

Significance
Classical atomistic molecular dynamics simulations and free-energy calculations performed on cytochrome c oxidase in an explicit membrane–solvent environment show that the water molecules in the nonpolar cavity near the active site of oxygen reduction reorientate according to the redox state of the enzyme, guiding the path of proton transfer and thereby effectively preventing short-circuit of the proton pump. The results highlight the important role of protein-bound water molecules in biological energy conversion––a notion shared with bacteriorhodopsin, the light-driven proton pump.
Keywords: cell respiration, atomistic molecular dynamics simulations, functional water molecules, free-energy calculations
Abstract
Molecular oxygen acts as the terminal electron sink in the respiratory chains of aerobic organisms. Cytochrome c oxidase in the inner membrane of mitochondria and the plasma membrane of bacteria catalyzes the reduction of oxygen to water, and couples the free energy of the reaction to proton pumping across the membrane. The proton-pumping activity contributes to the proton electrochemical gradient, which drives the synthesis of ATP. Based on kinetic experiments on the O–O bond splitting transition of the catalytic cycle (A → PR), it has been proposed that the electron transfer to the binuclear iron–copper center of O2 reduction initiates the proton pump mechanism. This key electron transfer event is coupled to an internal proton transfer from a conserved glutamic acid to the proton-loading site of the pump. However, the proton may instead be transferred to the binuclear center to complete the oxygen reduction chemistry, which would constitute a short-circuit. Based on atomistic molecular dynamics simulations of cytochrome c oxidase in an explicit membrane–solvent environment, complemented by related free-energy calculations, we propose that this short-circuit is effectively prevented by a redox-state–dependent organization of water molecules within the protein structure that gates the proton transfer pathway.
Life on Earth is supported by a constant supply of energy in the form of ATP. Cytochrome c oxidase (CcO) in the respiratory chains of mitochondria and bacteria catalyzes the exergonic reduction of molecular oxygen (O2) to water and uses the free energy of the reaction to pump protons across the membrane (1–3). The oxygen reduction reaction takes place at a highly conserved active site formed by two metal sites, heme a3 and CuB (Fig. 1 A and B), called the binuclear center (BNC). The electrons donated by the mobile electron carrier cytochrome c reach the BNC via two other conserved metal centers, CuA and heme a (Fig. 1A). The protons required for the chemistry of O2 reduction to water, and for proton pumping, are transported with the assistance of side chains of polar amino acids and conserved water molecules in the protein interior (4–6) (Fig. 1A). Two such proton transfer pathways have been described in the mitochondrial and bacterial A-type oxidases (to distinguish between different types of oxidases, see ref. 7), namely, the D and K channels (8, 9), the names of which are based on the conserved amino acid residues Asp91 and Lys319, respectively (Fig. 1A, amino acid numbering based on the bovine heart CcO). The D channel is responsible for the translocation of all of the pumped protons, and for the transfer of at least two of the four protons required for oxygen reduction chemistry, whereas the K channel supplies one or two protons to the BNC during the reductive phase of the catalytic cycle (8, 9). The D channel terminates at a highly conserved glutamic acid residue, Glu242, from where the protons are either transferred to the BNC for consumption, or to the proton-loading site (PLS) for pumping across the membrane (Fig. 1A). In 2003, Wikström et al. postulated a molecular mechanism in which water molecules in the nonpolar cavity above Glu242 would form proton-transferring chains, the orientation of which depends upon the redox state of the enzyme (10). They proposed that the reduction of the low-spin heme would result in transfer of a proton via a preorganized water chain from Glu242 to the d-propionate (Dprp) of the high-spin heme, whereas in the case when the electron has moved to the BNC, the water chain would reorientate and conduct protons from Glu242 to the BNC (Fig. 1A, and see below). Even though there is little direct experimental support available for such a water-gated mechanism, a recent FTIR study indeed suggests changes in water organization upon changes in the redox state of the enzyme (11). Many of the elementary steps that were postulated in the water-gated mechanism have gained support from experiments in the recent past (12, 13).
Fig. 1.
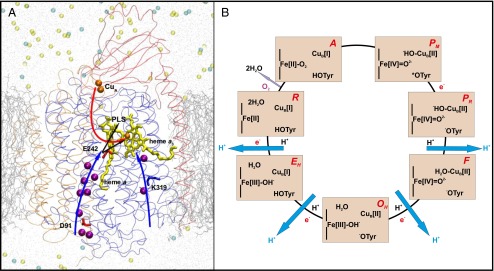
(A) A three-subunit (SU) CcO. SU I (blue), II (red), and III (orange) are displayed as transparent ribbons. The D and K channels of proton transfer are marked with blue arrows. Crystallographic water molecules present in these proton channels are shown in purple. Electron transfer (red arrow) takes place from CuA (orange) via heme a (yellow) to the binuclear center comprising heme a3 (yellow)–CuB (orange). Protons are transferred from Glu242 (E242) either to the PLS or to the binuclear center (black arrows). Lipid bilayer (silver lines), water (gray dots), and sodium (light yellow) and chloride (cyan) ions are also displayed. (B) The catalytic cycle of CcO. The states of heme a3, CuB, and the cross-linked tyrosine are displayed. Each light orange rectangle corresponds to a state of the BNC, the name of which is displayed in red (Upper Right). Pumped protons are shown in blue, black H+ indicates uptake of a proton for water formation, and e− indicates transfer of an electron from the low-spin heme a. Catalysis of O2 reduction occurs clockwise.
