

Significance
High-throughput DNA sequencing methods are revolutionizing our ability to census communities, but most analyses have focused on microbes. Using an environmental DNA sequencing approach based on cytochrome c oxidase subunit 1 primers, we document the enormous diversity and fine-scale geographic structuring of the cryptic animals living on oyster reefs, many of which are rare and very small. Sequence data reflected both the presence and relative abundance of organisms, but only 10.9% of the sequences could be matched to reference barcodes in public databases. These results highlight the enormous numbers of marine animal species that remain genetically unanchored to conventional taxonomy and the importance of standardized, genetically based biodiversity surveys to monitor global change.
Keywords: oyster reefs, operational taxonomic units, meiofauna, ARMS, cryptic species
Abstract
Documenting the diversity of marine life is challenging because many species are cryptic, small, and rare, and belong to poorly known groups. New sequencing technologies, especially when combined with standardized sampling, promise to make comprehensive biodiversity assessments and monitoring feasible on a large scale. We used this approach to characterize patterns of diversity on oyster reefs across a range of geographic scales comprising a temperate location [Virginia (VA)] and a subtropical location [Florida (FL)]. Eukaryotic organisms that colonized multilayered settlement surfaces (autonomous reef monitoring structures) over a 6-mo period were identified by cytochrome c oxidase subunit I barcoding (>2-mm mobile organisms) and metabarcoding (sessile and smaller mobile organisms). In a total area of ∼15.64 m2 and volume of ∼0.09 m3, 2,179 operational taxonomic units (OTUs) were recorded from 983,056 sequences. However, only 10.9% could be matched to reference barcodes in public databases, with only 8.2% matching barcodes with both genus and species names. Taxonomic coverage was broad, particularly for animals (22 phyla recorded), but 35.6% of OTUs detected via metabarcoding could not be confidently assigned to a taxonomic group. The smallest size fraction (500 to 106 μm) was the most diverse (more than two-thirds of OTUs). There was little taxonomic overlap between VA and FL, and samples separated by ∼2 m were significantly more similar than samples separated by ∼100 m. Ground-truthing with independent assessments of taxonomic composition indicated that both presence–absence information and relative abundance information are captured by metabarcoding data, suggesting considerable potential for ecological studies and environmental monitoring.
Understanding the diversity of life in the sea continues to challenge marine scientists because samples typically contain many rare species, most of them small and difficult to identify (1). Moreover, recent estimates suggest that between 33% and 91% of all marine species have never been named (2, 3). These constraints have limited our ability to investigate patterns of diversity beyond a few indicator groups (4), most often conspicuous macroinvertebrates and fish. For this reason, molecular methods, particularly high-throughput sequencing (HTS) approaches, hold considerable promise not only for fundamental understanding of diversity but also for biodiversity monitoring in the context of global change (5).
Molecular methods are particularly powerful when combined with standardized sampling, allowing for direct comparisons across space and through time. In the ocean, analyzing standard volumes of readily sampled material (e.g., seawater, sediments) has a long tradition, and, increasingly, HTS approaches are being applied to these samples (6). Complex hard substrates provide greater challenges for consistent sampling, which can be met either by collecting approximately standard volumes (e.g., of rubble) or by deploying settlement structures (e.g., ref. 7).
Here, we combine standardized sampling with molecular diversity assessments for samples from oyster reefs from one temperate location and one subtropical location on the US Atlantic Coast. In addition to their commercial value and their role in maintaining water quality, oyster beds shelter considerable diversity because of their 3D complexity, essentially the nontropical equivalent of coral reefs. They are also, like coral reefs, highly threatened, with up to 85% having been lost due to anthropogenic impacts (8).
We report analyses of a nested set of autonomous reef monitoring structures (ARMS), which provide surfaces and spaces for mobile and sessile organisms to settle on or shelter within (SI Text, section I and Fig. S1). ARMS were deployed for about 6 mo on the ocean side of the Eastern Shore of Virginia (VA) and in the Indian River Lagoon in Florida (FL). At each location, there were three replicates ∼2 m apart at each of three sites ∼100 m apart (total of 18 ARMS; Fig. S1A). Four fractions were analyzed separately: sessile organisms growing on the plates and three fractions of organisms retained by 2-mm, 500-μm, and 106-μm sieves. We sequenced the cytochrome c oxidase subunit I (COI) gene for each specimen of the >2-mm animals (barcoding). The remaining fractions were homogenized, and COI amplicons were analyzed from bulk samples using HTS (metabarcoding). Sequences were clustered in operational taxonomic units (OTUs) and identified to the lowest possible taxonomic level using nucleotide BLAST (BLASTn) searches against public databases or by phylogenetic assignment when no direct match could be found. The effectiveness of the metabarcoding approach was assessed for the sessile and 2-mm to 500-μm fractions by comparing numbers of sequences with point counts and estimates of total DNA per OTU, respectively. Noneukaryotic sequences were not analyzed.
