

Abstract
Recent advances in genome engineering technologies based on the CRISPR-associated RNA-guided endonuclease Cas9 are enabling the systematic interrogation of mammalian genome function. Analogous to the search function in modern word processors, Cas9 can be guided to specific locations within complex genomes by a short RNA search string. Using this system, DNA sequences within the endogenous genome and their functional outputs are now easily edited or modulated in virtually any organism of choice. Cas9-mediated genetic perturbation is simple and scalable, empowering researchers to elucidate the functional organization of the genome at the systems level and establish causal linkages between genetic variations and biological phenotypes. In this Review, we describe the development and applications of Cas9 for a variety of research or translational applications while highlighting challenges as well as future directions. Derived from a remarkable microbial defense system, Cas9 is driving innovative applications from basic biology to biotechnology and medicine.
Introduction
The development of recombinant DNA technology in the 1970s marked the beginning of a new era for biology. For the first time, molecular biologists gained the ability to manipulate DNA molecules, making it possible to study genes and harness them to develop novel medicine and biotechnology. Recent advances in genome engineering technologies are sparking a new revolution in biological research. Rather than studying DNA taken out of the context of the genome, researchers can now directly edit or modulate the function of DNA sequences in their endogenous context in virtually any organism of choice, enabling them to elucidate the functional organization of the genome at the systems level, as well as identify causal genetic variations.
Broadly speaking, genome engineering refers to the process of making targeted modifications to the genome, its contexts (e.g., epigenetic marks), or its outputs (e.g., transcripts). The ability to do so easily and efficiently in eukaryotic and especially mammalian cells holds immense promise to transform basic science, biotechnology, and medicine (Figure 1).
Figure 1. Applications of Genome Engineering.
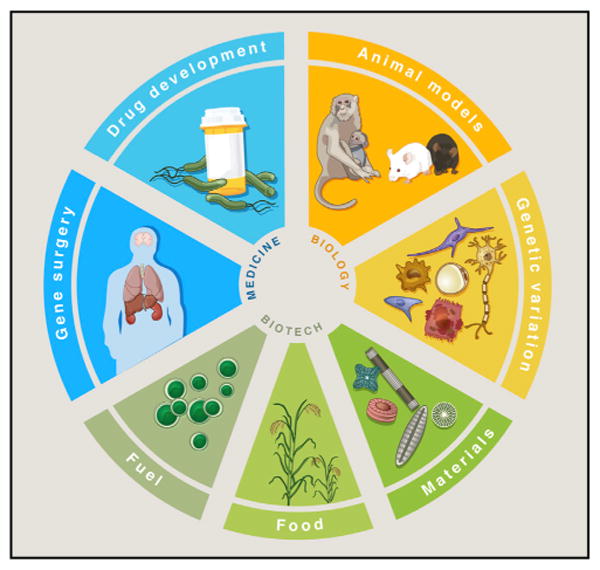
Genetic and epigenetic control of cells with genome engineering technologies is enabling a broad range of applications from basic biology to biotechnology and medicine. (Clockwise from top) Causal genetic mutations or epigenetic variants associated with altered biological function or disease phenotypes can now be rapidly and efficiently recapitulated in animal or cellular models (Animal models, Genetic variation). Manipulating biological circuits could also facilitate the generation of useful synthetic materials, such as algae-derived, silicabased diatoms for oral drug delivery (Materials). Additionally, precise genetic engineering of important agricultural crops could confer resistance to environmental deprivation or pathogenic infection, improving food security while avoiding the introduction of foreign DNA (Food). Sustainable and cost-effective biofuels are attractive sources for renewable energy, which could be achieved by creating efficient metabolic pathways for ethanol production in algae or corn (Fuel). Direct in vivo correction of genetic or epigenetic defects in somatic tissue would be permanent genetic solutions that address the root cause of genetically encoded disorders (Gene surgery). Finally, engineering cells to optimize high yield generation of drug precursors in bacterial factories could significantly reduce the cost and accessibility of useful therapeutics (Drug development).
For life sciences research, technologies that can delete, insert, and modify the DNA sequences of cells or organisms enable dissecting the function of specific genes and regulatory elements. Multiplexed editing could further allow the interrogation of gene or protein networks at a larger scale. Similarly, manipulating transcriptional regulation or chromatin states at particular loci can reveal how genetic material is organized and utilized within a cell, illuminating relationships between the architecture of the genome and its functions. In biotechnology, precise manipulation of genetic building blocks and regulatory machinery also facilitates the reverse engineering or reconstruction of useful biological systems, for example, by enhancing biofuel production pathways in industrially relevant organisms or by creating infection-resistant crops. Additionally, genome engineering is stimulating a new generation of drug development processes and medical therapeutics. Perturbation of multiple genes simultaneously could model the additive effects that underlie complex polygenic disorders, leading to new drug targets, while genome editing could directly correct harmful mutations in the context of human gene therapy (Tebas et al., 2014).
Eukaryotic genomes contain billions of DNA bases and are difficult to manipulate. One of the breakthroughs in genome manipulation has been the development of gene targeting by homologous recombination (HR), which integrates exogenous repair templates that contain sequence homology to the donor site (Figure 2A) (Capecchi, 1989). HR-mediated targeting has facilitated the generation of knockin and knockout animal models via manipulation of germline competent stem cells, dramatically advancing many areas of biological research. However, although HR-mediated gene targeting produces highly precise alterations, the desired recombination events occur extremely infrequently (1 in 106–109 cells) (Capecchi, 1989), presenting enormous challenges for large-scale applications of gene-targeting experiments.
Figure 2. Genome Editing Technologies Exploit Endogenous DNA Repair Machinery.
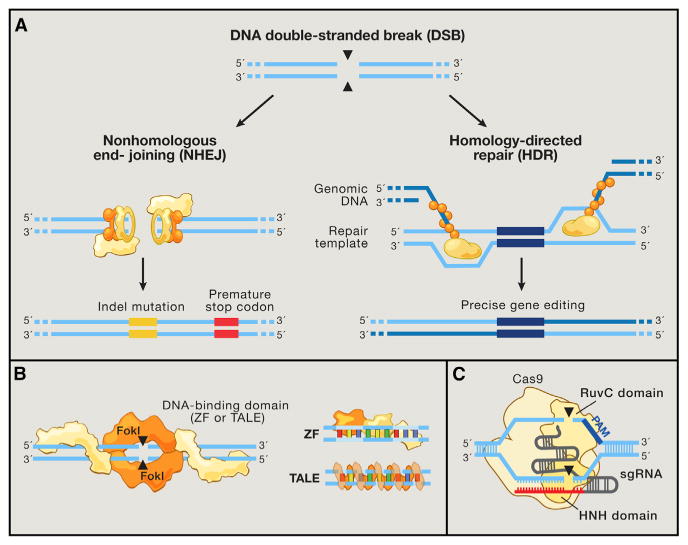
(A) DNA double-strand breaks (DSBs) are typically repaired by nonhomologous end-joining (NHEJ) or homology-directed repair (HDR). In the error-prone NHEJ pathway, Ku heterodimers bind to DSB ends and serve as a molecular scaffold for associated repair proteins. Indels are introduced when the complementary strands undergo end resection and misaligned repair due to micro-homology, eventually leading to frameshift mutations and gene knockout. Alternatively, Rad51 proteins may bind DSB ends during the initial phase of HDR, recruiting accessory factors that direct genomic recombination with homology arms on an exogenous repair template. Bypassing the matching sister chromatid facilitates the introduction of precise gene modifications.
(B) Zinc finger (ZF) proteins and transcription activator-like effectors (TALEs) are naturally occurring DNA-binding domains that can be modularly assembled to target specific sequences. ZF and TALE domains each recognize 3 and 1 bp of DNA, respectively. Such DNA-binding proteins can be fused to the FokI endonuclease to generate programmable site-specific nucleases.
(C) The Cas9 nuclease from the microbial CRISPR adaptive immune system is localized to specific DNA sequences via the guide sequence on its guide RNA (red), directly base-pairing with the DNA target. Binding of a protospacer-adjacent motif (PAM, blue) downstream of the target locus helps to direct Cas9-mediated DSBs.
To overcome these challenges, a series of programmable nuclease-based genome editing technologies have been developed in recent years, enabling targeted and efficient modification of a variety of eukaryotic and particularly mammalian species. Of the current generation of genome editing technologies, the most rapidly developing is the class of RNA-guided endonucleases known as Cas9 from the microbial adaptive immune system CRISPR (clustered regularly interspaced short palindromic repeats), which can be easily targeted to virtually any genomic location of choice by a short RNA guide. Here, we review the development and applications of the CRISPR-associated endonuclease Cas9 as a platform technology for achieving targeted perturbation of endogenous genomic elements and also discuss challenges and future avenues for innovation.
Programmable Nucleases as Tools for Efficient and Precise Genome Editing
A series of studies by Haber and Jasin (Rudin et al., 1989; Plessis et al., 1992; Rouet et al., 1994; Choulika et al., 1995; Bibikova et al., 2001; Bibikova et al., 2003) led to the realization that targeted DNA double-strand breaks (DSBs) could greatly stimulate genome editing through HR-mediated recombination events. Subsequently, Carroll and Chandrasegaran demonstrated the potential of designer nucleases based on zinc finger proteins for efficient, locus-specific HR (Bibikova et al., 2001, 2003). Moreover, it was shown in the absence of an exogenous homology repair template that localized DSBs can induce insertions or deletion mutations (indels) via the error-prone nonhomologous end-joining (NHEJ) repair pathway (Figure 2A) (Bibikova et al., 2002). These early genome editing studies established DSB-induced HR and NHEJ as powerful pathways for the versatile and precise modification of eukaryotic genomes.
To achieve effective genome editing via introduction of site-specific DNA DSBs, four major classes of customizable DNA-binding proteins have been engineered so far: meganucleases derived from microbial mobile genetic elements (Smith et al., 2006), zinc finger (ZF) nucleases based on eukaryotic transcription factors (Urnov et al., 2005; Miller et al., 2007), transcription activator-like effectors (TALEs) from Xanthomonas bacteria (Christian et al., 2010; Miller et al., 2011; Boch et al., 2009; Moscou and Bogdanove, 2009), and most recently the RNA-guided DNA endonuclease Cas9 from the type II bacterial adaptive immune system CRISPR (Cong et al., 2013; Mali et al., 2013a).
Meganuclease, ZF, and TALE proteins all recognize specific DNA sequences through protein-DNA interactions. Although meganucleases integrate its nuclease and DNA-binding domains, ZF and TALE proteins consist of individual modules targeting 3 or 1 nucleotides (nt) of DNA, respectively (Figure 2B). ZFs and TALEs can be assembled in desired combinations and attached to the nuclease domain of FokI to direct nucleolytic activity toward specific genomic loci. Each of these platforms, however, has unique limitations.
Meganucleases have not been widely adopted as a genome engineering platform due to lack of clear correspondence between meganuclease protein residues and their target DNA sequence specificity. ZF domains, on the other hand, exhibit context-dependent binding preference dueto crosstalk between adjacent modules when assembled into a larger array (Maeder et al., 2008). Although multiple strategies have been developed to account for these limitations (Gonzaelz et al., 2010; Sander et al., 2011), assembly of functional ZFPs with the desired DNA binding specificity remains a major challenge that requires an extensive screening process. Similarly, although TALE DNA-binding monomers are for the most part modular, they can still suffer from context-dependent specificity (Juillerat et al., 2014), and their repetitive sequences render construction of novel TALE arrays labor intensive and costly.