It is generally thought that the proton pump of CcO operates via the same mechanism in each of the 4 one-electron reduction steps of the catalytic cycle (Fig. 1B). However, kinetic data on two different transitions (A → PR and OH → EH) have suggested dissimilarities in some of the elementary steps (12, 13). Fully reduced enzyme reacts with oxygen and forms an oxygenated adduct A in ca. 10 µs, followed by splitting of the O–O bond leading to formation of the PR intermediate (in ∼25 μs) that is linked to loading of the PLS with a proton (3, 12). O–O bond splitting from state A in the absence of electrons in heme a or CuA yields the stable state PM without proton transfer to the PLS (3, 12). Therefore, it is the electron transfer from heme a into the BNC accompanying O–O bond scission during A → PR that is linked to the proton transfer to the PLS. The structure of the PR intermediate is well characterized with ferryl heme a3, cupric hydroxide, and tyrosinate (3, 14). In PM the tyrosine is almost certainly in the form of a neutral radical (3, 14), so the reaction PM → PR is a proton-coupled electron transfer reaction (PCET) that initiates the reactions of the proton pump (3, 12). Note that in the state PR the proton at the PLS partially neutralizes the electron in the BNC (3) in accordance with the charge-neutralization principle of the BNC (15). However, an important question arises: how can proton transfer from Glu242 to the BNC be prevented, which would short-circuit one step of proton pumping and form the next stable intermediate F? In the OH → EH transition of the catalytic cycle this short-circuit is minimized because reduction of the low-spin heme is thought to raise the pKa of the PLS sufficiently to lead to its protonation before transfer of the electron to the BNC (3, 10, 13, 16–18), and uncompensated proton transfer to the BNC is endergonic in nature (refs. 13,16,17; cf. ref. 19). In contrast, the likelihood of a proton leak in the A → PR transition increases manifold because the electron transfer from heme a to the BNC is required for loading of the PLS with a proton (3, 12). This facet is analyzed in the current work, and it is proposed that it is the orientation of the water molecules in the nonpolar cavity above Glu242 that effectively gates the pump and minimizes such a short-circuit.
Results
Classical all-atom MD simulations performed on certain key intermediate states of the catalytic cycle of CcO (Table 1) provide a detailed view of the organization and dynamics of water molecules in the nonpolar cavity just above the conserved glutamic acid Glu242 (Figs. 1 and 2). These water molecules may form continuous water chain connections from the proton donor Glu242 to two different proton acceptor groups, either the Dprp of the high-spin heme or an oxygenous metal ligand in the BNC. Depending upon the redox state of the enzyme, a preferential connection to either of the two proton acceptors has been observed (10), which is the “water-gated” phenomenon that is studied in detail here.
Table 1.
Description of model systems and simulation times
| State* | Heme a | Heme a3 | Copper | Tyrosine† | Time‡ |
| PM | Fe[III] | Fe[IV]=O2- | CuB[II]-OH− | TyrO* | ∼60 |
| PM' | Fe[II] | Fe[IV]=O2- | CuB[II]-OH− | TyrO* | ∼65 |
| PR | Fe[III] | Fe[IV]=O2- | CuB[II]-OH− | TyrO− | ∼65 |
| F | Fe[III] | Fe[IV]=O2- | CuB[II]-OH2 | TyrO− | ∼31 |
| F' | Fe[II] | Fe[IV]=O2- | CuB[II]-OH2 | TyrO− | ∼30 |
| FR | Fe[III] | Fe[III]-OH− | CuB[II]-OH− | TyrO− | ∼30 |
| FH' | Fe[II] | Fe[IV]=O2- | CuB[II] | TyrO− | ∼70 |
| FH,R | Fe[III] | Fe[IV]=O2- | CuB[I] | TyrO− | ∼70 |
| F'C | Fe[II] | Fe[IV]=O2- | CuB[II]-OH− | TyrOH | ∼30 |
| PM' | Fe[II] | Fe[IV]=O2- | CuB[II]-OH− | TyrO* | ∼30§ |
| PR | Fe[III] | Fe[IV]=O2- | CuB[II]-OH− | TyrO− | ∼30¶ |
Fig. 2.

The PUMP and CHEM configurations as snapshots from simulations. In the PUMP configuration a water wire forms between Glu242 (E242) and Dprp of heme a3. In the CHEM configuration an additional water wire connects Glu242 (E242) to the BNC (heme a3–CuB). Hydrogen atoms attached to nitrogen (blue) and oxygen (red) are shown in white, carbon in cyan, and Fe and Cu atoms in green and orange, respectively. The transparent ribbons show SU I (blue) and II (red).
