

Significance
Ubiquitin (Ub) conjugation triggers protein degradation by the proteasome. Here we describe an unexplored feature of the Ub conjugation system that entails differential activation of an E2-conjugating enzyme by its cognate E3 Ub ligases. In vitro and in vivo analyses of activity-reducing mutants of the yeast Ub-conjugating (Ubc) enzyme, Ubc7, demonstrated selective activation by one of its two cognate E3 ligases, Hrd1, but not by the other, Doa10. Supported by structural modeling of the RING:Ubc7∼Ub complex, these findings are consistent with a model whereby the E2∼Ub transition state depends on noncovalent interactions between helix α2 of Ubc7 and Ub that are differentially stabilized by the two E3 RING domains. This differential activation represents a previously unidentified mechanism for regulating protein ubiquitylation.
Keywords: ubiquitin-proteasome degradation system, ER-associated degradation, protein quality control, ubiquitin E3 ligase, ubiquitin conjugating enzyme
Abstract
A significant portion of ubiquitin (Ub)-dependent cellular protein quality control takes place at the endoplasmic reticulum (ER) in a process termed “ER-associated degradation” (ERAD). Yeast ERAD employs two integral ER membrane E3 Ub ligases: Hrd1 (also termed “Der3”) and Doa10, which recognize a distinct set of substrates. However, both E3s bind to and activate a common E2-conjugating enzyme, Ubc7. Here we describe a novel feature of the ERAD system that entails differential activation of Ubc7 by its cognate E3s. We found that residues within helix α2 of Ubc7 that interact with donor Ub were essential for polyUb conjugation. Mutagenesis of these residues inhibited the in vitro activity of Ubc7 by preventing the conjugation of donor Ub to the acceptor. Unexpectedly, Ub chain formation by mutant Ubc7 was restored selectively by the Hrd1 RING domain but not by the Doa10 RING domain. In agreement with the in vitro data, Ubc7 α2 helix mutations selectively impaired the in vivo degradation of Doa10 substrates but had no apparent effect on the degradation of Hrd1 substrates. To our knowledge, this is the first example of distinct activation requirements of a single E2 by two E3s. We propose a model in which the RING domain activates Ub transfer by stabilizing a transition state determined by noncovalent interactions between the α2 helix of Ubc7 and Ub and that this transition state may be stabilized further by some E3 ligases, such as Hrd1, through additional interactions outside the RING domain.
The ubiquitin (Ub) conjugation machinery employs three basic enzymatic activities, E1, E2, and E3, that work in concert to transfer Ub to client substrates and to form polyUb chains (1). Initially, an E1 Ub-activating enzyme forms a high-energy thioester bond with the C terminus of Ub, after which the Ub molecule is transferred to the active-site Cys of an E2 Ub-conjugating (Ubc) enzyme. The Ub-charged E2 binds to an E3 ligase and catalyzes the transfer of Ub to the ε-amino group of a Lys side chain within the substrate. Additional Ubs then can be ligated to the initial Ub molecule through sequential ubiquitylation cycles, ultimately forming a polyUb chain. Ub can be conjugated to itself via specific Lys residues, resulting in diverse types of chain linkages. Linkage through Lys48 is linked primarily to substrate degradation. Consequently, protein substrates carrying Lys48-linked polyUb chains bind to and are degraded by 26S proteasome.
Although it is well established that E3 ligases activate Ub ligation by E2s via their RING domains, very little is actually known about the underlying regulatory mechanism. Several recent studies determined the structure of RING domain complexes with Ub-charged UbcH5 (2–4). In one of these studies, the structure in solution of Ub-charged UbcH5c together with the mouse E3 ligase E4B U-box domain revealed that Ub can adopt an array of “open” and “closed” conformations (2). The productive closed conformation promotes a nucleophilic attack on the Ub∼E2 thioester by an incoming Lys (acceptor) residue (2). A similar closed conformation was identified in the structures of UbcH5a and UbcH5b, together with their cognate RING domains (3, 4). Taken together, these structural studies suggest that RING domains can catalyze Ub transfer by stabilizing a transition state of a closed conformation of the E2-bound (donor) Ub (5).
Among the fundamental intracellular functions of the Ub–proteasome system is maintenance of cellular protein quality control (PQC) by targeting a diverse array of transiently or permanently misfolded substrates for proteolysis. A central branch of PQC degradation takes place in the endoplasmic reticulum (ER) in a process termed “ER-associated degradation” (ERAD) (6). Despite the multitude of misfolded substrates, ERAD employs only a few E3–ligase complexes (7). In fact, the bakers' yeast S. cerevisiae ERAD system employs only two Ub-ligation complexes, specified by their E3 ligase components, Hrd1 and Doa10 (8–12). Importantly, each of the two E3 ligase complexes recognizes a distinct set of substrates, with minor overlaps (13).
Degradation by the yeast ERAD Ub-ligation system entails the combined activity of two E2 enzymes: Ubc6 and Ubc7 for the Doa10 pathway and Ubc1 and Ubc7 for the Hrd1 pathway (14, 15). The shared E2 enzyme, Ubc7, is a soluble cytosolic protein whose binding to either of the E3–ligase complexes at the ER membrane is mediated by the auxiliary ER membrane protein Cue1. Binding to Cue1 not only mediates the interaction with the E3–ligase complex but also protects Ubc7 from degradation and stimulates its Ub-transfer activity (16–20). Ubc7 is highly conserved in evolution, as evident from substantial sequence and structure similarities with its orthologs from other species (21). The human Ubc7 ortholog, Ube2g2 (21), functions together with several ER membrane-embedded E3 ligases, the best characterized of which is the tumor autocrine motility factor receptor, gp78 (22). Ubc7 and Ube2g2 are subjected to similar regulatory mechanisms: They bind to and are activated by the RING domains of their cognate E3s as well as by the E2-binding regions and CUE domains within Cue1 and gp78 (19, 20, 23–26). The evolutionarily conserved sequence, structure, and regulatory mechanisms of the Ubc7 E2s imply an essential physiological function.
In this study we explored the role of helix α2 of Ubc7 in enzyme activation. Based on our in vivo and in vitro observations and on the available structural information, we propose a mechanism whereby activation of Ubc7, mediated by noncovalent interaction with Ub at helix α2, is differentially affected by the RING domains of its cognate E3 ligases Hrd1 and Doa10.
Results
Lys118 Is Required for Ubc7 Activity.