Results
Patterns of Diversity and Abundance.
Organisms >2 mm had relatively low diversity, with FL samples having about 1.7-fold more OTUs in total than VA samples, and also more phyla represented. Barcoding revealed a total of 38 OTUs in VA (498 individuals) and 64 OTUs in FL (655 individuals). In both locations, the most abundant and species-rich higher taxon was the Arthropoda (16 OTUs in 385 individuals in VA and 30 OTUs in 583 individuals in FL; Fig. 1). Samples from both locations additionally contained representatives of the Annelida, Chordata, and Mollusca; FL samples also contained Platyhelminthes and Echinodermata. Over half of the sequences at both locations matched reference sequences (>97% similarity) in the GenBank sequence database (GenBank) or Barcode of Life Data Systems (BOLD) (Table 1 and Table S1).
Fig. 1.

OTU diversity and abundance in VA and FL. The category “Other animals” comprises Acoelomorpha, Chaetognatha, Ctenophora, Echiura, Entoprocta, Gastrotricha, Hemichordata, Nematoda, Nemertea, Platyhelminthes, Rotifera, Sipuncula, Tardigrada, and Xenacoelomorpha.
Table 1.
OTU diversity and abundance in VA and FL
| Diversity descriptors | VA | FL | ||||||||
| Barcoding | Metabarcoding | Barcoding | Metabarcoding | |||||||
| >2 mm | 2 mm to 500 μm | 500 to 106 μm | Sessile | Total | >2 mm | 2 mm to 500 μm | 500 to 106 μm | Sessile | Total | |
| No. of sequences | 498 | 256,147 | 97,439 | 218,704 | 572,290 | 655 | 155,232 | 86,350 | 168,031 | 409,613 |
| Total no. of OTUs | 38 | 651 | 828 | 436 | 1,204 | 64 | 821 | 976 | 591 | 1,391 |
| Mean no. of OTUs | 11.2 | 203.3 | 290.3 | 146.6 | 434.2 | 15.8 | 277.2 | 360.1 | 222.9 | 536.7 |
| Mean rarefied no. of OTUs | 8.2 | 117.1 | 229.7 | 85.7 | 333.5 | 9.6 | 202.6 | 312.1 | 157.4 | 484.4 |
| Chao1 | 46.0 | 1,075.6 | 1,204.9 | 638.7 | 1,711.4 | 104.8 | 1,183.0 | 1,486.0 | 858.0 | 1,945.7 |
| Chao1 (rarefied) | 49.2 | 552.7 | 917.8 | 451.5 | 1,174.0 | 144.7 | 866.5 | 1,213.9 | 562.1 | 1,483.7 |
| ACE | 47.6 | 1,062.8 | 1,223.0 | 628.3 | 1,743.0 | 108.6 | 1,197.8 | 1,483.1 | 819.38 | 1,982.0 |
| ACE (rarefied) | 57.6 | 515.4 | 975.7 | 459.1 | 1,217.8 | 91.6 | 837.7 | 1,213.2 | 556.02 | 1,521.3 |
| OTUs with match,* % | 60.5 | 14.1 | 10.6 | 16.2 | 10.2 | 57.8 | 15.7 | 11.8 | 16.9 | 11.9 |
| Unidentified OTUs, % | NA | 35.6 | 38.8 | 31.2 | 40.9 | NA | 26.8 | 27.1 | 23.8 | 28.3 |
| Singletons, % | 31.6 | 39.8 | 36.5 | 34.9 | 34.8 | 46.9 | 32.3 | 34.6 | 30.3 | 31.1 |
Additional data and calculation methods are provided in Table S1. NA, not applicable.
Greater than 97% similarity to GenBank or BOLD sequences.
In contrast, the sessile and two smaller sieved fractions had much higher diversity at the OTU and phylum levels, and a much lower proportion of OTUs matching sequences in public databases. In total, HTS detected 1,204 OTUs from 572,290 sequences and 1,391 OTUs from 409,613 sequences from VA and FL, respectively (Table 1). Matches to GenBank or BOLD sequences were low (<12%) and comparable at both locations. Several additional OTUs could be identified because they matched reference barcodes obtained from the >2-mm and 2-mm to 500-μm fractions that were characterized morphologically (3.2% in VA and 3.9% in FL). Many more of the remaining OTUs could be assigned to a higher taxonomic group using the Bayesian phylogenetic approach (45.7% in VA and 55.9% in FL). However, 40.9% (VA) and 28.3% (FL) of OTUs could not be confidently assigned to any taxonomic group using this approach. Taxonomic coverage was very broad, with 22 phyla of animals, five major groups of protists, two major groups of Fungi, and three major groups of Plantae. Among the identified OTUs, Arthropoda were again the most OTU-rich (21.5% in VA and 29% in FL; Fig. 1). In FL, Arthropoda also had the highest percentage of sequences (28.5%), but in VA, the largest number of sequences belonged to Cnidaria (38.7%).