Given the challenges associated with engineering of modular DNA-binding proteins, new modes of recognition would significantly simplify the development of custom nucleases. The CRISPR nuclease Cas9 is targeted by a short guide RNA that recognizes the target DNA via Watson-Crick base pairing (Figure 2C). The guide sequence within these CRISPR RNAs typically corresponds to phage sequences, constituting the natural mechanism for CRISPR antiviral defense, but can be easily replaced by a sequence of interest to retarget the Cas9 nuclease. Multiplexed targeting by Cas9 can now be achieved at unprecedented scale by introducing a battery of short guide RNAs rather than a library of large, bulky proteins. The ease of Cas9 targeting, its high efficiency as a site-specific nuclease, and the possibility for highly multiplexed modifications have opened up a broad range of biological applications across basic research to biotechnology and medicine.
The utility of customizable DNA-binding domains extends far beyond genome editing with site-specific endonucleases. Fusing them to modular, sequence-agnostic functional effector domains allows flexible recruitment of desired perturbations, such as transcriptional activation, to a locus of interest (Xu and Bestor, 1997; Beerli et al., 2000a; Konermann et al., 2013; Maeder et al., 2013a; Mendenhall et al., 2013). In fact, any modular enzymatic component can, in principle, be substituted, allowing facile additions to the genome engineering toolbox. Integration of genome- and epigenome-modifying enzymes with inducible protein regulation further allows precise temporal control of dynamic processes (Beerli et al., 2000b; Konermann et al., 2013).
CRISPR-Cas9: From Yogurt to Genome Editing
The recent development of the Cas9 endonuclease for genome editing draws upon more than a decade of basic research into understanding the biological function of the mysterious repetitive elements now known as CRISPR (Figure 3), which are found throughout the bacterial and archaeal diversity. CRISPR loci typically consist of a clustered set of CRISPR-associated (Cas) genes and the signature CRISPR array—a series of repeat sequences (direct repeats) interspaced by variable sequences (spacers) corresponding to sequences within foreign genetic elements (protospacers) (Figure 4). Whereas Cas genes are translated into proteins, most CRISPR arrays are first transcribed as a single RNA before subsequent processing into shorter CRISPR RNAs (crRNAs), which direct the nucleolytic activity of certain Cas enzymes to degrade target nucleic acids.
Figure 3. Key Studies Characterizing and Engineering CRISPR Systems.
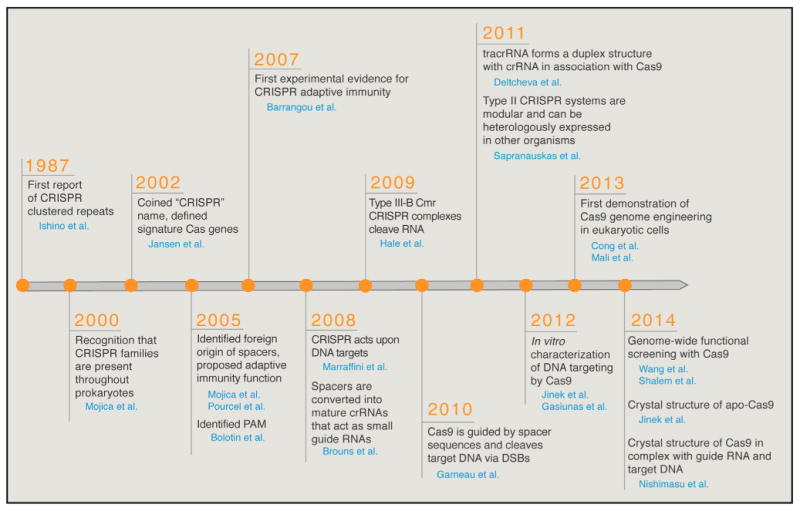
Cas9 has also been referred to as Cas5, Csx12, and Csn1 in literature prior to 2012. For clarity, we exclusively adopt the Cas9 nomenclature throughout this Review. CRISPR, clustered regularly interspaced short palindromic repeats; Cas, CRISPR-associated; crRNA, CRISPR RNA; DSB, double-strand break; tracrRNA, trans-activating CRISPR RNA.
Figure 4. Natural Mechanisms of Microbial CRISPR Systems in Adaptive Immunity.
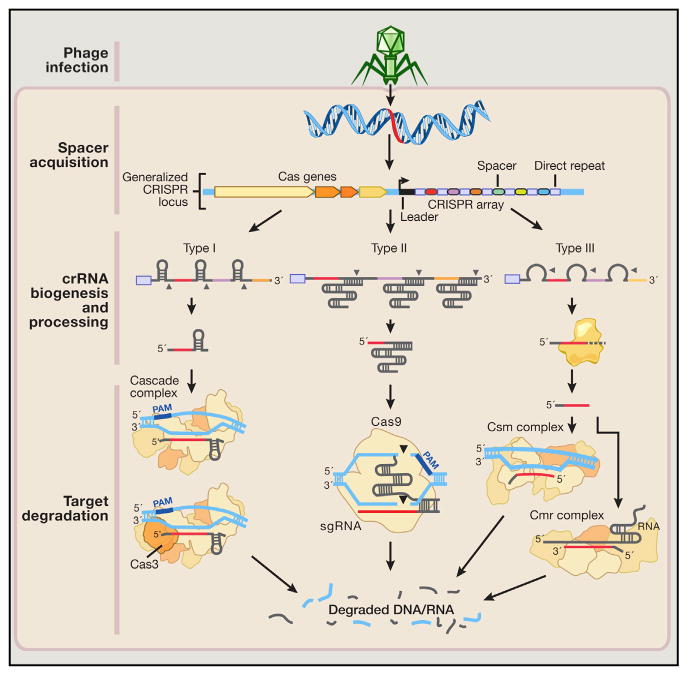
Following invasion of the cell by foreign genetic elements from bacteriophages or plasmids (step 1: phage infection), certain CRISPR-associated (Cas) enzymes acquire spacers from the exogenous protospacer sequences and install them into the CRISPR locus within the prokaryotic genome (step 2: spacer acquisition). These spacers are segregated between direct repeats that allow the CRISPR system to mediate self and nonself recognition. The CRISPR array is a noncoding RNA transcript that is enzymatically maturated through distinct pathways that are unique to each type of CRISPR system (step 3: crRNA biogenesis and processing).
In types I and III CRISPR, the pre-crRNA transcript is cleaved within the repeats by CRISPR-associated ribonucleases, releasing multiple small crRNAs. Type III crRNA intermediates are further processed at the 3′ end by yet-to-be-identified RNases to produce the fully mature transcript. In type II CRISPR, an associated trans-activating CRISPR RNA (tracrRNA) hybridizes with the direct repeats, forming an RNA duplex that is cleaved and processed by endogenous RNase III and other unknown nucleases. Maturated crRNAs from type I and III CRISPR systems are then loaded onto effector protein complexes for target recognition and degradation. In type II systems, crRNA-tracrRNA hybrids complex with Cas9 to mediate interference.
Both type I and III CRISPR systems use multiprotein interference modules to facilitate target recognition. In type I CRISPR, the Cascade complex is loaded with a crRNA molecule, constituting a catalytically inert surveillance complex that recognizes target DNA. The Cas3 nuclease is then recruited to the Cascade-bound R loop, mediating target degradation. In type III CRISPR, crRNAs associate either with Csm or Cmr complexes that bind and cleave DNA and RNA substrates, respectively. In contrast, the type II system requires only the Cas9 nuclease to degrade DNA matching its dual guide RNA consisting of a crRNA-tracrRNA hybrid.
The CRISPR story began in 1987. While studying the iap enzyme involved in isozyme conversion of alkaline phosphatase in E. coli, Nakata and colleagues reported a curious set of 29 nt repeats downstream of the iap gene (Ishino et al., 1987). Unlike most repetitive elements, which typically take the form of tandem repeats like TALE repeat monomers, these 29 nt repeats were interspaced by five intervening 32 nt nonrepetitive sequences. Over the next 10 years, as more microbial genomes were sequenced, additional repeat elements were reported from genomes of different bacterial and archaeal strains. Mojica and colleagues eventually classified interspaced repeat sequences as a unique family of clustered repeat elements present in >40% of sequenced bacteria and 90% of archaea (Mojica et al., 2000).
These early findings began to stimulate interest in such microbial repeat elements. By 2002, Jansen and Mojica coined the acronym CRISPR to unify the description of microbial genomic loci consisting of an interspaced repeat array (Jansen et al., 2002; Barrangou and van der Oost, 2013). At the same time, several clusters of signature CRISPR-associated (cas) genes were identified to be well conserved and typically adjacent to the repeat elements (Jansen et al., 2002), serving as a basis for the eventual classification of three different types of CRISPR systems (types I–III) (Haft et al., 2005; Makarova et al., 2011b). Types I and III CRISPR loci contain multiple Cas proteins, now known to form complexes with crRNA (CASCADE complex for type I; Cmr or Csm RAMP complexes for type III) to facilitate the recognition and destruction of target nucleic acids (Brouns et al., 2008; Hale et al., 2009) (Figure 4). In contrast, the type II system has a significantly reduced number of Cas proteins. However, despite increasingly detailed mapping and annotation of CRISPR loci across many microbial species, their biological significance remained elusive.
A key turning point came in 2005, when systematic analysis of the spacer sequences separating the individual direct repeats suggested their extrachromosomal and phage-associated origins (Mojica et al., 2005; Pourcel et al., 2005; Bolotin et al., 2005). This insight was tremendously exciting, especially given previous studies showing that CRISPR loci are transcribed (Tang et al., 2002) and that viruses are unable to infect archaeal cells carrying spacers corresponding to their own genomes (Mojica et al., 2005). Together, these findings led to the speculation that CRISPR arrays serve as an immune memory and defense mechanism, and individual spacers facilitate defense against bacteriophage infection by exploiting Watson-Crick base-pairing between nucleic acids (Mojica et al., 2005; Pourcel et al., 2005). Despite these compelling realizations that CRISPR loci might be involved in microbial immunity, the specific mechanism of how the spacers act to mediate viral defense remained a challenging puzzle. Several hypotheses were raised, including thoughts that CRISPR spacers act as small RNA guides to degrade viral transcripts in a RNAi-like mechanism (Makarova et al., 2006) or that CRISPR spacers direct Cas enzymes to cleave viral DNA at spacer-matching regions (Bolotin et al., 2005).
Working with the dairy production bacterial strain Streptococcus thermophilus at the food ingredient company Danisco, Horvath and colleagues uncovered the first experimental evidence for the natural role of a type II CRISPR system as an adaptive immunity system, demonstrating a nucleic-acid-based immune system in which CRISPR spacers dictate target specificity while Cas enzymes control spacer acquisition and phage defense (Barrangou et al., 2007). A rapid series of studies illuminating the mechanisms of CRISPR defense followed shortly and helped to establish the mechanism as well as function of all three types of CRISPR loci inadaptive immunity. By studying the type I CRISPR locus of Escherichia coli, van der Oost and colleagues showed that CRISPR arrays are transcribed and converted into small crRNAs containing individual spacers to guide Cas nuclease activity (Brouns et al., 2008). In the same year, CRISPR-mediated defense by a type III-A CRISPR system from Staphylococcus epidermidis was demonstrated to block plasmid conjugation, establishing the target of Cas enzyme activity as DNA rather than RNA (Marraffini and Sontheimer, 2008), although later investigation of a different type III-B system from Pyrococcus furiosus also revealed crRNA-directed RNA cleavage activity (Hale et al., 2009, 2012).
