

Significance
Systemic lupus erythematosus (SLE) is a chronic systemic autoimmune disease that presents with a diverse array of clinical symptoms and afflicts over 1.5 million Americans. Current treatments involve immunosuppressive regimens associated with debilitating and adverse effects. With the description of a role for innate signaling in SLE, safe and efficient therapies that block Toll-like receptors also have been stymied by the relative short in vivo half lives of known inhibitors and the dangerous outcome of complete MyD88 blockade. Key natural regulators of the disease process are not well described but are more likely to provide disease-specific therapeutics with fewer adverse effects. In this study, we have identified a novel function for Stimulator of interferon genes as a suppressor of disease and a target for future SLE therapeutics.
Keywords: STING, autoimmunity, IRF3, lupus, TLRs
Abstract
Cytosolic DNA-sensing pathways that signal via Stimulator of interferon genes (STING) mediate immunity to pathogens and also promote autoimmune pathology in DNaseII- and DNaseIII-deficient mice. In contrast, we report here that STING potently suppresses inflammation in a model of systemic lupus erythematosus (SLE). Lymphoid hypertrophy, autoantibody production, serum cytokine levels, and other indicators of immune activation were markedly increased in STING-deficient autoimmune-prone mice compared with STING-sufficient littermates. As a result, STING-deficient autoimmune-prone mice had significantly shorter lifespans than controls. Importantly, Toll-like receptor (TLR)-dependent systemic inflammation during 2,6,10,14-tetramethylpentadecane (TMPD)-mediated peritonitis was similarly aggravated in STING-deficient mice. Mechanistically, STING-deficient macrophages failed to express negative regulators of immune activation and thus were hyperresponsive to TLR ligands, producing abnormally high levels of proinflammatory cytokines. This hyperreactivity corresponds to dramatically elevated numbers of inflammatory macrophages and granulocytes in vivo. Collectively these findings reveal an unexpected negative regulatory role for STING, having important implications for STING-directed therapies.
The detection of microbial DNA and the production of type I IFNs and proinflammatory cytokines is a central component of host defense. The pattern recognition receptor Toll-like receptor (TLR) 9 detects DNA that accumulates in endolysosomal compartments. In addition, newly described cytosolic DNA sensors [cyclic guanosine monophosphate adenosine monophosphate synthase (cGAS) and IFN inducible protein 16 (IFI16), among others], detect DNA in the cytosol (1, 2). Each of these cytosolic receptors converges on the multipass transmembrane endoplasmic reticulum-associated protein, Stimulator of IFN genes (STING) (also known as “TMEM173,” “MPYS,” “MITA,” and “ERIS”) (3, 4). STING is considered a critical hub, in cytosolic DNA-sensing pathways, for innate responses directed against numerous bacterial, viral, and parasitic pathogens (4). STING activates host defense via induction of IFN-β through a well-characterized pathway involving TBK1 and interferon regulatory factor 3 (IRF3) (4) and the production of the NF-κB–driven cytokines TNF-α and IL-6 (3).
STING-dependent cytosolic sensors also detect endogenous ligands, as first shown by polymorphisms in DNaseIII (3′ repair exonuclease 1, Trex1) and related enzymes associated with DNA degradation in patients with autoimmune diseases (5–7). Trex1-deficient mice fail to degrade endogenous DNA, resulting in type I IFN and proinflammatory cytokine production and sterile inflammation (8, 9). Importantly, STING deficiency limits inflammation in the above model. However, its role in more common systemic autoimmune diseases such as systemic lupus erythematosus (SLE) has not yet been investigated.
To understand the impact of cytosolic DNA-sensing pathways on SLE, we intercrossed STING-deficient mice with the SLE-prone strain MRL.Faslpr and compared disease parameters in STING-deficient and STING-sufficient Faslpr littermates. Our analysis reveals a previously undescribed role for STING as a negative regulator of TLR-mediated systemic autoimmunity, as the STING−/− MRL.Faslpr mice developed more severe disease and accelerated mortality. STING−/− mice subjected to 2,6,10,14-tetramethylpentadecane (TMPD)-induced acute peritonitis also display aggravated inflammation. Both models are driven by endosomal TLRs; therefore it was particularly interesting to find that STING-deficient macrophages were hyperresponsive to TLR ligands. Collectively, our findings highlight unappreciated cross-talk between TLR and cytosolic nucleic acid-sensing pathways in maintaining immune homeostasis and raise a cautionary note about targeting the STING pathway in chronic inflammation.
Results
STING Deficiency Accelerates Lymphocyte Accumulation and Activation During Lupus.
We generated two cohorts of autoimmune-prone mice by intercrossing MRL.Faslpr mice with either STING- or IRF3-deficient strains. This strategy led to the generation of F2 mice that were Faslpr homozygotes and either STING−/− or IRF3−/− (hereafter referred to as “STING/lpr” and “IRF3/lpr,” respectively) as well as Faslpr/lpr STINGWT/WT or IRF3WT/WT (referred to as “WT/lpr”). MRL.Faslpr mice routinely develop clinical features of SLE associated with lymphoid hypertrophy by 20–24 wk of age. However, by 16 wk of age the STING/lpr offspring displayed markedly greater splenomegaly and lymphadenopathy and higher splenocyte numbers than their STING-sufficient WT/lpr littermates (Fig. 1 A and B and Fig. S1A). Faslpr/lpr STINGWT/− (Het) littermates resembled the WT/lpr cohort. In contrast, the IRF3/lpr mice did not show any alteration in disease progression compared with control WT/lpr mice, and the splenic compartments were identical in composition and size (Fig. 1 A and B). Moreover, the frequency and total number of the unusual CD4/CD8 double-negative (DN) TCRβ+ B220+ T-cell subset, characteristic of Faslpr/lpr mice (10), was increased significantly in the STING/lpr mice (Fig. 1C and Fig. S1B). STING/lpr mice also had larger numbers of total and activated CD44+ CD69+ B220− T cells and TCRβ− B cells (Fig. 1D and Fig. S1C). IRF3/lpr mice did not display any overt expansion of naive or activated T or B cells in their peripheral lymphoid organs (Fig. S1D). Importantly, STING−/− Fas-sufficient littermates (STING/WT) showed no evidence of lymphoid hypertrophy or immune activation. Additionally, the sex of the STING/lpr mice did not significantly alter the severity of disease, because both male and female STING/lpr mice showed similarly aggravated disease manifestations (Fig. S1E). Thus, STING restricts unchecked immune activation during systemic autoimmune disease and does so in an IRF3-independent manner.