As is common in many samples of diversity, there were a few common taxa and many rare taxa in all samples. The percentage of singletons was surprisingly uniform, ranging from 31.6–46.9% in the barcoded samples and from 30.3–39.8% in the metabarcoded samples (Table 1). The groups with the highest percentage of singletons were “unidentified” OTUs in both VA and FL (53.2% and 36% of singletons, respectively), followed by Arthropoda (14.6% and 23.3% of singletons, respectively). The most common OTUs, as measured by either the number of sequences (Fig. 2) or the number of samples where they occurred (Fig. S2), were more likely to match reference barcodes (>97% similarity).
Fig. 2.

Proportion of identified OTUs in a metabarcoding dataset according to the number of sequences. A match to the reference barcode was defined as >97% similarity.
Overall, total diversity was surprisingly high for such small samples, and statistical analysis revealed that sampling was far from exhaustive. The mean (±SD) number of taxa per ARMS was 439.3 ± 54.9 for VA (11.2 ± 4.1 barcoded and 434.2 ± 55.7 metabarcoded) and 545.8 ± 29.6 for FL (15.8 ± 4.9 barcoded and 536.7 ± 30.8 metabarcoded) (Table 1). Chao1, Chao2, and rarefaction estimates for increasing numbers of ARMS (Fig. 3) or sequences (Fig. S3) show numbers continuing to climb with increased sampling effort.
Fig. 3.

Sample-based rarefaction curves and Chao1 estimates of total OTU diversity.
Patterns of Community Composition.
For the >2-mm organisms, FL and VA were highly distinct, with just four of 98 OTUs in common. Principal component analysis (PCoA) shows FL and VA samples well separated in 2D space along an axis explaining 32.6% and 62.6% of the variation in community composition using Jaccard (presence–absence) and Bray–Curtis (relative abundance) indices, respectively (Fig. S4 A and C). VA and FL samples also partition on an unweighted pair group method with arithmetic mean (UPGMA) tree, with differences between locations well supported by jackknife subsampling (Fig. S4 B and D). Differences in community composition were also significant between locations (Jaccard: Fπ1,16 = 7.32, P < 0.001; Bray–Curtis: Fπ1,16 = 27.3, P < 0.001).
In contrast, the sessile and smaller sieved fractions from FL and VA were more similar, with 457 (21%) of the 2,138 OTUs detected via HTS shared. Nevertheless, VA and FL samples are consistently separated on PCoA based on incidence or abundance along a first axis explaining 21.0% and 30.4% of the variation in OTU composition, respectively (Fig. 4 A and C), and they partition into two distinct well-supported groups on a UPGMA tree (Fig. 4 B and D). With all three fractions aggregated, community composition was significantly different between ARMS from VA and FL (Jaccard: Fπ1,16 = 13.21, P < 0.001; Bray–Curtis: Fπ1,16 = 26.72, P < 0.001) with 15 OTUs contributing 50% of the difference between VA and FL based on relative abundance (Table S2).
Fig. 4.

Clustering analyses [PCoA (A and C) and jackknifed UPGMA trees (B and D)] depicting similarity in community composition based on OTU incidence (Jaccard; A and B) and relative abundance (Bray–Curtis; C and D). PC, principal component.
The sessile and smaller sieved fractions had distinct OTU composition in both VA and FL. Their separation is clearly seen in PCoA and jackknifed UPGMA trees in both FL and VA (Fig. 4). Fractions were significantly different at both locations based on either OTU presence–absence (VA: Fπ2,24 = 6.56, P < 0.001; FL: Fπ2,24 = 8.08, P < 0.001) or relative abundance (VA: Fπ2,24 = 14.82, P < 0.001; FL: Fπ2,24 = 13.78, P < 0.001). Although samples of the 2-mm to 500-μm and 500- to 106-μm fractions overlay on the 2D PCoA constructed using the Bray–Curtis metric (Fig. 4C), differences in OTU composition were significant at both locations (VA: Fπ1,15 = 13.05, P < 0.001; FL: Fπ1,15 = 7.82, P < 0.001). As expected, based on the plate appearances (Fig. S1C), Porifera and Chordata are primarily responsible for the distinctiveness of the sessile fraction in FL, whereas unidentified OTUs characterize the 500- to 106-μm fraction (Fig. S5).