As the pace of CRISPR research accelerated, researchers quickly unraveled many details of each type of CRISPR system (Figure 4). Building on an earlier speculation that protospacer adjacent motifs (PAMs) may direct the type II Cas9 nuclease to cleave DNA (Bolotin et al., 2005), Moineau and colleagues highlighted the importance of PAM sequences by demonstrating that PAM mutations in phage genomes circumvented CRISPR interference (Deveau et al., 2008). Additionally, for types I and II, the lack of PAM within the direct repeat sequence within the CRISPR array prevents self-targeting by the CRISPR system. In type III systems, however, mismatches between the 5′ end of the crRNA and the DNA target are required for plasmid interference (Marraffini and Sontheimer, 2010).
By 2010, just 3 years after the first experimental evidence for CRISPR in bacterial immunity, the basic function and mechanisms of CRISPR systems were becoming clear. A variety of groups had begun to harness the natural CRISPR system for various biotechnological applications, including the generation of phage-resistant dairy cultures (Quiberoni et al., 2010) and phylogenetic classification of bacterial strains (Horvath et al., 2008, 2009). However, genome editing applications had not yet been explored.
Around this time, two studies characterizing the functional mechanisms of the native type II CRISPR system elucidated the basic components that proved vital for engineering a simple RNA-programmable DNA endonuclease for genome editing. First, Moineau and colleagues used genetic studies in Streptococcus thermophilus to reveal that Cas9 (formerly called Cas5, Csn1, or Csx12) is the only enzyme within the cas gene cluster that mediates target DNA cleavage (Garneau et al., 2010). Next, Charpentier and colleagues revealed a key component in the biogenesis and processing of crRNA in type II CRISPR systems—a noncoding trans-activating crRNA (tracrRNA) that hybridizes with crRNA to facilitate RNA-guided targeting of Cas9 (Deltcheva et al., 2011). This dual RNA hybrid, together with Cas9 and endogenous RNase III, is required for processing the CRISPR array transcript into mature crRNAs (Deltcheva et al., 2011). These two studies suggested that there are at least three components (Cas9, the mature crRNA, and tracrRNA) that are essential for reconstituting the type II CRISPR nuclease system. Given the increasing importance of programmable site-specific nucleases based on ZFs and TALEs for enhancing eukaryotic genome editing, it was tantalizing to think that perhaps Cas9 could be developed into an RNA-guided genome editing system. From this point, the race to harness Cas9 for genome editing was on.
In 2011, Siksnys and colleagues first demonstrated that the type II CRISPR system is transferrable, in that transplantation of the type II CRISPR locus from Streptococcus thermophilus into Escherichia coli is able to reconstitute CRISPR interference in a different bacterial strain (Sapranauskas et al., 2011). By 2012, biochemical characterizations by the groups of Charpentier, Doudna, and Siksnys showed that purified Cas9 from Streptococcus thermophilus or Streptococcus pyogenes can be guided by crRNAs to cleave target DNA in vitro (Jinek et al., 2012; Gasiunas et al., 2012), in agreement with previous bacterial studies (Garneau et al., 2010; Deltcheva et al., 2011; Sapranauskas et al., 2011). Furthermore, a single guide RNA (sgRNA) can be constructed by fusing a crRNA containing the targeting guide sequence to a tracrRNA that facilitates DNA cleavage by Cas9 in vitro (Jinek et al., 2012).
In 2013, a pair of studies simultaneously showed how to successfully engineer type II CRISPR systems from Streptococcus thermophilus (Cong et al., 2013) and Streptococcus pyogenes (Cong et al., 2013; Mali et al., 2013a) to accomplish genome editing in mammalian cells. Heterologous expression of mature crRNA-tracrRNA hybrids (Cong et al., 2013) as well as sgRNAs (Cong et al., 2013; Mali et al., 2013a) directs Cas9 cleavage within the mammalian cellular genome to stimulate NHEJ or HDR-mediated genome editing. Multiple guide RNAs can also be used to target several genes at once. Since these initial studies, Cas9 has been used by thousands of laboratories for genome editing applications in a variety of experimental model systems (Sander and Joung, 2014). The rapid adoption of the Cas9 technology was also greatly accelerated through a combination of open-source distributors such as Addgene, as well as a number of online user forums such as http://www.genome-engineering.org and http://www.egenome.org.
Structural Organization and Domain Architecture of Cas9
The family of Cas9 proteins is characterized by two signature nuclease domains, RuvC and HNH, each named based on homology to known nuclease domain structures (Figure 2C). Though HNH is a single nuclease domain, the full RuvC domain is divided into three subdomains across the linear protein sequence, with RuvC I near the N-terminal region of Cas9 and RuvC II/III flanking the HNH domain near the middle of the protein. Recently, a pair of structural studies shed light on the structural mechanism of RNA-guided DNA cleavage by Cas9.
First, single-particle EM reconstructions of the Streptococcus pyogenes Cas9 (SpCas9) revealed a large structural rearrangement between apo-Cas9 unbound to nucleic acid and Cas9 in complex with crRNA and tracrRNA, forming a central channel to accommodate the RNA-DNA heteroduplex (Jinek et al., 2014). Second, a high-resolution structure of SpCas9 in complex with sgRNA and the complementary strand of target DNA further revealed the domain organization to comprise of an α-helical recognition (REC) lobe and a nuclease (NUC) lobe consisting of the HNH domain, assembled RuvC subdomains, and a PAM-interacting (PI) C-terminal region (Nishimasu et al., 2014) (Figure 5A and Movie S1).
Figure 5. Structural and Metagenomic Diversity of Cas9 Orthologs.
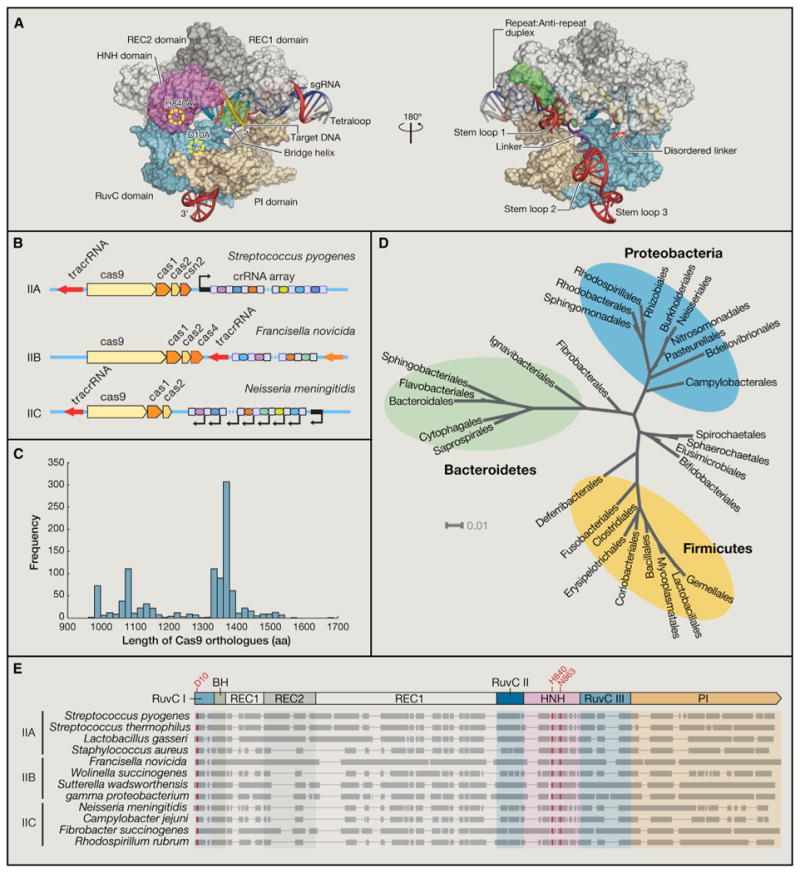
(A) Crystal structure of Streptococcus pyogenes Cas9 in complex with guide RNA and target DNA.
(B) Canonical CRISPR locus organization from type II CRISPR systems, which can be classified into IIA-IIC based on their cas gene clusters. Whereas type IIC CRISPR loci contain the minimal set of cas9, cas1, and cas2, IIA and IIB retain their signature csn2 and cas4 genes, respectively.
(C) Histogram displaying length distribution of known Cas9 orthologs as described in UniProt, HAMAP protein family profile MF_01480.
(D) Phylogenetic tree displaying the microbial origin of Cas9 nucleases from the type II CRISPR immune system. Taxonomic information was derived from greengenes 16S rRNA gene sequence alignment, and the tree was visualized using the Interactive Tree of Life tool (iTol).
(E) Four Cas9 orthologs from families IIA, IIB, and IIC were aligned by ClustalW (BLOSUM). Domain alignment is based on the Streptococcus pyogenes Cas9, whereas residues highlighted in red indicate highly conserved catalytic residues within the RuvC I and HNH nuclease domains.
Together, these two studies support the model that SpCas9 unbound to target DNA or guide RNA exhibits an autoinhibited conformation in which the HNH domain active site is blocked by the RuvC domain and is positioned away from the REC lobe (Jinek et al., 2014). Binding of the RNA-DNA heteroduplex would additionally be sterically inhibited by the orientation of the C-terminal domain. Asaresult, apo-Cas9 likely cannot bind nor cleave target DNA. Like many ribonucleoprotein complexes, the guide RNA serves as a scaffold around which Cas9 can fold and organize its various domains (Nishimasu et al., 2014).
The crystal structure of SpCas9 incomplex with an sgRNA and target DNA also revealed how the REC lobe facilitates target binding. An arginine-rich bridge helix (BH) within the REC lobe is responsible for contacting the 3′ 8–12 nt of the RNA-DNA heteroduplex (Nishimasu et al., 2014), which correspond with the seed sequence identified through guide sequence mutation experiments (Jinek et al., 2012; Cong et al., 2013; Fu et al., 2013; Hsu et al., 2013; Pattanayak et al., 2013; Mali et al., 2013b).
The SpCas9 structure also provides a useful scaffold for engineering or refactoring of Cas9 and sgRNA. Because the REC2 domain of SpCas9 is poorly conserved in shorter orthologs, domain recombination or truncation is a promising approach for minimizing Cas9 size. SpCas9 mutants lacking REC2 retain roughly 50% of wild-type cleavage activity, which could be partly attributed to their weaker expression levels (Nishimasu et al., 2014). Introducing combinations of orthologous domain recombination, truncation, and peptide linkers could facilitate the generation of a suite of Cas9 mutant variants optimized for different parameters such as DNA binding, DNA cleavage, or overall protein size.
Metagenomic, Structural, and Functional Diversity of Cas9
Cas9 is exclusively associated with the type II CRISPR locus and serves as the signature type II gene. Based on the diversity of associated Cas genes, type IICRISPR loci are further subdivided into three subtypes (IIA–IIC) (Figure 5B) (Makarova et al., 2011a; Chylinski et al., 2013). Type II CRISPR loci mostly consist of the cas9, cas1, and cas2 genes, as well as a CRISPR array and tracrRNA. Type IIC CRISPR systems contain only this minimal set of cas genes, whereas types IIA and IIB have an additional signature csn2 or cas4 gene, respectively (Chylinski et al., 2013).