Fig. 1.
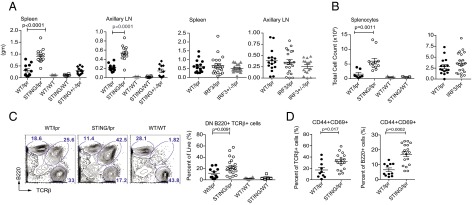
Lymphoid hypertrophy and increased immune activation in STING-deficient but not in IRF3-deficient autoimmune-prone mice. (A) Weights of spleens and largest axillary lymph nodes (LN) from F2 littermates. (B) Total number of splenic cells. (C, Left) Identification of splenic TCRβ− B220+, B220− TCRβ+, and B220+ TCRβ+ cells as determined by FACS analysis. (Right) Splenic percentages of B220+ TCRβ+ CD4− CD8− DN cells. (D) Percentage of activated splenic TCRβ+ T cells (Left) and B220+ B cells (Right) based on CD44 and CD69 activation markers. P values for STING/lpr vs. WT/lpr are noted (one-way ANOVA). Mice were 16 wk old unless otherwise noted.
STING Deficiency Leads to Increased Autoantibody Production.
Antinuclear antibodies (ANAs) are hallmarks of SLE (11). MRL.Faslpr mice normally make autoantibodies against both RNA- and DNA-associated autoantigens (12, 13). Importantly, STING/lpr, but not IRF3/lpr, mice had more B220+ CD138+ plasma cells than WT/lpr mice as well as a significantly greater incidence of IgG class-switched antibody-producing cells as determined by ELISPOT (Fig. 2A and Fig. S1F). To comprehensively compare autoantibody specificity and titers of the STING/lpr and WT/lpr mice, we used autoantigen microarrays (14). This analysis showed an overall increase in autoantibody titers in the STING/lpr sera, compared with WT/lpr littermates, especially with regard to autoantigens commonly associated with SLE such as DNA, histones, chromatin, small nuclear ribonucleoproteins, C1q, granulocyte markers, and extracellular matrix (Fig. 2B). Notably, the reactivity of sera from the 16-wk-old STING/lpr mice was markedly elevated compared with sera from age-matched WT/lpr littermates and more closely resembled the sera from 25-wk-old WT/lpr littermates.
Fig. 2.

Increased numbers of antibody-producing cells and accelerated autoantibody production in STING-deficient autoimmune-prone mice. (A, Left) Percentage of B220+ CD138+ plasma cells as determined by FACS analysis. (Right) Total number of antibody-forming cells (AFCs) quantified by IgG ELISPOT analysis. Data are presented as mean ± SEM. P values as noted (Student’s t test). (B) Autoantibody specificity and titers were determined by binding of sera from WT/lpr (n = 5) and STING/lpr (n = 9) littermates to a predefined autoantigen array. Data are presented as a heatmap summarizing IgM and IgG reactivities. Each columnar lane represents an individual mouse. Mice were 16 or 25 wk old; asterisks indicate 25-wk-old mice.
STING Deficiency but Not IRF3 Deficiency Leads to Accelerated Mortality, Lupus Nephritis, and Gross Morbidity in the MRL.Faslpr Model.
In agreement with their disease severity, STING/lpr mice exhibited more extensive renal disease than their WT/lpr littermates, as determined both by proteinuria and histological examination of the kidneys at 16 wk of age (Fig. 3 A and B). Histological examination of the STING/lpr kidneys showed a greater degree of monocytic and lymphocytic infiltrates (Fig. 3B, a–c), substantial protein deposition, and thickening of the basement membranes (Fig. 3B, d–f). Overall the STING/lpr mice had significantly higher nephritis scores (Fig. 3C), whereas the IRF3/lpr mice showed no signs of accelerated renal disease (Fig. 3D). Importantly, more severe nephritis was associated with significantly accelerated mortality in the STING/lpr mice. The median age at death was 17.5 wk for STING/lpr mice compared with 26 wk for the WT/lpr mice (P = 0.0037) (Fig. 3E).
Fig. 3.
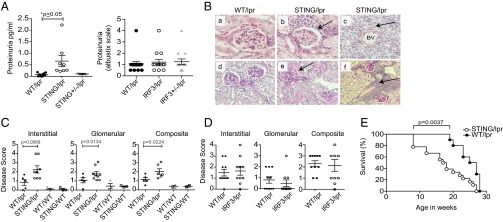
Accelerated lupus nephritis and mortality in STING-deficient autoimmune prone mice. (A) Urine albumin levels in STING/lpr, WT/lpr, and STING+/−/lpr mice by ELISA. Proteinuria in IRF3/lpr and WT/lpr mice was assessed by Siemen’s Albustix strips. P values were calculated by two-way ANOVA. (B) Representative H&E- (a–c) and PAS- (d–f) stained WT/lpr and STING/lpr glomeruli. Panels c and f are 40× fields of view from the samples in b and e. Mice were 16 wk old unless noted otherwise. BV, blood vessel. Arrows indicate inflammatory infiltrates. (C) Interstitial and glomerular renal disease was scored from 0 to 4 in a blinded manner for 16-wk-old WT/lpr (n = 5), STING/lpr (n = 6), WT/WT (n = 3) and STING/WT (n = 3) mice. The sum of interstitial and glomerular scores for each mouse was plotted to determine composite renal disease. (D) Similar scoring was performed for 22-wk-old IRF3/lpr (n = 9) and WT/lpr (n = 10) mice. P values for A–D were calculated using two-way ANOVA. (E) WT/lpr (n = 9) and STING/lpr (n = 12) mice were observed until the time of death. P values for WT/lpr vs. STING/lpr mice were calculated by log-rank test; P = 0.0037.
