

Significance
Small vessel disease (SVD) of the brain refers to a group of pathological processes leading to cerebral lesions, cognitive decline, and stroke. Despite the importance of SVD, there is no specific treatment, mainly due to a limited understanding of the disease pathogenesis. Using a recently developed mouse model of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy, a hereditary form of SVD, we determined the basis of altered brain artery function at an early stage of disease progression. We found that cerebrospecific up-regulation of the voltage-gated potassium channel, KV1, prevents intracerebral arterioles from constricting in response to physiological levels of intraluminal pressure. This impairment of a fundamental vascular function is expected to impact cerebral blood flow autoregulation and local dilation in response to neuronal activity (functional hyperemia).
Keywords: voltage-gated potassium channel, vascular smooth muscle cells, CADASIL, cerebral small vessel disease, channelopathy
Abstract
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), caused by dominant mutations in the NOTCH3 receptor in vascular smooth muscle, is a genetic paradigm of small vessel disease (SVD) of the brain. Recent studies using transgenic (Tg)Notch3R169C mice, a genetic model of CADASIL, revealed functional defects in cerebral (pial) arteries on the surface of the brain at an early stage of disease progression. Here, using parenchymal arterioles (PAs) from within the brain, we determined the molecular mechanism underlying the early functional deficits associated with this Notch3 mutation. At physiological pressure (40 mmHg), smooth muscle membrane potential depolarization and constriction to pressure (myogenic tone) were blunted in PAs from TgNotch3R169C mice. This effect was associated with an ∼60% increase in the number of voltage-gated potassium (KV) channels, which oppose pressure-induced depolarization. Inhibition of KV1 channels with 4-aminopyridine (4-AP) or treatment with the epidermal growth factor receptor agonist heparin-binding EGF (HB-EGF), which promotes KV1 channel endocytosis, reduced KV current density and restored myogenic responses in PAs from TgNotch3R169C mice, whereas pharmacological inhibition of other major vasodilatory influences had no effect. KV1 currents and myogenic responses were similarly altered in pial arteries from TgNotch3R169C mice, but not in mesenteric arteries. Interestingly, HB-EGF had no effect on mesenteric arteries, suggesting a possible mechanistic basis for the exclusive cerebrovascular manifestation of CADASIL. Collectively, our results indicate that increasing the number of KV1 channels in cerebral smooth muscle produces a mutant vascular phenotype akin to a channelopathy in a genetic model of SVD.
Small vessel disease (SVD) of the brain accounts for about one-fourth of ischemic strokes worldwide and is a leading cause of cognitive decline and disability (1). Alteration of cerebral arterioles, capillaries, or venules gives rise to white matter lesions and subcortical lacunar infarcts (2). A mix of genetic and environmental factors drives SVD, with both sporadic and inherited forms identified (1). Despite the importance of SVD, there is no specific treatment, mainly due to a limited understanding of the disease pathogenesis. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is an archetypal SVD that emerges as the most common heritable cause of stroke and vascular dementia in adults (3). It presents an autosomal dominant mode of transmission through stereotyped missense mutations in the extracellular domain (ECD) of NOTCH3 (NOTCH3ECD), a transmembrane receptor that undergoes proteolytic cleavage to release its ECD and an intracellular fragment, which translocates to the nucleus and acts as a transcription factor. NOTCH3 is predominantly expressed in vascular smooth muscle cells (SMCs) and is critically required for the maturation and function of small vessels (3, 4). Although mutant NOTCH3 is also expressed in SMCs of peripheral arteries, clinical manifestations are restricted to the brain (2, 3).
The NOTCH3ECD contains 34 epidermal growth factor receptor (EGFR) repeats, each of which contains six cysteine residues that are presumably critical structural determinants. Almost all CADASIL-associated NOTCH3 mutations identified to date involve substitutions that result in one or more EGFR repeats having an odd number of cysteines (3). A recently developed transgenic mouse model of CADASIL expressing the mutant protein NOTCH3(R169C), encoded by one such naturally occurring CADASIL mutation, has been shown to recapitulate the preclinical stages of the disease. In CADASIL, white matter lesions are the most consistent and earliest abnormalities, preceding the onset of other symptoms by 10–15 y (3). In the transgenic (Tg)Notch3R169C mouse model, white matter lesions are detectable at 12 mo, but impaired autoregulation of cerebral blood flow (CBF) and diminished contractile responses to intravascular pressure (myogenic tone) in isolated pial arteries appear as early as 6 mo (5, 6). Neurovascular coupling (NVC)—the process by which neuronal activity is linked to the microvasculature to promote the local vasodilation necessary to ensure an adequate supply of nutrients and oxygen to active neurons—is also disrupted in young (∼6-mo-old) TgNotch3R169C mice. Thus, cerebrovascular dysfunction emerges as an early event in the pathogenesis of SVD (5).
The topology of the brain vasculature creates distinct pathological predispositions. In contrast to the robust pial and capillary networks, which are rich in anastomoses, parenchymal arterioles (PAs) form a fragile, one-dimensional organization with limited collateral supply (7). This “stovepiping” creates a bottleneck effect that makes the column of brain parenchyma served by a given PA especially vulnerable to insults that affect flow through that PA (7). Thus, perhaps not surprisingly, PAs are major sites of vulnerability during the progression of vascular diseases (8, 9), including CADASIL and other sporadic forms of SVD (2, 10, 11).
Arteries and arterioles in vivo exhibit myogenic tone, a partially constricted state that allows further constriction or dilation in response to moment-to-moment fluctuations in blood pressure (9, 12). Myogenic tone is an intrinsic and fundamental feature of small arteries and arterioles that protects capillaries from disruptive high blood pressure; in the cerebral microcirculation, it provides the vasodilatory reserve necessary for effective NVC (13). The myogenic response is initiated by elevation of intravascular pressure, which causes a graded SMC membrane potential depolarization that activates voltage-dependent Ca2+ channels (VDCCs), increasing Ca2+ entry and promoting vasoconstriction (14–16). Large-conductance, Ca2+-sensitive K+ (BKCa) channels and voltage-gated K+ (KV) channels function as negative feedback elements that, when activated, serve as a brake on pressure-induced depolarization and constriction (15). Elevations in vasodilatory influences, such as nitric oxide (NO) and endothelial-dependent hyperpolarization, can also oppose the development of arteriolar tone (17). Although myogenic tone is reduced in pial arteries from TgNotch3R169C mice, it is not known whether similar effects manifest in PAs, the preferential target of the SVD process. Moreover, it is not known what molecular mechanism underlies this CADASIL-related vascular defect or why disease manifestations are restricted to the cerebrovascular circulation. Here, we sought to determine whether—and how—NOTCH3(R169C) produces the mutant cerebrovascular phenotype in PAs, focusing on the myogenic response, and explored the differential effects of this mutant in the peripheral and brain vasculature.
Results
PAs from TgNotch3R169C Mice Exhibit Diminished Constriction to Pressure in the Physiological Range.
