as described in Fig. 6C. Red arrow points to OHS2.
Discussion
The Frequency and Distribution of Mutations in the IGHV3-23*01 V Region Is Associated with a Very Intricate Distribution of DNA Sequence Motifs.
We have explored the relationship of local sequence context to the frequency of AID-induced mutations in the human IGHV3-23*01 in vivo, in vitro, and in Ramos B cells to gain a better understanding of how the V region sequence has evolved to generate antibody diversity and protective antibodies. We selected IGHV3-23*01 because it is one of the most highly used human V regions (41–43) and because a published database (45, 46) had a large subset of mutated V regions that were nonproductive and would reflect the inherent activity of AID and its associated factors in the absence of antigen selection. The most highly mutated sites in IGHV3-23*01 were overlapping AGCT and AGCA hotspot motifs (46, 61), which are found in CDR1 and CDR2 and at one location in the beginning of FW1 (Fig. 1). Mutations in the S regions are also focused on the AGCT and AGCA (38). However, the overlapping characteristic of these AID hotspot motifs alone is not sufficient to dictate the mutation pattern in the V region because the AGCT (OHS2) at the very 5′ end of CDR2 had a higher frequency of mutation than the other AGCTs and AGCAs and the TGCA overlapping hotspots were not highly mutated. This suggests that the broader sequence environment must also be important. Our analysis (Fig. 1 and Fig. S2) also reveals that there is a paucity of SYC and GRS cold spots and an increased abundance of AID hotspots and TA Polη hotspots in CDR1 and CDR2. Although there are many mutations in parts of FW3, the frequency is lower than in the CDRs. For example, there are many mutations in A:T around residue 280, where the A:Ts are between two simple AID hotspots that mutate at an intermediate frequency, and as in the CDR2, there are no AID cold spots. Mutations in FW3 have been observed previously (62), and recent studies (1, 30) have revealed the importance of mutations, and even deletions and insertions, in the FWs in the broadly neutralizing capacity of antibodies to HIV and influenza. In addition, it has been suggested that mutations in the FW are necessary to stabilize the structure of the CDRs (31). It is therefore quite likely that evolution of antibody V regions has also generated sequence contexts in the FWs that are more readily mutated and facilitate the generation of protective antibodies. Overall, these observations suggest that a complex local sequence environment, including the spatial relationship and relative distribution of both AID and Polη cold spots and hotspots, contributes significantly to the frequency and distribution of mutations throughout the Ig V region. This is further supported by our biochemical studies (Fig. 4) showing that the OHS1 and OHS2 AGCTs, and perhaps some of the neighboring sequences, were inherently more susceptible to AID deamination even when they were presented on single-stranded DNA to purified AID. In addition, the spatial interplay of AID hotspots and Polη hotspots is consistent with the functional, although not physical, interactions (63–66) between AID and MMR complexes. These considerations suggest it is important to use actual V regions rather than artificial synthetic sequences to study mechanistic issues such as the size of the patch that is excised during MMR to remove the G:U mismatch created by AID to introduce mutations in A:T.
Overlapping Hotspots Recruit and Regulate AID Activity to the IGHV3-23*01 V Region.
The very high frequency of mutation in OHS2, and to a lesser degree in the other overlapping hotspots, also suggested that in B cells expressing IGHV3-23*01, these overlapping hotspots might be the first sites to undergo AID mutation. It is unlikely that they are physical entry sites for the binding of AID because biochemical studies have shown that AID can bind equally well to single-stranded DNA that does and does not have Cs (67, 68). However, AID is a very inefficient enzyme deaminating only one of every 30 dCs it encounters in hotspot motifs (57), so it is possible that the AGCTs in IGHV3-23*01 are often the first site to be deaminated. This is consistent with their being the most frequent site of mutation, but they might sometimes also act as an activation site to extend AID mutation to other sites in some V regions. We examined this possibility for OHS2 first by separating the V regions into those that had undergone mutation in the dC on either both strands or either strand alone and compared them with the V regions that had not undergone mutation in the GC in OHS2. If we ignored the mutations that had occurred in GC in OHS2 in calculating the frequency of mutation, those V regions that had undergone mutations in OHS2 had 3.5 times more average mutations per V region than in those V regions where GC in OHS2 were not mutated and a threefold higher overall frequency of mutation throughout the V region (Figs. 2 and 3). This was also true of the AGCTs in OHS1 in CDR1, at 104 and 105 in CDR1, and at 9 and 10 in FW1, even though they had lower frequencies of mutation than OHS2 (Fig. 2). The biochemical mechanism responsible for higher frequencies of mutation of AGCT is unclear, but once this initial event occurs, it can lead to the recruitment of noncanonical base excision repair and MMR. The resulting frequency of mutation of A:T bases is dependent on the presence of Polη hotspots and their proximity to other motifs.