We found no evidence for fine-scale geographic structuring of the >2-mm samples based on PCoA (Fig. S4 A and C), UPGMA trees (Fig. S4 B and D), and permutational multivariate analysis of variance (PERMANOVA; Jaccard: Fπ5,12 = 2.33, P = 0.360; Bray–Curtis: Fπ5,12 = 6.40, P = 0.397). In contrast, fine-scale structuring in FL was apparent for all three fractions analyzed via metabarcoding on the jackknifed UPGMA tree (Fig. 4 B and D), with strong branch support for triplets representing adjacent ARMS. Although most adjacent sites in VA did not cluster as triplets on the UPGMA trees, differences between sites were highly significant in VA (Jaccard: Fπ2,24 = 1.07, P < 0.001; Bray–Curtis: Fπ2,24 = 1.41, P = 0.002) as well as in FL (Jaccard: Fπ2,24 = 1.77, P < 0.001; Bray–Curtis: Fπ2,24 = 2.11, P < 0.001).
The differences observed between locations, sites within locations, and fractions cannot be broadly attributed to differences in multivariate dispersion, because tests were insignificant except between fractions in FL (Jaccard: Fπ2,24 = 14.14, P < 0.001; Bray–Curtis: Fπ2,24 = 14.10, P < 0.001). Moreover, all differences in community composition remained significant after removing singletons from the metabarcoding dataset.
Validation of the Metabarcoding Approach.
Comparing HTS data against more direct assessments of community composition revealed that HTS is effective at detecting OTUs. A total of 91.2% (VA) and 77.1% (FL) of OTUs detected via barcoding of individual specimens isolated from the 2-mm to 500-μm fractions matched OTUs in the metabarcoding dataset from the same location. The proportion of matches between barcoding and metabarcoding datasets for individual ARMS ranged from 65.3–88.2% (i.e., 71.4% match between barcodes and metabarcodes of ARMS 5 in FL). A majority (50–100%) of undetected OTUs were represented by a single specimen, suggesting that they were rare or perhaps absent from the subsample (half of the total) crushed for DNA metabarcoding. Undetected OTUs belonged to several phyla, suggesting no particular taxonomic bias in OTU detection. Similarly, for sessile taxa, individual barcodes from subsamples of conspicuous taxa were always present in the overall metabarcoding dataset (n = 5 in VA and n = 13 in FL). The proportion of matches between barcoding and metabarcoding datasets for individual ARMS was 100% in VA and ranged from 90.9–91.7% in FL.
The relative abundance of sequences also showed good agreement with independent measures of relative abundance. In the 2-mm to 500-μm fractions, there was a significant positive relationship between the amount of DNA per OTU and the number of sequences per OTU for all ARMS from VA (ARMS 1: t22 = 3.95, P < 0.001; ARMS 4: t16 = 3.58, P < 0.001; ARMS 9: t18 = 6.64, P < 0.001; Fig. 5A) and FL (ARMS 1: t41 = 6.49, P < 0.001; ARMS 5: t34 = 4.54, P < 0.001; ARMS 9: t48 = 8.91, P < 0.001; Fig. 5B). At the phylum level (DNA and sequences per OTU pooled by phylum), there were too few points to compute statistics for ARMS individually, but there was an overall significant relationship between the amount of DNA and the number of sequences in VA (t10 = 6.14, P < 0.001; Fig. 5C) and FL (t20 = 3.72, P = 0.001; Fig. 5D). For sessile taxa in FL, measures of abundance based on point counts were significantly correlated with numbers of sequences in the metabarcoding dataset at both the OTU (ARMS 1: t10 = 3.70, P = 0.005; ARMS 5: t10 = 5.26, P < 0.001; ARMS 9: t12 = 3.60, P = 0.005; Fig. 5F) and phylum (t13 = 4.02, P = 0.002; Fig. 5H) levels. No statistics were calculated for the sessile OTUs from VA because of the limited number of data points (Fig. 5 E and G), but the pattern is similar to the pattern exhibited by the FL samples.
Fig. 5.
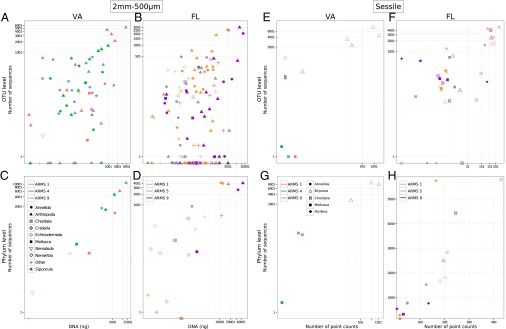
Relationship between the number of sequences per OTU (A, B, E, and F) or per phylum (C, D, G, and H) obtained via metabarcoding and the total amount of DNA (2-mm to 500-μm samples; A–D) or plate coverage (sessile taxa; E–H).