STING Deficiency but Not IRF3 Deficiency Leads to the Expansion of Unusual Inflammatory Myeloid and Dendritic Cell Populations.
Although STING is expressed ubiquitously, its expression in myeloid cells is closely tied to its role in innate responses (3, 15). Intriguingly, the number and activation status of myeloid cells (CD11b+ or CD11c+) in the STING/lpr spleens was increased greatly compared with WT/lpr littermates (Fig. 4 A and B and Fig. S2A). Within the myeloid compartment, CD11b+ Ly6Chi cells are considered a highly inflammatory class of monocytes, and the Ly6Ghi/Ly6Cint subset consists of inflammatory neutrophils (16). Both subsets are characterized by their association with an inflammatory milieu and are thought to give rise to cells that participate heavily in tissue destruction and remodeling (17, 18). Notably, STING/lpr mice showed a significant expansion and activation of both Ly6Chi inflammatory monocyte (R1) and Ly6Ghi neutrophil (R2) populations (Fig. 4C and Fig. S2 B and C). Activated F4/80+ macrophage populations were increased similarly (Fig. S2D). Importantly, neither macrophage nor neutrophil populations were altered significantly in the IRF3/lpr mice (Fig. S2E). There was also an increased frequency of F4/80− Ly6Chi CD11c+ inflammatory dendritic cells (iDC) (Fig. 4C and Fig. S2F). iDCs arise from the Ly6Chi inflammatory progenitor pools and are a major source of proinflammatory cytokines in models of tumor immunity (17, 18). The role of iDCs in SLE has not been noted or described previously, and therefore their presence in the accelerated pathology in STING/lpr was unexpected. The expansion of the Ly6C/G+ populations correlates with increased systemic levels of neutrophil- and macrophage-derived chemokines and cytokines including the M1-associated cytokines TNF-α and IL-6 and chemokines (CCL2, CCL5, CXCL1, CCL3, and CCL4) (Fig. 4D and Fig. S2G). Of note, IL-9 and IL-3 promote the proliferation and differentiation of hematopoietic stem cells into myeloid progenitor cells (19, 20), and increased systemic levels of IL-9 and IL-3 correlate directly with the myeloid expansion seen in STING/lpr mice (Fig. 4D). Together these data highlight the previously unrecognized role that STING normally plays in limiting the expansion and activity of pathogenic myeloid populations in systemic autoimmune diseases such as SLE.
Fig. 4.
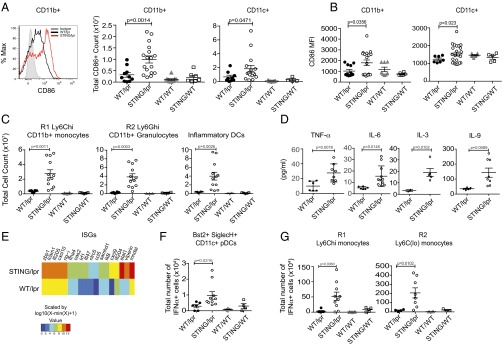
STING deficiency drives expansion and activation of the myeloid compartment and a robust IFN signature. (A) Identification and enumeration of total CD86+ splenic CD11b+ and CD11c+ cells from WT/lpr, STING/lpr, WT/WT, and STING/WT mice. (B) Mean fluorescence intensity (MFI) of CD86 staining on CD11b+ and CD11c+ cells. All mice were 16 wk old (see also Fig. S2A). (C) Total numbers of Ly6Chi CD11b+ inflammatory monocytes (R1) and Ly6Cint Ly6Ghi CD11b+ N1 neutrophils (R2) as well as CD11c+ F4/80− Ly6Chi inflammatory DCs (also see Fig. S2B). (D) Serum levels of cytokines determined by Luminex-multiplex array. P values were determined by Student’s t test. (E) mRNA profiling of ISGs in splenic tissue from WT/lpr and STING/lpr mice using nanostring technology is summarized as a heatmap. The analysis compares the mean intensities of signals in WT/lpr (n = 6) and STING/lpr (n = 6) mice and is transformed by a log10 function. (F and G) Quantitation of cells positive for intracellular staining of IFN-α within the CD11c+ Bst-2+ SiglecH+ pDCs (F) and Ly6Chi CD11b+ inflammatory monocytes and Ly6Clo CD11b+ monocytes (G). R1 and R2 gates were used to designate Ly6Chi and Ly6C+/lo cells. (Also see Fig. S3). All analysis was done on 16-wk-old mice unless noted otherwise.
STING Deficiency Results in the Increased Expression of IFN-Stimulated Genes and IFN-α–Secreting Cell Populations.