Although these findings are consistent with the individual AGCTs sometimes acting as an initiation or activation site, they are merely correlations and could also reflect a different potential for SHM in different B cells. For example, a memory B cell might have come from a clone that expressed lower levels of AID or some of its associated proteins at the time when mutations were occurring, and then it would have fewer mutated V regions. Distinguishing between these two different models is especially difficult because it has not been possible to establish how many mutations occur during each cell division (25, 53–55) or to find convincing experimental evidence that AID is processive in vivo (53). It is even possible that AID does not always behave the same way on each V region at each cell division. For example, AID might occasionally increase its efficiency, pause for a longer period, or remain associated with the transcriptional apparatus to generate multiple mutations during a single cell cycle to mutate more distant sites. This could occur, for example, in B cells that go through multiple sequential divisions in the dark zone germinal center and that undergo more mutations than the majority of the B cells that shuttle back forth at every cell division between the dark and the light zone (25).
To examine this question further, we used site-directed mutagenesis to substitute neutral or cold spots for the AGCTs and their neighboring sequences in CDR1 and CDR2 and then examined these modified sequences in a biochemical system and in Ramos cells (Figs. 4 and 5). In the biochemical system, on the top strand of the modified V region, there are significantly fewer mutations elsewhere in the modified V region compared with in the WT V region. These studies confirmed that purified AID had a preference for the AGCT, even on single-stranded DNA. However, it is difficult to extrapolate the in vitro findings to the in vivo events because in the biochemical system, AID both is limiting and is processive through a slide and jump mechanism (56, 57). The Ramos cells provide a good cellular model for the initial mutational events that occur in vivo because AID is constantly expressed and the relative susceptibility of the different AID hotspots to mutation is similar to that seen in mice (69). However, the frequency of AID-induced mutation is 20–50 times lower in the Ramos cells than is estimated to occur in vivo (4, 58–60). Because the frequency of mutation was so much lower than in vivo and almost all of the mutated V regions had only one mutation, it was easier to see that the OHS2 had an ∼10 times higher frequency of mutation than the other AGCTs and an ∼100 times higher frequency of mutation than at many other sites in the V region (Fig. 5). When the 3-23 V regions from which the overlapping hotspots had been removed were inserted into the endogenous locus in Ramos cells, there was an approximately twofold reduction in the percentage of V regions mutated, and the overall frequency of mutation was significantly reduced (Fig. 6). When both OHS2 and OHS1 or only OHS2 was replaced, almost all of the loss of mutations was restricted to the sites that had been changed. However, there was also a significant decrease in mutation elsewhere in the V region, which is consistent with the results of the memory B-cell database analysis.
These studies suggest that most of the initial AID-induced mutations in IGHV3-23*01 are the result of a largely stochastic process that greatly favors mutations at AGCT and AGCA overlapping hotspot motifs. The relative frequency of these mutations depends on the motif itself and on the local sequence environment, including the presence of other motifs. In vivo, the initial AID-induced mutation in each of the AGCTs is associated with greater increases in AID mutations throughout the V region. It is not clear why this effect is much greater in vivo than in Ramos cells, but it is possible that the higher levels of AID or the different relative expression of other factors regulated by the germinal center environment either increase the likelihood of AID acting processively or change the susceptibility of other parts of the V region to mutation.
Potential Effect of the Mutations in the Overlapping Hotspots on the Binding of Antigen.