We first studied the PM → PR transition of the catalytic cycle (2, 3) in three different states (Table 1): PM, where heme a is oxidized; PM', where heme a is reduced; and PR, where the BNC initially in state PM has just received the electron from heme a. The data presented in Table 2 show that immediately before the proton-coupled electron transfer (state PM'), a proton-conducting water chain connection forms from Glu242 to Dprp (PUMP configuration, Fig. 2) with over 200 times higher propensity compared with the corresponding connection from Glu242 to the BNC (CHEM configuration, Fig. 2). In contrast, after the electron has moved to the BNC (forming PR), the tendency to form a water chain from Glu242 to the BNC increased nearly 50 times, and the probability of the PUMP configuration decreased by nearly sixfold. The same data are displayed in Fig. 3, but highlighting the stability of the two configurations over the course of the simulation in different redox states. Note especially that upon reduction of the low-spin heme (PM') the PUMP configuration is abundant whereas the CHEM configuration vanishes almost completely. In the case where the electron has been transferred to the BNC (PR), the CHEM configuration resurfaces and stabilizes together with the PUMP configuration, forming a fork-like structure (Fig. 2). Inspection of the data in Fig. 3 also reveals that the PUMP configuration is persistent for longer time periods in the PM' state relative to PM and PR, where its probability approaches zero at the longest times. Most importantly, the CHEM configuration vanishes altogether after ∼25 ns in the PM' state (Fig. 3). The data in Table S1 show that a similar result is obtained even if the number of water molecules above and below Glu242 is varied.
Table 2.
Orientation of water chains in the nonpolar cavity of the enzyme in different redox states
| Redox state | PUMP* | CHEM* |
| PM | 16,963 (28) | 2,520 (4) |
| PM' | 29,488 (46) | 143 (0.2)/3.8† |
| PR | 4,826 (7) | 7,418 (11) |
| F | 764 (2.5) | 0 (0) |
| F' | 10,756 (36) | 3 (0.01)/5.6† |
| FR | 4,261 (14) | 3,343 (11) |
| FH' | 16,304 (24) | 279 (0.4)/3.4† |
| FH,R | 142 (0.2) | 12,219 (18) |
| F'C | 4,356 (14) | 5,868 (19)/1.0† |
| F' ‡ | 12,160 (41) | 0 (0) |
PUMP denotes a complete water wire from Glu242 to the Dprp of the high-spin heme, and CHEM denotes a corresponding water wire to the binuclear site (Fig. 2), based on the hydrogen bonding criteria described in Materials and Methods. The total number of frames in each configuration is given, along with the percentage of the observed water chain out of all frames, in parentheses.
Energy cost [being described as –kB T ln P(CHEM) in units of kcal/mol] associated with the formation of the CHEM configuration in the states when heme a is reduced.
Data based on charge parameterization used in Yang and Cui (ref. 24, and references therein) but with the structure of the F' state as described in the current work.
Fig. 3.

Persistence of the PUMP and CHEM water chain configurations over the course of simulation in three different redox states (PM, PM', and PR) smoothened by a running average of 100 simulation snapshots. Abscissa is simulation time in nanoseconds, and ordinate describes whether a PUMP or a CHEM path exists (#1) or not (#0) based on the H-bonding criteria discussed in Materials and Methods.
Similar statistics are also observed for the intermediate states F, F', and FR. F is the state of the BNC after protonation of the PR state (Fig. 1B and Table 1). In the state F' the low-spin heme is further reduced and now the PUMP configuration of the water molecules is stabilized by nearly 4 orders of magnitude relative to the CHEM configuration (Table 2). In contrast, in the FR state, when the electron has moved over to the BNC, the probability of the CHEM configuration increased by three orders of magnitude, being now roughly equally probable as the PUMP configuration.
To further test the reversibility of the water chain organization, we performed ∼30-ns simulations starting from a preformed PUMP or CHEM configuration, but reversing the redox states in which they had been formed (Table 1). The data in Fig. 4 show that when started from a preformed PUMP configuration typical of the PM' state, and switching to the PR redox state, the PUMP configuration was destabilized in ∼15 ns. At around the same time the CHEM configuration became much more stable, in agreement with the data shown in Table 2 and Fig. 3. On the other hand, when started from a preformed CHEM configuration, the PUMP configuration rapidly stabilized in the PM' redox state, and lasted for about 5 ns, after which water molecules exited the nonpolar cavity (SI Text and Fig. S1). The CHEM configuration remained slightly unstable relative to the PUMP configuration in the PM' redox state, and was completely eliminated at ∼4 ns (Fig. 4).
Fig. 4.

Temporal behavior of the preformed PUMP and CHEM water chain configurations in the PR (preformed PUMP) and PM' (preformed CHEM) redox states. The plots are smoothened by a running average of 100 simulation snapshots. Abscissa is time of simulation in nanoseconds, and ordinate describes whether a PUMP or a CHEM path exists (#1) or not (#0) based on the H-bonding criteria discussed in Methods section.