We have shown previously that when the Ubc7 level exceeds that of its binding partner Cue1, Ubc7 undergoes auto-polyubiquitylation at the active-site Cys residue and subsequent degradation (18). However, a small fraction of Cue1-bound, monoubiquitylated Ubc7 was dithiothreitol (DTT) resistant, suggesting an isopeptide bond with an internal Lys residue(s) (Fig. S1A). Restoring individual Lys residues in a Lys-less Ubc7 (Ubc7KO) and testing for the reappearance of monoUb-Ubc7 detected a preferred single-ubiquitylation site at Lys118 (Fig. S1B). To investigate whether Lys118 was important for Ubc7’s Ub-conjugating activity, we measured the stability of the Doa10 substrate Ura3-CL1, composed of a fusion between the URA3 gene product orotidine-5′-phosphate decarboxylase (Ura3) and the CL1 degradation signal (27, 28), in cells expressing either wild-type or mutant Ubc7 (Fig. 1A). Ura3-CL1 was barely detected in cells with intact Ubc7. Because Ura3-CL1 was expressed from a strong inducible promoter, its low steady-state level likely reflected its rapid degradation rate. Indeed, upon longer exposure of the immunoblot reaction, Ura3-CL1 was visible in all tested strains (Fig. S2). Cycloheximide (CHX)-chase degradation analysis revealed a substantial increase in the Ura3-CL1 steady-state level at time 0 that remained almost constant throughout the CHX-chase period in cells expressing either Ubc7KO or a Lys118-to-Arg Ubc7 (Ubc7R118) mutant. Restoration of Lys118 alone to Ubc7KO was sufficient to reinstate the low steady-state level at time 0 and complete disappearance thereafter. These findings imply that Lys118 of Ubc7 plays an essential role in Ub-mediated degradation.
Fig. 1.

Mutations in helix α2 inhibit Ubc7 activity. (A) Ura3-CL1 degradation in cells expressing Ubc7 mutants. Cells were collected at the indicated time periods after the addition of the translation inhibitor CHX. Ura3-CL1 was visualized by immunoblotting with anti-HA Abs. Probing the blot with anti-G6PD Abs provided a loading control. (B) Coomassie staining of purified Ubc7 or Ubc7R118 shows equal amounts of copurified Cue1. Ubc7-2HA and Cue1 were expressed in bacteria from a bicistronic plasmid and subsequently were purified using a chitin-intein tag (Materials and Methods). (C) Self-monoubiquitylation of Ubc7. Purified Ubc7:Cue1 dimers were incubated with E1, ATP, and FlagUbR48 for 45 min at 30 °C. Proteins were resolved by SDS/PAGE under reducing conditions and were detected by immunoblotting with anti-Flag (Ub) or anti-HA (Ubc7) Abs. (D) PolyUb chain formation by wild-type Ubc7 or Ubc7R118. The ubiquitylation reaction was performed as described in C using FlagUb. (E) A structural model for the Doa10 RING:Ubc7∼Ub complex. The model is based on a superimposition of Ubc7 (PDB ID code 4JQU), Doa10 RING (PDB ID code 2M6M), and the RNF4 RING:UbcH5B∼Ub complex (PDB ID code 4AP4). The area within the rectangle is enlarged below. (F and G) Formation of polyUb chains by the indicated Ubc7 mutants.
To test the effect of Lys118 mutations on the intrinsic activity of Ubc7 directly, wild-type and mutant Ubc7 were coexpressed in bacteria together with Cue1 lacking the transmembrane domain (Cue1ΔTM) (23) and was purified at an approximately 1:1 ratio (Fig. 1B). The Ubc7:Cue1 heterodimers subsequently were incubated in an in vitro ubiquitylation reaction that revealed DTT-resistant Ubc7–monoUb conjugates that, in contrast to the selective monoubiquitylation in vivo, were equally present both in wild-type Ubc7 and Ubc7R118 (Fig. 1C). However, compared with wild-type Ubc7, Ubc7R118 assembled polyUb chains very poorly after a 45-min ubiquitylation reaction (Fig. 1D). Therefore, we set out to explore the possibility that Ubc7R118 inhibition is independent of Lys118 monoubiquitylation and instead is dependent on an alternative mechanism.
Determination of the Role of Lys118 of Ubc7 in Catalysis.
Previous analyses of E2 enzymes activity considered Lys residues spatially adjacent to the active site as potential effectors of the enzymatic reaction (29, 30). The Ubc7 crystal structure indicates that Lys118 is located in helix α2 that is remote from the active site (31). However, Lys118 was both essential and sufficient for Doa10-mediated degradation (Fig. 1A). Apparently, the UbcH5∼Ub structure also reveals a noncovalent interaction between helix α2 and the donor Ub (2–4) that possibly exposes the Ub thioester to nucleophilic attack by an incoming amine group and thus promotes Ub transfer (2).
To understand further the role of Lys118 in E2 catalysis, we generated an in silico tertiary model of Ubc7, Doa10, RING, and Ub (Fig. 1E). The model was based on a superimposition of the Ubc7 crystal structure [Protein Data Bank (PDB) ID code 4JQU] and a solution structure of the Doa10 RING domain (PDB ID code 2M6M) with the structure of the RING finger protein 4 (RNF4) RING:UbcH5B∼Ub complex (PDB ID code 4AP4) (3, 19). The resulting in silico model predicted a hydrogen bond between the side chain of Lys118 and the backbone carbonyl of Leu8 of Ub (Fig. 1E). The model also predicted a hydrophobic interaction between the side chains of Leu121 of Ubc7 and Val70 of Ub. Parallel mutations in respective residues of UbcH5, Lys101 and Leu104, to Ala inhibited the E2 activity, likely by abolishing a critical Ub interaction with helix α2 (2–4).
We thus hypothesized that, as with UbcH5, the reduced activity of Ubc7R118 was caused by aberrant helix α2∼Ub interaction and that additional mutations in this structure also might be deleterious. Indeed, replacing Leu121 with Ala, (equivalent to the UbcH5A104 mutation) substantially inhibited Ubc7A121 activity as compared with wild-type Ubc7 (Fig. 1F). Furthermore, according to the structure prediction (Fig. 1E), a critical hydrogen bond that stabilizes the closed conformation exists between the ε amine of Lys118 and Leu8 in the Ub backbone. To test the requirement for this putative interaction, we examined the activity of additional single-amino acid substitutions, representing distinct side-chain properties, at position 118. As predicted, all tested amino acid substitutions substantially reduced the activity of Ubc7 relative to the wild type (Fig. 1G), underscoring the strict requirement for a Lys residue at position 118.
Ubc7R118 Is an Intrinsic Activity-Reducing Mutant.
The E2-mediated Ub transfer comprises two partial activities: initial formation of a high-energy Ub thioester at the active site (charging) and subsequent formation of an isopeptide bond with an acceptor Lys (transfer). Consequently, to understand the role of Lys118 in catalysis, we compared the partial activities of wild-type and mutant Ubc7. To measure Ubc7 charging, we used a Ub derivative labeled with fluorescein (UbFL) on Cys48 that replaces the original Lys residue at this position. Thus, while serving as a tag, the fluorescein attached to Lys48 prevents the subsequent formation of a Lys48-linked polyUb chain, ensuring a single charge cycle. Consequently, the E2 was incubated with E1 and UbFL for various time periods; then a Ubc7∼UbFL adduct was monitored. The kinetics of Ubc7∼UbFL formation (Fig. 2A) detected a charged E2 after 15 s, reaching a maximum after 2 min. Importantly, no differences in UbFL charging between wild-type Ubc7 and Ubc7R118 were observed, indicating that the Arg118 mutation did not affect Ubc7 charging.