To investigate the effects of NOTCH3(R169C) expression on cerebrovascular myogenic responses, we measured changes in the diameter of precapillary segments of PAs from 6-mo-old TgNotch3R169C mice in response to changes in intraluminal pressure (Fig. 1 A and B). Transgenic mice overexpressing WT NOTCH3 (TgNotch3WT) and nontransgenic (Non-Tg) mice were used as controls. Elevation of intraluminal pressure to 20 mmHg constricted PAs from Non-Tg, TgNotch3WT, and TgNotch3R169C mice to a similar extent (∼20%). However, at higher pressures, TgNotch3R169C arterioles constricted significantly less than control arterioles. At 40 mmHg, the estimated pressure experienced in vivo by cerebral arterioles of this size (18), TgNotch3R169C PAs constricted 38% less than controls (Fig. 1 A and B). These results indicate that the mutant protein affects a process that it is triggered by elevated pressure. Consistent with our previous report (5), we found that myogenic response to elevation of intravascular pressure to 60 mmHg was similarly diminished in posterior cerebral arteries (PCAs) from TgNotch3R169C mice (Table S1). Myogenic responses in both PCAs and PAs from TgNotch3WT mice were not significantly different from those in Non-Tg mice (Table S1 and Fig. 1 A and B), indicating that overexpression of the WT receptor did not affect myogenic tone.
Fig. 1.

PAs from CADASIL mice constrict less to pressures above 30 mmHg. (A) Typical recordings of PA internal diameter during myogenic constriction in response to increasing intraluminal pressure. (B) Summary myogenic tone data expressed as means ± SEM (**P < 0.01, ***P < 0.001; one-way ANOVA). (C) Representative traces showing dilation and constriction of pressurized (40 mmHg) PAs induced by elevation of extracellular K+. (D) Summary data expressed as means ± SEM (n = number of animals).
The development of arteriolar tone, even at lower pressures, depends on Ca2+ influx via activated VDCCs, a process initiated by membrane potential depolarization (16, 19, 20). Conversely, membrane potential hyperpolarization can rapidly dilate cerebral arteries by deactivating VDCCs. The lack of difference in myogenic tone at 20 mmHg suggested that VDCC activity was unaltered in TgNotch3R169C PAs. To confirm this and assess the integrity of the contractile apparatus, we manipulated the membrane potential by increasing external K+ concentration ([K+]o) and measured arteriolar diameter. Modest elevations of [K+]o (to <20 mM) cause membrane potential hyperpolarization by activating strong inward rectifier K+ (Kir) channels (21), whereas elevations of [K+]o above 25 mM cause membrane potential depolarization. Although there was an apparent trend toward reduced dilation in TgNotch3R169C mice, this nonsignificant difference is an artifact, reflecting the presentation method, which expressed dilation relative to baseline to allow both constriction and dilation to be shown on the same graphs, and the fact that PAs from these animals had less tone compared with controls. When expressed relative to the diameter of fully dilated vessels, elevation of extracellular K+ concentration from 3 mM to 20 mM caused almost a maximal dilation of arterioles in all groups. Therefore, at 40 mmHg, the relationship between [K+]o and PA diameter for TgNotch3R169C mice was essentially the same as that for TgNotch3WT and Non-Tg control mice (Fig. 1 C and D), indicating that the mutant NOTCH3(R169C) protein does not affect Kir-mediated dilation, VDCC-dependent constriction, or the contractile response of PAs.
Membrane Potential Hyperpolarization Underlies the Diminished Arteriolar Tone in TgNotch3R169C Mice.
One likely explanation for the diminished myogenic response in TgNotch3R169C mice is blunted membrane potential depolarization to pressure. We have previously shown that the relationship between membrane potential and diameter/Ca2+ of pial arteries (16) and parenchymal arterioles (19) is very steep and that membrane depolarization from −60 mV to −30 mV causes an artery to transition from full dilation to full constriction. This relationship predicts that a 9-mV hyperpolarization (equivalent to blunting depolarization by 9 mV) could account for the diminished tone in arterioles from TgNotch3R169C mice at a pressure of 40 mmHg (19). At a holding pressure of 10 mmHg, SMC membrane potentials were −54 ± 2 mV, −52 ± 1 mV, and −53 ± 1 mV in isolated PAs from TgNotch3R169C, TgNotch3WT, and Non-Tg mice, respectively (Fig. 2 A and B), consistent with the similar degree of constriction observed at this pressure. In contrast, increasing intraluminal pressure to 40 mmHg depolarized the membrane potential of SMCs in arterioles of TgNotch3WT and Non-Tg animals by 16.5 mV and 18.3 mV, respectively, but depolarized SMCs in arterioles from TgNotch3R169C mice by only 9.4 mV—a difference very close to the predicted value. To further examine this relationship, we fitted a linear regression to the relationship between the percentage of myogenic tone and membrane potential, using values obtained from Non-Tg and TgNotch3WT animals (Fig. 2C). At −44.2 mV—the membrane potential observed in PAs from TgNotch3R169C mice pressurized to 40 mmHg—this linear equation predicted a myogenic tone of 24%. Remarkably, the average tone observed in PAs from TgNotch3R169C mice—22% ± 2%—was very close to the predicted value and in fact was contained within the 95% confidence interval of the linear regression. Therefore, the blunted depolarization to pressure is sufficient to account for the reduction in myogenic tone.
Fig. 2.

The membrane potential of PAs from CADASIL mice are more hyperpolarized at 40 mmHg. (A) Membrane potential (mV) of SMCs in pressurized PAs at 10 mmHg (upper traces) and 40 mmHg (lower traces). (B) Membrane potential summary data at 10 mmHg and 40 mmHg, expressed as means ± SEM recorded in three different cells (**P < 0.01; one-way ANOVA). The number of animals is shown in parentheses. (C) Percentage of myogenic tone induced by 10 mmHg and 40 mmHg and plotted as a function of membrane potential. Values from TgNotch3R169C animals are consistent with the linear regression obtained from Non-Tg and TgNotch3WT animals, showing a similar relationship between tone and membrane potential.
The Diminished Myogenic Tone in PAs from TgNotch3R169C Mice Is Not Attributable to Increases in Endothelial Vasodilator Influences or Smooth Muscle BK Channel Activity.
The endothelium exerts a tonic dilatory influence on the diameter of PAs that is mediated, in part, by the activity of endothelial small- and intermediate-conductance, calcium-sensitive K+ channels (SK and IK, respectively) (22). Thus, one potential explanation for the decreased depolarization to pressure in TgNotch3R169C PAs could be hyperpolarization of the smooth muscle membrane potential due to increased SK and/or IK channel activity. However, the vasoconstriction induced by inhibition of endothelial SK channels with 100 nM apamin or IK channels with 300 nM charybdotoxin (in the presence of the BK channel blocker paxilline) was not different between TgNotch3R169C and control (TgNotch3WT and Non-Tg) PAs (Fig. 3 A and B). The synthetic IK and SK channel agonist, NS309 (1 µM), which shifts Ca2+ sensitivity to lower concentrations, caused a rapid and reversible dilation that was prevented by inhibition of IK and SK channels (Fig. 3). Vasoconstrictor responses to inhibition of endothelial nitric oxide synthase (eNOS) with l-NAME (100 µM) were similarly unaffected in TgNotch3R169C PAs (Fig. 3 A and B). Collectively, these results indicate that elevated endothelial function is not responsible for the decreased myogenic tone observed in TgNotch3R169C PAs.