Overall, the water dynamics data (Table 2 and Table S1, and Figs. 3 and 4) from 12 independent simulations representing the PM to PR, and the F to FR transitions of the catalytic cycle suggest that upon reduction of heme a, the propensity to form a water wire from Glu242 to the Dprp of the high-spin heme is at least about two orders of magnitude higher than forming the configuration that would assist proton transfer from Glu242 to the BNC. This result is supported by umbrella sampling calculations (SI Text and Fig. S2), which show that the energetic cost associated with formation of the CHEM configuration in the state PM' is 3.14 ± 0.33 kcal/mol, in agreement with the equilibrium simulation data (Table 2).
The possible physical origins of the observed water wire rearrangement call for a brief note. In the original work it was already suggested that the electric field induced by an electron on heme a, or in the BNC, could influence the water molecule organization due to charge–dipole interactions (ref. 10; cf. ref. 20). The redox-state–dependent water dynamics presented here in a much more comprehensive system consolidates this point. However, it is also likely that stabilization of local hydrogen bonds assists in further stabilization of water chains upon changes in redox states. For instance, the partial charge on the oxygen atom ligating CuB changes from −0.66e to −0.80e upon reduction (PM' → PR) (21), which may strengthen the hydrogen bond with the nearest water molecule. This stabilization would then affect the entire water chain up to the terminal proton donor Glu242 due to cooperative hydrogen bonding effects among water molecules (22, 23).
Recently, Yang and Cui (24) investigated the validity of the water-gated mechanism by performing ∼20-ns MD simulations in an explicit membrane environment. In contrast with the work presented here, their equivalent of the F' state had a hydroxyl ligand of CuB and a protonated tyrosine (state F'C in Table 1). They found that the OH ligand of CuB always acts as a H-bond acceptor from the nearest water molecule, and concluded that such a water wire to the BNC compromises the water-gated mechanism. We decided to investigate this issue in more detail here. First, note that FTIR data have shown that the cross-linked tyrosine is deprotonated in the state F (25). Therefore, a water molecule rather than a hydroxyl ion most likely ligates the CuB ion in this state (Table 1; see also ref. 26). This difference is found to be very important and is assessed below. In agreement with Yang and Cui (24), we also observed that the OH ligand of CuB may indeed act as a preferential H-bond donor to the oxygenous ligand of heme a3 for ca. 83% of the simulation length (averaged over all PM → PR simulations), suggesting that it can act as a primary H-bond acceptor from the nearest water molecule. However, our data on the P states clearly show that a stable water wire connection to the OH ligand of CuB forms only in the state when the electron has moved over to the BNC, even though the OH ligand of CuB is present in all of the three states simulated (PM, PM', and PR). To further test whether the presence of a H-bond acceptor in the BNC compromises the water-gated mechanism, we performed fairly long (∼70-ns) MD simulations in yet another version of the F states (FH' and FH,R). These states are similar to F' and FR, except that the water ligand of CuB has been removed (26), which leaves the ferryl iron-oxo group as a strong hydrogen bond acceptor in the BNC. The data presented in Table 2 show a similar trend as for the other intermediates. The PUMP configuration is some 60-fold more stable than the CHEM configuration before the PCET (FH' state). Hence, the water-gated mechanism operates independently of the identity of the hydrogen bond acceptor group in the BNC.
The original simulation studies proposing the water-gated mechanism used a much simpler model system (10), and the simulations were performed in vacuum or gas phase (27). To compare those results with the current simulations, we conducted test calculations by running 10-ns vacuum simulations in two different redox states (FH' and FH,R). In agreement with previous studies (10, 27), we observed that in the FH' state the energy cost associated with forming the “wrong” CHEM configuration is nearly 7 kcal/mol, whereas in the state FH,R a water wire to BNC (CHEM configuration) readily forms (Table S2). The energy is ca. 3 kcal/mol less (lower limit) when the occupancy of water chains is obtained from the current protein–lipid–solvent simulations (Table 2). The reasons for this difference are most likely due to the differences in simulation methodologies. Nevertheless, it is clear that the water dynamics in the nonpolar cavity follows the water-gated principle qualitatively in the same way irrespective of the model system used.
Yang and Cui (24) claimed that the water-gated principle is the result of the previously used simplified computational models, implying the lack of a membrane–solvent environment. Our current data demonstrate that this conclusion is incorrect. On the other hand, we agree with these authors that the correct results of such analyses require careful consideration of the ligand state of the metal sites and the protonation states of key residues. As shown in Table 2 and Table S2, the distinction between the PUMP and CHEM configurations of the water molecules vanishes entirely in the structure of the state F' used by Yang and Cui (24), where the tyrosine rather than the OH ligand of CuB is protonated (state F'C in Tables 1 and 2 and Table S2). Finally, to test how sensitive our results are to the assigned partial charges, we repeated the test of the F' state but using the charges used by Yang and Cui (ref. 24, and references therein). As shown in Table 2, the result is in good agreement with the water-gate model, proving that the failure to observe water-gated behavior by these authors was due to the incorrect F'C structure.