Fig. 2.

The reduced catalytic activity of Ubc7R118 is partially rescued by the Hrd1 RING domain. (A) Kinetics of Ubc7∼Ub thioester formation. The Ubc7:Cue1 dimer was incubated with E1 and Ub48FL for the indicated time periods at 30 °C. Proteins were separated by SDS/PAGE with or without DTT, after which fluorescence was detected using a Typhoon reader (GE Healthcare). (B, Left) Flowchart for single-round Ub-turnover experiments. Ubc7 and the Ubc7:Cue1 dimer were charged separately with Ub and FlagUbR48, respectively, in the presence of E1 and ATP. Both reactions were terminated by the addition of Apyrase to deplete ATP. Equal volumes of each reaction then were mixed for further incubation with either buffer or an E3 ligase; then diUb (Ub–FlagUbR48) formation was determined by immunoblot analysis with anti-Flag Abs. (Right) Graphic depiction of the single-round Ub-turnover reaction. (C) Rates of Ub–FlagUbR48 formation by Ubc7 and Ubc7R118, determined in a single-round Ub-turnover assay in the absence of E3. The E2∼Ub thioesters were combined and incubated at 30 °C for the indicated time periods. The reaction was terminated by the addition of SDS sample buffer. Ubiquitylated proteins were resolved by SDS/PAGE and visualized by immunoblot analysis with anti-Flag Abs. Arrow indicates the position of diUb. (D and E) Activation of polyUb chain formation by E3 RING domains. Either Ubc7 or Ubc7R118 was incubated in an ubiquitylation reaction mixture, as described in Materials and Methods in the presence of equal amounts of recombinant Doa10 or Hrd1 RING domains. Ub chains were resolved by SDS/PAGE and visualized by an immunoblot analysis with anti-Flag Abs.
Next, we tested whether the Ub transfer activity of Ubc7 was affected. To this end, we set up a single-round Ub-turnover experiment, a modification of a similar assay previously used to determine the activity of the mammalian Ubc7 ortholog, Ube2g2, that facilitates Ub conjugation by engaging donor and acceptor Ub molecules (Fig. 2B) (32, 33). Consequently, to ensure a single-round Ub transfer, we used preformed thiol esters of Ubc7:Cue1 with FlagUbR48 as donor Ub species, and Ubc7 (without Cue1), charged with wild-type Ub, as acceptor Ub species. Because FlagUbR48 can be conjugated to an acceptor Ub but then blocks further Lys48‐linked conjugation, it serves only as a donor, whereas in the absence of Cue1, Ubc7 serves solely as an acceptor (19), because it cannot transfer the active-site bound Ub (23, 26). Accordingly, purified Ubc7 or Ubc7R118 in complex with Cue1 initially was charged with FlagUbR48 and, in parallel wild-type Ubc7 purified in the absence of Cue1, was charged with wild-type Ub (Fig. 2B). Both Ub-charging reactions were terminated by ATP depletion and then were coincubated further for various time periods to allow Ub transfer. Measurement of the diUb, Ub–FlagUbR48, dimer formation (Fig. 2C) revealed that when Ubc7∼FlagUbR48 was used as a donor, the diUb was detected after 10 min (marked by an arrow), whereas only a faint Ub–FlagUbR48 signal was observed after 30 min in the presence of donor Ubc7R118∼FlagUbR48. The diminished Ub transfer activity of Ubc7R118 thus explains the substantial difference between the activity of the wild-type and the mutant enzyme in the formation of the polyUb chain.
Differential Requirements for Ubc7 Activation by the Doa10 and Hrd1 RING Domains.
Doa10 and Hrd1 activate Ubc7, but, unlike other E2s, Ubc7 can form polyUb chains in vitro even in their absence (Fig. 1 D, F, and G) (19, 20, 23). We next tested whether the RING domains of either Doa10 or Hrd1 can still activate polyUb chain formation by Ubc7R118. To this end, we analyzed the E2 activity in the presence of purified Doa10 RING domain (Fig. 2D and Fig. S3). The Doa10 RING domain activated Ubc7 in a time-dependent manner (Fig. 2D) but failed to enhance Ubc7R118 activity. Unexpectedly, the Hrd1 RING domain (Hrd1 RING) enhanced wild-type as well as Ubc7R118 activities, although the latter were enhanced to a lesser extent (Fig. 2E). Notably, incubating Ubc7 with Hrd1 RING produced high-molecular-weight Ub adducts, considerably larger than those produced in the absence of RING E3 or upon incubation with Doa10 RING. The copurification of these high-molecular-weight Ub species with GST-tagged Hrd1 RING (Fig. S4) confirmed that they were primarily polyUb–Hrd1 RING conjugates, as previously shown (5).
A Selective Effect of the Lys118-to-Arg Mutation in Ubc7 on ERAD.
It was possible that the apparent inability of Doa10 to activate Ubc7R118 compared with Hrd1 was caused by the inherently lower in vitro activity of Doa10 relative to that of Hrd1. To exclude this possibility, we tested the ability of increasing concentrations of Doa10 to activate ubiquitylation. The results presented in Fig. S3 showed that, indeed, increasing the Doa10 concentration enhanced wild-type Ubc7-mediated polyubiquitylation but did not affect the Ubc7R118 activity whatsoever. We further investigated the differential activation of Ubc7 by the two E3 ligases by comparing the stability of a battery of Doa10 and Hrd1 substrates (Fig. 3A) (10, 12, 13, 34–37) in cells expressing either wild-type Ubc7 or Ubc7R118. To this end, we initially tested whether the R118 mutation affected Ubc7 stability. We found no significant differences in the steady-state levels of Ubc7, Ubc7R118, and Ubc7A89 that were expressed chromosomally under control of the Ubc7 promoter and no changes in Ubc7 levels during a CHX-chase degradation experiment (Fig. 3B). These results indicated that the R118 mutation did not destabilize Ubc7 and excluded the possibility that different E2 levels might affect the stability of the E3 substrate in subsequent experiments. Consequently we next tested whether Lys118 was essential for the in vivo degradation of Doa10 and Hrd1 substrates (Fig. 3 C and D). Determining the steady-state levels of the Doa10 substrate Deg1–Vma12–Ura3 (28) revealed that, among the various Lys-to-Arg mutants, only Ubc7R118 had a stabilizing effect, although to a lesser extent than the active-site mutant Ubc7A89 (Fig. 3C). In contrast, none of the Lys-to-Arg Ubc7 mutants had a stabilizing effect on the Hrd1 substrate mutant carboxypeptidase Y (CPY*) (Fig. 3D and Fig. S5) (12). Thus, among the 11 Lys residues within Ubc7, only Lys118 was essential and sufficient for substrate degradation by the Doa10 pathway but dispensable for degradation by the Hrd1 pathway. These in vivo degradation studies correlated well with the in vitro activities of wild-type and mutant Ubc7.
Fig. 3.