Subtype classification of type II CRISPR loci is based on the architecture and organization of each CRISPR locus. For example, type IIA and IIB loci usually consist of four cas genes, whereas type IIC loci only contain three cas genes. However, this classification does not reflect the structural diversity of Cas9 proteins, which exhibit sequence homology and length variability irrespective of the subtype classification of their parental CRISPR locus. Of >1,000 Cas9 nucleases identified from sequence databases (UniProt) based on homology, protein length israther heterogeneous, roughly ranging from 900 to 1600 amino acids (Figure 5C). The length distribution of most Cas9 proteins can be divided into two populations centered around 1,100 and 1,350 amino acids in length. It is worth noting that a third population of large Cas9 proteins belonging to subtype IIA, formerly called Csx12, typically contain around 1500 amino acids.
Despite the apparent diversity of protein length, all Cas9 proteins share similar domain architecture (Makarova et al., 2011a; Chylinski et al., 2013, 2014; Fonfara et al., 2014), consisting of the RuvC and HNH nuclease domains and the REC domain, an α-helix-rich region with an Arg-rich bridge helix. Unlike type I and III CRISPR systems, which are found in both bacteria and archaea, type II CRISPRs have so far only been found in bacterial strains (Chylinski et al., 2013). The majority of Cas9 orthologs in fact belong to the phyla of Bacteroidetes, Proteobacteria, and Firmicutes (Figure 5D).
The length difference among Cas9 proteins largely results from variable conservation of the REC domain (Figure 5E), which associates with the sgRNA and target DNA. For example, the type IIC Actinomyces naeslundii Cas9, which is more compact than its Streptococcus pyogenes ortholog, has a much smaller REC lobe with substantially different orientation (Jinek et al., 2014).
Protospacer Adjacent Motif: Cas9 Target Range and Search Mechanism
A critical feature of the Cas9 system is the protospacer-adjacent motif (PAM), which flanks the 3′ end of the DNA target site (Figure 2C) and dictates the DNA target search mechanism of Cas9. In addition to facilitating self versus non-self discrimination by Cas9 (Shah et al., 2013), because direct repeats do not contain PAM sites, biochemical and structural characterization of SpCas9 suggested that PAM recognition is involved in triggering the transition between Cas9 target binding and cleavage conformations (Sternberg et al., 2014; Jinek et al., 2014; Nishimasu et al., 2014).
Single-molecule imaging indicated that Cas9-crRNA-tracrRNA complexes first associate with PAM sequences throughout the genome, allowing Cas9 to initiate DNA strand separation via unknown mechanisms (Sternberg et al., 2014). DNA competitor cleavage assays additionally suggested that formation of the RNA-DNA heteroduplex is initiated at the PAM site before proceeding PAM distally by interrogating the target site upstream of the PAM for guide sequence complementarity (Sternberg et al., 2014). Binding of the PAM and a matching target then triggers Cas9 nuclease activity by activating the HNH and RuvC domains, supported by the observation of HNH domain flexibility within the Cas9-sgRNA-DNA ternary complex (Nishimasu et al., 2014).
The complexity of the PAM sequences also determines the overall DNA targeting space of Cas9. For example, the 5′-NGG of SpCas9 allows it to target, on average, every 8 bp within the human genome (Cong et al., 2013; Hsu et al., 2013). Additionally, SpCas9 can target sites flanked by 5′-NAG PAMs (Jiang et al., 2013; Hsu et al., 2013), albeit at a lower efficiency, further expanding its editing versatility. The PAM is specific to each Cas9 ortholog, even within the same species, such as 5′-NNA GAAW for Streptococcus thermophilus CRISPR1 (Deveau et al., 2008) and 5′-NGGNG for Streptococcus thermophilus CRISPR3 (Horvath et al., 2008). Another Cas9 from Neisseria meningitidis with a 5′-NNNNGATT PAM requirement (Zhang et al., 2013) was recently applied in human pluripotent stem cells (Hou et al., 2013).
Computational (Chylinski et al., 2013, 2014; Fonfara et al., 2014) or metagenomic analysis of bacteria and archaea containing CRISPR loci could lead to the discovery of Cas9 nucleases with additional PAMs to expand the targeting range of the Cas9 toolkit. Delivery of multiple Cas9 proteins with different PAM requirements facilitates orthogonal genome engineering, in which independent but simultaneous functions are applied at different loci within the same cell or cell population. NmCas9 and SpCas9, for example, can be employed for independent transcriptional repression and nuclease activity (Esvelt et al., 2013).
PAM specificity can also be modified. For instance, orthologous replacement of the PAM-interacting (PI) domain from the Streptococcus thermophilus CRISPR3 Cas9 with the corresponding domain from Streptococcus pyogenes Cas9 successfully altered PAM recognition from 5′-NGGNG to 5′-NGG (Nishimasu et al., 2014). PAM engineering strategies could also be exploited to generate short Cas9 orthologs with flexible 5′-NGG or 5′-NG PAM domains.
Genome Editing Using CRISPR-Cas9 in Eukaryotic Cells
To date, the Streptococcus pyogenes Cas9 (SpCas9) has been used broadly to achieve efficient genome editing in a variety of species and cell types, including human cell lines, bacteria, zebrafish, yeast, mouse, fruit fly, roundworm, rat, common crops, pig, and monkey (see Sander and Joung [2014] for a detailed list). SpCas9 is also dramatically expanding the catalog of genetically tractable model organisms, for example, by enabling the introduction of multiplex mutations in cynomolgus monkeys (Niu et al., 2014).
SpCas9 can be targeted either with a pair of crRNA and tracrRNA (Cong et al., 2013) or with a chimeric sgRNA (Cong et al., 2013; Mali et al., 2013a; Cho et al., 2013; Jinek et al., 2013), as the crRNA or sgRNA contains a 20 nt guide sequence that directly matches the target site. The only requirement for the selection of Cas9 target sites is the presence of a protospacer-adjacent motif (PAM) immediately downstream of the target site.
An early discrepancy in the use of SpCas9 editing of the human genome was the drastically higher levels of NHEJ-induced indels given the same target site, when using the engineered dual guide RNA system (Cong et al., 2013) compared to the engineered sgRNA(+48) scaffold, which only contained up to the 48th base of tracrRNA. Although sgRNA(+48) is sufficient for cleaving DNA in vitro (Jinek et al., 2012), extension of the 3′ tracrRNA sequence preserved several hairpin structures (sgRNA(+72) and sgRNA(+84)) that were critical for effective sgRNA-mediated genome editing in vivo (Mali et al., 2013a; Hsu et al., 2013). The additional stem loops enhance the stability of the sgRNA (Hsu et al., 2013) and are important for proper Cas9-sgRNA-DNA ternary complex formation (Nishimasu et al., 2014). These analyses of the sgRNA structure and function indicate that careful sgRNA design is critical for optimal Cas9 activity, especially for testing novel Cas9 candidates derived from metagenomic analysis.
One hallmark of the natural CRISPR-Cas9 system is its inherent ability to efficiently cleave multiple distinct target sequences in parallel (Barrangou et al., 2007; Garneau et al., 2010) by converting a pre-crRNA transcript containing many spacers into individual guide RNAs duplexes (mature crRNA and tracrRNA) through hybridization with tracrRNA (Deltcheva et al., 2011). Harnessing this unique aspect of CRISPR interference would enable highly scalable multiplex perturbations. Indeed, coexpression of a CRISPR array containing spacers targeting different genes (Cong et al., 2013) or a battery of several sgRNAs (Mali et al., 2013a; Wang et al., 2013) together with SpCas9 has led to efficient multiplex editing in mammalian cells. Surprisingly, CRISPR arrays containing direct repeats interspaced by designer spacers were processed into mature guide RNAs without the introduction of bacterial RNase III. Because RNase III is required for crRNA maturation in prokaryotic cells (Deltcheva et al., 2011), it is likely that endogenous mammalian RNases play compensatory roles (Cong et al., 2013).
Specificity of Cas9 Nucleases
Because genome editing leads to permanent modifications within the genome, the targeting specificity of Cas9 nucleases is of particular concern, especially for clinical applications and gene therapy. A combination of in vitro and in vivo assays has been typically used to characterize the specificity of ZFNs and TALENs (Gabriel et al., 2011), but systematic analysis has remained challenging due to difficulties in synthesizing large libraries of proteins with varying sequence specificity. However, Cas9 target recognition is dictated by the Watson-Crick base-pairing interactions of an RNA guide with its DNA target, enabling experimentally tractable and systematic evaluation of the effect of guide RNA-target DNA mismatches on Cas9 activity.
Streptococcus pyogenes Cas9 specificity has been extensively characterized by multiple groups using mismatched guide RNA libraries, in vitro selection, and reporter assays (Fu et al., 2013; Hsu et al., 2013; Mali et al., 2013b; Pattanayak et al., 2013). In contrast to previous studies that suggested a seed sequence model for Cas9 specificity, wherein the first 8–12 PAM-proximal guide sequence bases determine specificity (Jinek et al., 2012; Cong et al., 2013), these studies collectively demonstrate that Cas9 tolerates mismatches throughout the guide sequence in a manner that is sensitive to the number, position, and distribution of the mismatches (Fu et al., 2013; Hsu et al., 2013; Mali et al., 2013b; Pattanayak et al., 2013). Although the PAM-distal bases of the guide sequence are less important for specificity, meaning that mismatches at those positions often do not abolish Cas9 activity, all positions within the guide contribute to overall specificity. Importantly, off-target sites followed by the 5′-NAG PAM can also lead to off-target cleavage, demonstrating the importance of considering both NGG and NAG PAMs in off-target analysis.
Interestingly, Cas9 requires extensive homology between the guide RNA and target DNA in order to cleave but can remain semi-transiently bound with only a short stretch of complementary sequence between the guide RNA and target DNA. These observations suggest that Cas9 has many off-target binding sites but cleaves only a small fraction of them (Wu et al., 2014). Thus, concerns about off-target activity could vary widely given a desired application that exploits Cas9 for its DNA binding or cleavage capabilities.
Enzymatic concentration is also an important factor in determining Cas9 off-target mutagenesis. This is particularly important because Cas9 can tolerate even five mismatches within the target site (Fu et al., 2013). Mismatches appear to be better tolerated when Cas9 is present at high concentrations (Hsu et al., 2013; Pattanayak et al., 2013), leading to higher off-target activity; decreasing Cas9 concentration significantly improves the on- to off-target ratio at the expense of the efficiency of on-target cleavage (Hsu et al., 2013). The duration of Cas9 expression is likely an additional factor that tunes off-target activity, though its contributions remain to be carefully investigated.
While potential off-target sites have typically been computationally determined by searching for genomic sequences with high sequence similarity to the desired target locus, wholegenome sequencing or other unbiased ways of labeling DNA DSBs genome-wide may illuminate off-target sites that are not predictable by first-order sequence comparison. Unbiased genome-wide characterizations have been previously used to characterize ZFN off-target mutagenesis (Gabriel et al., 2011) and could easily be adapted for Cas9 nuclease activity. Such data, perhaps in combination with thermodynamic characterization of guide RNA and target DNA hybridization, will likely provide a quantitative framework for assessing and predicting the off-target activity of Cas9. Multiple groups now provide Cas9 target selection tools (e.g., http://tools.genome-engineering. org, http://zifit.partners.org, and http://www.e-crisp.org).