Discussion
In the A → PR transition of the catalytic cycle, electron transfer from the low-spin heme to the BNC is coupled to the loading of the proton pump by proton transfer from Glu242 to the PLS (3, 12). Here, the possibility of premature electron coupled proton transfer directly to the BNC is a serious risk, which would inevitably lead to short-circuit and loss of proton pumping. As shown here, the water-gated mechanism becomes crucial in this scenario. The data in Table 2 show that before the proton-coupled electron transfer (PCET), when the low-spin heme is reduced, the PUMP configuration of the water molecules is preferred over the CHEM configuration by a factor of at least 60 and up to nearly 4,000, depending on the precise structure of the binuclear center. This stabilization of the PUMP configuration effectively prevents the short-circuit, and ensures a stoichiometric proton-pumping efficiency of at least 98%. The proposed protein-bound water rearrangement, which shares similarity with the water dynamics observed for the light-induced proton pump, bacteriorhodopsin (28), supports the general view that protein-bound water molecules play very important roles in enzyme function. Time-resolved FTIR studies and/or high-resolution X-ray structures of intermediate states would be required to test the proposal experimentally.
The PCET event is reversible. The water chain from Glu242 to the Dprp of heme a3 (PUMP orientation) remains formed after the PCET so that the proton may be transferred back to the Glu242, and the risk of short-circuiting the pump by transfer of the proton to the BNC must be considered. First, the strong proton–electron coupling in the PCET requires such proton transfer back to Glu242 to be coupled to transfer of the electron back to heme a by nanosecond electron tunneling (29). Secondly, as discussed previously (16, 17, 30), the Dprp is almost certainly not identical to the PLS, so that the protonation of the former is likely to be only transient.
The fact that the CHEM configuration does form before the PCET, albeit very rarely compared with the PUMP configuration (Table 2 and Fig. 3), is a more serious problem. The rate-limiting step for the P→F transition, for example, takes ca. 100 μs (3), which seems to be long enough to allow for short-circuiting proton transfer to the BNC by the CHEM pathway, even though the latter forms rarely on a subnanosecond time scale (Table 2). This problem may be solved by the gating properties of Glu242 (31–33). Glu242 undergoes side-chain isomerization between an “up” position where it interacts with water molecules in the apolar cavity (Fig. 2), and the “down” position seen in all crystal structures where it interacts with water molecules at the end of the D pathway (Fig. 1A). After donating its proton to the PLS via the PUMP configuration of water molecules, the deprotonated Glu242 anion flips to the down position remarkably quickly (∼1 ps; refs. 31, 32), which eliminates protonic connectivity between the PLS and the BNC. Moreover, the down position of the Glu242 anion is preferred over the up position by a factor of at least 104 (31). The Glu242 anion is subsequently protonated via the D pathway. At a pH of 7 on the aqueous N side of the membrane and assuming a pKa of 9 for Glu242 (3), there is further stabilization by a factor of 102. The very fast isomerization of the Glu242 anion to the down position thus provides a kinetic trap that minimizes the possibility of a protonic leak to the BNC. From a thermodynamic viewpoint the isomerization and acid–base properties of Glu242 destabilize the state before PCET by a factor of ≥106. Together with the ≥102-fold preference of the PUMP configuration over CHEM, these properties are sufficient to effectively minimize short-circuit of the proton pump, as verified by mathematical modeling (33). Thus, the water orientation is essential to prevent proton leakage, but it is not alone sufficient to achieve the high efficiency observed for the proton pump of CcO. Finally, because the anionic Glu242 in the down conformation is known to accumulate water molecules to stabilize its negative charge (31, 32), the nonpolar cavity may transiently dry out and prevent any protonic equilibration between the protonated PLS and the reduced BNC. Water is constantly produced at the BNC during turnover, and exits the nonpolar cavity (27, 34). Indeed, in the simulations presented here, water molecules are observed to leave the nonpolar cavity, and exchange with the crystallographic water molecules present in the hydrophilic domain above the propionates (SI Text).
It may be of interest to compare the present results with the sequence of proton-pumping events proposed for one-electron reduction of the ferric–cupric enzyme (state OH) (13, 16–18). In that case the reduction of heme a has been proposed to lead to protonation of the PLS from the N side of the membrane before and independent of electron transfer to the BNC. Premature proton transfer to the BNC is then thought to be prevented for thermodynamic reasons, and gating by water molecules would seem to be of less importance. However, previous density functional theory and electrostatic calculations have led to the suggestion of a strongly coupled PCET (19, 35) also in this step of the catalytic cycle, in which case the water-gated mechanism would gain even higher significance. The notion of an identical principle of proton pumping in the different steps of the catalytic cycle would support this view, and further experimentation is required to settle this matter.
Conclusions
Atomistic MD simulations on a three-subunit CcO complex in an explicit lipid–solvent environment, and comprising two different reaction steps of the catalytic cycle, suggest that water-gated proton transfer is an essential albeit not sufficient feature to prevent short-circuit of the proton-pumping mechanism. The results show that the water-gated principle is independent of the identity of the proton acceptor group in the binuclear center, but strongly dependent on its protonation state, and corroborate the role of water molecules in achieving high efficiency in biological energy transduction.