Fig. 3.

Endothelial-dependent influences on myogenic tone and BK channel activity are unaltered in PAs from TgNotch3R169C mice. (A, a–c) Representative traces showing constriction of pressurized (40 mmHg) PAs induced by the SK channel blocker apamin (300 nM), the BK channel blocker paxilline (1 µM), the IK channel blocker charybdotoxin (100 nM), and the eNOS inhibitor L-NAME (100 µM). (B) Summary data expressed as means ± SEM. N.S. indicates not significantly different (P > 0.05; one-way ANOVA). The number of animals is shown in parentheses. (C) Typical recording of the internal diameter of PAs during myogenic constriction in response to increasing intraluminal pressure in the presence of the BK channel blocker paxilline (1 µM). (D) Summary myogenic tone data (means ± SEM) in the presence and absence of paxilline.
Although BK channels exert powerful negative feedback regulation of membrane potential and tone in pressurized pial arteries (23), these channels do not appear to play a significant role in PAs under physiological conditions (22, 24). Consistent with this, the myogenic response of TgNotch3R169C PAs measured over a range of intravascular pressures was the same in the presence and absence of the BK channel blocker paxilline (1 µM) (Fig. 3 C and D), suggesting that a change in BK channel activity is not likely the common denominator.
PAs from TgNotch3R169C Mice Are Hypersensitive to 4-Aminopyridine.
The compound 4-aminopyridine (4-AP) preferentially blocks members of the KV1 family (25), which exert powerful negative feedback control over vasoconstriction in pial arteries (26) and PAs (27). In particular, molecular evidence indicates that KV1.2 and KV1.5 channels underlie the 4-AP sensitivity of K+ currents, membrane potential, and tone in arteries (28–32) and PAs (27). Here, we found that 4-AP (1 mM and 5 mM) induced a concentration-dependent constriction of PAs from mice of all genotypes (Fig. 4A, a–c). Importantly, the degree of vasoconstriction was greater in TgNotch3R169C PAs, suggesting higher KV1 activity (Fig. 4B). Moreover, with KV1 activity inhibited (1 mM 4-AP), PAs from all three genotypes constricted to the same level in response to pressure (40% ± 2%, 40% ± 4%, and 46% ± 5% for TgNotch3R169C, TgNotch3WT, and Non-Tg mice, respectively; P = 0.5), indicating that the remaining ionic conductances likely drive the membrane potential to similar values. Stromatoxin (20 nM) (33), an inhibitor of KV2 channels, did not significantly constrict PAs from TgNotch3R169C or Non-Tg mice (Fig. S1 A and B), further supporting the interpretation that changes in KV1 activity underlie the observed hypersensitivity of TgNotch3R169C PAs to 4-AP.
Fig. 4.

The 4-AP–induced constriction is elevated in PAs from TgNotch3R169C mice. (A, a–c) Representative traces showing constriction of pressurized (40 mmHg) PAs induced by the KV channel blocker 4-AP (1 mM and 5 mM). (B) Summary data expressed as means ± SEM (*P < 0.05; one-way ANOVA). The number of animals is shown in parentheses.
The regulation of myogenic tone differs between PAs and larger pial arteries on the surface of the brain (22, 24). However, myogenic tone is similarly attenuated in PAs and pial arteries (5) (Table S1), suggesting a common mechanism. To confirm this, we applied the pharmacologic approach described above to pressurized pial arteries from TgNotch3R169C and control (TgNotch3WT and Non-Tg) mice. Consistent with the findings from PAs, sensitivity to 4-AP (1 mM) was increased in pressurized (60 mmHg) pial arteries from TgNotch3R169C mice (Fig. S2 A and B). Furthermore, blocking BK channels or IK and SK channels, or inhibiting eNOS, had the same effect in all three genotypes (Fig. 2), and inhibition of KV2 channels with stromatoxin (20 nM) constricted pial arteries from TgNotch3R169C and Non-Tg to the same extent (−14% ± 2% and −14% ± 3%, respectively; Fig. S1 C and D). Taken together, these results suggest that KV1 channel activity is increased in both pial arteries and PAs of TgNotch3R169C mice.
KV Channel Current Density Is Increased in Cerebral Artery Myocytes from TgNotch3R169C Mice.
An increase in KV activity could occur through a change in channel gating properties, recruitment of new KV channel family members, or an increase in channel number. To investigate these possibilities, we directly measured KV currents in myocytes freshly isolated from cerebral arteries of TgNotch3R169C mice, as described in SI Materials and Methods. Currents were recorded in response to 250-ms voltage steps from −70 mV to +60 mV (holding potential, −80 mV) in 10-mV increments (Fig. 5A). Myocytes from TgNotch3R169C, TgNotch3WT, and Non-Tg animals had similar cell capacitances, indicating a similar membrane surface area. KV current density in myocytes from TgNotch3R169C mice was significantly greater than that in control cells at all voltage steps from −30 mV to +60 mV (Fig. 5 A and B); at 0 mV, KV current density was ∼60% greater in TgNotch3R169C myocytes than in TgNotch3WT myocytes. In contrast, activation time constants (τact), determined from an exponential fit of individual voltage-evoked current traces (20-mV increments), were the same in cerebral myocytes from TgNotch3R169C, TgNotch3WT, and Non-Tg mice, suggesting that channel gating was unchanged (Fig. 5C and Table S2). Deactivation time constants (τdeact) of the current decay (“tail currents”) after stepping from a test potential to −40 mV, obtained by exponential fits, were the same in myocytes from all three genotypes (Fig. 5D and Table S2). Half-maximal activation voltage (V0.5) and slope (k), determined by fitting normalized peak tail currents to the Boltzmann equation, were similarly indistinguishable among genotypes (Fig. 5E and Table S2). The absence of a difference in τact, V0.5, k, or τdeact indicates that the increase in current density in TgNotch3R169C myocytes is attributable to an increase in the number of channels rather than a change in their gating properties or subtype composition. We then compared the effect of 4-AP on the current density in TgNotch3R169C, TgNotch3WT, and Non-Tg myocytes. Consistent with an increase in KV1 channel density, 4-AP (1 mM)–sensitive currents were significantly increased in cerebral SMCs from TgNotch3R169C myocytes (Fig. S3). Using the Goldman–Hodgkin–Katz constant field equation and a single-channel conductance of 15 pS (34), we estimated that the average number of functional KV channels per myocyte was 4,970 in TgNotch3R169C mice, compared with 3,275 in TgNotch3WT mice and 3,060 in Non-Tg mice.
Fig. 5.