Discussion
Our intensive survey of the marine diversity of a small area (a total of 7.82 m2 and 0.05 m3 per locality) yielded a surprising amount of diversity: 1,218 OTUs in VA and 1,421 OTUs in FL. Although more than half of the barcode-based OTUs from invertebrates and fish >2 mm matched barcodes in public libraries, only 10–12% (VA/FL) of the metabarcode-based OTUs in the sessile and smaller sieved fractions matched GenBank or BOLD barcodes. As a result, identification of OTUs detected via metabarcoding relied mostly on phylogenetic assignments to taxonomic groups represented in GenBank, a database still lacking COI references for numerous families of marine invertebrates. Moreover, numerous OTUs remained unidentified, which may be due to the scarcity of representative COI sequences for entire branches of the tree of life, COI barcode misidentifications in GenBank, or limitations in using the hypervariable COI region for phylogenetic assignments. Methodological artifacts (e.g., PCR and sequencing errors or amplification of pseudogenes) are also a possibility, but they likely account for a minor proportion of unidentified OTUs, given our stringent quality filtering based on amino acid translation.
Regardless of taxonomy, our data provide a unique opportunity to investigate local and regional patterns of diversity across size fractions of mobile and sessile taxa. As expected, the >2-mm fraction is much less diverse than smaller mobile fractions (over an order of magnitude in VA and FL), a difference partly inherent to the sequencing approach used. Although barcoding provides specimen level data, metabarcoding captures not only “free-living” forms but also parasites, gut contents, and other forms of environmental DNA. We found the smallest sized organisms (500 to 106 μm) to have a 1.96- to 1.54-fold greater rarefied diversity than the 2-mm to 500-μm organisms in VA and FL, respectively, and the sessile fraction had the lowest rarefied OTU richness of the three metabarcoded fractions at both sites (1.37- to 1.29-fold smaller than 2-mm to 500-μm organisms, respectively). A higher local diversity in smaller sized organisms is consistent with the literature (9). However, the small sieve (106 μm) is likely accumulating debris and body parts shedding from sessile and larger motile animals (retained by coarser sieves) during field processing, thereby “artificially” increasing diversity in the 500- to 106-μm fraction [e.g., some sessile OTUs (i.e., Porifera, Bryozoa) are major contributors to differences between sieved fractions in both VA and FL; Table S2].
Also, as expected [(10) and ubiquity hypothesis (11)], the larger organisms showed a greater difference in estimated diversity between the temperate (37.6°N) and subtropical (27.4°N) locations (2.3-fold greater diversity in FL than VA) than did organisms belonging to the smaller sized fractions (1.1- to 1.4-fold greater in FL than VA for the two smaller mobile fractions). (The sessile fraction is composed of large and small organisms, and so cannot be separated in this fashion.) The latitudinal inflation in diversity of the >2-mm fraction is comparable to the latitudinal inflation in diversity observed for Western Atlantic coastal fishes from these latitudes (a two- to threefold difference) and somewhat less than the latitudinal inflation in diversity observed for decapod crustaceans (five- to sixfold difference) (figure 2 of ref. 12).
Our data showed no evidence for fine-scale spatial structuring in larger animal communities but demonstrated community partitioning at the 100-m scale for assemblages of sessile and microscopic taxa. More limited postsettlement dispersal abilities make these communities sensitive to local scale differences in environmental factors (13, 14). Moreover, because numerous microscopic animals may have specific associations with sessile taxa, spatial structuring in sessile assemblages may amplify differences between communities of small mobile taxa.
Finally, the assessment of the robustness of the metabarcoding approach targeting the COI gene suggests this method can be used reliably to detect OTU presence–absence, and it provides useful information on OTU relative abundance as well. Notably, the reliability improves as one moves to coarser groupings, because we have shown a remarkable fidelity at the level of the phylum, which suggests limited PCR bias among distant taxonomic groups. This finding is noteworthy because many ecological assessments work at the level of functional groups rather than at the level of species. Alternatively, PCR-free shotgun metagenomic approaches will be less prone to bias but require much higher sequencing depth (15), therefore increasing the cost for sufficient replication. Taxonomic coverage among animals will also increase with sequencing multiple independent markers [i.e., 18S, 16S (16)], but targeting nonprotein-coding genes may increase the probability of including sequencing artifacts. Moreover, alternative barcode genes would provide a more comprehensive survey of fungi [i.e., internal transcribed spacer (17)] and protists [i.e., 18S (18)]. As marine monitoring moves from the use of indicator groups to more comprehensive community-level analysis of alpha and beta diversity, this study provides support to encourage more routine use of a metabarcoding approach.