Ubc7R118 differentially influences the in vivo degradation of Doa10 and Hrd1 substrates. (A) Schematic presentation of the different ERAD substrates used in this study. (B) Determination of Ubc7 stability in cells expressing wild-type Ubc7, Ubc7A89, or Ubc7R118. Cells at logarithmic phase were collected at time 0 or after incubation with CHX for the indicated time periods and were lysed by incubation with 0.1 N NaOH for 5 min, followed by boiling in sample buffer containing 50 mg/mL DTT. Ubc7 was visualized by immunoblot using anti-HA Abs. (C–H) Determination of the stability of the indicated Doa10 and Hrd1 substrates (shown in A) in cells expressing wild-type Ubc7, Ubc7A89, or Ubc7R118. Cells were treated and as described in B. Proteins were visualized by immunoblotting with anti-Ubc6 Abs, anti-FLAG Abs (for visualizing Flag-tagged Deg1-Vma12-Ura3 and Vma12-DegAB), or anti-HA Abs (for visualizing CPY*, KHN, or KWW). In all experiments G6PD staining was used for loading control. (I) Induction of UPR in Ubc7 mutant cells. The indicated cells, expressing a LacZ reporter under the control of the UPR-activated Kar2 promoter (UPRE-LacZ), were lysed, and β-galactosidase steady-state levels were determined by an activity assay using ortho-Nitrophenyl-β-galactosidase as a substrate. Data shown are the mean ± SE of three or more independent experiments.
To ascertain further the selective inhibition of the Doa10 pathway by Ubc7R118, we measured the degradation kinetics of additional ERAD substrates by CHX-chase (Fig. 3 E and F). Both Doa10 substrates, Ubc6 (10) and Vma12–DegAB (36), were markedly stabilized in cells expressing Ubc7R118 (Fig. 3 E and F), whereas the degradation of the Hrd1 substrates KHN, containing the yeast Kar2 signal sequence, fused to the simian virus 5 HA-neuraminidase ectodomain (37), and KWW, composed of the KHN luminal domain, fused to the single transmembrane ER protein Wsc1 (13), remained unaffected (Fig. 3 G and H). Noteworthy was the shift in the molecular weight of KHN and KWW caused by the O-linked glycosylation of the proteins that, having evaded ERAD, were modified in the Golgi apparatus (13, 37). Evidently, the Lys-to-Arg substitution at position 118 was an activity-reducing rather than an inactivating mutation, because its effect varied between Doa10 substrates and the Lys-to-Arg mutant was consistently and substantially more active than a Cys-to-Ala catalytic site mutant.
To confirm that the reduced degradation of Doa10 substrates in cells expressing Ubc7R118 was indeed caused by a defect in ubiquitylation, we used Vma12–DegABDD, a variant of the Doa10 substrate Vma12–DegAB that normally is ubiquitylated by Doa10 but is not degraded (38). Consistent with the previous degradation results, an immunoblot analysis revealed a reduction of polyubiquitylated Vma12–DegABDD in cells expressing Ubc7R118 to the levels observed in the Ubc7KO cells (Fig. S6). Importantly, an analysis of isolated Ubc7–Doa10 complexes showed no differences between wild-type and mutant Ubc7 in their association with Doa10 (Fig. S7), suggesting that the observed differences in Ubc7 activity were conferred by the Ub-conjugation activity of the E2. Taken together, these results indicate that the activation of Ubc7R118 by its cognate E3s was selective: The activation profoundly disrupts the degradation of Doa10 substrates but had little effect on Hrd1 substrates.
The ERAD pathway is a key mechanism for protein quality control, as evidenced by the induction of the unfolded protein response (UPR) when the ER is overloaded with misfolded proteins (39) or when ERAD components are compromised (10, 14, 39). Because the tested helix α2 mutants selectively inhibited Doa10- but not Hrd1-mediated degradation, we predicted that their expression might induce a substantial UPR, albeit a milder one than that induced by the complete abolishment of Ubc7 activity. Indeed, expression of a LacZ reporter under the control of the UPR-activated Kar2 promoter (UPRE-LacZ) (40) indicated that the UPR was increased by ∼75% over basal levels in cells expressing helix α2 mutants of Ubc7, compared with an ∼200% increase in ubc7Δ cells or in cells expressing Ubc7A89 (Fig. 3I). We attribute the induction of UPR by helix α2 mutants to their inability to ubiquitylate the physiological ERAD substrates of Doa10.
Hrd1 Activation of Ubc7 Requires Alignment of Helix α2 with Ub.
The UbcH5 helix α2:Ub interactions determine the transition state of the Ub transfer reaction (2–4). The effect of the Lys118 mutation suggests a similar transition state for Ubc7-mediated Ub transfer. Nevertheless, our findings that Hrd1 tolerated Arg at position 118 suggested that it either compensated for suboptimal interactions at the Ubc7∼Ub interface or that it employed a spatially distinct Ubc7∼Ub transition state. If activation by Hrd1 requires the same helix α2:Ub alignment, then a local repulsion force in this interface would be expected to inhibit Ub transfer. To test this expectation, we replaced Lys118 with Glu, introducing an opposite negative charge. This substitution would be expected to repel the carbonyl of Leu8 at the Ub backbone, which is predicted to form an electrostatic bond with Lys118 (Fig. 1E). Indeed, Hrd1 activated Ubc7R118- but not Ubc7E118-dependent Ub transfer and polyubiquitylation (Fig. 4 A and B), although Ubc7E118 was able to form a thioester with Ub efficiently (Fig. 4C). Notably, several high-molecular-weight bands that were present in the Ub transfer reaction corresponded to Hrd1 RING polyUb conjugates. Their formation correlated well with the efficiency of the Ub transfer reaction, as determined by the formation of diUb conjugates (Fig. 4A). In agreement with these findings, in vivo expression of Ubc7E118 completely stabilized a Hrd1 substrate (Fig. 4D). Together, these results indicated that activation of Ubc7 by Hrd1 was through the closed (helix α2:Ub) conformation of the donor Ub∼Ubc7.
Fig. 4.

Hrd1 RING is incapable of activating Ubc7E118. (A) Hrd1-mediated activation of diUb formation. A single-round Ub-turnover assay was performed as described in Fig. 2C using Ubc7, Ubc7R118, or Ubc7E118 as Ub donors in the presence of Hrd1 RING. The reaction was incubated for 30 min at 30 °C; then diUb formation was determined. Arrow indicates the migration distance of diUb. (B) Hrd1 activation of polyUb chain formation by the indicated Ubc7 variants. The ubiquitylation reaction was carried out as described in Fig. 2D and was terminated after 30 min. (C) Kinetics of Ub∼Ubc7 thiol ester formation. The Ubc7:Cue1 dimer was incubated at 30 °C with E1 and FlagUbR48 for the indicated time periods. Proteins were resolved on SDS/PAGE and were subjected to immunoblot with anti-Flag Abs. (D) Degradation of KHN in cells expressing the indicated Ubc7 variants was determined by CHX-chase as described in Fig. 1A.