Improving Cas9 Target Recognition Fidelity
Cas9 nucleases cleave DNA through the activity of their RuvC and HNH nuclease domains, each of which nicks a strand of DNA to generate a blunt-ended DSB (Figure 2C). SpCas9 can be converted into a DNA “nickase” that creates a single-stranded break (SSB) by catalytically inactivating the RuvC or HNH nuclease domains (Gasiunas et al., 2012; Jinek et al., 2012; Sapranauskas et al., 2011) via point mutations (Figure 6A). Because DNA single-strand breaks are repaired via the high-fidelity base excision repair (BER) pathway (Dianov and Hübscher, 2013), Cas9 nickases can be exploited for more specific NHEJ as well as HR.
Figure 6. Applications of Cas9 as a Genome Engineering Platform.
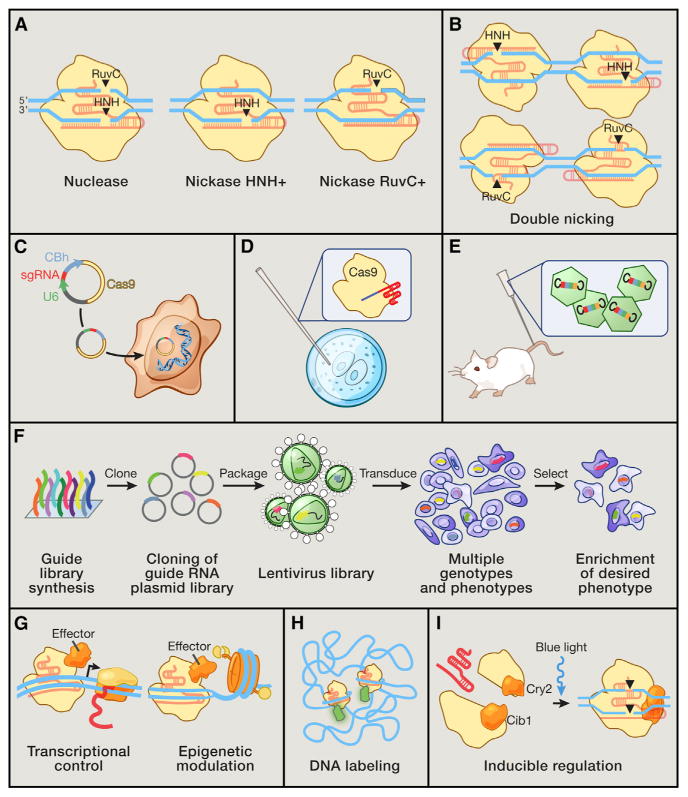
(A) The Cas9 nuclease cleaves DNA via its RuvC and HNH nuclease domains, each ofwhich nicks a DNA strand to generate blunt-end DSBs. Either catalytic domain can be inactivated to generate nickase mutants that cause single-strand DNA breaks.
(B) Two Cas9 nickase complexes with appropriately spaced target sites can mimic targeted DSBs via cooperative nicks, doubling the length of target recognition without sacrificing cleavage efficiency.
(C) Expression plasmids encoding the Cas9 gene and a short sgRNA cassette driven by the U6 RNA polymerase III promoter can be directly transfected into cell lines of interest.
(D) Purified Cas9 protein and in vitro transcribed sgRNA can be microinjected into fertilized zygotes for rapid generation of transgenic animal models.
(E) For somatic genetic modification, high-titer viral vectors encoding CRISPR reagents can be transduced into tissues or cells of interest.
(F) Genome-scale functional screening can be facilitated by mass synthesis and delivery of guide RNA libraries.
(G) Catalytically dead Cas9 (dCas9) can be converted into a general DNA-binding domain and fused to functional effectors such as transcriptional activators or epigenetic enzymes. The modularity of targeting and flexible choice of functional domains enable rapid expansion of the Cas9 toolbox.
(H) Cas9 coupled to fluorescent reporters facilitates live imaging of DNA loci for illuminating the dynamics of genome architecture.
(I) Reconstituting split fragments of Cas9 via chemical or optical induction of heterodimer domains, such as the cib1/cry2 system from Arabidopsis, confers temporal control of dynamic cellular processes.
To improve on-target DSB specificity, a double-nicking approach analogous to dimeric ZFNs or TALENs can be used to increase the overall number of bases that are specifically recognized in the target DNA. Using pairs of guide RNAs (Mali et al., 2013b; Ran et al.,2013)and an SpCas9 HNH+/RuvC− nickase mutant (D10A), properly spaced cooperative nicks can mimic DSBs and mediate efficient indel formation (Figure 6B). Because off-target nick sites are precisely repaired, this multiplexed nicking strategy can improve specificity by up to 1,500× relative to the wild-type Cas9 (Ran et al., 2013).
Because Cas9 nuclease or multiplex nicking activity both stimulate NHEJ, a population of cells cotargeted with a homology donor would eventually possess a mix of indel mutants and donor integrants. Single DNA nicks, however, are also able to mediate donor recombination, albeit at a lower level than with DSBs (Hsu et al., 2013). Cas9 nickases with single sgRNAs can thus be used to mediate HR rather than NHEJ. Furthermore, off-target integration is highly unlikely due to long homology arms flanking the donor cassette.
In addition to the double-nicking strategy, sgRNAs truncated by 2 or 3 nt have been reported to significantly increase targeting specificity of SpCas9, potentially due to greater mismatch sensitivity (Fu et al., 2014). These truncated sgRNAs can be combined with multiplex nicking to further reduce off-target mutagenesis (Fu et al., 2014). Future structure-function analyses and Cas9 and protein engineering via rational design or directed evolution may lead to further improvements in Cas9 specificity.
Applications of Cas9 in Research, Medicine, and Biotechnology
Cas9 can be used to facilitate a wide variety of targeted genome engineering applications. The wild-type Cas9 nuclease has enabled efficient and targeted genome modification in many species that have been intractable using traditional genetic manipulation techniques. The ease of retargeting Cas9 by simply designing a short RNA sequence also enables large-scale unbiased genome perturbation experiments to probe gene function or elucidate causal genetic variants. In addition to facilitating co-valent genome modifications, the wild-type Cas9 nuclease can also be converted into a generic RNA-guided homing device (dCas9) by inactivating the catalytic domains. The use of effector fusions can greatly expand the repertoire of genome engineering modalities achievable using Cas9. For example, a variety of proteins or RNAs can be tethered to Cas9 or sgRNA to alter transcription states of specific genomic loci, monitor chromatin states, or even rearrange the three-dimensional organization of the genome.
Rapid Generation of Cellular and Animal Models
Cas9-mediated genome editing has enabled accelerated generation of transgenic models and expands biological research beyond traditional, genetically tractable animal model organisms (Sander and Joung, 2014). By recapitulating genetic mutations found in patient populations, CRISPR-based editing could be used to rapidly model the causal roles of specific genetic variations instead of relying on disease models that only phenocopy a particular disorder. This could be applied to develop novel transgenic animal models (Wang et al., 2013; Niu et al., 2014), to engineer isogenic ES and iPS cell disease models with specific mutations introduced or corrected, respectively, or in vivo and ex vivo gene correction (Schwank et al., 2013; Wu et al., 2013).
For generation of cellular models, Cas9 can be easily introduced into the target cells using transient transfection of plasmids carrying Cas9 and the appropriately designed sgRNA (Figure 6C). Additionally, the multiplexing capabilities of Cas9 offer a promising approach for studying common human diseases—such as diabetes, heart disease, schizophrenia, and autism—that are typically polygenic. Large-scale genome-wide association studies (GWAS), for example, have identified haplotypes that show strong association with disease risk. However, it is often difficult to determine which of several genetic variants in tight linkage disequilibrium with the haplotype or which of several genes in the region are responsible for the phenotype. Using Cas9, one could study the effect of each individual variant or test the effect of manipulating each individual gene on an isogenic background by editing stem cells and differentiating them into cell types of interest.
For generation of transgenic animal models, Cas9 protein and transcribed sgRNA can be directly injected into fertilized zygotes to achieve heritable gene modification at one or multiple alleles in models such as rodents and monkeys (Wang et al., 2013; Li et al., 2013; Yang et al., 2013; Niu et al., 2014) (Figure 6D). By bypassing the typical ES cell targeting stage in generating transgenic lines, the generation time for mutant mice and rats can be reduced from more than a year to only several weeks. Such advances will facilitate cost-effective and large-scale in vivo mutagenesis studies in rodent models and can be combined with highly specific editing (Fu et al., 2014; Ran et al., 2013) to avoid confounding off-target mutagenesis. Successful multiplex targeting in cynomolgus monkey models was also recently reported (Niu et al., 2014), suggesting the potential for establishing more accurate modeling of complex human diseases such as neuropsychiatric disorders using primate models. Additionally, Cas9 could be harnessed for direct modification of somatic tissue, obviating the need for embryonic manipulation (Figure 6E) as well as enabling therapeutic use for gene therapy.
One outstanding challenge with transgenic animal models generated via zygotic injection of CRISPR reagents is genetic mosaicism, partly due to a slow rate of nuclease-induced mutagenesis. Studies to date have typically relied on the injection of Cas9 mRNA into zygotes (fertilized embryos at the single-cell stage). However, because transcription and translation activity is suppressed in the mouse zygote, Cas9 mRNA translation into active enzymatic form is likely delayed until after the first cell division (Oh et al., 2000). Because NHEJ-mediated repair is thought to introduce indels of random length, this translation delay likely plays a major role in contributing to genetic mosaicism in CRISPR-modified mice. To overcome this limitation, Cas9 protein and sgRNA could be directly injected into single-cell fertilized embryos. The high rate of nonmutagenic repair by the NHEJ process may additionally contribute to undesired mosaicism because introducing indels that mutate the Cas9 recognition site would then have to compete with zygotic division rates. To increase the mutagenic activity of NHEJ, a pair of sgRNAs flanking a small fragment of the target gene may be used to increase the probability of gene disruption.
Functional Genomic Screens
The efficiency of genome editing with Cas9 makes it possible to alter many targets in parallel, thereby enabling unbiased genome-wide functional screens to identify genes that play an important role in a phenotype of interest. Lentiviral delivery of sgRNAs directed against all genes (either together with Cas9 or to cells already expressing Cas9) can be used to perturb thousands of genomic elements in parallel. Recent papers have demonstrated the ability to perform robust negative and positive selection screens in human cells (Wang et al., 2014; Shalem et al., 2014) by introducing loss-of-function mutations into early, constitutive coding exons of a different gene in each cell (Figure 6F). Genome-wide loss-of-function screens have previously used RNAi, but this approach leads to only partial knockdown, has extensive off-target effects, and is limited to transcribed (and usually protein-coding) genes. By contrast, Cas9-mediated pooled sgRNA screens have been shown to provide increased screening sensitivity as well as consistency and can be designed to target nearly any DNA sequence (Shalem et al., 2014).
Future applications of single sgRNA libraries may also enable the perturbation of noncoding genetic elements, while multiplex sgRNA delivery may be used to dissect the function of large genomic regions through tiled microdeletions. For example, systematic targeting of gene regulatory regions could facilitate the discovery of distant enhancers, general promoter architectures, and any additional regulatory elements that have an effect on protein levels. An additional application could be to dissect large, uncharacterized genomic regions that are implicated in sequencing studies or GWAS.
Tethering dCas9 to different effector domains may also facilitate genomic screens beyond loss-of-function phenotypes. dCas9 fused to epigenetic modifiers, for instance, could be used to study the effects of methylation or certain chromatin states on cellular differentiation or disease pathologies, whereas transcriptional activators allow screening for gain-of-function phenotypes. Using truncated sgRNAs or building redundancy with several sgRNAs targeting each locus would be important design principles for filtering out false positive signals and improving the interpretability of screening data.