Materials and Methods
ARMS Deployment, Collection, and Sampling.
ARMS were deployed subtidally adjacent to natural oyster reefs in VA and FL for ∼6 mo (Fig. S1 and SI Text, section II). Upon retrieval, each plate was kept submerged in seawater. Plates were photographed on both sides, and representative sessile taxa were individually tissue-sampled for DNA barcoding. Sessile organisms growing on the plates were then scraped into a tray and homogenized using a kitchen blender, and ∼45 g of tissue was preserved in DMSO buffer (SI Text, sections II.D and III). Water holding ARMS was filtered through 2-mm, 500-μm, and 106-μm sieves. Mobile specimens retained by the 2-mm sieve were photographed alive, identified to the lowest taxon level possible based on morphology, and individually preserved in 95% ethanol (EtOH). The two smaller sieved fractions were initially bulk-preserved in 95% EtOH, and the organic fraction was later separated from sediments by decantation (SI Text, section IV). Each organic fraction was split in half by weight; the first half was crushed using a mortar and pestle to be analyzed via DNA metabarcoding, and the second half was archived in 95% EtOH (a summary of the protocol is provided in Fig. S1D). The biomass and amount of sediment are provided in Table S3.
DNA Barcoding and Metabarcoding.
For barcoding, tissue was subsampled from each specimen and placed individually in 96-well Costar plates (Corning) for phenol DNA extraction. DNA amplification and Sanger sequencing used standard protocols (SI Text, section V.A) and previously published primers (19, 20). For metabarcoding, DNA was extracted from 10 g of homogenized sessile tissue and the crushed half of the 2-mm to 500-μm and 500- to 106-μm samples using the MO-BIO Powermax Soil DNA Isolation Kit (SI Text, section V.B). Three replicate PCR assays were performed to amplify an ∼313-bp COI fragment for each of the 54 bulk samples (SI Text, section V.B). We used a hierarchical tagging approach for sample multiplexing [combination of tailed PCR primers and Ion Xpress (Life Technologies) barcode adapters; SI Text, section V.B and Table S4]. Amplicons were sequenced on the Ion Torrent platform (Life Technologies) following the manufacturer’s instructions (SI Text, section V.B). Barcode sequences were deposited in GenBank (accession nos. KP253982–KP255345) and BOLD (doi: dx.doi.org/10.5883/DS-ARMS), and the metabarcode datasets were deposited in the Dryad Digital Repository (doi: doi.org/10.5061/dryad.d0r79).
Sequence Analysis of Barcodes and Metabarcodes.
For barcodes, forward and reverse sequences were assembled, checked for stop codons or frame shifts, and edited in Geneious (Biomatters). We used the Bayesian clustering algorithm implemented in clustering 16S rRNA for OTU prediction (CROP) (21) to delineate OTUs based on the natural distribution of sequence dissimilarity in the dataset (SI Text, section VI.A). CROP outputs a representative sequence per OTU that was used for taxonomic identification. For metabarcodes, higher quality reads prefiltered by Torrent Suite Software version 4.0.2 (Life Technologies) were assigned to samples based on the combination primer tail-Ion Xpress barcode. Additional sequences were removed if they did not meet several criteria (SI Text, section VI.B). We then took advantage of the coding property of the COI gene to improve the quality and reliability of our dataset further by discarding reads with any anomaly in their amino acid translation using Multiple Alignment of Coding Sequences (MACSE) (22) (SI Text, section VI.B). Finally, reads were screened for chimeras using UCHIME (23). Remaining reads were clustered in OTUs using CROP following the parameters detailed in SI Text, section VI.A. For taxonomic assignments, we performed BLASTn searches of OTU representative sequences of the barcoding and metabarcoding datasets against full GenBank and BOLD databases. We accepted a species level match when similarity to the reference barcode was >97%. In the absence of a direct match, we used a phylogenetic approach implemented in the Statistical Assignment Package (24) to assign a higher taxonomic level (SI Text, section VI.C).
Statistical Analyses.