KV channel current density is elevated in SMCs from TgNotch3R169C mice. (A) Families of KV currents from isolated cerebral arterial and arteriolar SMCs elicited by voltage pulses from −70 mV to +60 mV in the presence of 1 µM paxilline (included to block BK channel currents). (B) Graph summarizing the current density, calculated by dividing membrane current amplitude at the end of the pulse by cell capacitance (*P < 0.05; one-way ANOVA). (C) Activation time constants (τact) were determined from an exponential fit of individual voltage-evoked (20-mV steps) current traces in the presence and absence of 4-AP (5 mM). (D) Deactivation time constants (τdeact), obtained from an exponential fit of tail currents at −40 mV. (E) Steady-state activation properties of KV currents measured from normalized tail currents. The voltage for half-maximal activation (V0.5) and the factor k were obtained from a fit of the data to the Boltzman equation.
Heparin-Binding EGF–Induced Reduction in the Number of Functional KV1 Channels Restores Myogenic Tone in PAs from TgNotch3R169C Mice.
It has previously been reported that KV1 channel activity in cerebral arteries is suppressed by activation of the receptor tyrosine kinase EGFR with the agonist heparin-binding EGF-like growth factor (HB-EGF) (35), an effect that reflects enhanced KV1 channel endocytosis (36). To provide further support for the concept that an increase in KV1 channel number in cerebral artery SMCs is responsible for the impaired myogenic tone in TgNotch3R169C cerebral microvessels, we tested whether directly reducing the surface expression of KV channels by treatment with HB-EGF restored myogenic tone in PAs from TgNotch3R169C mice. Consistent with previous studies in wild-type cerebral arteries, HB-EGF induced a concentration-dependent vasoconstriction of TgNotch3R169C PAs (Fig. 6A). Using the relationship between HB-EGF concentration and percentage of increase in tone, we determined that an HB-EGF concentration of 30 ng/mL produced the 60% increase in tone necessary to reach TgNotch3WT levels at 40 mmHg (Fig. 6B). At this concentration, HB-EGF significantly reduced KV current densities in TgNotch3R169C cerebral myocytes (recorded at +30 mV) from 24 pA/pF to 19 pA/pF (Fig. 6 C and D), a density not significantly different from that observed in TgNotch3WT cerebral myocytes (18 pA/pF; Fig. 5B). Importantly, it also normalized myogenic tone (Fig. 6E). Collectively, these results confirm that up-regulation of KV1 channel number plays a pivotal role in the altered myogenic phenotype of TgNotch3R169C cerebral arteries/arterioles.
Fig. 6.

HB-EGF–induced suppression of KV1 channels restores myogenic response in PAs from TgNotch3R169C mice. (A) Typical recording of the internal diameter of pressurized PAs (40 mmHg) from a TgNotch3R169C mouse during the vasoconstriction induced by cumulative addition of HB-EGF (0.003–100 ng/mL). (B) Summary data showing EC50 value. (C) Family of KV currents, recorded from isolated TgNotch3R169C cerebral arterial SMCs (10.3 pF) with and without HB-EGF (30 ng/mL), in the presence of 1 µM paxilline to block BK channel currents. (D) Graph summarizing the current density of eight myocytes from five different animals of each genotype (*P < 0.05; one-way ANOVA). (E) Summary myogenic tone data for PAs from TgNotch3R169C mice during myogenic constriction in response to increasing intraluminal pressure, with and without 30 ng/mL HB-EGF (n = 8) expressed as means ± SEM (**P < 0.01; one-way ANOVA).
Vascular Function and KV Current Densities in Peripheral Resistance Arteries Are Unaffected by the NOTCH3(R169C) Mutant.
NOTCH3 is expressed by SMCs throughout the vasculature (4), and deposits of NOTCH3ECD, the pathological hallmark of CADASIL, are detected in both cerebral and peripheral vessels in patients (37, 38). However, despite this, the clinical manifestations of the disease are restricted to the central nervous system (3). As a first step toward identifying differences between the cerebral and peripheral circulation in CADASIL that might reveal pathological mechanisms, we compared the effects of the R169C mutation in the cerebral circulation with those in the systemic circulation, using third-order branches of mesenteric resistance arteries, which also express KV1 and KV2 channel family members (31, 39–41). Immunostaining with an antibody against NOTCH3ECD confirmed that mesenteric arteries from TgNotch3R169C mice displayed the characteristic granular accumulation of NOTCH3ECD at the plasma membrane of SMCs (Fig. S4). However, in contrast to cerebral arteries, mesenteric arteries from TgNotch3R169C mice showed no difference in myogenic response compared with those from Non-Tg mice (Fig. 7 A and B), and phenylephrine-induced constriction was also unaltered (Fig. 7 C and D). Moreover, 4-AP (1 mM and 5 mM) caused a concentration-dependent constriction of mesenteric arteries that was similar in both genotypes (Fig. 7 C and E). Interestingly, HB-EGF, which effectively suppressed KV1 currents in PA myocytes from TgNotch3R169C mice, did not affect KV current density or promote constriction in mesenteric arteries from either genotype (Fig. 7 C, F, and G), suggesting that the HB-EGF–dependent mechanism of KV1 channel regulation does not operate in mesenteric arteries.
Fig. 7.

Resistance mesenteric arteries from TgNotch3R169C mice display normal myogenic responses. (A) Typical recordings of the internal diameter of third-order mesenteric arteries during myogenic constriction in response to increasing intraluminal pressure. (B) Summary myogenic tone data (means ± SEM). (C) Typical recordings of pressurized (80 mmHg) third-order mesenteric arteries from Non-Tg and TgNotch3R169C mice showing induction of vasoconstriction by phenylephrine (cumulative addition of 1–10 µM) and 4-AP (1 mM and 5 mM), but not by HB-EGF (100 ng/mL). (D) Summary data showing EC50 values of phenylephrine-induced constriction. (E) Summary data showing percentage of increase in tone (means ± SEM). The number of animals is shown in parentheses. (F) Family of KV currents recorded in the presence of 1 µM paxilline (to block BK channel currents) from isolated mesenteric artery SMCs elicited by voltage pulses from −70 mV to +60 mV. (G) Graph summarizing the current density, calculated by dividing membrane current amplitude at the end of the pulse by cell capacitance.
KV channel currents in SMCs from mesenteric arteries exhibited properties (V0.5, k, τact, or τdeact) similar to those of SMCs from cerebral arteries (Table S2). These data, combined with expression data (31, 39–41) and the functional effects of 4-AP, suggest that the same KV channels regulate membrane potential in cerebral and mesenteric arteries. However, unlike the case in cerebral arteries, KV current density was virtually identical in mesenteric artery SMCs from Non-Tg and TgNotch3R169C mice (22 pA/pF at +30 mV; Fig. 7G). Interestingly, KV channel current density in mesenteric myocytes from Non-Tg and TgNotch3R169C mice was similar to that in cerebral artery myocytes from TgNotch3R169C mice (24 pA/pF at +30 mV; Fig. 5B). These results suggest that KV channels are already expressed at elevated levels in SMCs in the peripheral circulation, consistent with the hypothesized absence of a functional EGFR-mediated KV1 endocytosis mechanism in these peripheral vessels.