Type I IFNs have been shown to play a significant role in SLE (21). Therefore it was essential to determine whether the IFN signature was altered in the STING/lpr mice. Gene-expression profiling of STING/lpr and WT/lpr splenocytes showed a significant increase in the expression of IFN-stimulated genes (ISGs) in STING/lpr spleens compared with WT/lpr littermates, as is consistent with disease severity but paradoxical with STING deficiency (Fig. 4E). An up-regulated ISG signature generally is indicative of increased levels of type I IFNs and IFN-producing cells in the periphery. Plasmacytoid dendritic cells (pDCs) are thought to be the major type I IFN-producing cell (IPC) in both murine models of SLE and in human patients, although other myeloid and lymphoid lineage cells also are known contributors (22, 23). Importantly, the number of pDCs identified as CD11cintSiglec-H+Bst2+ (Fig. S3A) was increased significantly in the spleens from STING/lpr mice but not in the IRF3/lpr spleens (Fig. S3B). By using intracellular staining, we identified not only increased numbers of IFN-α–producing pDCs but also IFN-α–producing Ly6Chi and Ly6Clo monocytes in STING/lpr mice (Fig. 4 F and G and Fig. S3C). IFN-α–producing Ly6Chi monocytes have been found at the site of viral infections as well as in inflammatory diseases such as colitis (18). However, the increase in IFN-α+ Ly6Clo cells was surprising (Fig. 4G), because these subsets have not been identified previously as sources of type I IFN in SLE or other diseases. This finding suggests that typical and atypical IFN-α–producing pathways are significantly dysregulated in the absence of STING.
TMPD-Induced Peritonitis Is Amplified by STING Deficiency.
Several key features of SLE are recapitulated by the i.p. injection of the hydrocarbon oil TMPD (also known as “pristane”). These features include a type I IFN signature, ANAs, and glomerulonephritis (24, 25). Importantly, TMPD-induced autoimmunity is dependent upon TLR7 and type I IFNs (26). Pristane injection of C57/Bl6 mice results in an early and persistent peritoneal inflammation and the recruitment of TLR7hi Ly6Chi monocytes, a potential source of type I IFNs, in this model (25). We thus sought to determine if TMPD-induced, TLR-dependent inflammation is also modulated by STING. The extravasation of inflammatory monocytes and granulocytes into the peritoneal cavity following TMPD administration was entirely dependent on endosomal TLRs, because no evidence of inflammation occurred in pristane-treated Unc93b3d/3d mice (Fig. 5A). In comparison, STING−/− mice showed greater accumulation of total cells, CD11b+ myeloid cells, and especially CD11b+ Ly6Chi inflammatory monocytes. The expression of ISGs (IRF7 and Mx1) was also increased in the peritoneal exudate cells (PECs) from STING−/− mice but not Unc93b3d/3d mice (Fig. 5B), again implicating type I IFN in pathology. Although there was no significant increase in the number of Ly6Cint Ly6Ghi granulocytes in the peritoneum, the percentage of Ly6Cint Ly6Ghi granulocytes was significantly higher in the spleens of the TMPD-injected STING−/− mice than in controls (Fig. 5C). The accumulation of inflammatory granulocytes in the spleens of pristane-treated mice at 6 mo postinjection has been reported previously (16); however, to our knowledge, this is the first report of such an early signature of systemic inflammation. These data document a second example in which STING negatively regulates a TLR-dependent inflammatory response.
Fig. 5.
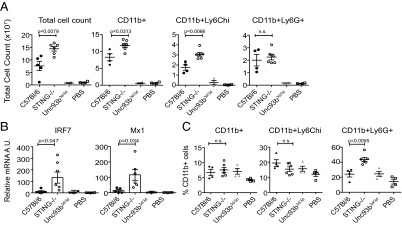
STING deficiency exacerbates TMPD-induced TLR and type I IFN-dependent inflammation. (A) Comparison of the total numbers of PECs and cellular subsets 2 wk after TMPD treatment in C57BL/6, STING−/−, Unc93b3d/3d, and PBS-injected mice. Total numbers of CD11b+, Ly6Chi, and Ly6Ghi cells in the peritoneal lavage fluid. (B) RT-PCR analysis of IRF7 and Mx1 expression in total PECs. P values were determined by Student’s t test. (C) Percentages of splenic CD11b+, Ly6Chi, and Ly6Ghi cell types in TMPD-treated mice. P values in A and C were determined for C57BL/6 vs. STING−/− mice (two-way ANOVA).
Macrophages Derived from STING-Deficient Mice Are Hyperresponsive to TLR7 and TLR9 Ligands.
In all SLE models evaluated to date, immune activation depends on the ability of endogenous TLRs to recognize nucleic acid-associated autoantigens (25, 27). Therefore, we reasoned that STING/lpr-derived macrophages could be inherently more reactive to such autoantigens via TLR7- and/or TLR9-dependent pathways. To test this possibility directly, bone marrow-derived macrophages (BMMs) from STING/lpr and WT/lpr mice were stimulated with TLR7 and TLR9 ligands. As expected, STING/lpr macrophages failed to produce the proinflammatory cytokines TNF-α and IL-6 in response to immune-stimulatory DNA (ISD) known to activate STING-dependent cytosolic DNA sensors (3). However, these same cells were hyperresponsive to the TLR9 ligand CpGB ODN and the TLR7 ligand CLO97. Additionally, macrophages from STING/WT littermates were similarly hyperresponsive to TLR7 and TLR9 ligands (Fig. 6A). To confirm that this increase actually was caused by STING deficiency alone and not by other MRL-associated genes, we also compared STING-deficient and STING-sufficient C57BL/6 BMMs (Fig. S4A). Again the STING−/− BMMs were hyperresponsive to TLR7 and TLR9 ligands, despite impaired responses to ISD or B-form DNA (pdAdT). Moreover, STING−/− BMMs made significantly more IL-6 in response to limiting concentrations of TLR7/9 ligands (Fig. 6B) as well as additional TLR ligands (Fig. S4B). In addition, TLR3 stimulation of STING−/− BMMs resulted in elevated levels of IFN-β (Fig. S4C). Thus, more than one pathway, downstream of TLRs, is modified by STING deficiency.