Next we examined the role of the critical helix α2 in vivo by measuring the stability of Doa10 and Hrd1 substrates in cells expressing a different helix α2 mutation, Ubc7A121. Consistent with our previous results, this Ubc7 mutant stabilized the Doa10 substrate Ubc6 (10) (Fig. 5A), but it had no affect on the degradation of the Hrd1 substrate KHN (Fig. 5B). Similar results were obtained subsequently with additional E3-specific substrates, GFP-DegAB and CPY* (Fig. S8), confirming that Hrd1 did not require optimal helix α2:Ub interactions.
Fig. 5.

Hrd1 overcomes mutations disrupting helix α2:Ub interaction. (A and B) Degradation of Ubc6 (Doa10 substrate) (A) and KHN (Hrd1 substrate) (B) in cells expressing wild-type Ubc7, Ubc7R118, or Ubc7A121. Protein degradation was determined by CHX-chase, followed by immunoblotting using anti-Ubc6 or anti-HA (for KHN) Abs. The G6PD staining provides a loading control for Ubc6. The asterisk indicates a cross-reacting protein that serves as a loading control for KHN. (C) PolyUb chain formation by Ubc7 in the presence of Hrd1. The ubiquitylation assay was performed as described in Fig. 2D using Ub, UbA8, or UbA70, as indicated. PolyUb conjugates were visualized by immunoblot analysis using anti-Ub Abs. (D) Hrd1-mediated activation of diUb formation. A single-round Ub-turnover assay was performed as described in Materials and Methods and in the legend of Fig. 2C, using a mixture of wild-type and mutant Ub as donors and acceptors, as indicated. Arrow indicates the migration distance of diUb.
To confirm this conclusion further, we investigated the ability of Hrd1 to stimulate Ubc7-mediated Ub transfer and polyubiquitylation activities in vitro in the presence of Ala substitutions of either Leu8 or Val70 Ub. According to the structural model (Fig. 1E), Leu8 and Val70 interact with Lys118 and Leu121, respectively, and thus are expected to have an effect on Hrd1 activity similar to that of the respective helix α2 mutations. Indeed, the Hrd1 RING activated both Ala8 and Ala70 polyUb chain formation, the latter to a much greater extent (Fig. 5C). To demonstrate specifically that Hrd1 RING enhances Ub transfer by stabilizing the transition state, we employed the single-turnover Ub-transfer assay using Ub mutants as donors, acceptors, or both (Fig. 5D). In agreement with the polyubiquitylation results, in which most of the observed polyUb chains were conjugated to Hrd1 RING (Fig. S4), the Hrd1 RING stimulated transfer of both Ub mutants as either donors or acceptors. However, Hrd1 self-ubiquitylation was substantially reduced in the presence of Ala8 as a donor.
Discussion
The present study employs Ubc7 mutants to explore a possibly differential activation of Ubc7 by its two cognate E3 ligases, Doa10 and Hrd1. Initially Lys118 was identified as an essential residue for the degradation of a Doa10 substrate (Fig. 1A). The in vitro studies further revealed that the Lys residue at position 118 of Ubc7, rather than ubiquitylation at this site, was critical (Fig. 1 C and D). The in silico structure prediction (Fig. 1E) implicated helix α2 of the enzyme, where Lys118 is located, to the Ub transfer reaction. This hypothesis was confirmed by examination of additional amino acid substitutions at this site, all of which impaired Ubc7 activity. To identify the functional significance of Lys118, a single-round Ub-transfer reaction was designed using wild-type or mutant Ubc7 as the donor E2. These experiments revealed that Lys118 was essential for the donor E2 activity (Fig. 2C), consistent with previous observations that polyUb chain formation by Ube2g2, the mammalian ortholog of Ubc7, requires distinct activities of donor and acceptor E2∼Ub species (32, 33). Incubation of wild-type and mutant Ubc7 with either Doa10 or Hrd1 RING produced an unexpected result: Although Hrd1 RING activated polyubiquitylation by both Ubc7 variants, Doa10 activated only the wild-type E2 (Fig. 2 D and E). The physiological relevance of these in vitro activity studies was established by showing a selective requirement for helix α2 for the Doa10 and Hrd1 degradation pathways in intact yeast cells. In accordance with the in vitro Ubc7 activity results, the degradation of Doa10 substrates was markedly inhibited in cells expressing selected helix α2 mutants, whereas the Hrd1 substrates were essentially unaffected (Fig. 3). Furthermore, Ubc7 helix α2 mutations induced an intermediate UPR response because misfolded substrates are expected to accumulate from partial inhibition of ERAD (Fig. 3I). It is noteworthy that the tested helix α2 mutations are not inactivating but rather are activity reducing, as evidenced by the intermediate levels of substrate stabilization they confer, compared with the full stabilization conferred by active-site C89A mutation. Inhibition of Hrd1 activation of Ubc7E118 (Fig. 4) indicated that Hrd1 uses a similar helix α2:Ub transition state. However, the ability of Hrd1 to tolerate mutations that weakened the helix α2:Ub interaction suggested that it stabilized the transition state by additional interactions, most likely outside the RING domain.
Based on the data regarding the in vitro and in vivo Ubc7 activity presented in this study and on the established structure of RING domain complexes with Ub-charged UbcH5 (2–4), we propose a model for the underlying RING:Ubc7∼Ub interactions (Fig. 6). According to the model, Ub binding at the active site is in a loose (open) conformation. Because Ub wobbles, it only infrequently attains the transition state that ensures optimal positioning of the donor Ub for a nucleophilic attack by an acceptor Ub. Hence, Ub transfer in the absence of RING is relatively inefficient. Binding of a cognate RING domain to Ubc7∼Ub stabilizes the transition state (closed conformation) through noncovalent interaction of helix α2 with the active site-bound Ub. These Helix α2/Ub interactions are critical for Doa10 and are less critical for Hrd1, which can compensate for suboptimal Ubc7:Ub interactions by stabilizing the transition state for effective Ub transfer by additional interactions with regions flanking the RING domain.
Fig. 6.
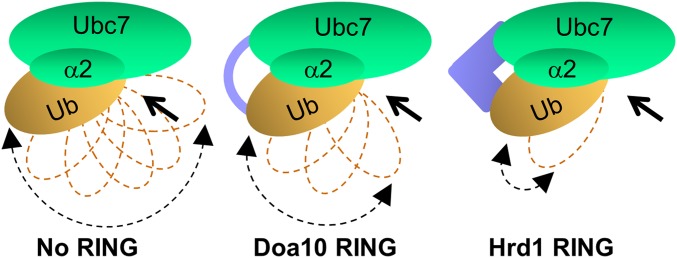
A proposed model for the activation of Ubc7 by the RING domains of Doa10 and Hrd1. Ub bound to Ubc7 at the active site is largely in a loose (open) conformation. Both Doa10 and Hrd1 RING promote the closed transition state conformation by stabilizing noncovalent interaction of Ub with helix α2. The Hrd1 RING domain can form additional interactions that stabilize the closed conformation and thus can overcome compromised helix α2:Ub interactions. Arrows indicate the direction of nucleophilic attack by an acceptor Lys. Joined arrowheads indicate the degree to which the donor Ub is free to wobble.
