

Abstract
The copper influx transporter CTR1 is also a major influx transporter for cisplatin (cDDP) in tumor cells. It influences the cytotoxicity of cDDP both in vivo and in vitro. Whereas Cu triggers internalization of CTR1 from the plasma membrane, cDDP does not. To investigate the mechanisms of these effects, myc-tagged forms of wild type hCTR1 and variants in which Y103 was converted to alanine, C189 was converted to serine, or the K178/K179 dilysine motif was converted to alanines were re-expressed in mouse embryo cells in which both alleles of CTR1 had been knocked out and also in HEK293T cells. The Y103A mutation and to a lesser extent the C189S mutation reduced internalization of CTR1 induced by Cu while the K178A/K179A had little effect. Both Y103 and C189 were required for Cu and cDDP transport whereas the K178/K179 motif was not. While Y103 lies in an YXXM motif that, when phosphorylated, is a potential docking site for phosphatidylinositol 3-kinase and other proteins involved in endocytosis, Western blot analysis of immunoprecipitated myc-CTR1, and proteomic analysis of peptides derived from CTR1, failed to identify any basal or Cu-induced phosphorylation. However, proteomic analysis did identify an interaction of CTR1 with IRS-4 and this was confirmed by co-immunoprecipitation from HEK cells expressing either FLAG-CTR1 or myc-CTR1. The interaction was greater in the Y103A-expressing cells. We conclude that Y103 is required for the internalization of hCTR1 in response to Cu, that this occurs by a mechanism other than phosphorylation and that mutation of Y103 modulates the interaction with IRS-4.
Keywords: Cisplatin, Copper transporter 1, Insulin receptor substrate 4
1. Introduction
Copper transporter 1 (CTR1) is a high-affinity Cu influx transporter in mammalian cells. It is of interest to oncologists because it also mediates the uptake of the platinum-containing chemotherapeutic agents in tumor cells [1–7]. It regulates sensitivity to the cytotoxic effect of cDDP both in vitro and in vivo. Knockout of both alleles of CTR1 reduces initial cDDP influx [6], enhances efflux [8] and renders cells resistant to Cu, cDDP, carboplatin and oxaliplatin [1,2,5,6,9]. Re-expression of hCTR1 in cells lacking endogenous CTR1 restores cDDP uptake and sensitivity [6], as does forced over-expression in cells containing endogenous CTR1 [9].
As shown in Fig. 1, hCTR1 contains 190 amino acids and consists of an extracellular N-terminal domain, three transmembrane domains and two intracellular domains, the first being a loop connecting the first and second transmembrane domains and the second being the 15 amino acid C-terminal tail region. CTR1 exists as a homotrimer on the cell surface and electron crystallography and molecular modeling suggest that it forms a pore consisting of 4 stacked rings of 3 methionines each with a narrow entrance at the extracellular end and a vestibule at the intracellular end [10–12]. Both Cu1+ and cDDP are soft Lewis acids that readily form weak bonds with methionine, and it has been suggested that CTR1 transports Cu1+ through a series of transchelation reactions that pass the Cu1+ from one ring of methionines to the next and eventually to the ring of cysteines at the bottom of the pore [10,11,13]. The ability of CTR1 to transport Cu1+ but not Cu2+ may result from the tighter binding of Cu2+ to the methionines such that it cannot be handed through the pore by transchelation [13]. While the C-terminal C189 amino acids are quite far away from the methionine rings above them, biochemical studies indicate that the cysteines bind Cu1+ and do influence Cu transport [14]. It has been proposed that the cysteines function as a gate that controls movement through the other parts of the pore [11,15–17].
Fig. 1.

Schematic diagram of the amino acid sequence of hCTR1. Boxes highlight the Y103, C189 and K178/K179 residues.
High concentrations of Cu trigger the internalization of CTR1, and in some types of cells this is accompanied by degradation [18–20] whereas in others it is not [7,21]. It has been hypothesized that degradation may serve to limit accumulation of toxic levels of the metal [9,21–23]. In yeast the degradation of CTR1 requires the E3 ubiquitin ligase Rsp5 [23].
There are three sites within hCTR1 that are of interest with respect to control of its internalization. The first of these is the tyrosine at position 103 that lies in the intracellular loop between trans-membrane domains one and two. Tyrosines in the cytosolic domains of proteins are often involved in endocytosis [24]. The Y103 site is of particular interest because the YXXM motif within which it resides is a potential phosphorylation site that, once phosphorylated, is a candidate for binding to the SH2 domain of the p85 subunit of PI3K or other proteins involved in the endocytosis of PDGFR and other surface proteins [25]. A second site of interest is the next to last amino acid which is a cysteine at position 189 that resides in an HCH motif at the C-terminal end of hCTR1. In the case of hCTR1, C189 appears to be important for the proper assembly of the homotrimer in the membrane [26]. In yeast cysteines in the C-terminal tail appear to function as a switch that prevents further Cu transport through the pore [17]. A third site of interest is the pair of lysines at positions 178 and 179. Prior studies have demonstrated that yCTR1 and mCTR1 become ubiquitinylated in response to Cu or cDDP exposure [23,27] and the K178/K179 is a potential ubiquitinylation site that is solvent exposed in the all-atom model of CTR1 [12].