Discussion
In this study, we explored the basis of altered brain artery function in a genetic model of SVD. We found that pressure-induced vasoconstriction of brain arterioles from a TgNotch3R169C mouse model of CADASIL is diminished at a very early stage of the disease (i.e., 6 mo before formation of white matter lesions). Consistent with this weaker myogenic response, membrane potential measurements revealed abnormal hyperpolarization in PAs from these mice. Importantly, we provide to our knowledge the first insight into the molecular basis of the cerebral artery dysfunction associated with CADASIL, demonstrating that up-regulation of KV1 channel number is responsible for blunted membrane potential depolarization and diminished myogenic response at physiological pressures in PAs, as well as pial arteries, from TgNotch3R169C mice (Fig. 8).
Fig. 8.
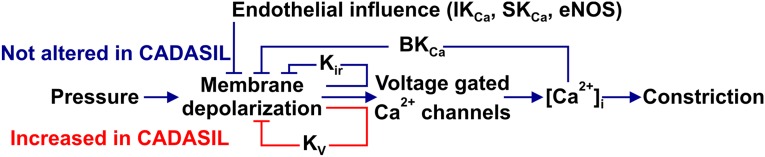
Proposed mechanism leading to altered membrane potential and myogenic tone in CADASIL. The number of functional KV1 channels in the smooth muscle cell membrane increases by about 60%, thereby enhancing the negative feedback response to an elevation in intravascular pressure. No other functional feature of the parenchymal arterioles was affected at this early stage in disease progression.
KV current density in TgNotch3R169C mice was increased by ∼60% without a change in KV kinetics or activation properties, suggesting that NOTCH3(R169C) expression leads to an increase in the number of KV1 channels in the plasma membrane. Consistent with this interpretation, functional KV1 channel numbers were estimated to increase by 60% in myocytes from TgNotch3R169C mice. Moreover, constriction to the preferential KV1 channel blocker, 4-AP, was augmented by NOTCH3(R169C) expression, with electrophysiology data indicating that the KV1.5 subtype is likely up-regulated. This interpretation is consistent with previous reports that heteromultimeric KV1.2/1.5 channels underlie the effect of 4-AP on KV currents and arterial tone (28–31), and KV1.5, whose current properties most closely match native KV currents in cerebral and mesenteric myocytes (Table S2), is a key player in myogenic tone regulation (32). In contrast, pharmacological data and biophysical properties were not consistent with a role for KV2 or KV7 channels (Table S2). Moreover, vasodilatory influences of the endothelium (IK/SK, NO) and the BK channel-dependent negative feedback loop were unchanged in both pial arteries and PAs of TgNotch3R169C mice.
The fact that NOTCH3(R169C) did not affect constriction of PAs to low intravascular pressure (20 mmHg) or the ability of PAs to constrict to 60 mM external K+, but greatly reduced constriction to physiological pressures (40 mmHg) (18) indicates that the defect emerges in association with increased intravascular pressure and SMC membrane potential depolarization. The reduced myogenic tone was not due to a change in the relationship between membrane potential and myogenic tone. Interestingly, the greater constriction induced by 4-AP in cerebral arteries/arterioles from TgNotch3R169C mice eliminated the difference in myogenic tone, supporting the concept that the abnormal membrane potential is directly responsible for the difference in tone. Finally, decreasing KV current density in TgNotch3R169C cerebral SMCs to the levels measured in TgNotch3WT mice through HB-EGF–induced KV channel endocytosis was sufficient to restore a WT-like myogenic response in PAs from TgNotch3R169C mice. Taken together, these results indicate that the elevation in KV channel number resulting from the presence of the R169C mutation fully accounts for the impaired myogenic response. To our knowledge, this is the first report of abnormal membrane potential regulation in CADASIL, specifically, or cerebral SVD generally.
The myogenic response was normal in third-order mesenteric arteries from TgNotch3R169C mice, and 4-AP–induced constriction and KV channel activity were unchanged in this peripheral vascular bed, despite expression of the mutated protein and accumulation of NOTCH3ECD. These observations are consistent with previous reports that the phenotypic manifestations of CADASIL are essentially restricted to the cerebral circulation and reinforce the validity of TgNotch3R169C mice as a preclinical model of CADASIL. The mechanistic basis of the brain-specific consequences of NOTCH3(R169C) expression will require more extensive investigation, but KV channel up-regulation is clearly central to this tissue-restricted phenotype. Although the NOTCH3 signaling pathway has been reported to regulate KV1.5 gene expression (42), it seems unlikely that this mechanism could account for the cerebrovascular specific manifestations of the NOTCH3(R169C) mutant because this mutant would be expected to have a global effect on the vasculature, yet KV current densities were unchanged in mesenteric artery myocytes. More importantly, HB-EGF completely normalized KV current density and myogenic tone in cerebral arteries, implying that altered membrane trafficking of KV1.5 is capable of fully accounting for the up-regulation of KV channels in TgNotch3R169C mice. The similar biophysical properties of KV channels in cerebral and peripheral vasculature (Table S2) also argue that differences in subunit composition are not responsible for the differential effects of the NOTCH3(R169C) mutant in the two vascular beds, an argument buttressed by a pharmacological profile consistent with a KV1.2/1.5 channel subtype and literature support for the expression of KV1 family members in both cerebral and mesenteric arteries (28–31, 39–41). Differences in the caliber of PAs and mesenteric arteries are also not likely a factor insofar as the effects of NOTCH3(R169C) expression on pial arteries, which have an internal diameter similar to that of the third-order mesenteric arteries used here, were essentially identical to those on PAs. However, the fact that HB-EGF normalized KV current density and restored myogenic tone in cerebral arteries from TgNotch3R169C mice, but had no effect on mesenteric arteries from these mice, suggests a possible mechanistic rationale for the cerebrovascular-specific effects of CADASIL. According to this scenario, the peripheral vasculature does not respond to HB-EGF because it lacks the EGFR-mediated pathway that promotes KV channel endocytosis in cerebral arteries. This formulation can be extended to account for up-regulation of KV channels in SMCs of pial arteries and PAs in TgNotch3R169C mice by postulating that expression of NOTCH3(R169C) suppresses the EGFR pathway in these cells, resulting in decreased EGFR-mediated endocytosis (and thus increased surface expression) of KV channels. In the context of animal models of subarachnoid hemorrhage (SAH), where this EGFR-mediated endocytosis pathway was first characterized (35, 36), KV channels are down-regulated due to increased matrix metalloproteinase (MMP)-mediated cleavage of pro–HB-EGF and enhanced shedding of HB-EGF. Interestingly, Joutel and coworkers recently reported (43) increased levels and activity of the MMP inhibitor, tissue inhibitor of metalloproteinase 3 (TIMP3) in the characteristic NOTCH3ECD aggregates that accumulate around small cerebral vessels in CADASIL patients and TgNotch3R169C mice early in the progression of the disease, a finding that would be consistent with diminished HB-EGF shedding and the postulated suppression of EGFR-mediated KV channel endocytosis. Indeed, TIMP3 specifically inhibits ADAM17 (44), also known as TNFα-converting enzyme (TACE), which is involved in the shedding of several EGFR ligands, including HB-EGF (45, 46).