The physiological significance of selective Ubc7 activation in the ERAD pathway is still unclear. Although both Doa10 and Hrd1 are situated at the ER membrane, each recognizes a discrete set of substrates with a distinct membrane topology (13). The faster conjugation rate observed for the Cue1:Ubc7:Hrd1 complex relative to its Doa10 counterpart may reflect a requirement for the extraction of luminal Hrd1substrates into the cytosol. For example, ubiquitylation of luminal ERAD substrates recruits the Cdc48 ATPase complex, the binding of which prevents reverse translocation of the substrate into the ER lumen (41, 42). Accordingly, rapid ubiquitylation of the substrate may be an important factor to ensure efficient extraction of the degradation substrate. This stringent requirement for ubiquitylation may be redundant for Doa10 substrates, the majority of which are cytosolic and nuclear proteins.
The activation of polyUb chain elongation by Hrd1 in the presence of Ala8 and Ala70 Ub variants was surprising in the light of studies showing that the mutant Ub is largely inactive (2, 3, 43). Notably, unlike most RING E3s, Hrd1 RING requires cytosolic flanking regions for its in vitro ubiquitylation activity (23). These regions may be those that are required for stabilization of the α2:Ub transition state and thus also compensate for the omission of otherwise missing critical interactions, such as the one absent between Lys118 of Ubc7 and Leu8 of Ub. We attempted to test the role of these regions by creating a hybrid enzyme containing the Doa10 RING domain flanked by the Hrd1 cytosolic regions, assuming that they may elicit RING Doa10 activity in the presence mutant Ubc7, similar to that of Hrd1. However, this hybrid protein was completely inactive. The observed differential activation of polyUbA8 and polyUbA70 chain formation (Fig. 5C) is puzzling, because most studies indicate no activation of these mutants (2, 3, 43). The more severe effect of the Ala8 mutation may suggest that this residue creates additional essential interactions with RING E3s (2–4) that, when disrupted by the mutation, cannot be compensated for, even by Hrd1.
More than three decades after the discovery of the Ub ligation system, it now appears more complex than initially described (44). Although the system is still perceived as a linear, sequential process, apparently there are multiple levels of regulation. The distinct requirements for Ubc7 activation by its cognate E3 ligases Doa10 and Hrd1 provide evidence of a potential retrograde regulatory mechanism of ERAD and possibly of other Ub-conjugation systems.
Materials and Methods
Plasmids, Yeast Strains, and Antibodies.
Yeast strains and plasmids used in this study are listed in Tables S1 and S2. Details of yeast and plasmid preparation are provided in SI Materials and Methods. The following Abs were used: anti-GFP (Roche), anti–glucose-6-phosphate dehydrogenase (G6PD) (Sigma-Aldrich), anti-Flag (Sigma-Aldrich), anti -HA (Roche), anti-myc (9E10) (Covance), anti-GST (Santa Cruz Biotechnology), anti-Doa10 (45), anti-Cue1 (raised against Cue1ΔTM), and anti-Ubc6 antiserum (raised against the N-terminal 225-residue fragment of Ubc6).
Site-Directed Mutagenesis and Transfer PCR.
Site-directed mutagenesis was performed using PfuUltra polymerase (Stratagene) according to the manufacturer’s instructions. All mutations were confirmed by sequencing. Transfer PCR was performed using Phusion polymerase (New England Biolabs), according to Erijman et al. (46). All mutations were confirmed by sequencing.
CHEX-Chase and Immunoblot.
CHEX-chase was performed as previously described (35).
Immunoprecipitation.
Determination of Ubc7–Ub conjugates.
ubc7Δ cells coexpressing chromosomally integrated Ubc7-3HA and CUP1p-induced UbA76 were incubated with 100 uM CuSO4 until late logarithmic phase. Cells lysis, protein separation, and immunoblotting were done essentially as described (18).
Determination of Doa10:Ubc7 complexes.
Doa10 immune complexes were isolated from a preparation of crude microsomal fraction treated with 1% digitonin, as previously described (47). Myc-Doa10:Ubc7-3HA complexes were isolated from microsomal extracts by immunoprecipitation with anti-myc Abs, followed by binding to protein A beads. The immunocomplexes were subjected to immunoblotting after separation on SDS/PAGE using anti-myc and anti-HA Abs.
In Vivo Ubiquitylation.
In vivo ubiquitylation analysis was performed as previously described (35).
β-Galactosidase Activity.
The UPR was measured in yeast cells expressing a LacZ reporter under the control of the UPR-activated Kar2 promoter UPRE-LacZ. Cells were harvested, and β-galactosidase activity was measured as previously described (48).
Protein Purification.
Ubc7 and Cue1, coexpressed in a bicistronic plasmid, were purified from bacteria using a chitin-intein tag as previously described (23). The tag was removed by incubating the purified protein with 50 mM DTT, followed by dialysis (23). RING domains of Hrd1 (amino acids 287–531) or Doa10 (amino acids 1–128) coupled to GST were purified from bacteria using glutathione affinity resin beads (Novagene) and were eluted with 50 mM glutathione. Ub variants were purified by precipitation in perchloric acid according to Beal et al. (49). Briefly, 500 mL of isopropyl β-d-1-thiogalactopyranoside–induced BL21 cells were lysed using a microfluidizer, and Ub was precipitated on ice by the slow addition of 1% (vol/vol) of 70% perchloric acid (Sigma-Aldrich) to the stirring extract. The soluble fraction containing Ub was dialyzed and concentrated using Amicon Ultra centrifugal filters (3 K molecular weight cutoff). Synthesis of UbFL was done according to Berndsen et al. (50). The purity of the isolated proteins was checked by separating on 15% SDS/PAGE after Coomassie brilliant blue staining.
In Vitro Ubiquitylation.
The in vitro ubiquitylation assay was done as described by Bazirgan et al. (23). Briefly, 1 μM wild-type or mutant Ubc7, 1 μM Cue1, 1 μM Doa10 or Hrd1 RING, 50 nM E1, 4 μM FlagUb, and reaction buffer containing 5 mM MgCl2, 4 mM ATP, and 50 mM Tris (pH 7.6) were incubated at 30 °C for the indicated times, resolved by 5–15% SDS/PAGE, and immunoblotted with anti-Flag or anti-Ub Abs, as indicated.
Charging of Ubc7 at the Active Site.
Ubc7 charging with Ub on the active site was done using UbFL or a Flag-tagged UbR48 mutant. For charging with UbFL, Ub Lys48 first was replaced with Cys by site-directed mutagenesis. Next, fluorescein was attached to Cys48, as described by Berndsen et al. (50), to obtain UbFL. Ubc7 in the Ubc7:Cue1 complex was charged with UbFL at the active site by incubation at 30 °C for the indicated time periods. The reaction mixture contained 1 μM Ubc7:Cue1, 80 μM UbFL, 1 μM E1, 10 mM MgCl2, 100 mM NaCl, 200 μM ATP, and 30 mM Hepes (pH 7.6). Reactions were terminated by adding SDS/PAGE loading buffer with or without DTT. Proteins were separated on 15% SDS/PAGE, and images were taken using a Typhoon laser scanner (GE Healthcare). For charging with FlagUbR48, the Ubc7:Cue1 complex was charged with FlagUbR48 in a reaction mixture containing 1 μM Ubc7:Cue1, 4 μM FlagUbR48, 1 μM E1, 5 mM MgCl2, 4 mM ATP, and 50 mM Tris (pH 7.6). Reactions were resolved as above, and immunoblotting was done using anti-Flag Abs.