We have concluded that the V region is a highly evolved collection of DNA motifs that allow AID and the error-prone MMR that it recruits to preferentially mutate parts of the V region to provide useful antibodies. The IGHV3-23*01 germ line DNA sequence and the mutations that arise from it in the absence of antigen selection allow us to think about the starting point for the generation of antibodies to many different antigens. Examination of the crystal structure of IGHV3-23*01 (Fig. S3) revealed that OHS2 is at the very base of CDR2 and that the alanine 50 encoded by it is in close proximity both to the tyrosine 59, which is at the C-terminal end of CDR2, and the serine 35, which is encoded by the overlapping hotspots at residue 104 and 105, which are at the C-terminal end of CDR1. The unselected mutations that are recorded at G in OHS2 in the database (46) are all replacement mutations. Fifty percent of those mutations were transitions that would create a threonine, which, based on modeling with the Pymol program, would potentially create clashing of the side chains and destabilize the base of CDR2. The rest of the mutations are transversions that generate a serine that would have little effect on the protein structure or a proline that could have large effects. In addition, 65% of the mutations at residue 104, which encodes the serine in the C-terminal end of CDR1, are transitions that would create an alanine, and the rest are transversions that would create a threonine or isoleucine; according to the Pymol program, these changes could also create instability at the bases of both CDR2 and CDR1. These mutations are reminiscent of the mutations at the bases of the CDRs in anti-HIV and anti-influenza antibodies that have no direct contact with the antigen but change the stability of the CDR loops in some cases, converting the antibodies from affinity-matured highly neutralizing antibodies with restricted specificity into broadly neutralizing and protective antibodies to those viruses (1, 70, 71). The glutamine and leucine encoded by the highly mutated AGCT at residues 9 and 10 (Fig. S3) are interesting because this AGCT is present in many different human V regions and is spatially close to CDR1. According to the Pymol program, the amino acid changes that arise there will not affect the structure of CDR1 but could themselves be interacting with antigen, and perhaps contribute to the docking of the antibody. In addition, there are regions in parts of FW3 that have DNA motifs that allow them to undergo relatively high frequencies of mutations (Fig. 1) that in other V regions contribute to the broadly neutralizing capacity of antibodies to HIV (1) and influenza (30) by stabilizing or modifying the CDR structures.
In conclusion, the association of mutations with the very intricate organization of various motifs in the IGHV3-23*01 region establishes that the DNA sequence is highly evolved to determine the frequency of AID and error-prone MMR mutations in this particular V region. A similar analysis of other antigen selected and unselected human V regions should reveal why different V regions often dominate the response to different antigens and why we need so many V regions. Our findings also suggest that part of the process of developing new vaccine strategies should include the identification of motifs in those V regions that are likely to be initially be targeted AID and to use that information in planning which antigens should be used in sequential vaccines (1, 30, 72–74). Similar genomewide analysis could also increase our understanding of the how AID is targeted to V and SRs and to certain off-target sites and contributes to our understanding of the biochemical mechanisms of these processes.
Methods
Cell Culture.
The Ramos Burkitt’s lymphoma cell line with RMCE was generated (58) and grown in Iscove’s modified Dulbecco’s medium (BioWhittaker) supplemented with 10% (vol/vol) FBS (Atlanta Biologicals), 100 U/mL penicillin-streptomycin (Mediatech), and 0.6 mg/mL hygromycin (Calbiochem). Soft agar cloning was performed and Ramos 6.25 subclone was used as IgM+ control.
RMCE Replacement Plasmids Construction.
The pUCVDJ1EμSμ replacement plasmids created as described in ref. 58 were digested with AgeI and EcoRI, and the IGHV4-34 promoter and VDJ region were cut out from the construct, leaving behind the 2L and L3 for exchange and Eμ and Sμ region. The germ line IGHV3-23*01 gene from promoter to J6 was amplified with primers (forward: ATACCGGTGAGAAGTCAGGTCCAGTGGTG; reverse: GAATTCGGCTCCCAGATCCTCAAGGCAC) by using genomic DNA from a patient with chronic lymphocytic leukemia provided by Nicholas Chiarozzi’s laboratory as a template. PCR products were digested with AgeI and EcoRI and ligated into the previously AgeI- and EcoRI-digested pUCVDJ1EμSμ, and then pUC3-23WTEμSμ was made. The sequence-modified IGHV3-23*01 DNA fragments (3-23Mod and 3-23AATT) were generated by site-directed mutagenesis. The modified IGHV3-23*01 DNA fragments were digested with AgeI and EcoRI and ligated into previously digested pUCVDJ1EμSμ, and then pUC3-23ModEμSμ and pUC3-23AATTEμSμ were constructed.
Plasmid Transfection, Fluorescence Analysis, and Sorting.
The Ramos cells with RMCE were electroporated with replacement plasmids pUC3-23WTEμSμ, pUC3-23ModEμSμ, and pUC3-23AATTEμSμ, as described in ref. 58. After transfection, the Ramos cells were stained with anti-human IgM-fluorescein isothiocyanate, and IgM-fluorescein isothiocyanate-positive cells were sorted and subcloned by soft agar cloning and then cultured in Iscove’s modified Dulbecco’s medium for a total of 3 mo.