Transcriptional Modulation
dCas9 binding alone to DNA elements may repress transcription by sterically hindering RNA polymerase machinery (Qi et al., 2013), likely by stalling transcriptional elongation. This CRISPR-based interference, or CRISPRi, works efficiently in prokaryotic genomes but is less effective in eukaryotic cells (Gilbert et al., 2013). The repressive function of CRISPRi can be enhanced by tethering dCas9 to transcriptional repressor domains such as KRAB or SID effectors, which promote epigenetic silencing (Gilbert et al., 2013; Konermann et al., 2013). However, dCas9-mediated transcriptional repression needs to be further improved—in the current generation of dCas9-based eukaryotic transcription repressors, even the addition of helper functional domains results in only partial transcriptional knockdown (Gilbert et al., 2013; Konermann et al., 2013).
Cas9 can also be converted into a synthetic transcriptional activator by fusing it to VP16/VP64 or p65 activation domains (Figure 6G). In general, targeting Cas9 activators with a single sgRNA to a particular endogenous gene promoter leads to only modest transcriptional upregulation (Konermann et al., 2013; Maeder et al., 2013b; Perez-Pinera et al., 2013; Mali et al., 2013b). By tiling a promoter with multiple sgRNAs, several groups have reported strong synergistic effects with nonlinear increases in activation (Perez-Pinera et al., 2013; Maeder et al., 2013b; Mali et al., 2013b). Although the requirement for multiple sgRNAs to achieve efficient transcription activation is potentially advantageous for increased specificity, screening applications employing libraries of sgRNAs will require highly efficient and specific transcriptional control using individual guide RNAs.
Epigenetic Control
Complex genome functions are defined by the highly dynamic landscape of epigenetic states. Epigenetic modifications that tune histones are thus crucial for transcriptional regulation and play important roles in a variety of biological functions. These marks, such as DNA methylation or histone acetylation, are established and maintained in mammalian cells by a variety of enzymes that are recruited to specific genomic loci either directly or indirectly through scaffolding proteins.
Previously, zinc finger proteins and TAL effectors have been used in a small number of proof-of-concept studies to achieve locus-specific targeting of epigenetic modifying enzymes (Beerli et al., 2000a; Konermann et al., 2013; Maeder et al., 2013a; Mendenhall et al., 2013). Cas9 epigenetic effectors (epiCas9s) that can artificially install or remove specific epigenetic marks at specific loci would serve as a more flexible platform to probe the causal effects of epigenetic modifications in shaping the regulatory networks of the genome (Figure 6G). Of course, the potential for off-target activity or crosstalk between effector domains and endogenous epigenetic complexes would need to be carefully characterized. One solution could be to harness prokaryotic epigenetic enzymes to develop orthogonal epigenetic regulatory systems that minimize crosstalk with endogenous proteins.
Live Imaging of the Cellular Genome
The spatial organization of functional and structural elements within the cell contribute to the functional output of genomes, which can be amplified or suppressed dynamically. However, the way that genomes are modified and how their structural organization in vivo modulates functional output remain unclear. Genomic loci located megabases apart or on entirely different chromosomes could be brought into close proximity given appropriate chromosomal organization, thus mediating long-range trans interactions.
Studying the interactions of specific genes given changing chromatin states would require a robust method to visualize DNA in living cells. Traditional techniques for labeling DNA, such as fluorescence in situ hybridization (FISH), require sample fixation and are thus unable to capture live processes. Fluorescently tagged Cas9 labeling of specific DNA loci was recently developed as a powerful live-cell-imaging alternative to DNA-FISH (Chen et al., 2013) (Figure 6H). Advances in orthogonal Cas9 proteins or modified sgRNAs will build out multi-color and multi-locus capabilities to enhance the utility of CRISPR-based imaging for studying complex chromosomal architecture and nuclear organization.
Inducible Regulation of Cas9 Activity
By exploiting the bilobed structure of Cas9, it may be possible to split the protein into two units and control their reassembly via small-molecule or light-inducible heterodimeric domains (Figure 6I). Small-molecule induction would facilitate systemic control of Cas9 in patients or animal models, whereas optical regulation enables more spatially precise perturbation. For example, the light-inducible dimerization domains CIB1 and CRY2 or chemically inducible analogs ABI and PYL, which have been successfully used to construct inducible TALEs (Konermann et al., 2013), may be adapted to engineer inducible Cas9 systems.
Future Development of Cas9-Based Genome Engineering Technologies
Unbiased Analysis of Cas9 Binding and Cleavage
Despite the rapid adoption of Cas9 as a platform technology for genetic and epigenetic perturbation and significant progress in understanding and improving Cas9 specificity, its on- and off-target DNA binding and cleavage profiles still need to be thor oughly evaluated. Studies to date characterizing Cas9 off-target activity have relied on in silico computational prediction or in vitro selection. As a result, they have been unable to account for the likelihood of Cas9 activity that is unpredictable by sequence ho-mology to the sgRNA guide sequence.
Because the off-target activity of dCas9 binding for effector domain localization may be much more extensive than Cas9-mediated genome editing, unbiased profiling methods are needed to refine our understanding of “true positive” Cas9 off-target activity that actually leads to undesired functional outcomes. Cas9-based chromatin immunoprecipitation sequencing (ChIP-seq) analysis at multiple target sites could be a high-throughput solution for understanding binding degen eracy (Wu et al., 2014), whereas techniques for detecting and labeling double-strand breaks (Crosetto et al., 2013) will help to achieve a comprehensive map of Cas9-induced off-target indels.
These data together will help to generate predictive models for minimizing off-target activity in gene therapeutics or other applications requiring high levels of precision. Understanding Cas9 binding and cleavage in the context of chromatin accessibility and epigenetic states will also inform better computational evaluation of guide RNA specificity. For example, particular sgRNAs could be evaluated based on the genomic nature of its off-target sites, which would vary by guide sequence. Degenerate targeting of transcriptionally silent genes for a cell type or tissue of interest would likely be preferred to off-target sites in the coding region of essential housekeeping genes.
Although it is still unclear how Cas9 is affected by chromatin accessibility and heterochromatin versus euchromatin, dCas9 transcriptional activators can upregulate transcription at sites lacking DNase I hypersensitivity sites, indicating successful binding to inaccessible chromatin (Perez-Pinera et al., 2013) (Figure 6G). CpG methylation does not appear to affect DNA cleavage in vitro, and Cas9 could introduce indels at a highly methylated promoter in vivo (Hsu et al., 2013). It will be important to evaluate Cas9 binding and cleavage of genomic loci in relevant primary cells with different chromatin states, ideally in post-mitotic cells in which genomic architecture is stably defined.
Overall, these efforts aimed at improving our understanding of Cas9 binding and cleavage specificity will complement existing methods (Mali et al., 2013b; Ran et al., 2013; Fu et al., 2014) as well as future protein engineering and metagenomic mining efforts to improve Cas9 specificity and the selection of guide RNA target sites.
Development of Versatile Delivery and Expression Systems for Applications of Cas9
Viral vectors such as adeno-associated virus (AAV) or lentivirus are commonly used for delivering genes of interest in vivo or into cell types resistant to common transfection methods, such as immune cells. AAV vectors have been commonly used for attractive candidates for efficient gene delivery in vivo because of their low immunogenic potential, reduced oncogenic risk from host-genome integration, and well-characterized serotype specificity (Figure 6E). However, the most commonly used Cas9 nuclease-encoding gene from Streptococcus pyogenes is >4 kb in length, which is difficult to transduce using AAV due to its 4.7 kb packaging capacity. Non-viral approaches for introducing CRISPR reagents in vivo present a fertile ground for developing novel delivery strategies, from liposomes and aptamers to cell-penetrating peptides and the molecular Trojan horse (Niewoehner et al., 2014).
However, viral approaches are still highly desirable due to their low immunogenicity and wide array of characterized tropisms. The size constraints of viral vectors can be sidestepped by using significantly smaller Cas9 orthologs derived from metagenomic discovery, several of which have already been characterized and validated in human cells (Cong et al., 2013; Hou et al., 2013; Esvelt et al., 2013). Interestingly, short Cas9 variants reported to date recognize much longer PAM sequences than SpCas9 (5′-NNAGAAW from Streptococcus thermophilus CRISPR1 or 5′-NNNNGATT from Neisseria meningitidis) (Zhang et al., 2013; Garneau et al., 2010), whereas some longer orthologs have more relaxed PAMs (5′-NG from Francisella novicida) (Fonfara et al., 2014). Although the effect of PAM restriction on DNA targeting specificity remains to be investigated, the more limited overall targeting range of short Cas9 variants may be partially compensated for by decreasing the number of potential off-target substrates genome-wide.
Moving beyond Endogenous Cellular Repair
The current generation of genome editing technologies depends on the endogenous DNA repair machinery to introduce loss-of-function mutations or precise modifications (Figure 2A). Although Cas9 can be used to generate indel mutations via NHEJ with high efficiency, the absolute rate of HDR remains relatively low. Although it is sufficient for the generation of cell lines, especially when paired with drug selection or FACS enrichment, poor rates of recombination greatly limit the practical utility of Cas9-mediated targeted gene insertion in fertilized zygotes or somatic tissue. Homologous recombination proteins are also mainly expressed in the G2 phase of the cell cycle, making HDR-based gene editing difficult in postmitotic cells such as neurons or cardiac myocytes. As a result, methods for stimulating HDR-based repair or alternative strategies for efficient gene insertion are urgently needed. For instance, the highly efficient DNA damage repair system in Deinococcus radiodurans (Zahradka et al., 2006) may be exploited to enable efficient genome editing in mitotic as well as postmitotic cells.
Furthermore, the majority of CRISPR-based technology development has focused on the signature Cas9 nuclease from type II CRISPR systems. However, there remains a wide diversity of CRISPR types and functions. Cas RAMP module (Cmr) proteins identified in Pyrococcus furiosus and Sulfolobus solfataricus (Hale et al., 2012) constitute an RNA-targeting CRISPR immune system, forming a complex guided by small CRISPR RNAs that target and cleave complementary RNA instead of DNA. Cmr protein homologs can be found throughout bacteria and archaea, typically relying on a 5′ site tag sequence on the target-matching crRNA for Cmr-directed cleavage.
Unlike RNAi, whichis targeted largely by a 6 nt seed region and to a lesser extent 13 other bases, Cmr crRNAs contain 30–40 nt of target complementarity. Cmr-CRISPR technologies for RNA targeting are thus a promising target for orthogonal engineering and minimal off-target modification. Although the modularity of Cmr systems for RNA-targeting in mammalian cells remains to be investigated, Cmr complexes native to P. furiosus have already been engineered to target novel RNA substrates (Hale et al., 2009, 2012).
Cas9 as a Therapeutic Molecule for Treating Genetic Disorders
Although Cas9 has already been widely used as a research tool, a particularly exciting future direction is the development of Cas9 as a therapeutic technology for treating genetic disorders. For a monogenic recessive disorder due to loss-of-function mutations (such as cystic fibrosis, sickle-cell anemia, or Duchenne muscular dystrophy), Cas9 may be used to correct the causative mutation. This has many advantages over traditional methods of gene augmentation that deliver functional genetic copies via viral vector-mediated overexpression—particularly that the newly functional gene is expressed in its natural context. For dominant-negative disorders in which the affected gene is haplosufficient (such as transthyretin-related hereditary amyloidosis or dominant forms of retinitis pigmentosum), it may also be possible to use NHEJ to inactivate the mutated allele to achieve therapeutic benefit. For allele-specific targeting, one could design guide RNAs capable of distinguishing between single-nucleotide polymorphism (SNP) variations in the target gene, such as when the SNP falls within the PAM sequence.