Single-Round Ub Turnover.
The single-round Ub-turnover assay is based on the method described by the Liu et al. (33) for studying the activity of the Ubc7 human homolog Ube2g2 (33). Purified Ubc7 (2 μM), without Cue1, was charged with untagged Ub for 15min, as described above. In parallel, 2 μM of the Ubc7:Cue1 complex was charged with FlagUbR48 for 15 min. Then 5,000 mU/mL Apyrase (New England Biolabs) was added to deplete the ATP. Equal volumes of both mixtures (with or without Cue1) were mixed together with 2.5 μM GST‐Doa10 or Hrd1 RING and were incubated at 30 °C for the indicated times. Reactions were stopped by SDS/PAGE loading buffer with 50 mg/mL DTT and immunoblotted with anti-Flag, anti-GST, or anti-Cue1 Abs.
Supplementary Material
Acknowledgments
We thank A. Ciechanover, R. Hampton, M. Hochstrasser, R. Kulka, and D. Ng for plasmids and strains, Dr. A. Shiber for technical assistance, and Dr. W. Breuer for critically reviewing the manuscript. This work was supported by Israel Science Foundation Grant 786/08.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1415621112/-/DCSupplemental.
References
- 1.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 2.Pruneda JN, et al. Structure of an E3:E2∼Ub complex reveals an allosteric mechanism shared among RING/U-box ligases. Mol Cell. 2012;47(6):933–942. doi: 10.1016/j.molcel.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Plechanovová A, Jaffray EG, Tatham MH, Naismith JH, Hay RT. Structure of a RING E3 ligase and ubiquitin-loaded E2 primed for catalysis. Nature. 2012;489(7414):115–120. doi: 10.1038/nature11376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dou H, Buetow L, Sibbet GJ, Cameron K, Huang DT. BIRC7-E2 ubiquitin conjugate structure reveals the mechanism of ubiquitin transfer by a RING dimer. Nat Struct Mol Biol. 2012;19(9):876–883. doi: 10.1038/nsmb.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Metzger MB, Pruneda JN, Klevit RE, Weissman AM. RING-type E3 ligases: Master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim Biophys Acta. 2014;1843(1):47–60. doi: 10.1016/j.bbamcr.2013.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brodsky JL, Skach WR. Protein folding and quality control in the endoplasmic reticulum: Recent lessons from yeast and mammalian cell systems. Curr Opin Cell Biol. 2011;23(4):464–475. doi: 10.1016/j.ceb.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ravid T, Hochstrasser M. Diversity of degradation signals in the ubiquitin-proteasome system. Nat Rev Mol Cell Biol. 2008;9(9):679–690. doi: 10.1038/nrm2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carvalho P, Goder V, Rapoport TA. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell. 2006;126(2):361–373. doi: 10.1016/j.cell.2006.05.043. [DOI] [PubMed] [Google Scholar]
- 9.Denic V, Quan EM, Weissman JS. A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell. 2006;126(2):349–359. doi: 10.1016/j.cell.2006.05.045. [DOI] [PubMed] [Google Scholar]
- 10.Swanson R, Locher M, Hochstrasser M. A conserved ubiquitin ligase of the nuclear envelope/endoplasmic reticulum that functions in both ER-associated and Matalpha2 repressor degradation. Genes Dev. 2001;15(20):2660–2674. doi: 10.1101/gad.933301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hampton RY, Gardner RG, Rine J. Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol Biol Cell. 1996;7(12):2029–2044. doi: 10.1091/mbc.7.12.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bordallo J, Plemper RK, Finger A, Wolf DH. Der3p/Hrd1p is required for endoplasmic reticulum-associated degradation of misfolded lumenal and integral membrane proteins. Mol Biol Cell. 1998;9(1):209–222. doi: 10.1091/mbc.9.1.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vashist S, Ng DT. Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J Cell Biol. 2004;165(1):41–52. doi: 10.1083/jcb.200309132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friedlander R, Jarosch E, Urban J, Volkwein C, Sommer T. A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat Cell Biol. 2000;2(7):379–384. doi: 10.1038/35017001. [DOI] [PubMed] [Google Scholar]
- 15.Chen P, Johnson P, Sommer T, Jentsch S, Hochstrasser M. Multiple ubiquitin-conjugating enzymes participate in the in vivo degradation of the yeast MAT alpha 2 repressor. Cell. 1993;74(2):357–369. doi: 10.1016/0092-8674(93)90426-q. [DOI] [PubMed] [Google Scholar]
- 16.Biederer T, Volkwein C, Sommer T. Role of Cue1p in ubiquitination and degradation at the ER surface. Science. 1997;278(5344):1806–1809. doi: 10.1126/science.278.5344.1806. [DOI] [PubMed] [Google Scholar]
- 17.Bazirgan OA, Hampton RY. Cue1p is an activator of Ubc7p E2 activity in vitro and in vivo. J Biol Chem. 2008;283(19):12797–12810. doi: 10.1074/jbc.M801122200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ravid T, Hochstrasser M. Autoregulation of an E2 enzyme by ubiquitin-chain assembly on its catalytic residue. Nat Cell Biol. 2007;9(4):422–427. doi: 10.1038/ncb1558. [DOI] [PubMed] [Google Scholar]
- 19.Metzger MB, et al. A structurally unique E2-binding domain activates ubiquitination by the ERAD E2, Ubc7p, through multiple mechanisms. Mol Cell. 2013;50(4):516–527. doi: 10.1016/j.molcel.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bagola K, et al. Ubiquitin binding by a CUE domain regulates ubiquitin chain formation by ERAD E3 ligases. Mol Cell. 2013;50(4):528–539. doi: 10.1016/j.molcel.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 21.Katsanis N, Fisher EM. Identification, expression, and chromosomal localization of ubiquitin conjugating enzyme 7 (UBE2G2), a human homologue of the Saccharomyces cerevisiae ubc7 gene. Genomics. 1998;51(1):128–131. doi: 10.1006/geno.1998.5263. [DOI] [PubMed] [Google Scholar]
- 22.Fang S, et al. The tumor autocrine motility factor receptor, gp78, is a ubiquitin protein ligase implicated in degradation from the endoplasmic reticulum. Proc Natl Acad Sci USA. 2001;98(25):14422–14427. doi: 10.1073/pnas.251401598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bazirgan OA, Garza RM, Hampton RY. Determinants of RING-E2 fidelity for Hrd1p, a membrane-anchored ubiquitin ligase. J Biol Chem. 2006;281(51):38989–39001. doi: 10.1074/jbc.M608174200. [DOI] [PubMed] [Google Scholar]