The defect in myogenic tone appears in both pial arteries and parenchymal arterioles of TgNotch3R169C mice, but the pathological issues that present with SVD in patients occur preferentially in arterioles within the brain (1, 9). Although it is difficult to predict the consequences of an “early” defect in myogenic tone in PAs, the intraparenchymal circulation clearly constitutes a unique vascular bed with specific features—shared by human and rodent brains (47, 48)—that might contribute to later manifestations of SVD. Notable in this context, the limited collateral blood supply between PAs creates a “bottleneck” effect (7) that segregates perfused territories and therefore worsens the impact of PA lesions on nervous tissue (8, 9). Moreover, because PAs are the last vessels upstream of the capillary bed, the decreased resistance created by the reduction in myogenic tone would ultimately hamper the ability of PAs to protect capillaries from disruptive effects of acute elevations of arterial pressure. Our observations may also help account for other aspects of the CADASIL phenotype observed in the TgNotch3R169C mouse model (5). For example, the less constricted basal state of PAs in this model implies a diminution in the vasodilatory reserve, potentially decreasing NVC efficiency. A substantial part of this hemodynamic response is mechanistically linked to PA dilation (13), which is preconditioned by the resting tone of the vessel (49, 50). Thus, the diminished myogenic responsiveness of the cerebral microvasculature of TgNotch3R169C mice—a direct consequence of KV1 channel up-regulation—would be expected to impair NVC. This is in fact the case (5).
Our results support the concept that the level of myogenic tone depends on surface expression of the KV channel—a major negative feedback element that controls vascular smooth muscle membrane potential. The fact that altered surface membrane expression of KV channels in cerebral arteries is the key feature underlying disparate pathologies—CADASIL (present study) and SAH (35, 36)—suggests a high degree of plasticity in KV channel regulatory mechanisms. In both pathologies, a change in the gain of the KV1-mediated negative feedback circuit would be expected to affect CBF and the responses of arteries to pressure fluctuations. Importantly, because no other features of smooth muscle or endothelial cells appeared to be altered in the TgNotch3R169C mouse model of SVD, our findings suggest that up-regulation of KV1 channels is directly responsible for the observed cerebrovascular dysfunction. Others have previously reported channelopathies in which the identified channel mutation altered cell surface expression without affecting the intrinsic properties of the channel (51). Our current results, taken together with these previous observations, argue that the concept of channelopathy should be broadened to include mutations in nonchannel proteins that produce pathologies by altering the number of functional channels without affecting their intrinsic properties. Our findings further suggest that the KV1 channel and the pathways that regulate its surface expression are potential therapeutic targets in SVD.
Materials and Methods
Animal Model.
The Tg mouse lines, TgNotch3WT and TgNotch3R169C, have been previously described (5). Non-Tg mice are nontransgenic littermates obtained during breeding of TgNotch3WT and TgNotch3R169C mice and were used as wild-type mice. Six-month-old animals were euthanized by i.p. injection of sodium pentobarbital (100 mg/kg) followed by rapid decapitation. We focused on mice at this age because this is well in advance (6 mo) of the development of significant white matter lesion burden and for comparison with our previous study (5, 6). TgNotch3WT and TgNotch3R169C mice (on an FVB/N background) overexpress rat wild-type NOTCH3 and the CADASIL-causing NOTCH3(R169C) mutant protein, respectively, to a similar degree (approximately fourfold) compared with the levels of endogenous NOTCH3 in Non-Tg mice (5). Expression of CADASIL-causing mutations at normal endogenous levels does not produce a CADASIL-like phenotype, likely because the slowly developing mutant phenotype is unable to manifest during the short lifespan of a mouse (52). Overexpression of the mutant protein overcomes this constraint and is thus a key feature of this model. All experimental protocols used in this study were in accord with institutional guidelines approved by the Institutional Animal Care and Use Committee of the University of Vermont.
Diameter Measurements.
Precapillary segments of PAs that arise from the middle cerebral artery M1 region and perfuse the neocortex, PCAs, and third-order mesenteric arteries (∼100 μm internal diameter) were dissected free of surrounding tissue, cannulated on glass micropipettes (with one end occluded for PAs) in an organ chamber (University of Vermont Instrumentation and Model Facility), and pressurized using an arteriograph system (Living Systems Instrumentation). Vessel internal diameter was continuously monitored using a CCD camera and edge-detection software (IonOptix). Drugs were applied by addition to the superfusate.
Arteriolar Smooth Muscle Membrane Potential Recordings.
PAs were pressurized as described above. Membrane potential was recorded in myocytes, using glass microelectrodes filled with 0.5 M KCl (tip resistance, 150–250 MΩ) (19).
SMC Isolation and Perforated Patch-Clamp Experiments.
Single SMCs were isolated from cerebral arteries (anterior, middle, and posterior) and third-order mesenteric arteries by enzymatic digestion followed by gentle trituration. Outward K+ currents were recorded from single cells, using the perforated-cell configuration of the patch-clamp technique, in the presence of 1 µM paxilline to block BK currents. The relationship between myocyte membrane voltage and the amplitudes of the tail currents (I) was fitted to a Boltzmann equation,
where Imax is the measured peak tail current, to allow determination of the half-maximal activation potential (V0.5) and slope (k).
Estimation of the Number of Channels.
In symmetrical, high extracellular K+ ([K+]o), and intracellular K+ ([K+]i) solutions, the Goldman–Hodgkin–Katz flux equation (53) for K+ predicts a linear relationship between channel current amplitude i and membrane potential:
With [K+]o = [K+]i the equation becomes
where PK is the permeability to K+ of a single channel (in centimeters per second); E is the membrane potential (volts); F, R, and T have their usual meanings; and the K+ concentration is given in moles per milliliter. Because K+ single-channel conductance is defined as , permeability can be defined as
Using a single-channel conductance of 15 pS recorded in inside-out patches with symmetrical [K+] = 140 mM (34), we calculated PK = 2.83 × 10−14 cm/s at 23 °C. The single-channel current amplitude, i, was then estimated using PK and the Goldman–Hodgkin–Katz flux equation at a given voltage (−40 mV) with [K+]o = 3 mM and [K+]i = 140 mM. Finally, the number of channels (N) was determined using the macroscopic current amplitude (I) equation, I = iNPo, with Po = 0.014 at −40 mV (34).
Statistical Analysis.
At least two arteries and arterioles per animal were used in diameter measurements, with “n” indicating the number of animals. In perforated patch-clamp experiments, at least seven myocytes from five different animals were used per group. Data are expressed as means ± SEM. The significance of differences between two means was determined using Student’s t test. Multiple comparisons were evaluated by one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test. Statistical significance was accepted at the 95% (P < 0.05) confidence level.
For detailed information on diameter measurements, membrane potential recordings, SMCs isolation, perforated patch-clamp experiments, estimation of the number of channels, NOTCH3ECD immunohistochemistry, solutions, and drugs, please refer to SI Materials and Methods.