Some monogenic diseases also result from duplication of genomic sequences. For these diseases, the multiplexing capability of Cas9 may be exploited for deletion of the duplicated elements. For example, trinucleotide repeat disorders could be treated using two simultaneous DSBs to excise the repeat region. The success of this strategy will likely be higher for diseases such as Friedreich's ataxia, in which duplications occur in noncoding regions of the target gene, because NHEJ-mediated repair may lead to imperfect or frameshifted repair junctions.
In addition to repairing mutations underlying inherited disorders, Cas9-mediated genome editing might be used to introduce protective mutations in somatic tissues to combat nongenetic or complex diseases. For example, NHEJ-mediated inactivation of the CCR5 receptor in lymphocytes (Lombardo et al., 2007) may be a viable strategy for circumventing HIV infection, whereas deletion of PCSK9 (Cohenet al., 2005) orangiopoietin (Musunuru et al., 2010) may provide therapeutic effects against statin-resistant hypercholesterolemia or hyperlipidemia. Although these targets may be also addressed using siRNA-mediated protein knockdown, a unique advantage of NHEJ-mediated gene inactivation is the ability to achieve permanent therapeutic benefit without the need for continuing treatment. As with all gene therapies, it will of course be important to establish that each proposed therapeutic use has a favorable benefit-risk ratio.
Cas9 could be used beyond the direct genome modification of somatic tissue, such as for engineering therapeutic cells. Chimeric antigen receptor (CAR) T cells can be modified ex vivo and reinfused into a patient to specifically target certain cancers (Couzin-Frankel, 2013). The ease of design and testing of Cas9 may also facilitate the treatment of highly rare genetic variants through personalized medicine. Supporting these tremendous possibilities are a number of animal model studies as well as clinical trials using programmable nucleases that already provide important insights into the future development of Cas9-based therapeutics.
Recently, hydrodynamic delivery of plasmid DNA encoding Cas9 and sgRNA along with a repair template into the liver of an adult mouse model of tyrosinemia was shown to be able to correct the mutant Fah gene and rescue expression of the wild-type Fah protein in ∼1 out of 250 cells (Yin et al., 2014). In addition, clinical trials successfully used ZF nucleases to combat HIV infection by ex vivo knockout of the CCR5 receptor. In all patients, HIV DNA levels decreased, and in one out of four patients, HIV RNA became undetectable (Tebas et al., 2014). Both of these results demonstrate the promise of programmable nucleases as a new therapeutic platform.
However, numerous challenges still lie ahead. Most importantly, successful clinical translation will depend on appropriate and efficacious delivery systems to target specific disease tissues. To achieve high levels of therapeutic efficacy and simultaneously address a broad spectrum of genetic disorders, homologous recombination efficiency will need to be significantly improved. Although permanent genome modification has advantages over monoclonal antibody or siRNA treatments, which require repeated administration of the therapeutic molecule, the long-term implications remain unclear. As researchers further develop and test Cas9 toward clinical translation, it will be paramount to thoroughly characterize the safety as well as physiological effects of Cas9 using a variety of preclinical models.
Conclusions
The story of how a mysterious prokaryotic viral defense system became one of the most powerful and versatile platforms for engineering biology highlights the importance of basic science research. Just as recombinant DNA technology benefited from basic investigation of the restriction enzymes that are central to warfare between phage and bacteria, the latest generation of Cas9-based genome engineering tools are also based on components from the microbial antiphage defense system. It is highly likely that the future solutions for efficient and precise gene modification will be found in as of yet unexplored corners of the rich biological diversity of nature.
Supplementary Material
Acknowledgments
We gratefully acknowledge Sigrid Knemeyer for help with illustration; Ian Slay-maker for structural guidance; Chengwei Luo for expertise in phylogenetic analysis; Emmanuelle Charpentier, Philippe Horvath, Charles Jennings, Ellen Law, Luciano Marraffini, Francisco Mojica, Hiroshi Nishimasu, Virginijus Siksnys, and Alexandro Trevino for discussion and comments; and the CRISPR community for this beautiful story. P.D.H. is a James Mills Pierce Fellow. This work is supported by the NIMH through a NIH Director's Pioneer Award (DP1-MH100706), the NINDS through a NIH Transformative R01 grant (R01-NS 07312401), NSF, the Keck, McKnight, Damon Runyon, Searle Scholars, Klingenstein, Vallee, Merkin, and Simons Foundations, and Bob Metcalfe. CRISPR reagents are available to the academic community through Addgene, and associated protocols, support forum, and computational tools are available via the Zhang lab website (http://www.genome-engineering.org).
Footnotes
Supplemental Information Supplemental Information includes one movie and can be found with this article at http://dx.doi.org/10.1016/j.cell.2014.05.010.
References
- Barrangou R, van der Oost J. CRISPR-Cas Systems: RNA-Mediated Adaptive Immunity in Bacteria and Archaea. Heidelberg, Germany: Springer; 2013. [Google Scholar]
- Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- Beerli RR, Dreier B, Barbas CF., 3rd Positive and negative regulation of endogenous genes by designed transcription factors. Proc Natl Acad Sci USA. 2000a;97:1495–1500. doi: 10.1073/pnas.040552697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerli RR, Schopfer U, Dreier B, Barbas CF., 3rd Chemically regulated zinc finger transcription factors. J Biol Chem. 2000b;275:32617–32627. doi: 10.1074/jbc.M005108200. [DOI] [PubMed] [Google Scholar]
- Bibikova M, Carroll D, Segal DJ, Trautman JK, Smith J, Kim YG, Chandrasegaran S. Stimulation of homologous recombination through targeted cleavage by chimeric nucleases. Mol Cell Biol. 2001;21:289–297. doi: 10.1128/MCB.21.1.289-297.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibikova M, Golic M, Golic KG, Carroll M. Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics. 2002;161:1169–1175. doi: 10.1093/genetics/161.3.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibikova M, Beumer K, Trautman JK, Carroll D. Enhancing gene targeting with designed zinc finger nucleases. Science. 2003;300:764. doi: 10.1126/science.1079512. [DOI] [PubMed] [Google Scholar]
- Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, Lahaye T, Nickstadt A, Bonas U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326:1509–1512. doi: 10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- Bolotin A, Quinquis B, Sorokin A, Ehrlich SD. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extra-chromosomal origin. Microbiology. 2005;151:2551–2561. doi: 10.1099/mic.0.28048-0. [DOI] [PubMed] [Google Scholar]
- Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, van der Oost J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321:960–964. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capecchi MR. Altering the genome by homologous recombination. Science. 1989;244:1288–1292. doi: 10.1126/science.2660260. [DOI] [PubMed] [Google Scholar]
- Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W, Li GW, Park J, Blackburn EH, Weissman JS, Qi LS, Huang B. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 2013;155:1479–1491. doi: 10.1016/j.cell.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SW, Kim S, Kim JM, Kim JS. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol. 2013;31:230–232. doi: 10.1038/nbt.2507. [DOI] [PubMed] [Google Scholar]
- Choulika A, Perrin A, Dujon B, Nicolas JF. Induction of homologous recombination inmammalian chromosomesby using the I-SceI system of Saccharomyces cerevisiae. Mol Cell Biol. 1995;15:1968–1973. doi: 10.1128/mcb.15.4.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, Bogdanove AJ, Voytas DF. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics. 2010;186:757–761. doi: 10.1534/genetics.110.120717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chylinski K, Le Rhun A, Charpentier E. The tracrRNA and Cas9 families of type II CRISPR-Cas immunity systems. RNA Biol. 2013;10:726–737. doi: 10.4161/rna.24321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chylinski K, Makarova KS, Charpentier E, Koonin EV. Classification and evolution of type II CRISPR-Cas systems. Nucleic Acids Res. 2014 doi: 10.1093/nar/gku241. Published online Apr 11, 2014. http://dx.doi.org/10.1093/nar/gku241. [DOI] [PMC free article] [PubMed]
- Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet. 2005;37:161–165. doi: 10.1038/ng1509. [DOI] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science. 2013;342:1432–1433. doi: 10.1126/science.342.6165.1432. [DOI] [PubMed] [Google Scholar]
- Crosetto N, Mitra A, Silva MJ, Bienko M, Dojer N, Wang Q, Karaca E, Chiarle R, Skrzypczak M, Ginalski K, et al. Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat Methods. 2013;10:361–365. doi: 10.1038/nmeth.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471:602–607. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveau H, Barrangou R, Garneau JE, Labonté J, Fremaux C, Boyaval P, Romero DA, Horvath P, Moineau S. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol. 2008;190:1390–1400. doi: 10.1128/JB.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dianov GL, Hübscher U. Mammalian base excision repair: the forgotten archangel. Nucleic Acids Res. 2013;41:3483–3490. doi: 10.1093/nar/gkt076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esvelt KM, Mali P, Braff JL, Moosburner M, Yaung SJ, Church GM. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat Methods. 2013;10:1116–1121. doi: 10.1038/nmeth.2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonfara I, Le Rhun A, Chylinski K, Makarova KS, Lécrivain AL, Bzdrenga J, Koonin EV, Charpentier E. Phylogeny of Cas9 determines functional exchangeability of dual-RNA and Cas9 among orthologous type II CRISPR-Cas systems. Nucleic Acids Res. 2014;42:2577–2590. doi: 10.1093/nar/gkt1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32:279–284. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel R, Lombardo A, Arens A, Miller JC, Genovese P, Kaeppel C, Nowrouzi A, Bartholomae CC, Wang J, Friedman G, et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat Biotechnol. 2011;29:816–823. doi: 10.1038/nbt.1948. [DOI] [PubMed] [Google Scholar]
- Garneau JE, Dupuis ME, Villion M, Romero DA, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadán AH, Moineau S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010;468:67–71. doi: 10.1038/nature09523. [DOI] [PubMed] [Google Scholar]
- Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci USA. 2012;109:E2579–E2586. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzaelz B, Schwimmer LJ, Fuller RP, Ye Y, Asawapornmongkol L, Barbas CF. Modular system for the construction of zinc-finger libraries and proteins. Nat Protoc. 2010;5:791–810. doi: 10.1038/nprot.2010.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haft DH, Selengut J, Mongodin EF, Nelson KE. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput Biol. 2005;1:e60. doi: 10.1371/journal.pcbi.0010060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale CR, Zhao P, Olson S, Duff MO, Graveley BR, Wells L, Terns RM, Terns MP. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell. 2009;139:945–956. doi: 10.1016/j.cell.2009.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale CR, Majumdar S, Elmore J, Pfister N, Compton M, Olson S, Resch AM, Glover CV, 3rd, Graveley BR, Terns RM, Terns MP. Essential features and rational design of CRISPR RNAs that function with the Cas RAMP module complex to cleave RNAs. Mol Cell. 2012;45:292–302. doi: 10.1016/j.molcel.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath P, Romero DA, Coûté -Monvoisin AC, Richards M, Deveau H, Moineau S, Boyaval P, Fremaux C, Barrangou R. Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J Bacteriol. 2008;190:1401–1412. doi: 10.1128/JB.01415-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath P, Coûté -Monvoisin AC, Romero DA, Boyaval P, Fremaux C, Barrangou R. Comparative analysis of CRISPR loci in lactic acid bacteria genomes. Int J Food Microbiol. 2009;131:62–70. doi: 10.1016/j.ijfoodmicro.2008.05.030. [DOI] [PubMed] [Google Scholar]