In Vitro AID Deamination Assay.
IGHV3-23*01 gene fragments were amplified from a vector containing germ line IGHV3-23*01 cDNA (provided by Nicholas Chiorazzi) by using 30 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 1 min. The primers used were 5′-TAATGCATTTGCCTTTCTCTCCACAGGT-3′ (forward) and 5′-TAATGCATTTCGGGGAAGTAGTCCTTGA-3′ (reverse). PCR products were cloned into the PstI site of a derivative of the M13 mp2 phage vector described previously (75) in both orientations, with either the top or bottom strand inserted downstream of the lacZ gene. Closed circular DNA gapped substrates were prepared as previously described (6). Deamination reactions using 500 ng gapped DNA substrate and GST-AID protein purified from SF9 insect cells (6) were performed as described in ref. 75. ssDNAs from white M13 phage plaques were isolated using the QIAprep Spin M13 Kit (Qiagen) and sequenced by using Sanger sequencing machine and primer P7: 5′-ATGTGAGCGAGTAACAAC-3′. Deaminations were scored by C-to-T transition mutations, as described in refs. 6 and 75. Sequences were analyzed using the SHMtool (76). Modified IGHV3-23*01 gene fragments for making the 3-23Mod M13 mp2 phage were generated by site-direct mutagenesis and then amplified with the same primers and conditions used for generating 3-23WT DNA fragments. The same procedures for making the constructs and deamination analysis were used for 3-23Mod as for 3-23WT.
Ilumina Mi seq Sequencing Library Preparation.
After transfection of Ramos (RMCE) cells with replacement vectors, the transfectants were carried for 3 mo, and genomic DNAs were extracted from Ramos 1 × 106 cells using a DNeasy kit from QIAGEN. A 360-bp fragment in the V region was amplified using the oligonucleotides (forward: TTTGCAGGTGTCCAGTGT; reverse: CCAGTAGTCAAAGTAGTAGATCCC) with 2 U pfu Turbo Cx Hotstart polymerase (Stratagene) in a 25-µL reaction under the following conditions: 94 °C for 5 min, followed by 26 cycles at 94 °C for 20 s, 60 °C for 20 s, and 72 °C for 30 s. DNA fragments were processed through successive enzymatic treatments of end repair, dA tailing, and ligation to adapters according to the manufacturer’s instructions (Illumina Truseq kit). The adapter contains the barcode sequences. Adapter-ligated libraries were further amplified by PCR with Phusion High-Fidelity DNA Polymerase (Finnzymes), using Illumina PE primers for 10 cycles. The resulting purified DNA libraries were multiplexed and applied to an Illumina flow cell for cluster generation and sequenced on the Genome Analyzer according to the manufacturer’s protocols in the Einstein Epigenetics Facility.
Analysis of Deep Sequencing Data.
The 250 × 250 paired-end deep-sequencing data were analyzed using customized R scripts. Barcodes were initially removed using the Illumina Basespace pipeline. For quality filtering, reads with mean quality Q < 25 were removed. Within the remaining reads, individual sites with quality Q < 30 were also removed by restoring them to the consensus. Further filtering was used to remove sequences with three or more mismatches, which we considered unlikely to happen in Ramos, given the usual low mutation frequencies. We found empirically that this filter also removes almost all sequences with indels, which were not considered in this analysis. Within the overlap region of the two paired-end reads, the higher-quality site call was used if quality scores were different; otherwise, if the call was also different, the site was restored to consensus.
Supplementary Material
Acknowledgments
This work was supported by NIH Research Grants 5R01CA072649 and 9R01AI112335 (to M.D.S.), NIH Grant 1R01GM111741-01A1 (to T.M.), Royal Society Grant RG2014 R1 (to R.C.), NIH Grant ES013192 (to M.F.G.), and NIH Grants R01CA164468 and R01DA033788 (to A.B.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1500788112/-/DCSupplemental.