Fig. 6.
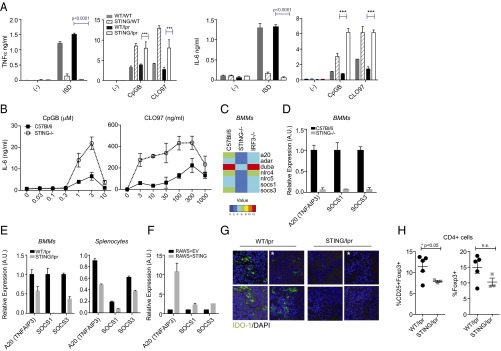
STING regulates responsiveness to TLR ligands and controls the expression of critical negative regulators of innate signaling. (A) Levels of TNF-α and IL-6 secreted by BMMs from WT/lpr, STING/lpr, WT/WT, and STING/WT mice in response to TLR9 (CpGB, 5 μM), TLR7 (CLO97, 300 ng/mL), and ISD (3 μM) ligands. Supernatants were collected 24 h after stimulation. (B) Levels of IL-6 from C57BL/6 and STING−/− BMMs treated with a range of concentrations of CpGB (TLR9) and CLO97 (TLR7) for 24 h. Concentrations and units are noted. (C) Nanostring mRNA profiling of key negative regulators of innate signaling in BMMs derived from C57BL/6, STING−/−, and IRF3−/− mice after stimulation with ISD for 3 h, represented as a heatmap. The analysis compares the mean intensities of gene expression in C57BL/6, STING−/−, and IRF3−/− BMMs and is transformed by a log2 function. (D) Real-time PCR analysis of A20, SOCS1, and/or SOCS3 mRNA from unstimulated C57BL/6 (black bars) and STING−/− (gray bars) BMMs. (E, Left) Real-time PCR analysis of A20, SOCS1, and/or SOCS3 mRNA WT/lpr (black bars) and STING/lpr (gray bars) BMMs. (Right) Nanostring mRNA profiling of splenic tissue from WT/lpr and STING/lpr mice. The analysis compares relative basal expression levels of A20, SOCS1, and SOCS3 in WT/lpr mice (n = 4; black bars) and STING/lpr (n = 6; gray bars) mice. (F) RT-PCR analysis of A20, SOCS1, and SOCS3 expression in RAW 264.7 cell lines stably expressing either an empty vector (RAWS+EV) or STING-HA-citrine (RAWS+STING). (G) Immunofluorescence of splenic tissue from WT/lpr and STING/lpr mice stained for IDO-1 (green) and nuclei (DAPI; blue). Shown are three representative images from 16-wk-old STING/lpr mice and from WT/lpr mice, one image from a 25-wk-old WT/lpr mouse, and one image from a 25-wk-old STING/lpr mouse. The asterisks indicate the images from the 25-wk-old mice. Total numbers of mice analyzed were n = 5 (WT/lpr) and n = 9 (STING/lpr). (H) Percentages of CD25+ Foxp3+ and total Foxp3+ (Treg cells) T cells gated on live CD4+ T cells from the axillary lymph node of WT/lpr and STING/lpr mice. P values in A–E were calculated using Student’s t test; P values in G were calculated using two-way ANOVA.
STING Deficiency but Not IRF3 Deficiency Results in Reduced Levels of Known Negative Regulators of TLR Signaling.
STING deficiency could lead to amplified activation of TLR-dependent macrophage responses either by increasing the level of TLR expression or by decreasing negative regulation of TLR signaling. We compared basal levels of endosomal TLRs in WT, STING−/−, WT/lpr, and STING/lpr BMMs but found no difference in TLR3, TLR7, or TLR9 expression (Fig. S5A). Surprisingly, unstimulated STING−/− BMMs had dramatically decreased expression of several well-characterized negative regulators of TLR signaling, including A20 and suppressor of cytokine signaling 1 (SOCS1) and 3 (SOCS3), compared with WT controls (Fig. 6 C and D). Importantly, these genes are not regulated through an IRF3-dependent mechanism, because IRF3−/− BMMs did not display any change in basal expression levels of these critical negative regulators (Fig. 6C). Levels of A20, SOCS1, and SOCS3 were also decreased substantially in activated STING−/− BMMs (Fig. S5B). Similar analysis of STING/lpr- and WT/lpr-derived BMMs revealed significantly attenuated A20, SOCS1, and SOCS3 levels basally and in response to TLR ligands (Fig. 6E and Fig. S5C). Furthermore, gene profiling of STING/lpr and WT/lpr splenic tissue also revealed a significant decrease in the expression of these negative regulators (Fig. 6E). To confirm that the expression of these negative regulators was regulated directly by STING, we determined the effect of STING overexpression on these genes. We found that stable overexpression of STING in RAW264.7 macrophage cells resulted in elevated basal levels of A20, SOCS1, and SOCS3, further demonstrating STING-dependent regulation of these components (Fig. 6G). Moreover, ectopic expression of STING reduced the production of TNF-α and IL-6 in response to TLR ligands but not in response to cytosolic DNA ligands (Fig. S5D).