Supplementary Material
Acknowledgments
The authors thank Drs. Albert Gonzales, Masayo Koide, Thomas Longden, Nathan Tykocki, and George Wellman for helpful advice and the “CADASIL, Together We Have Hope” nonprofit organization for their support and promoting awareness of CADASIL. This work was supported by National Institutes of Health Grants R37DK053832, P01HL095488, R01HL44455, and R01HL121706; the Totman Trust for Medical Research (M.T.N.); postdoctoral fellowships from the Lundbeck Foundation (to C.K. and T.D.); the Fondation Leducq Transatlantic Network of Excellence on the Pathogenesis of Small Vessel Disease of the Brain (A.J. and M.T.N.); the United Leukodystrophy Foundation CADASIL research grant (to F.D.); and the National Research Agency, France (Grants ANR Genopath 2009-RAE09011HSA and ANR Blanc 2010-RPV11011HHA) (to A.J.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. S.M. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1420765112/-/DCSupplemental.
References
- 1.Pantoni L. Cerebral small vessel disease: From pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010;9(7):689–701. doi: 10.1016/S1474-4422(10)70104-6. [DOI] [PubMed] [Google Scholar]
- 2.Wardlaw JM, Smith C, Dichgans M. Mechanisms of sporadic cerebral small vessel disease: Insights from neuroimaging. Lancet Neurol. 2013;12(5):483–497. doi: 10.1016/S1474-4422(13)70060-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chabriat H, Joutel A, Dichgans M, Tournier-Lasserve E, Bousser M-G. Cadasil. Lancet Neurol. 2009;8(7):643–653. doi: 10.1016/S1474-4422(09)70127-9. [DOI] [PubMed] [Google Scholar]
- 4.Domenga V, et al. Notch3 is required for arterial identity and maturation of vascular smooth muscle cells. Genes Dev. 2004;18(22):2730–2735. doi: 10.1101/gad.308904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Joutel A, et al. Cerebrovascular dysfunction and microcirculation rarefaction precede white matter lesions in a mouse genetic model of cerebral ischemic small vessel disease. J Clin Invest. 2010;120(2):433–445. doi: 10.1172/JCI39733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cognat E, Cleophax S, Domenga-Denier V, Joutel A. Early white matter changes in CADASIL: Evidence of segmental intramyelinic oedema in a pre-clinical mouse model. Acta Neuropathol Commun. 2014;2:49. doi: 10.1186/2051-5960-2-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nishimura N, Schaffer CB, Friedman B, Lyden PD, Kleinfeld D. Penetrating arterioles are a bottleneck in the perfusion of neocortex. Proc Natl Acad Sci USA. 2007;104(1):365–370. doi: 10.1073/pnas.0609551104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shih AY, et al. The smallest stroke: Occlusion of one penetrating vessel leads to infarction and a cognitive deficit. Nat Neurosci. 2013;16(1):55–63. doi: 10.1038/nn.3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80(4):844–866. doi: 10.1016/j.neuron.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duering M, et al. Incident lacunes preferentially localize to the edge of white matter hyperintensities: Insights into the pathophysiology of cerebral small vessel disease. Brain. 2013;136(Pt 9):2717–2726. doi: 10.1093/brain/awt184. [DOI] [PubMed] [Google Scholar]
- 11.Joutel A, Faraci FM. Cerebral small vessel disease: Insights and opportunities from mouse models of collagen IV-related small vessel disease and cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Stroke. 2014;45(4):1215–1221. doi: 10.1161/STROKEAHA.113.002878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faraci FM, Heistad DD. Regulation of large cerebral arteries and cerebral microvascular pressure. Circ Res. 1990;66(1):8–17. doi: 10.1161/01.res.66.1.8. [DOI] [PubMed] [Google Scholar]
- 13.Fernández-Klett F, Offenhauser N, Dirnagl U, Priller J, Lindauer U. Pericytes in capillaries are contractile in vivo, but arterioles mediate functional hyperemia in the mouse brain. Proc Natl Acad Sci USA. 2010;107(51):22290–22295. doi: 10.1073/pnas.1011321108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bayliss WM. On the local reactions of the arterial wall to changes of internal pressure. J Physiol. 1902;28(3):220–231. doi: 10.1113/jphysiol.1902.sp000911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol. 1995;268(4 Pt 1):C799–C822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- 16.Knot HJ, Nelson MT. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol. 1998;508(Pt 1):199–209. doi: 10.1111/j.1469-7793.1998.199br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Faraci FM, Heistad DD. Regulation of the cerebral circulation: Role of endothelium and potassium channels. Physiol Rev. 1998;78(1):53–97. doi: 10.1152/physrev.1998.78.1.53. [DOI] [PubMed] [Google Scholar]
- 18.Baumbach GL, Sigmund CD, Faraci FM. Cerebral arteriolar structure in mice overexpressing human renin and angiotensinogen. Hypertension. 2003;41(1):50–55. doi: 10.1161/01.hyp.0000042427.05390.5c. [DOI] [PubMed] [Google Scholar]
- 19.Nystoriak MA, et al. Fundamental increase in pressure-dependent constriction of brain parenchymal arterioles from subarachnoid hemorrhage model rats due to membrane depolarization. Am J Physiol Heart Circ Physiol. 2011;300(3):H803–H812. doi: 10.1152/ajpheart.00760.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davis MJ, Hill MA. Signaling mechanisms underlying the vascular myogenic response. Physiol Rev. 1999;79(2):387–423. doi: 10.1152/physrev.1999.79.2.387. [DOI] [PubMed] [Google Scholar]
- 21.Knot HJ, Zimmermann PA, Nelson MT. Extracellular K(+)-induced hyperpolarizations and dilatations of rat coronary and cerebral arteries involve inward rectifier K(+) channels. J Physiol. 1996;492(Pt 2):419–430. doi: 10.1113/jphysiol.1996.sp021318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hannah RM, Dunn KM, Bonev AD, Nelson MT. Endothelial SK(Ca) and IK(Ca) channels regulate brain parenchymal arteriolar diameter and cortical cerebral blood flow. J Cereb Blood Flow Metab. 2011;31(5):1175–1186. doi: 10.1038/jcbfm.2010.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brayden JE, Nelson MT. Regulation of arterial tone by activation of calcium-dependent potassium channels. Science. 1992;256(5056):532–535. doi: 10.1126/science.1373909. [DOI] [PubMed] [Google Scholar]
- 24.Dabertrand F, Nelson MT, Brayden JE. Acidosis dilates brain parenchymal arterioles by conversion of calcium waves to sparks to activate BK channels. Circ Res. 2012;110(2):285–294. doi: 10.1161/CIRCRESAHA.111.258145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gutman GA, et al. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol Rev. 2005;57(4):473–508. doi: 10.1124/pr.57.4.10. [DOI] [PubMed] [Google Scholar]