References
- 1.West AP, Jr, et al. Structural insights on the role of antibodies in HIV-1 vaccine and therapy. Cell. 2014;156(4):633–648. doi: 10.1016/j.cell.2014.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Keim C, Kazadi D, Rothschild G, Basu U. Regulation of AID, the B-cell genome mutator. Genes Dev. 2013;27(1):1–17. doi: 10.1101/gad.200014.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rogozin IB, Diaz M. Cutting edge: DGYW/WRCH is a better predictor of mutability at G:C bases in Ig hypermutation than the widely accepted RGYW/WRCY motif and probably reflects a two-step activation-induced cytidine deaminase-triggered process. J Immunol. 2004;172(6):3382–3384. doi: 10.4049/jimmunol.172.6.3382. [DOI] [PubMed] [Google Scholar]
- 4.Zhang W, et al. Clonal instability of V region hypermutation in the Ramos Burkitt’s lymphoma cell line. Int Immunol. 2001;13(9):1175–1184. doi: 10.1093/intimm/13.9.1175. [DOI] [PubMed] [Google Scholar]
- 5.Golding GB, Gearhart PJ, Glickman BW. Patterns of somatic mutations in immunoglobulin variable genes. Genetics. 1987;115(1):169–176. doi: 10.1093/genetics/115.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pham P, Bransteitter R, Petruska J, Goodman MF. Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature. 2003;424(6944):103–107. doi: 10.1038/nature01760. [DOI] [PubMed] [Google Scholar]
- 7.Jolly CJ, et al. The targeting of somatic hypermutation. Semin Immunol. 1996;8(3):159–168. doi: 10.1006/smim.1996.0020. [DOI] [PubMed] [Google Scholar]
- 8.Di Noia J, Neuberger MS. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature. 2002;419(6902):43–48. doi: 10.1038/nature00981. [DOI] [PubMed] [Google Scholar]
- 9.Di Noia JM, Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Annu Rev Biochem. 2007;76:1–22. doi: 10.1146/annurev.biochem.76.061705.090740. [DOI] [PubMed] [Google Scholar]
- 10.Stavnezer J, et al. Differential expression of APE1 and APE2 in germinal centers promotes error-prone repair and A:T mutations during somatic hypermutation. Proc Natl Acad Sci USA. 2014;111(25):9217–9222. doi: 10.1073/pnas.1405590111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peled JU, et al. The biochemistry of somatic hypermutation. Annu Rev Immunol. 2008;26:481–511. doi: 10.1146/annurev.immunol.26.021607.090236. [DOI] [PubMed] [Google Scholar]
- 12.Bransteitter R, Pham P, Scharff MD, Goodman MF. Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase. Proc Natl Acad Sci USA. 2003;100(7):4102–4107. doi: 10.1073/pnas.0730835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peters A, Storb U. Somatic hypermutation of immunoglobulin genes is linked to transcription initiation. Immunity. 1996;4(1):57–65. doi: 10.1016/s1074-7613(00)80298-8. [DOI] [PubMed] [Google Scholar]
- 14.Sun J, Rothschild G, Pefanis E, Basu U. Transcriptional stalling in B-lymphocytes: A mechanism for antibody diversification and maintenance of genomic integrity. Transcription. 2013;4(3):127–135. doi: 10.4161/trns.24556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nambu Y, et al. Transcription-coupled events associating with immunoglobulin switch region chromatin. Science. 2003;302(5653):2137–2140. doi: 10.1126/science.1092481. [DOI] [PubMed] [Google Scholar]
- 16.Pavri R, et al. Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell. 2010;143(1):122–133. doi: 10.1016/j.cell.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Willmann KL, et al. A role for the RNA pol II-associated PAF complex in AID-induced immune diversification. J Exp Med. 2012;209(11):2099–2111. doi: 10.1084/jem.20112145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jeevan-Raj BP, et al. Epigenetic tethering of AID to the donor switch region during immunoglobulin class switch recombination. J Exp Med. 2011;208(8):1649–1660. doi: 10.1084/jem.20110118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Michael N, et al. The E box motif CAGGTG enhances somatic hypermutation without enhancing transcription. Immunity. 2003;19(2):235–242. doi: 10.1016/s1074-7613(03)00204-8. [DOI] [PubMed] [Google Scholar]
- 20.Orthwein A, Di Noia JM. Activation induced deaminase: How much and where? Semin Immunol. 2012;24(4):246–254. doi: 10.1016/j.smim.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 21.Woo CJ, Martin A, Scharff MD. Induction of somatic hypermutation is associated with modifications in immunoglobulin variable region chromatin. Immunity. 2003;19(4):479–489. doi: 10.1016/s1074-7613(03)00261-9. [DOI] [PubMed] [Google Scholar]