Secreted negative regulators of immune responses also contribute to self-tolerance. Recent studies have shown that myeloid cell production of Indoleamine 2,3-deoxygenase-1 (IDO-1), particularly by dendritic cells (DCs), promotes the conversion and expansion of regulatory T cells (28). When spleen sections from 16-wk-old STING/lpr and WT/lpr littermates were stained with anti–IDO-1 antibody, spleens from STING/lpr mice had greatly reduced IDO-1 levels compared with age-matched littermate controls (Fig. 6G). Because IDO-1 is thought to control the generation of Treg cells, we determined the number of Treg cells in STING/lpr and control mice. We found that STING/lpr mice had significantly lowered percentages of activated CD4+ CD25+ FOXP3+ Treg cells and also tended toward lowered percentages of CD4+ FOXP3+ Treg cells in axillary lymph nodes (Fig. 6H). These data, along with other recent studies (29), explain how immune suppression in STING−/− animals may be systemically compromised. Overall, these data strongly indicate that in the context of an SLE autoimmune-prone background, STING-deficient myeloid cells mount an amplified response to nucleic acid-associated autoantigens caused by the reduced expression of molecules that directly or indirectly limit immune activation.
Discussion
Foreign DNA can trigger both endosomal TLR9 and cytosolic STING-dependent DNA sensors, leading to the robust production of type I IFNs and other proinflammatory cytokines important in the clearance of microbial pathogens. Chronic stimulation of these sensors by endogenous ligands also provokes debilitating autoinflammatory or autoimmune diseases such as seen in mice or humans with compromised capacities to degrade DNA. In contrast to experimental settings where STING provokes inflammation, our studies show that STING can attenuate inflammation in both a chronic systemic autoimmune disease and in an acute inflammatory response that is dependent on TLR-mediated detection of endogenous ligands. STING-deficient MRL/lpr mice develop a dramatically accelerated disease profile characterized by the expansion and activation of both the lymphoid and myeloid compartments, increased numbers of type I IFN-producing cells, elevated autoantibody titers, and a shorter lifespan. Similar to our findings in the MRL/lpr model, STING deficiency exacerbated TMPD-induced inflammation, a condition also associated with type I IFNs and TLR-activated Ly6Chi monocytes (24, 26).
Nonautoimmune-prone STING-deficient mice do not exhibit any evidence of systemic autoimmunity. Therefore, the exacerbated disease phenotype of the STING-deficient MRL/lpr mice likely results from the failure to constrain aberrantly activated TLR signaling cascades responsible for disease. Intriguingly, A20-, SOCS1-, or SOCS3-deficient mice exhibit phenotypes similar to our STING/lpr mice (30, 31). In addition to impaired expression of these negative regulators of TLR activation, STING/lpr mice also failed to express the secreted immunosuppressive protein IDO-1. Previous studies documented IDO-1 expression in the spleens of prediseased MRL.Faslpr mice and its reduced expression with disease progression (32). Moreover, treatment with an IDO-1 inhibitor 1-methyl tryptophan (1-MT) accelerates disease in the MRL.Faslpr mice, implicating IDO-1 in the negative regulation of immune activation associated with autoimmunity. Because IDO-1 is potently induced by TLR-dependent and -independent pathways and regulates the expansion of Tregs, it is a critical factor in limiting disease severity (28). Recently, Huang et al. (33) showed that IDO-1 expression in DCs is attenuated in the absence of STING, leading to increased IL-6 production by STING−/− DCs in response to apoptotic cells. We now show decreased IDO-1 levels and a corresponding decrease in Treg cells in STING-deficient SLE-prone mice. These conditions are likely to contribute to the exacerbated disease phenotype.
A seemingly unrelated outcome of disease in the STING/lpr mice is the large number of monocytic cells in secondary lymphoid tissues. However, monocyte egress from the bone marrow and local proliferation is a known consequence of TLR engagement (34). Whether increased TLR activity also contributes to the excess production of the hematopoiesis-modulating cytokines IL-9 and IL-3, thereby further promoting myeloid expansion in STING/lpr animals, remains to be determined.
Although an increase in proinflammatory cytokines in response to apoptotic cells and TLR9 and TLR3 ligands has been noted previously in STING−/− macrophages by other investigators (33, 35), the significance of this dysregulation of TLR signaling was not addressed further or extended to other TLRs. Previous studies have focused largely on the ability of STING to engage the TBK1–IRF3 signaling pathway. It is critical to note that, thus far, IRF3 has been implicated only in the transcription of type I IFNs (36). Furthermore, IRF3 deficiency does not affect the disease course of MRL/lpr, and therefore it is likely that a STING-dependent but IRF3-independent pathway mediates the effects we observed. Intriguingly, some evidence exists for the role of IRF3/7 heterodimers in inducing type I IFNs downstream of STING (37). However, a definitive role for IRF7 has not yet been evaluated in lupus and may contribute to lupus pathology downstream of both STING and TLRs. Prior studies have shown that cytosolic DNA can induce the expression of proinflammatory cytokines through the activation of an NF-κB–dependent pathway downstream of STING (3). Intriguingly, we show that the basal expression of negative regulators of TLR signaling such as A20, SOCS1, and SOCS3, known NF-κB target genes, is down-regulated significantly in STING−/− cells but not in IRF3−/− cells. These data further highlight the contribution of NF-κB signaling downstream of STING under homeostatic conditions and point to a constitutive role of this pathway in immune regulation. It is possible that other transcription factors, such as IRFs and STATs, may be engaged downstream of STING, but likely candidates IRF5, IRF1, STAT1, and STAT6 have previously been shown to promote lupus pathology (38–42), in contrast to the suppressive role of STING documented in this study. Thus, it is unlikely that transcriptional activity of any of these factors contributes to a STING-dependent suppressive pathway.