We summarized barcoding and metabarcoding data in separate OTU tables. Sample-based rarefaction curves and nonparametric species richness estimators were computed in EstimateS (25). Each OTU table was rarefied to the lowest number of sequences using Quantitative Insights into Microbial Ecology (QIIME) (26) to account for differences in abundance. We used Jaccard (presence–absence) and Bray–Curtis (relative abundance) metrics to calculate pairwise community distance matrices and examine differences in beta diversity. Patterns of sample dissimilarity were visualized using PCoA. Hierarchical cluster trees were also constructed using UPGMA with jackknife support to examine the robustness of sample clustering. We examined differences in community composition between locations and sites using PERMANOVA (27). We also tested whether the average distance to the group centroid is equivalent among groups [multivariate dispersion: PERMDISP (28)]. To account for the stratified structure of the design, we constrained permutations within factors when necessary (i.e., within locality to test for differences among sites). Metabarcoding data were initially aggregated per ARMS to test for overall differences in OTU composition and dispersion between locations and sites. We then partitioned the OTU table between locations to test for differences between fractions and sites. We repeated all statistical analysis after removing singletons from the metabarcoding OTU table to test for the robustness of the patterns. Finally, we conducted a similarity of percentage analysis to determine which OTUs were major contributors to differences between locations and fractions based on relative abundance. All tests were computed in the R package Vegan (29) and significance-tested using 1,000 permutations.
Assessment of Reliability of Metabarcoding Approach.
One ARMS from each of the three sites at each location was randomly chosen. Archived bulk samples were resuspended in a graduated beaker containing 100 mL of 95% EtOH and homogenized with a spatula, and 20 mL was immediately collected using a Hensel–Stempel pipette. All specimens were isolated and identified to the lowest taxonomic level; the entire specimen was used for phenol DNA extraction, and the mitochondrial COI gene was sequenced for OTU delineation. The amount of DNA in each individual extract was measured with a Qubit fluorometer (dsDNA HS Assay kit; Invitrogen), enabling the calculation of the total amount of DNA represented by each OTU and subsequent comparison with the number of reads obtained for the same OTU via metabarcoding (SI Text, section VII.A). For the sessile organisms, we individually sampled and barcoded morphologically distinctive taxa to identify matching OTUs in the metabarcoding dataset. The number of reads per OTU was then compared with their estimated cover on each ARMS as measured by a point count approach implemented in Coral Point Count with Excel extensions (CPCe) (30) (SI Text, section VII.B).
The relationship between the number of reads in metabarcoding dataset and the amount of DNA or number of point counts (for 2-mm to 500-μm and sessile specimens, respectively) was tested using a generalized linear model. Given the nature and significant overdispersion of the data, we fitted a quasi-Poisson model with log link function.
Supplementary Material
Acknowledgments
We thank Natalia Agudelo, Sherry Reed, Woody Lee, and Sean Fate for help in the field; the Smithsonian Laboratory of Analytical Biology staff for assistance; and Daryl Hurley II for allowing research to be conducted on his oyster reef in VA. Research Permit SAJ-2012-02893(NW-SLR) was provided by the US Army Corps of Engineers in FL. Financial support was provided by the Sant Chair and Smithsonian Tennenbaum Marine Observatories Network, for which this paper is Contribution 1.
Footnotes
The authors declare no conflict of interest.
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos. KP253982–KP255345), the Barcode Of Life Data Systems (doi: dx.doi.org/10.5883/DS-ARMS), and the Dryad Digital Repository (doi: doi.org/10.5061/dryad.d0r79).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1424997112/-/DCSupplemental.
References
- 1.Bouchet P, Lozouet P, Maestrati P, Heros V. Assessing the magnitude of species richness in tropical marine environments: Exceptionally high numbers of molluscs at a New Caledonia site. Biol J Linn Soc Lond. 2002;75(4):421–436. [Google Scholar]
- 2.Appeltans W, et al. The magnitude of global marine species diversity. Curr Biol. 2012;22(23):2189–2202. doi: 10.1016/j.cub.2012.09.036. [DOI] [PubMed] [Google Scholar]
- 3.Mora C, Tittensor DP, Adl S, Simpson AGB, Worm B. How many species are there on Earth and in the ocean? PLoS Biol. 2011;9(8):e1001127. doi: 10.1371/journal.pbio.1001127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tittensor DP, et al. Global patterns and predictors of marine biodiversity across taxa. Nature. 2010;466(7310):1098–1101. doi: 10.1038/nature09329. [DOI] [PubMed] [Google Scholar]