- 22.Kodgire P, Mukkawar P, North JA, Poirier MG, Storb U. Nucleosome stability dramatically impacts the targeting of somatic hypermutation. Mol Cell Biol. 2012;32(10):2030–2040. doi: 10.1128/MCB.06722-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang L, Wuerffel R, Feldman S, Khamlichi AA, Kenter AL. S region sequence, RNA polymerase II, and histone modifications create chromatin accessibility during class switch recombination. J Exp Med. 2009;206(8):1817–1830. doi: 10.1084/jem.20081678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Faili A, et al. AID-dependent somatic hypermutation occurs as a DNA single-strand event in the BL2 cell line. Nat Immunol. 2002;3(9):815–821. doi: 10.1038/ni826. [DOI] [PubMed] [Google Scholar]
- 25.Gitlin AD, Shulman Z, Nussenzweig MC. Clonal selection in the germinal centre by regulated proliferation and hypermutation. Nature. 2014;509(7502):637–640. doi: 10.1038/nature13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rouaud P, et al. The IgH 3′ regulatory region controls somatic hypermutation in germinal center B cells. J Exp Med. 2013;210(8):1501–1507. doi: 10.1084/jem.20130072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buerstedde JM, Alinikula J, Arakawa H, McDonald JJ, Schatz DG. Targeting of somatic hypermutation by immunoglobulin enhancer and enhancer-like sequences. PLoS Biol. 2014;12(4):e1001831. doi: 10.1371/journal.pbio.1001831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu M, et al. Two levels of protection for the B cell genome during somatic hypermutation. Nature. 2008;451(7180):841–845. doi: 10.1038/nature06547. [DOI] [PubMed] [Google Scholar]
- 29.Yamane A, et al. Deep-sequencing identification of the genomic targets of the cytidine deaminase AID and its cofactor RPA in B lymphocytes. Nat Immunol. 2011;12(1):62–69. doi: 10.1038/ni.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pappas L, et al. Rapid development of broadly influenza neutralizing antibodies through redundant mutations. Nature. 2014;516(7531):418–422. doi: 10.1038/nature13764. [DOI] [PubMed] [Google Scholar]
- 31.Wang F, et al. Somatic hypermutation maintains antibody thermodynamic stability during affinity maturation. Proc Natl Acad Sci USA. 2013;110(11):4261–4266. doi: 10.1073/pnas.1301810110. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Zheng NY, Wilson K, Jared M, Wilson PC. Intricate targeting of immunoglobulin somatic hypermutation maximizes the efficiency of affinity maturation. J Exp Med. 2005;201(9):1467–1478. doi: 10.1084/jem.20042483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wagner SD, Milstein C, Neuberger MS. Codon bias targets mutation. Nature. 1995;376(6543):732. doi: 10.1038/376732a0. [DOI] [PubMed] [Google Scholar]
- 34.Shapiro GS, Aviszus K, Murphy J, Wysocki LJ. Evolution of Ig DNA sequence to target specific base positions within codons for somatic hypermutation. J Immunol. 2002;168(5):2302–2306. doi: 10.4049/jimmunol.168.5.2302. [DOI] [PubMed] [Google Scholar]
- 35.Yu K, Huang FT, Lieber MR. DNA substrate length and surrounding sequence affect the activation-induced deaminase activity at cytidine. J Biol Chem. 2004;279(8):6496–6500. doi: 10.1074/jbc.M311616200. [DOI] [PubMed] [Google Scholar]
- 36.Zarrin AA, et al. An evolutionarily conserved target motif for immunoglobulin class-switch recombination. Nat Immunol. 2004;5(12):1275–1281. doi: 10.1038/ni1137. [DOI] [PubMed] [Google Scholar]
- 37.Tashiro J, Kinoshita K, Honjo T. Palindromic but not G-rich sequences are targets of class switch recombination. Int Immunol. 2001;13(4):495–505. doi: 10.1093/intimm/13.4.495. [DOI] [PubMed] [Google Scholar]
- 38.Han L, Masani S, Yu K. Overlapping activation-induced cytidine deaminase hotspot motifs in Ig class-switch recombination. Proc Natl Acad Sci USA. 2011;108(28):11584–11589. doi: 10.1073/pnas.1018726108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang ZZ, et al. The strength of an Ig switch region is determined by its ability to drive R loop formation and its number of WGCW sites. Cell Reports. 2014;8(2):557–569. doi: 10.1016/j.celrep.2014.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matsuda F, et al. The complete nucleotide sequence of the human immunoglobulin heavy chain variable region locus. J Exp Med. 1998;188(11):2151–2162. doi: 10.1084/jem.188.11.2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barbas SM, et al. Human autoantibody recognition of DNA. Proc Natl Acad Sci USA. 1995;92(7):2529–2533. doi: 10.1073/pnas.92.7.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brezinschek HP, Brezinschek RI, Lipsky PE. Analysis of the heavy chain repertoire of human peripheral B cells using single-cell polymerase chain reaction. J Immunol. 1995;155(1):190–202. [PubMed] [Google Scholar]