- 26.Knot HJ, Nelson MT. Regulation of membrane potential and diameter by voltage-dependent K+ channels in rabbit myogenic cerebral arteries. Am J Physiol. 1995;269(1 Pt 2):H348–H355. doi: 10.1152/ajpheart.1995.269.1.H348. [DOI] [PubMed] [Google Scholar]
- 27.Straub SV, et al. Regulation of intracerebral arteriolar tone by K(v) channels: Effects of glucose and PKC. Am J Physiol Cell Physiol. 2009;297(3):C788–C796. doi: 10.1152/ajpcell.00148.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Albarwani S, et al. Voltage-gated K+ channels in rat small cerebral arteries: Molecular identity of the functional channels. J Physiol. 2003;551(Pt 3):751–763. doi: 10.1113/jphysiol.2003.040014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thorneloe KS, et al. Molecular composition of 4-aminopyridine-sensitive voltage-gated K(+) channels of vascular smooth muscle. Circ Res. 2001;89(11):1030–1037. doi: 10.1161/hh2301.100817. [DOI] [PubMed] [Google Scholar]
- 30.Kerr PM, et al. Heteromultimeric Kv1.2-Kv1.5 channels underlie 4-aminopyridine-sensitive delayed rectifier K(+) current of rabbit vascular myocytes. Circ Res. 2001;89(11):1038–1044. doi: 10.1161/hh2301.100803. [DOI] [PubMed] [Google Scholar]
- 31.Plane F, et al. Heteromultimeric Kv1 channels contribute to myogenic control of arterial diameter. Circ Res. 2005;96(2):216–224. doi: 10.1161/01.RES.0000154070.06421.25. [DOI] [PubMed] [Google Scholar]
- 32.Chen TT, Luykenaar KD, Walsh EJ, Walsh MP, Cole WC. Key role of Kv1 channels in vasoregulation. Circ Res. 2006;99(1):53–60. doi: 10.1161/01.RES.0000229654.45090.57. [DOI] [PubMed] [Google Scholar]
- 33.Escoubas P, Diochot S, Célérier M-L, Nakajima T, Lazdunski M. Novel tarantula toxins for subtypes of voltage-dependent potassium channels in the Kv2 and Kv4 subfamilies. Mol Pharmacol. 2002;62(1):48–57. doi: 10.1124/mol.62.1.48. [DOI] [PubMed] [Google Scholar]
- 34.Aiello EA, Malcolm AT, Walsh MP, Cole WC. Beta-adrenoceptor activation and PKA regulate delayed rectifier K+ channels of vascular smooth muscle cells. Am J Physiol. 1998;275(2 Pt 2):H448–H459. doi: 10.1152/ajpheart.1998.275.2.H448. [DOI] [PubMed] [Google Scholar]
- 35.Koide M, Penar PL, Tranmer BI, Wellman GC. Heparin-binding EGF-like growth factor mediates oxyhemoglobin-induced suppression of voltage-dependent potassium channels in rabbit cerebral artery myocytes. Am J Physiol Heart Circ Physiol. 2007;293(3):H1750–H1759. doi: 10.1152/ajpheart.00443.2007. [DOI] [PubMed] [Google Scholar]
- 36.Ishiguro M, et al. Oxyhemoglobin-induced suppression of voltage-dependent K+ channels in cerebral arteries by enhanced tyrosine kinase activity. Circ Res. 2006;99(11):1252–1260. doi: 10.1161/01.RES.0000250821.32324.e1. [DOI] [PubMed] [Google Scholar]
- 37.Joutel A, et al. The ectodomain of the Notch3 receptor accumulates within the cerebrovasculature of CADASIL patients. J Clin Invest. 2000;105(5):597–605. doi: 10.1172/JCI8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Joutel A, et al. Skin biopsy immunostaining with a Notch3 monoclonal antibody for CADASIL diagnosis. Lancet. 2001;358(9298):2049–2051. doi: 10.1016/S0140-6736(01)07142-2. [DOI] [PubMed] [Google Scholar]
- 39.Xu C, Lu Y, Tang G, Wang R. Expression of voltage-dependent K(+) channel genes in mesenteric artery smooth muscle cells. Am J Physiol. 1999;277(5 Pt 1):G1055–G1063. doi: 10.1152/ajpgi.1999.277.5.G1055. [DOI] [PubMed] [Google Scholar]
- 40.Fountain SJ, et al. Functional up-regulation of KCNA gene family expression in murine mesenteric resistance artery smooth muscle. J Physiol. 2004;556(Pt 1):29–42. doi: 10.1113/jphysiol.2003.058594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cox RH. Molecular determinants of voltage-gated potassium currents in vascular smooth muscle. Cell Biochem Biophys. 2005;42(2):167–195. doi: 10.1385/CBB:42:2:167. [DOI] [PubMed] [Google Scholar]
- 42.Fouillade C, et al. Transcriptome analysis for Notch3 target genes identifies Grip2 as a novel regulator of myogenic response in the cerebrovasculature. Arterioscler Thromb Vasc Biol. 2013;33(1):76–86. doi: 10.1161/ATVBAHA.112.251736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Monet-Leprêtre M, et al. Abnormal recruitment of extracellular matrix proteins by excess Notch3 ECD: A new pathomechanism in CADASIL. Brain. 2013;136(Pt 6):1830–1845. doi: 10.1093/brain/awt092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Amour A, et al. TNF-alpha converting enzyme (TACE) is inhibited by TIMP-3. FEBS Lett. 1998;435(1):39–44. doi: 10.1016/s0014-5793(98)01031-x. [DOI] [PubMed] [Google Scholar]
- 45.Lee DC, et al. TACE/ADAM17 processing of EGFR ligands indicates a role as a physiological convertase. Ann N Y Acad Sci. 2003;995:22–38. doi: 10.1111/j.1749-6632.2003.tb03207.x. [DOI] [PubMed] [Google Scholar]
- 46.Sahin U, et al. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol. 2004;164(5):769–779. doi: 10.1083/jcb.200307137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blinder P, Shih AY, Rafie C, Kleinfeld D. Topological basis for the robust distribution of blood to rodent neocortex. Proc Natl Acad Sci USA. 2010;107(28):12670–12675. doi: 10.1073/pnas.1007239107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lauwers F, Cassot F, Lauwers-Cances V, Puwanarajah P, Duvernoy H. Morphometry of the human cerebral cortex microcirculation: General characteristics and space-related profiles. Neuroimage. 2008;39(3):936–948. doi: 10.1016/j.neuroimage.2007.09.024. [DOI] [PubMed] [Google Scholar]
- 49.Blanco VM, Stern JE, Filosa JA. Tone-dependent vascular responses to astrocyte-derived signals. Am J Physiol Heart Circ Physiol. 2008;294(6):H2855–H2863. doi: 10.1152/ajpheart.91451.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Longden TA, Dabertrand F, Hill-Eubanks DC, Hammack SE, Nelson MT. Stress-induced glucocorticoid signaling remodels neurovascular coupling through impairment of cerebrovascular inwardly rectifying K+ channel function. Proc Natl Acad Sci USA. 2014;111(20):7462–7467. doi: 10.1073/pnas.1401811111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xie G, et al. A new Kv1.2 channelopathy underlying cerebellar ataxia. J Biol Chem. 2010;285(42):32160–32173. doi: 10.1074/jbc.M110.153676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ayata C. CADASIL: Experimental insights from animal models. Stroke. 2010;41(10) Suppl:S129–S134. doi: 10.1161/STROKEAHA.110.595207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hodgkin AL, Katz B. The effect of sodium ions on the electrical activity of giant axon of the squid. J Physiol. 1949;108(1):37–77. doi: 10.1113/jphysiol.1949.sp004310. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.