Materials and Methods
The three subunit enzyme [subunits I, II, and III; Protein Data Bank ID code 1V54 (36)] was used to construct the simulation system shown in Fig. 1. All crystallographic water molecules that were present in these subunits were retained for model construction. In addition, four or five water molecules were explicitly modeled in the nonpolar cavity above Glu242. Previously, up to four water molecules have been predicted to reside in this cavity (10, 37, 38). To assess the robustness of the results we performed 15–25-ns-long simulations with three more water molecules modeled in the cavity below Glu242 in different redox states (Table S1). The redox states of the metal centers in different simulation states are shown in Table 1.
The protonation states of BNC ligands were decided on the basis of available experimental data (3) and previous density functional theory (DFT) calculations performed on cluster systems (3, 21, 26). It is known based on spectroscopic measurements that all P and F variants have a ferryl heme a3 (3, 14) except for the postulated state FR, which has ferric heme a3 ligated by a hydroxyl ion (26). CuB, on the other hand, is known to be ligated either by an OH in all P, or by a water molecule in all F states (14), with the exception of state FR (ref. 26; see also Table 1). CuB is not ligated by any oxygenous ligand in the FH' and FH,R states (ref. 26; see also Table 1).
The crucial amino acid residue Glu242 was modeled protonated in all simulations based on data available from kinetic and FTIR experiments (3). Asp364, which forms a hydrogen bond with the A-propionate of high-spin heme, was also kept protonated based on DFT calculation data (21, 30). The cross-linked tyrosine, which supplies a proton and an electron in the early phase of the catalytic cycle (Fig. 1B), was modeled as a neutral radical in the PM and PM' states, but was tyrosinate in all other states, in agreement with FTIR data (ref. 25, cf. ref. 24; see also Table 1). Lys319 in the K channel was protonated in all of the simulations based on DFT–electrostatic calculations (39), as well as His207 in subunit III due to its ion pairing with Glu90 in the same subunit. All other amino acid residues were kept in their standard protonation states.
The charges and parameters of standard amino acid residues were obtained from the Chemistry at Harvard Macromolecular Mechanics (CHARMM) force field (40), and for metal centers, charges and parameters were obtained from previous studies (21, 26). After construction of the protein system, it was immersed in a lipid bilayer comprising cardiolipin (CL), phosphatidylcholine (PC), and phosphatidylethanolamine (PE) molecules. The ratio of CL:PC:PE molecules in the simulation system was 1:3.38:3.05, in accordance with previously published data (41). The charges and parameters of lipid molecules were taken from ref. 41, but were modified according to the more recent and accurate C36 CHARMM force field (42). The whole system was solvated with TIP3 water molecules, and ions (Na+ and Cl−) were added to neutralize the system and to give a salt concentration of 100 mM. The model system consisted of nearly 280,000 atoms in total.
The MD simulations were performed using the NAMD (43) program. The time step of 1 fs was used in all simulations. The temperature and pressure were maintained at 310 K and 1.0132 bar using the Langevin thermostat and barostat, respectively. A cutoff of 12 Å was applied, with a switching distance of 10 Å for nonbonded interactions. Electrostatics was evaluated with the particle mesh Ewald method implemented in NAMD (43). An initial energy minimization of ca. 2,000–4,000 steps was performed before all simulations, which were performed without any constraints. The protonated Glu242 modeled in its down crystallographic conformation (Fig. 1A) spontaneously flipped up in simulations of all states (Fig. 2). The data were saved every picosecond (every 1,000 steps). The simulation times of individual runs in different redox states are given in Table 1. The simulation times and the frequency of writing coordinates to trajectory files are sufficiently balanced with respect to the dynamics of water molecules, which is known to be rapid (28, 44). Overall, the total simulation time of all simulation runs is ca. 0.76 µs. All analysis on simulation trajectories was done with the help of Tcl scripting available in VMD (45). The H-bonding criteria of distance(D-H...A) < 4 Å and angle(D-H...A) > 150° were used during the analysis in accordance with the previous studies (46). A detailed description of the free energy calculations is given in SI Text.
Supplementary Material
Acknowledgments
We acknowledge stimulating discussions with Dr. Gerhard Hummer (Max Planck Institute for Biophysics, Frankfurt, Germany). V.S. acknowledges postdoctoral researcher funding from the Academy of Finland. M.W. acknowledges support from the Academy of Finland, Biocentrum Helsinki, and the Sigrid Jusélius Foundation. We also acknowledge financial support from the Academy of Finland Center of Excellence program (to I.V., T.R., and V.S.) and support awarded by the European Research Council [Advanced Grant CROWDED-PRO-LIPIDS (I.V.)], and the Sigrid Jusélius Foundation (to I.V.). We are grateful to Center for Scientific Computing (CSC)–IT Center for Science (Espoo, Finland) for computing resources.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1409543112/-/DCSupplemental.