- 43.Jiang N, et al. Lineage structure of the human antibody repertoire in response to influenza vaccination. Sci Transl Med. 2013;5(171):171ra119. doi: 10.1126/scitranslmed.3004794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dal-Bo M, et al. B-cell receptor, clinical course and prognosis in chronic lymphocytic leukaemia: The growing saga of the IGHV3 subgroup gene usage. Br J Haematol. 2011;153(1):3–14. doi: 10.1111/j.1365-2141.2010.08440.x. [DOI] [PubMed] [Google Scholar]
- 45.Ohm-Laursen L, Nielsen M, Larsen SR, Barington T. No evidence for the use of DIR, D-D fusions, chromosome 15 open reading frames or VH replacement in the peripheral repertoire was found on application of an improved algorithm, JointML, to 6329 human immunoglobulin H rearrangements. Immunology. 2006;119(2):265–277. doi: 10.1111/j.1365-2567.2006.02431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ohm-Laursen L, Barington T. Analysis of 6912 unselected somatic hypermutations in human VDJ rearrangements reveals lack of strand specificity and correlation between phase II substitution rates and distance to the nearest 3′ activation-induced cytidine deaminase target. J Immunol. 2007;178(7):4322–4334. doi: 10.4049/jimmunol.178.7.4322. [DOI] [PubMed] [Google Scholar]
- 47.Chahwan R, Edelmann W, Scharff MD, Roa S. AIDing antibody diversity by error-prone mismatch repair. Semin Immunol. 2012;24(4):293–300. doi: 10.1016/j.smim.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.North B, Lehmann A, Dunbrack RL., Jr A new clustering of antibody CDR loop conformations. J Mol Biol. 2011;406(2):228–256. doi: 10.1016/j.jmb.2010.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu TT, Kabat EA. An analysis of the sequences of the variable regions of Bence Jones proteins and myeloma light chains and their implications for antibody complementarity. J Exp Med. 1970;132(2):211–250. doi: 10.1084/jem.132.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spencer J, Dunn-Walters DK. Hypermutation at A-T base pairs: The A nucleotide replacement spectrum is affected by adjacent nucleotides and there is no reverse complementarity of sequences flanking mutated A and T nucleotides. J Immunol. 2005;175(8):5170–5177. doi: 10.4049/jimmunol.175.8.5170. [DOI] [PubMed] [Google Scholar]
- 51.Zivojnovic M, et al. Somatic hypermutation at A/T-rich oligonucleotide substrates shows different strand polarities in Ung-deficient or -proficient backgrounds. Mol Cell Biol. 2014;34(12):2176–2187. doi: 10.1128/MCB.01452-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao Y, et al. Mechanism of somatic hypermutation at the WA motif by human DNA polymerase η. Proc Natl Acad Sci USA. 2013;110(20):8146–8151. doi: 10.1073/pnas.1303126110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Storb U, Shen HM, Nicolae D. Somatic hypermutation: Processivity of the cytosine deaminase AID and error-free repair of the resulting uracils. Cell Cycle. 2009;8(19):3097–3101. doi: 10.4161/cc.8.19.9658. [DOI] [PubMed] [Google Scholar]
- 54.McKean D, et al. Generation of antibody diversity in the immune response of BALB/c mice to influenza virus hemagglutinin. Proc Natl Acad Sci USA. 1984;81(10):3180–3184. doi: 10.1073/pnas.81.10.3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jacob J, Przylepa J, Miller C, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. III. The kinetics of V region mutation and selection in germinal center B cells. J Exp Med. 1993;178(4):1293–1307. doi: 10.1084/jem.178.4.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pham P, Calabrese P, Park SJ, Goodman MF. Analysis of a single-stranded DNA-scanning process in which activation-induced deoxycytidine deaminase (AID) deaminates C to U haphazardly and inefficiently to ensure mutational diversity. J Biol Chem. 2011;286(28):24931–24942. doi: 10.1074/jbc.M111.241208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mak CH, Pham P, Afif SA, Goodman MF. A mathematical model for scanning and catalysis on single-stranded DNA, illustrated with activation-induced deoxycytidine deaminase. J Biol Chem. 2013;288(41):29786–29795. doi: 10.1074/jbc.M113.506550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baughn LB, et al. Recombinase-mediated cassette exchange as a novel method to study somatic hypermutation in Ramos cells. MBio. 2011;2(5):e00186–11. doi: 10.1128/mBio.00186-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sale JE, Neuberger MS. TdT-accessible breaks are scattered over the immunoglobulin V domain in a constitutively hypermutating B cell line. Immunity. 1998;9(6):859–869. doi: 10.1016/s1074-7613(00)80651-2. [DOI] [PubMed] [Google Scholar]