Dysregulation of TLR signaling is a recurrent theme in the development and severity of systemic autoimmune diseases such as SLE. Both TLR7 and TLR9 activation have been linked to the pathogenesis of SLE (43). However, despite similar tissue expression and signaling pathways, deficiency of these TLRs on an MRL.Faslpr background, as well as other autoimmune-prone strains, has revealed opposing roles for these TLRs in SLE. Although opposing roles of TLR7/9 in autoimmunity have been known for some time, mechanistic insight into their cross-regulation is still lacking. In many ways the exacerbated disease we observed in STING/lpr mice resembles that reported for TLR9/lpr mice; both strains show elevated expression of ISGs and marked expansion and activation of lymphocyte compartments. Our studies now suggest that modulation of negative regulators of signaling may be a common mechanism of cross-regulation applicable to TLRs as well as other innate pathways during autoimmunity.
It is conceivable that the negative regulatory role of STING we observed is independent of its role in DNA sensing and represents a homeostatic role for STING resulting from stochastic dimerization. Alternatively, low levels of cytosolic DNA, perhaps through reactivated retro-elements, may account for the homeostatic involvement of STING. The precise mechanism by which STING regulates homeostasis remains an open question. Although loss-of-function polymorphisms for STING are known, they have not yet been linked to any human diseases (44). Loss-of-function mutations in Trex1, presumably leading to hyperactivation of STING, are associated with Aicardi–Goutieres syndrome. Moreover, gain-of-function mutations in STING are now linked to a class of vascular and pulmonary inflammatory syndromes as well as familial systemic diseases (45, 46). Together, these clinical observations have identified the STING pathway as a potential therapeutic target. Our data now raise a cautionary note regarding the use of such therapeutics, because they may have unintended consequences and perturb a carefully orchestrated balance between cytosolic and endosomal signaling cascades.
Experimental Procedures
Generation and Characterization of Autoimmune STING-Deficient Mice.
The University of Massachusetts Medical School and Yale Institutional Animal Care and Use Committees approved all animal work.
STING−/− mice (3) (from G. Barber, University of Miami, Coral Gables, FL) and IRF3−/− mice (47) were crossed to Fas-deficient, lupus-prone MRL/Mplpr/lpr mice (Jackson Laboratory). A controlled F2 intercross between heterozygote STING+/− lpr/+ littermates was used to generate STING-deficient lupus-prone mice homozygous for deficiency in Fas as well as STING (STING/lpr, n = 17–20) or wild type for STING (WT/lpr, n = 9–15). The heterogeneous genetic composition of the F2 generation was controlled by the use of large cohorts of littermates. Most mice were analyzed at 16 wk of age; three STING/lpr mice and three WT/lpr mice were analyzed at 25 wk of age.
Statistical Analysis.
Experiments are reported as the means ± SEM/SD. Statistical comparisons were made using a Student t test and GraphPad Prism software (GraphPad Software) for bivariate studies and one-way ANOVA for multivariate studies. P values for all criteria measured are reported, and P values <0.05 were considered statistically significant.
Please see SI Experimental Procedures for more information on the methods used in this study.
Supplementary Material
Acknowledgments
We thank Brian G. Monks, Zhaozhao Jiang, Tara Robidoux, and Numana Bhat for contributions at critical phases of experimentation. This work was supported by Alliance for Lupus Research Grant ALR 260146 (to S.S.), National Institutes of Health (NIH) Grants AI093752 (to K.A.F.), AR050256 (to A.M.-R.), and AI101048 (to L.J.B.). Support for A.M.C. was provided by NIH Grant 2T32GM07205. Support for J.C., S.A.S., and G.M.O. was provided by NIH Grant 5T32A1095213.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1420217112/-/DCSupplemental.
References
- 1.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339(6121):786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Unterholzner L, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol. 2010;11(11):997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455(7213):674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burdette DL, Vance RE. STING and the innate immune response to nucleic acids in the cytosol. Nat Immunol. 2013;14(1):19–26. doi: 10.1038/ni.2491. [DOI] [PubMed] [Google Scholar]
- 5.Shin HD, et al. DNase II polymorphisms associated with risk of renal disorder among systemic lupus erythematosus patients. J Hum Genet. 2005;50(3):107–111. doi: 10.1007/s10038-004-0227-3. [DOI] [PubMed] [Google Scholar]
- 6.Crow YJ, et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. Nat Genet. 2006;38(8):917–920. doi: 10.1038/ng1845. [DOI] [PubMed] [Google Scholar]
- 7.Lee-Kirsch MA, et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet. 2007;39(9):1065–1067. doi: 10.1038/ng2091. [DOI] [PubMed] [Google Scholar]
- 8.Kawane K, et al. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature. 2006;443(7114):998–1002. doi: 10.1038/nature05245. [DOI] [PubMed] [Google Scholar]
- 9.Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134(4):587–598. doi: 10.1016/j.cell.2008.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seong CS, Varela-Ramirez A, Aguilera RJ. DNase II deficiency impairs innate immune function in Drosophila. Cell Immunol. 2006;240(1):5–13. doi: 10.1016/j.cellimm.2006.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Craft JE. Dissecting the immune cell mayhem that drives lupus pathogenesis. Sci Transl Med. 2011;3(73):ps9. doi: 10.1126/scitranslmed.3002138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Christensen SR, et al. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med. 2005;202(2):321–331. doi: 10.1084/jem.20050338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christensen SR, et al. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25(3):417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 14.Li L, et al. Murine lupus strains differentially model unique facets of human lupus serology. Clin Exp Immunol. 