- Hou Z, Zhang Y, Propson NE, Howden SE, Chu LF, Sontheimer EJ, Thomson JA. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc Natl Acad Sci USA. 2013;110:15644–15649. doi: 10.1073/pnas.1313587110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol. 1987;169:5429–5433. doi: 10.1128/jb.169.12.5429-5433.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen R, Embden JD, Gaastra W, Schouls LM. Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol. 2002;43:1565–1575. doi: 10.1046/j.1365-2958.2002.02839.x. [DOI] [PubMed] [Google Scholar]
- Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol. 2013;31:233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. RNA-programmed genome editing in human cells. eLife. 2013;2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Jiang F, Taylor DW, Sternberg SH, Kaya E, Ma E, Anders C, Hauer M, Zhou K, Lin S, et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science. 2014;343:1247997. doi: 10.1126/science.1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juillerat A, Dubois G, Valton J, Thomas S, Stella S, Maréchal A, Langevin S, Benomari N, Bertonati C, Silva GH, et al. Comprehensive analysis of the specificity of transcription activator-like effector nucleases. Nucleic Acids Res. 2014;42:5390–5402. doi: 10.1093/nar/gku155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Hsu PD, Heidenreich M, Cong L, Platt RJ, Scott DA, Church GM, Zhang F. Optical control of mammalian endogenous transcription and epigenetic states. Nature. 2013;500:472–476. doi: 10.1038/nature12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Teng F, Li T, Zhou Q. Simultaneous generation and germline transmission of multiple gene mutations in rat using CRISPR-Cas systems. Nat Biotechnol. 2013;31:684–686. doi: 10.1038/nbt.2652. [DOI] [PubMed] [Google Scholar]
- Lombardo A, Genovese P, Beausejour CM, Colleoni S, Lee YL, Kim KA, Ando D, Urnov FD, Galli C, Gregory PD, et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol. 2007;25:1298–1306. doi: 10.1038/nbt1353. [DOI] [PubMed] [Google Scholar]
- Maeder ML, Thibodeau-Beganny S, Osiak A, Wright DA, Anthony RM, Eichtinger M, Jiang T, Foley JE, Winfrey RJ, Townsend JA, et al. Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol Cell. 2008;31:294–301. doi: 10.1016/j.molcel.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder ML, Angstman JF, Richardson ME, Linder SJ, Cascio VM, Tsai SQ, Ho QH, Sander JD, Reyon D, Bernstein BE, et al. Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins. Nat Biotechnol. 2013a;31:1137–1142. doi: 10.1038/nbt.2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder ML, Linder SJ, Cascio VM, Fu Y, Ho QH, Joung JK. CRISPR RNA-guided activation of endogenous human genes. Nat Methods. 2013b;10:977–979. doi: 10.1038/nmeth.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Grishin NV, Shabalina SA, Wolf YI, Koonin EV. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct. 2006;1:7. doi: 10.1186/1745-6150-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Aravind L, Wolf YI, Koonin EV. Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPR-Cas systems. Biol Direct. 2011a;6:38. doi: 10.1186/1745-6150-6-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, Moineau S, Mojica FJ, Wolf YI, Yakunin AF, et al. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol. 2011b;9:467–477. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013a;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S, Yang L, Church GM. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol. 2013b;31:833–838. doi: 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA, Sontheimer EJ. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science. 2008;322:1843–1845. doi: 10.1126/science.1165771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA, Sontheimer EJ. Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature. 2010;463:568–571. doi: 10.1038/nature08703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendenhall EM, Williamson KE, Reyon D, Zou JY, Ram O, Joung JK, Bernstein BE. Locus-specific editing of histone modifications at endogenous enhancers. Nat Biotechnol. 2013;31:1133–1136. doi: 10.1038/nbt.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JC, Holmes MC, Wang J, Guschin DY, Lee YL, Rupniewski I, Beausejour CM, Waite AJ, Wang NS, Kim KA, et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol. 2007;25:778–785. doi: 10.1038/nbt1319. [DOI] [PubMed] [Google Scholar]
- Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ, et al. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol. 2011;29:143–148. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- Mojica FJ, Díez-Villaseñor C, Soria E, Juez G. Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria. Mol Microbiol. 2000;36:244–246. doi: 10.1046/j.1365-2958.2000.01838.x. [DOI] [PubMed] [Google Scholar]
- Mojica FJ, Díez-Villaseñor C, García-Martínez J, Soria E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol. 2005;60:174–182. doi: 10.1007/s00239-004-0046-3. [DOI] [PubMed] [Google Scholar]
- Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326:1501. doi: 10.1126/science.1178817. [DOI] [PubMed] [Google Scholar]
- Musunuru K, Pirruccello JP, Do R, Peloso GM, Guiducci C, Sougnez C, Garimella KV, Fisher S, Abreu J, Barry AJ, et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N Engl J Med. 2010;363:2220–2227. doi: 10.1056/NEJMoa1002926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewoehner J, Bohrmann B, Collin L, Urich E, Sade H, Maier P, Rueger P, Stracke JO, Lau W, Tissot AC, et al. Increased brain penetration and potency of a therapeutic antibody using amonovalent molecular shuttle. Neuron. 2014;81:49–60. doi: 10.1016/j.neuron.2013.10.061. [DOI] [PubMed] [Google Scholar]
- Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, Ishitani R, Zhang F, Nureki O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156:935–949. doi: 10.1016/j.cell.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu Y, Shen B, Cui Y, Chen Y, Wang J, Wang L, Kang Y, Zhao X, Si W, Li W, et al. Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell. 2014;156:836–843. doi: 10.1016/j.cell.2014.01.027. [DOI] [PubMed] [Google Scholar]
- Oh B, Hwang S, McLaughlin J, Solter D, Knowles BB. Timely translation during the mouse oocyte-to-embryo transition. Development. 2000;127:3795–3803. doi: 10.1242/dev.127.17.3795. [DOI] [PubMed] [Google Scholar]
- Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol. 2013;31:839–843. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Pinera P, Kocak DD, Vockley CM, Adler AF, Kabadi AM, Polstein LR, Thakore PI, Glass KA, Ousterout DG, Leong KW, et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat Methods. 2013;10:973–976. doi: 10.1038/nmeth.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plessis A, Perrin A, Haber JE, Dujon B. Site-specific recombination determined by I-SceI, a mitochondrial group I intron-encoded endonuclease expressed in the yeast nucleus. Genetics. 1992;130:451–460. doi: 10.1093/genetics/130.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourcel C, Salvignol G, Vergnaud G. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology. 2005;151:653–663. doi: 10.1099/mic.0.27437-0. [DOI] [PubMed] [Google Scholar]
- Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiberoni A, Moineau S, Rousseau GM, Reinheimer J, Ackermann HW. Streptococcus thermophilus bacteriophages. Int Dairy J. 2010;20:657–664. [Google Scholar]
- Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, Zhang F. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouet P, Smih F, Jasin M. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol Cell Biol. 1994;14:8096–8106. doi: 10.1128/mcb.14.12.8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin N, Sugarman E, Haber JE. Genetic and physical analysis of double-strand break repair and recombination in Saccharomyces cerevisiae. Genetics. 1989;122:519–534. doi: 10.1093/genetics/122.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32:347–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander JD, Dahlborg EJ, Goodwin MJ, Cade L, Zhang F, Cifuentes D, Curtin SJ, Blackburn JS, Thibodeau-Beganny S, Qi Y, et al. Selection-free zinc-finger-nuclease engineering by context-dependent assembly (CoDA) Nat Methods. 2011;8:67–69. doi: 10.1038/nmeth.1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapranauskas R, Gasiunas G, Fremaux C, Barrangou R, Horvath P, Siksnys V. The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res. 2011;39:9275–9282. doi: 10.1093/nar/gkr606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwank G, Koo BK, Sasselli V, Dekkers JF, Heo I, Demircan T, Sasaki N, Boymans S, Cuppen E, van der Ent CK, et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell. 2013;13:653–658. doi: 10.1016/j.stem.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Shah SA, Erdmann S, Mojica FJ, Garrett RA. Protospacer recognition motifs: mixed identities and functional diversity. RNA Biol. 2013;10:891–899. doi: 10.4161/rna.23764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J, Grizot S, Arnould S, Duclert A, Epinat JC, Chames P, Prieto J, Redondo P, Blanco FJ, Bravo J, et al. A combinatorial approach to create artificial homing endonucleases cleaving chosen sequences. Nucleic Acids Res. 2006;34:e149. doi: 10.1093/nar/gkl720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507:62–67. doi: 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang TH, Bachellerie JP, Rozhdestvensky T, Bortolin ML, Huber H, Drungowski M, Elge T, Brosius J, Hüttenhofer A. Identification of 86 candidates for small non-messenger RNAs from the archaeon Archaeoglobus fulgidus. Proc Natl Acad Sci USA. 2002;99:7536–7541. doi: 10.1073/pnas.112047299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebas P, Stein D, Tang WW, Frank I, Wang SQ, Lee G, Spratt SK, Surosky RT, Giedlin MA, Nichol G, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med. 2014;370:901–910. doi: 10.1056/NEJMoa1300662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S, Jamieson AC, Porteus MH, Gregory PD, Holmes MC. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435:646–651. doi: 10.1038/nature03556. [DOI] [PubMed] [Google Scholar]
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Liang D, Wang Y, Bai M, Tang W, Bao S, Yan Z, Li D, Li J. Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell. 2013;13:659–662. doi: 10.1016/j.stem.2013.10.016. [DOI] [PubMed] [Google Scholar]
- Wu X, Scott DA, Kriz AJ, Chiu AC, Hsu PD, Dadon DB, Cheng AW, Trevino AE, Konermann S, Chen S, et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nature Biotechnol. 2014 doi: 10.1038/nbt.2889. Published online Apr 20, 2014. http://dx.doi.org/10.1038/nbt.2889. [DOI] [PMC free article] [PubMed]
- Xu GL, Bestor TH. Cytosine methylation targetted to pre-determined sequences. Nat Genet. 1997;17:376–378. doi: 10.1038/ng1297-376. [DOI] [PubMed] [Google Scholar]
- Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 2013;154:1370–1379. doi: 10.1016/j.cell.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Xue W, Chen S, Bogorad RL, Benedetti E, Grompe M, Koteliansky V, Sharp PA, Jacks T, Anderson DG. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nature Biotechnol. 2014 doi: 10.1038/nbt.2884. Published online Mar 30, 2014. http://dx.doi.org/10.1038/nbt.2884. [DOI] [PMC free article] [PubMed]
- Zahradka K, Slade D, Bailone A, Sommer S, Averbeck D, Petranovic M, Lindner AB, Radman M. Reassembly of shattered chromosomes in Deinococcus radiodurans. Nature. 2006;443:569–573. doi: 10.1038/nature05160. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Heidrich N, Ampattu BJ, Gunderson CW, Seifert HS, Schoen C, Vogel J, Sontheimer EJ. Processing-independent CRISPR RNAs limit natural transformation in Neisseria meningitidis. Mol Cell. 2013;50:488–503. doi: 10.1016/j.molcel.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.