References
- 1.Wikström MK. Proton pump coupled to cytochrome c oxidase in mitochondria. Nature. 1977;266(5599):271–273. doi: 10.1038/266271a0. [DOI] [PubMed] [Google Scholar]
- 2.Ferguson-Miller S, Babcock GT. Heme-copper terminal oxidases. Chem Rev. 1996;96(7):2889–2908. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 3.Kaila VRI, Verkhovsky MI, Wikström M. Proton-coupled electron transfer in cytochrome oxidase. Chem Rev. 2010;110(12):7062–7081. doi: 10.1021/cr1002003. [DOI] [PubMed] [Google Scholar]
- 4.Tsukihara T, et al. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 A. Science. 1996;272(5265):1136–1144. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- 5.Yoshikawa S, et al. Redox-coupled crystal structural changes in bovine heart cytochrome c oxidase. Science. 1998;280(5370):1723–1729. doi: 10.1126/science.280.5370.1723. [DOI] [PubMed] [Google Scholar]
- 6.Iwata S, Ostermeier C, Ludwig B, Michel H. Structure at 2.8 A resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature. 1995;376(6542):660–669. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 7.Pereira MM, Santana M, Teixeira M. A novel scenario for the evolution of haem-copper oxygen reductases. Biochim Biophys Acta. 2001;1505(2-3):185–208. doi: 10.1016/s0005-2728(01)00169-4. [DOI] [PubMed] [Google Scholar]
- 8.Konstantinov AA, Siletsky S, Mitchell D, Kaulen A, Gennis RB. The roles of the two proton input channels in cytochrome c oxidase from Rhodobacter sphaeroides probed by the effects of site-directed mutations on time-resolved electrogenic intraprotein proton transfer. Proc Natl Acad Sci USA. 1997;94(17):9085–9090. doi: 10.1073/pnas.94.17.9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wikström M, Jasaitis A, Backgren C, Puustinen A, Verkhovsky MI. The role of the D- and K-pathways of proton transfer in the function of the haem-copper oxidases. Biochim Biophys Acta. 2000;1459(2-3):514–520. doi: 10.1016/s0005-2728(00)00191-2. [DOI] [PubMed] [Google Scholar]
- 10.Wikström M, Verkhovsky MI, Hummer G. Water-gated mechanism of proton translocation by cytochrome c oxidase. Biochim Biophys Acta. 2003;1604(2):61–65. doi: 10.1016/s0005-2728(03)00041-0. [DOI] [PubMed] [Google Scholar]
- 11.Maréchal A, Rich PR. Water molecule reorganization in cytochrome c oxidase revealed by FTIR spectroscopy. Proc Natl Acad Sci USA. 2011;108(21):8634–8638. doi: 10.1073/pnas.1019419108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belevich I, Verkhovsky MI, Wikström M. Proton-coupled electron transfer drives the proton pump of cytochrome c oxidase. Nature. 2006;440(7085):829–832. doi: 10.1038/nature04619. [DOI] [PubMed] [Google Scholar]
- 13.Belevich I, Bloch DA, Belevich N, Wikström M, Verkhovsky MI. Exploring the proton pump mechanism of cytochrome c oxidase in real time. Proc Natl Acad Sci USA. 2007;104(8):2685–2690. doi: 10.1073/pnas.0608794104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wikström M. Active site intermediates in the reduction of O(2) by cytochrome oxidase, and their derivatives. Biochim Biophys Acta. 2012;1817(4):468–475. doi: 10.1016/j.bbabio.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 15.Rich PR. Towards an understanding of the chemistry of oxygen reduction. Aust J Plant Physiol. 1995;22(3):479–486. [Google Scholar]
- 16.Wikström M, Verkhovsky MI. Mechanism and energetics of proton translocation by the respiratory heme-copper oxidases. Biochim Biophys Acta. 2007;1767(10):1200–1214. doi: 10.1016/j.bbabio.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 17.Siegbahn PEM, Blomberg MR. Energy diagrams and mechanism for proton pumping in cytochrome c oxidase. Biochim Biophys Acta. 2007;1767(9):1143–1156. doi: 10.1016/j.bbabio.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 18.Konstantinov AA. Cytochrome c oxidase: Intermediates of the catalytic cycle and their energy-coupled interconversion. FEBS Lett. 2012;586(5):630–639. doi: 10.1016/j.febslet.2011.08.037. [DOI] [PubMed] [Google Scholar]
- 19.Popović DM, Stuchebrukhov AA. Coupled electron and proton transfer reactions during the O→E transition in bovine cytochrome c oxidase. Biochim Biophys Acta. 2012;1817(4):506–517. doi: 10.1016/j.bbabio.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blomberg MR, Siegbahn PE. The mechanism for proton pumping in cytochrome c oxidase from an electrostatic and quantum chemical perspective. Biochim Biophys Acta. 2012;1817(4):495–505. doi: 10.1016/j.bbabio.2011.09.014. [DOI] [PubMed] [Google Scholar]
- 21.Johansson MP, Kaila VRI, Laakkonen L. Charge parameterization of the metal centers in cytochrome c oxidase. J Comput Chem. 2008;29(5):753–767. doi: 10.1002/jcc.20835. [DOI] [PubMed] [Google Scholar]
- 22.Steiner T. The hydrogen bond in the solid state. Angew Chem Int Ed Engl. 2002;41(1):49–76. doi: 10.1002/1521-3773(20020104)41:1<48::aid-anie48>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 23.Rozas I. On the nature of hydrogen bonds: An overview on computational studies and a word about patterns. Phys Chem Chem Phys. 2007;9(22):2782–2790. doi: 10.1039/b618225a. [DOI] [PubMed] [Google Scholar]