- 24.Das R, et al. Allosteric activation of E2-RING finger-mediated ubiquitylation by a structurally defined specific E2-binding region of gp78. Mol Cell. 2009;34(6):674–685. doi: 10.1016/j.molcel.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen B, et al. The activity of a human endoplasmic reticulum-associated degradation E3, gp78, requires its Cue domain, RING finger, and an E2-binding site. Proc Natl Acad Sci USA. 2006;103(2):341–346. doi: 10.1073/pnas.0506618103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kostova Z, Mariano J, Scholz S, Koenig C, Weissman AM. A Ubc7p-binding domain in Cue1p activates ER-associated protein degradation. J Cell Sci. 2009;122(Pt 9):1374–1381. doi: 10.1242/jcs.044255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gilon T, Chomsky O, Kulka RG. Degradation signals for ubiquitin system proteolysis in Saccharomyces cerevisiae. EMBO J. 1998;17(10):2759–2766. doi: 10.1093/emboj/17.10.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Metzger MB, Maurer MJ, Dancy BM, Michaelis S. Degradation of a cytosolic protein requires endoplasmic reticulum-associated degradation machinery. J Biol Chem. 2008;283(47):32302–32316. doi: 10.1074/jbc.M806424200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Machida YJ, et al. UBE2T is the E2 in the Fanconi anemia pathway and undergoes negative autoregulation. Mol Cell. 2006;23(4):589–596. doi: 10.1016/j.molcel.2006.06.024. [DOI] [PubMed] [Google Scholar]
- 30.Lin Y, Hwang WC, Basavappa R. Structural and functional analysis of the human mitotic-specific ubiquitin-conjugating enzyme, UbcH10. J Biol Chem. 2002;277(24):21913–21921. doi: 10.1074/jbc.M109398200. [DOI] [PubMed] [Google Scholar]
- 31.Cook WJ, Martin PD, Edwards BF, Yamazaki RK, Chau V. Crystal structure of a class I ubiquitin conjugating enzyme (Ubc7) from Saccharomyces cerevisiae at 2.9 angstroms resolution. Biochemistry. 1997;36(7):1621–1627. doi: 10.1021/bi962639e. [DOI] [PubMed] [Google Scholar]
- 32.Li W, Tu D, Brunger AT, Ye Y. A ubiquitin ligase transfers preformed polyubiquitin chains from a conjugating enzyme to a substrate. Nature. 2007;446(7133):333–337. doi: 10.1038/nature05542. [DOI] [PubMed] [Google Scholar]
- 33.Liu W, et al. Dimeric Ube2g2 simultaneously engages donor and acceptor ubiquitins to form Lys48-linked ubiquitin chains. EMBO J. 2014;33(1):46–61. doi: 10.1002/embj.201385315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Metzger MB, Michaelis S. Analysis of quality control substrates in distinct cellular compartments reveals a unique role for Rpn4p in tolerating misfolded membrane proteins. Mol Biol Cell. 2009;20(3):1006–1019. doi: 10.1091/mbc.E08-02-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Furth N, et al. Exposure of bipartite hydrophobic signal triggers nuclear quality control of Ndc10 at the endoplasmic reticulum/nuclear envelope. Mol Biol Cell. 2011;22(24):4726–4739. doi: 10.1091/mbc.E11-05-0463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shiber A, Breuer W, Brandeis M, Ravid T. Ubiquitin conjugation triggers misfolded protein sequestration into quality control foci when Hsp70 chaperone levels are limiting. Mol Biol Cell. 2013;24(13):2076–2087. doi: 10.1091/mbc.E13-01-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vashist S, et al. Distinct retrieval and retention mechanisms are required for the quality control of endoplasmic reticulum protein folding. J Cell Biol. 2001;155(3):355–368. doi: 10.1083/jcb.200106123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alfassy OS, Cohen I, Reiss Y, Tirosh B, Ravid T. Placing a disrupted degradation motif at the C terminus of proteasome substrates attenuates degradation without impairing ubiquitylation. J Biol Chem. 2013;288(18):12645–12653. doi: 10.1074/jbc.M113.453027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jonikas MC, et al. Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum. Science. 2009;323(5922):1693–1697. doi: 10.1126/science.1167983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mori K, et al. A 22 bp cis-acting element is necessary and sufficient for the induction of the yeast KAR2 (BiP) gene by unfolded proteins. EMBO J. 1992;11(7):2583–2593. doi: 10.1002/j.1460-2075.1992.tb05323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eisele F, Schäfer A, Wolf DH. Ubiquitylation in the ERAD Pathway. Subcell Biochem. 2010;54:136–148. doi: 10.1007/978-1-4419-6676-6_11. [DOI] [PubMed] [Google Scholar]
- 42.Claessen JH, Kundrat L, Ploegh HL. Protein quality control in the ER: Balancing the ubiquitin checkbook. Trends Cell Biol. 2012;22(1):22–32. doi: 10.1016/j.tcb.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wickliffe KE, Lorenz S, Wemmer DE, Kuriyan J, Rape M. The mechanism of linkage-specific ubiquitin chain elongation by a single-subunit E2. Cell. 2011;144(5):769–781. doi: 10.1016/j.cell.2011.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hershko A, Heller H, Elias S, Ciechanover A. Components of ubiquitin-protein ligase system. Resolution, affinity purification, and role in protein breakdown. J Biol Chem. 1983;258(13):8206–8214. [PubMed] [Google Scholar]
- 45.Kreft SG, Wang L, Hochstrasser M. Membrane topology of the yeast endoplasmic reticulum-localized ubiquitin ligase Doa10 and comparison with its human ortholog TEB4 (MARCH-VI) J Biol Chem. 2006;281(8):4646–4653. doi: 10.1074/jbc.M512215200. [DOI] [PubMed] [Google Scholar]
- 46.Erijman A, Shifman JM, Peleg Y. A single-tube assembly of DNA using the transfer-PCR (TPCR) platform. Methods Mol Biol. 2014;1116:89–101. doi: 10.1007/978-1-62703-764-8_7. [DOI] [PubMed] [Google Scholar]
- 47.Kreft SG, Hochstrasser M. An unusual transmembrane helix in the Doa10 ERAD ubiquitin ligase modulates degradation of its cognate E2. J Biol Chem. 2011 doi: 10.1074/jbc.M110.196360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Plemper RK, Böhmler S, Bordallo J, Sommer T, Wolf DH. Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature. 1997;388(6645):891–895. doi: 10.1038/42276. [DOI] [PubMed] [Google Scholar]
- 49.Beal R, Deveraux Q, Xia G, Rechsteiner M, Pickart C. Surface hydrophobic residues of multiubiquitin chains essential for proteolytic targeting. Proc Natl Acad Sci USA. 1996;93(2):861–866. doi: 10.1073/pnas.93.2.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Berndsen CE, Wiener R, Yu IW, Ringel AE, Wolberger C. A conserved asparagine has a structural role in ubiquitin-conjugating enzymes. Nat Chem Biol. 2013;9(3):154–156. doi: 10.1038/nchembio.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.