- 5.Bourlat SJ, et al. Genomics in marine monitoring: New opportunities for assessing marine health status. Mar Pollut Bull. 2013;74(1):19–31. doi: 10.1016/j.marpolbul.2013.05.042. [DOI] [PubMed] [Google Scholar]
- 6.Fonseca VG, et al. Metagenetic analysis of patterns of distribution and diversity of marine meiobenthic eukaryotes. Glob Ecol Biogeogr. 2014;23(11):1293–1302. [Google Scholar]
- 7.Plaisance L, Caley MJ, Brainard RE, Knowlton N. The diversity of coral reefs: What are we missing? PLoS ONE. 2011;6(10):e25026. doi: 10.1371/journal.pone.0025026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beck MW, et al. Oyster reefs at risk and recommendations for conservation, restoration, and management. Bioscience. 2011;61(2):107–116. [Google Scholar]
- 9.Azovsky A. Size-dependent species-area relationships in benthos: Is the world more diverse for microbes? Ecography. 2002;25(3):273–282. [Google Scholar]
- 10.Hillebrand H. On the generality of the latitudinal diversity gradient. Am Nat. 2004;163(2):192–211. doi: 10.1086/381004. [DOI] [PubMed] [Google Scholar]
- 11.Fenchel T, Finlay BJ. The ubiquity of small species: Patterns of local and global diversity. Bioscience. 2004;54(8):777–784. [Google Scholar]
- 12.Macpherson E. Large-scale species-richness gradients in the Atlantic Ocean. Proc Biol Sci. 2002;269(1501):1715–1720. doi: 10.1098/rspb.2002.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Green JL, et al. Spatial scaling of microbial eukaryote diversity. Nature. 2004;432(7018):747–750. doi: 10.1038/nature03034. [DOI] [PubMed] [Google Scholar]
- 14.Curini-Galletti M, et al. Patterns of diversity in soft-bodied meiofauna: Dispersal ability and body size matter. PLoS ONE. 2012;7(3):e33801. doi: 10.1371/journal.pone.0033801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou X, et al. Ultra-deep sequencing enables high-fidelity recovery of biodiversity for bulk arthropod samples without PCR amplification. Gigascience. 2013;2(1):4. doi: 10.1186/2047-217X-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gibson J, et al. Simultaneous assessment of the macrobiome and microbiome in a bulk sample of tropical arthropods through DNA metasystematics. Proc Natl Acad Sci USA. 2014;111(22):8007–8012. doi: 10.1073/pnas.1406468111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schoch CL, et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci USA. 2012;109(16):6241–6246. doi: 10.1073/pnas.1117018109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pawlowski J, et al. CBOL protist working group: Barcoding eukaryotic richness beyond the animal, plant, and fungal kingdoms. PLoS Biol. 2012;10(11):e1001419. doi: 10.1371/journal.pbio.1001419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geller J, Meyer C, Parker M, Hawk H. Redesign of PCR primers for mitochondrial cytochrome c oxidase subunit I for marine invertebrates and application in all-taxa biotic surveys. Mol Ecol Resour. 2013;13(5):851–861. doi: 10.1111/1755-0998.12138. [DOI] [PubMed] [Google Scholar]
- 20.Leray M, et al. A new versatile primer set targeting a short fragment of the mitochondrial COI region for metabarcoding metazoan diversity: Application for characterizing coral reef fish gut contents. Front Zool. 2013;10:34. doi: 10.1186/1742-9994-10-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hao X, Jiang R, Chen T. Clustering 16S rRNA for OTU prediction: A method of unsupervised Bayesian clustering. Bioinformatics. 2011;27(5):611–618. doi: 10.1093/bioinformatics/btq725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ranwez V, Harispe S, Delsuc F, Douzery EJP. MACSE: Multiple Alignment of Coding SEquences accounting for frameshifts and stop codons. PLoS ONE. 2011;6(9):e22594. doi: 10.1371/journal.pone.0022594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27(16):2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Munch K, Boomsma W, Huelsenbeck JP, Willerslev E, Nielsen R. Statistical assignment of DNA sequences using Bayesian phylogenetics. Syst Biol. 2008;57(5):750–757. doi: 10.1080/10635150802422316. [DOI] [PubMed] [Google Scholar]
- 25.Colwell RK. 2006 EstimateS: Statistical Estimation of Species Richness and Shared Species from Samples. Version 9.1. Available at purl.oclc.org/estimates. Accessed August 15, 2014.
- 26.Caporaso JG, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001;26:32–46. [Google Scholar]
- 28.Anderson MJ, Ellingsen KE, McArdle BH. Multivariate dispersion as a measure of beta diversity. Ecol Lett. 2006;9(6):683–693. doi: 10.1111/j.1461-0248.2006.00926.x. [DOI] [PubMed] [Google Scholar]
- 29.Oksanen J, et al. 2009 Vegan: Community ecology package. R package version 2.0-10. Available at cran.r-project.org/package=vegan. Accessed August 15, 2014.
- 30.Kohler KE, Gill SM. Coral Point Count with Excel extensions (CPCe): A Visual Basic program for the determination of coral and substrate coverage using random point count methodology. Comput Geosci. 2006;32(9):1259–1269. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.