2012;168(2):178–185. doi: 10.1111/j.1365-2249.2012.04568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahn J, Gutman D, Saijo S, Barber GN. STING manifests self DNA-dependent inflammatory disease. Proc Natl Acad Sci USA. 2012;109(47):19386–19391. doi: 10.1073/pnas.1215006109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bossaller L, et al. Overexpression of membrane-bound fas ligand (CD95L) exacerbates autoimmune disease and renal pathology in pristane-induced lupus. J Immunol. 2013;191(5):2104–2114. doi: 10.4049/jimmunol.1300341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rivollier A, He J, Kole A, Valatas V, Kelsall BL. Inflammation switches the differentiation program of Ly6Chi monocytes from antiinflammatory macrophages to inflammatory dendritic cells in the colon. J Exp Med. 2012;209(1):139–155. doi: 10.1084/jem.20101387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11(11):762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goswami R, Kaplan MH. A brief history of IL-9. J Immunol. 2011;186(6):3283–3288. doi: 10.4049/jimmunol.1003049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reddy EP, Korapati A, Chaturvedi P, Rane S. IL-3 signaling and the role of Src kinases, JAKs and STATs: A covert liaison unveiled. Oncogene. 2000;19(21):2532–2547. doi: 10.1038/sj.onc.1203594. [DOI] [PubMed] [Google Scholar]
- 21.Theofilopoulos AN. TLRs and IFNs: Critical pieces of the autoimmunity puzzle. J Clin Invest. 2012;122(10):3464–3466. doi: 10.1172/JCI63835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pascual V, Farkas L, Banchereau J. Systemic lupus erythematosus: All roads lead to type I interferons. Curr Opin Immunol. 2006;18(6):676–682. doi: 10.1016/j.coi.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 23.Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity. 2006;25(3):383–392. doi: 10.1016/j.immuni.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 24.Reeves WH, Lee PY, Weinstein JS, Satoh M, Lu L. Induction of autoimmunity by pristane and other naturally occurring hydrocarbons. Trends Immunol. 2009;30(9):455–464. doi: 10.1016/j.it.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee PY, et al. TLR7-dependent and FcgammaR-independent production of type I interferon in experimental mouse lupus. J Exp Med. 2008;205(13):2995–3006. doi: 10.1084/jem.20080462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Savarese E, et al. Requirement of Toll-like receptor 7 for pristane-induced production of autoantibodies and development of murine lupus nephritis. Arthritis Rheum. 2008;58(4):1107–1115. doi: 10.1002/art.23407. [DOI] [PubMed] [Google Scholar]
- 27.Green NM, Marshak-Rothstein A. Toll-like receptor driven B cell activation in the induction of systemic autoimmunity. Semin Immunol. 2011;23(2):106–112. doi: 10.1016/j.smim.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013;34(3):137–143. doi: 10.1016/j.it.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lemos H, et al. Activation of the STING adaptor attenuates experimental autoimmune encephalitis. J Immunol. 2014;192(12):5571–5578. doi: 10.4049/jimmunol.1303258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coornaert B, Carpentier I, Beyaert R. A20: Central gatekeeper in inflammation and immunity. J Biol Chem. 2009;284(13):8217–8221. doi: 10.1074/jbc.R800032200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dalpke A, Heeg K, Bartz H, Baetz A. Regulation of innate immunity by suppressor of cytokine signaling (SOCS) proteins. Immunobiology. 2008;213(3-4):225–235. doi: 10.1016/j.imbio.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 32.Ravishankar B, et al. Tolerance to apoptotic cells is regulated by indoleamine 2,3-dioxygenase. Proc Natl Acad Sci USA. 2012;109(10):3909–3914. doi: 10.1073/pnas.1117736109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang L, et al. Cutting edge: DNA sensing via the STING adaptor in myeloid dendritic cells induces potent tolerogenic responses. J Immunol. 2013;191(7):3509–3513. doi: 10.4049/jimmunol.1301419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi C, et al. Bone marrow mesenchymal stem and progenitor cells induce monocyte emigration in response to circulating toll-like receptor ligands. Immunity. 2011;34(4):590–601. doi: 10.1016/j.immuni.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461(7265):788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol. 2008;26:535–584. doi: 10.1146/annurev.immunol.26.021607.090400. [DOI] [PubMed] [Google Scholar]
- 37.Sharma S, et al. Innate immune recognition of an AT-rich stem-loop DNA motif in the Plasmodium falciparum genome. Immunity. 2011;35(2):194–207. doi: 10.1016/j.immuni.2011.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Niewold TB, et al. Association of the IRF5 risk haplotype with high serum interferon-alpha activity in systemic lupus erythematosus patients. Arthritis Rheum. 2008;58(8):2481–2487. doi: 10.1002/art.23613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tada Y, et al. Interferon regulatory factor 5 is critical for the development of lupus in MRL/lpr mice. Arthritis Rheum. 2011;63(3):738–748. doi: 10.1002/art.30183. [DOI] [PubMed] [Google Scholar]
- 40.Yu HH, et al. Interleukin 4 and STAT6 gene polymorphisms are associated with systemic lupus erythematosus in Chinese patients. Lupus. 2010;19(10):1219–1228. doi: 10.1177/0961203310371152. [DOI] [PubMed] [Google Scholar]
- 41.Singh RR, et al. Differential contribution of IL-4 and STAT6 vs STAT4 to the development of lupus nephritis. J Immunol. 2003;170(9):4818–4825. doi: 10.4049/jimmunol.170.9.4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reilly CM, et al. Interferon regulatory factor-1 gene deletion decreases glomerulonephritis in MRL/lpr mice. Eur J Immunol. 2006;36(5):1296–1308. doi: 10.1002/eji.200535245. [DOI] [PubMed] [Google Scholar]
- 43.Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol. 2006;6(11):823–835. doi: 10.1038/nri1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jin L, et al. Identification and characterization of a loss-of-function human MPYS variant. Genes Immun. 2011;12(4):263–269. doi: 10.1038/gene.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jeremiah N, et al. Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J Clin Invest. 2014;124(12):5516–5520. doi: 10.1172/JCI79100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu Y, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. 2014;371(6):507–518. doi: 10.1056/NEJMoa1312625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sato M, et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 2000;13(4):539–548. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.