- 60.Kano C, Wang JY. High levels of AID cause strand bias of mutations at A versus T in Burkitt’s lymphoma cells. Mol Immunol. 2013;54(3-4):397–402. doi: 10.1016/j.molimm.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 61.Weiser AA, et al. Affinity maturation of B cells involves not only a few but a whole spectrum of relevant mutations. Int Immunol. 2011;23(5):345–356. doi: 10.1093/intimm/dxr018. [DOI] [PubMed] [Google Scholar]
- 62.Kehoe JM, Capra JD. Localization of two additional hypervariable regions in immunoglobulin heavy chains. Proc Natl Acad Sci USA. 1971;68(9):2019–2021. doi: 10.1073/pnas.68.9.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wilson TM, et al. MSH2-MSH6 stimulates DNA polymerase eta, suggesting a role for A:T mutations in antibody genes. J Exp Med. 2005;201(4):637–645. doi: 10.1084/jem.20042066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ranjit S, et al. AID recruits UNG and Msh2 to Ig switch regions dependent upon the AID C terminus [corrected] J Immunol. 2011;187(5):2464–2475. doi: 10.4049/jimmunol.1101406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li Z, et al. The mismatch repair protein Msh6 influences the in vivo AID targeting to the Ig locus. Immunity. 2006;24(4):393–403. doi: 10.1016/j.immuni.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 66.Frieder D, Larijani M, Collins C, Shulman M, Martin A. The concerted action of Msh2 and UNG stimulates somatic hypermutation at A. T base pairs. Mol Cell Biol. 2009;29(18):5148–5157. doi: 10.1128/MCB.00647-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Larijani M, et al. AID associates with single-stranded DNA with high affinity and a long complex half-life in a sequence-independent manner. Mol Cell Biol. 2007;27(1):20–30. doi: 10.1128/MCB.00824-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mu Y, Prochnow C, Pham P, Chen XS, Goodman MF. A structural basis for the biochemical behavior of activation-induced deoxycytidine deaminase class-switch recombination-defective hyper-IgM-2 mutants. J Biol Chem. 2012;287(33):28007–28016. doi: 10.1074/jbc.M112.370189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Larijani M, Frieder D, Basit W, Martin A. The mutation spectrum of purified AID is similar to the mutability index in Ramos cells and in ung(-/-)msh2(-/-) mice. Immunogenetics. 2005;56(11):840–845. doi: 10.1007/s00251-004-0748-0. [DOI] [PubMed] [Google Scholar]
- 70.Lingwood D, et al. Structural and genetic basis for development of broadly neutralizing influenza antibodies. Nature. 2012;489(7417):566–570. doi: 10.1038/nature11371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xu H, et al. Key mutations stabilize antigen-binding conformation during affinity maturation of a broadly neutralizing influenza antibody lineage. Proteins. December 18, 2014 doi: 10.1002/prot.24745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schmidt AG, et al. Preconfiguration of the antigen-binding site during affinity maturation of a broadly neutralizing influenza virus antibody. Proc Natl Acad Sci USA. 2013;110(1):264–269. doi: 10.1073/pnas.1218256109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Haynes BF, Kelsoe G, Harrison SC, Kepler TB. B-cell-lineage immunogen design in vaccine development with HIV-1 as a case study. Nat Biotechnol. 2012;30(5):423–433. doi: 10.1038/nbt.2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jardine J, et al. Rational HIV immunogen design to target specific germline B cell receptors. Science. 2013;340(6133):711–716. doi: 10.1126/science.1234150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.MacCarthy T, et al. V-region mutation in vitro, in vivo, and in silico reveal the importance of the enzymatic properties of AID and the sequence environment. Proc Natl Acad Sci USA. 2009;106(21):8629–8634. doi: 10.1073/pnas.0903803106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Maccarthy T, Roa S, Scharff MD, Bergman A. SHMTool: A webserver for comparative analysis of somatic hypermutation datasets. DNA Repair (Amst) 2009;8(1):137–141. doi: 10.1016/j.dnarep.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.