- 24.Yang S, Cui Q. Glu-286 rotation and water wire reorientation are unlikely the gating elements for proton pumping in cytochrome C oxidase. Biophys J. 2011;101(1):61–69. doi: 10.1016/j.bpj.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gorbikova EA, Wikström M, Verkhovsky MI. The protonation state of the cross-linked tyrosine during the catalytic cycle of cytochrome c oxidase. J Biol Chem. 2008;283(50):34907–34912. doi: 10.1074/jbc.M803511200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharma V, Karlin KD, Wikström M. Computational study of the activated O(H) state in the catalytic mechanism of cytochrome c oxidase. Proc Natl Acad Sci USA. 2013;110(42):16844–16849. doi: 10.1073/pnas.1220379110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wikström M, et al. Gating of proton and water transfer in the respiratory enzyme cytochrome c oxidase. Proc Natl Acad Sci USA. 2005;102(30):10478–10481. doi: 10.1073/pnas.0502873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Freier E, Wolf S, Gerwert K. Proton transfer via a transient linear water-molecule chain in a membrane protein. Proc Natl Acad Sci USA. 2011;108(28):11435–11439. doi: 10.1073/pnas.1104735108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jasaitis A, Johansson MP, Wikström M, Vos MH, Verkhovsky MI. Nanosecond electron tunneling between the hemes in cytochrome bo3. Proc Natl Acad Sci USA. 2007;104(52):20811–20814. doi: 10.1073/pnas.0709876105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaila VRI, Sharma V, Wikström M. The identity of the transient proton loading site of the proton-pumping mechanism of cytochrome c oxidase. Biochim Biophys Acta. 2011;1807(1):80–84. doi: 10.1016/j.bbabio.2010.08.014. [DOI] [PubMed] [Google Scholar]
- 31.Kaila VRI, Verkhovsky MI, Hummer G, Wikström M. Glutamic acid 242 is a valve in the proton pump of cytochrome c oxidase. Proc Natl Acad Sci USA. 2008;105(17):6255–6259. doi: 10.1073/pnas.0800770105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woelke AL, et al. Exploring the possible role of Glu286 in CcO by electrostatic energy computations combined with molecular dynamics. J Phys Chem B. 2013;117(41):12432–12441. doi: 10.1021/jp407250d. [DOI] [PubMed] [Google Scholar]
- 33.Kim YC, Hummer G. Proton-pumping mechanism of cytochrome c oxidase: A kinetic master-equation approach. Biochim Biophys Acta. 2012;1817(4):526–536. doi: 10.1016/j.bbabio.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmidt B, McCracken J, Ferguson-Miller S. A discrete water exit pathway in the membrane protein cytochrome c oxidase. Proc Natl Acad Sci USA. 2003;100(26):15539–15542. doi: 10.1073/pnas.2633243100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hammes-Schiffer S, Stuchebrukhov AA. Theory of coupled electron and proton transfer reactions. Chem Rev. 2010;110(12):6939–6960. doi: 10.1021/cr1001436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsukihara T, et al. The low-spin heme of cytochrome c oxidase as the driving element of the proton-pumping process. Proc Natl Acad Sci USA. 2003;100(26):15304–15309. doi: 10.1073/pnas.2635097100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Riistama S, et al. Bound water in the proton translocation mechanism of the haem-copper oxidases. FEBS Lett. 1997;414(2):275–280. doi: 10.1016/s0014-5793(97)01003-x. [DOI] [PubMed] [Google Scholar]
- 38.Tuukkanen A, Kaila VRI, Laakkonen L, Hummer G, Wikström M. Dynamics of the glutamic acid 242 side chain in cytochrome c oxidase. Biochim Biophys Acta. 2007;1767(9):1102–1106. doi: 10.1016/j.bbabio.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 39.Tuukkanen A, Verkhovsky MI, Laakkonen L, Wikström M. The K-pathway revisited: A computational study on cytochrome c oxidase. Biochim Biophys Acta. 2006;1757(9-10):1117–1121. doi: 10.1016/j.bbabio.2006.05.041. [DOI] [PubMed] [Google Scholar]
- 40.MacKerell AD, Jr, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102(18):3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 41.Postila PA, et al. Key role of water in proton transfer at the Qo-site of the cytochrome bc1 complex predicted by atomistic molecular dynamics simulations. Biochim Biophys Acta. 2013;1827(6):761–768. doi: 10.1016/j.bbabio.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 42.Klauda JB, et al. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J Phys Chem B. 2010;114(23):7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Phillips JC, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26(16):1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grudinin S, Büldt G, Gordeliy V, Baumgaertner A. Water molecules and hydrogen-bonded networks in bacteriorhodopsin—molecular dynamics simulations of the ground state and the M-intermediate. Biophys J. 2005;88(5):3252–3261. doi: 10.1529/biophysj.104.047993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 46.Sharma V, Wikström M, Kaila VRI. Dynamic water networks in cytochrome cbb3 oxidase. Biochim Biophys Acta. 2012;1817(5):726–734. doi: 10.1016/j.bbabio.2011.09.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.