In order to further elucidate how Cu and cDDP differ with respect to their interaction with CTR1, we constructed vectors expressing N-terminally myc-tagged variant forms of hCTR1 in which eitherY103 was converted to alanine, C189 was converted to a serine or both lysines in the K178/K179 motif were converted to alanine. These variants were expressed in mouse embryo fibroblast cells in which both alleles of mCTR1 had been knocked out, and in human HEK293T cells. We report here that whereas Y103 and C189 are required for Cu and cDDP transport, the K178/K179 motif is not. We found that Y103 is required for the Cu-induced internalization of CTR1 from the plasma membrane, but that neither Y103 nor serines or threonines are phosphorylated under either basal conditions or after exposure to Cu. To identify partners that interact with CTR1 during Cu-induced internalization we conducted a proteomic analysis of proteins that immunoprecipitated with CTR1 and report here that the identification of a new interacting protein, insulin receptor substrate 4 (IRS-4).
2. Materials and methods
2.1. Drugs and reagents
cDDP was purchased as Platinol™ from the pharmacy at the Moores Cancer Center; it contains 3.33 mM cDDP in 0.9% NaCl. The cDDP was diluted into DMEM-RS Reduced Serum Media (HyClone, Logan, UT). Bradford reagent and detergent-compatible protein assay kit, DC™ Protein Assay were purchased from BioRad Laboratories, Inc. (Hercules, CA). Anti-myc primary antibody 9B11, anti-Na+/K+ ATPase antibody, anti-phosphotyrosine monoclonal antibody and anti-IRS-2 antibody were purchased from Cell Signaling Technology, Inc. (Danvers, MA). Anti-FLAG antibody was purchased from Sigma Aldrich (St. Louis, MO). Anti-CTR1 primary antibody was purchased from Abcam (Cambridge, UK), anti-IRS-4 antibody and anti-phosphoserine/threonine/tyrosine antibody from Abcam (Cambridge, UK), and IR fluorophore-conjugated secondary antibodies were obtained from Li-Cor Biosciences (Lincoln, NE). DSP (dithiobis[succinimidylpropionate]) was purchased from Thermo Scientific (Pittsburg, PA).
2.2. Cell types, culture and engineering
Mouse embryonic fibroblasts containing wild type alleles of CTR1 (CTR1+/+) and a line in which both copies of CTR1 had been somatically knocked out (CTR1−/−) were graciously provided by Thiele [28]. The absence of CTR1 mRNA in the CTR1−/− cells has been repeatedly documented by qRT-PCR and the DDP-resistant phenotype by testing for cytotoxicity [6,8,29–32]. HEK293T cells were obtained from the American Type Tissue Culture Collection. The myc-CTR1−/−/WT and HEK293/myc-CTR1WT sublines were constructed by infecting the CTR1−/− and HEK293T cells with a lentivirus expressing wild type human CTR1 cDNA, N-terminally tagged with the myc epitope, using the ViraPower Lentiviral Induction kit (Invitrogen, Carlsbad, CA). Point mutations to the myc-tagged wild type CTR1 molecule were generated with the GeneTailor Site-Directed Mutagenesis Kit (Invitrogen, Carlsbad, CA) using the following primers: for the myc-CTR1−/−/Y103A mutation (gcgtaagtcacaagtcagcattcgcgccaattccatgcctgtcccaggacca, tggtcctgggacaggcatggaattggcgcgaatgctgacttgtgacttacgc), for the myc-CTR1−/−/C189S mutation (atggaacaaaaacttatttc, tcaatggctatgctctgtgatatc). The Forward and reverse PCR primers for this production of the K178A/K179A mutation were, respectively, 5′-cctatgaaggagaagtcgacccgtcgtcgtcaccatcacctatagtgtctcgtaacg-3′ and 5′-cgttacgagacactataggtgatggtgacgacgacgggtcgacttctccttcatagg-3′. Sublines expressing these mutant forms of CTR1 were generated by infecting the CTR1−/− and HEK293T cells with lentivirus expressing the mutants using the ViraPower system, as described for the myc-CTR1−/−/WT and HEK293/myc-CTRWT cells above. HEK293 containing tetracycline-regulated wild type hCTR1 gene with N-terminally tagged with the FLAG epitope (HEK293 FLP-In T-Rex cell) was provided by Dr. Jack Kaplan, 1 μg/mL tetracycline was used to induce the over-expression on wild-type hCTR1.
2.3. Biotinylation and immunoblotting
Mouse embryo fibroblasts and HEK293 cells were surface biotinylated with EZ-LINK sulfo-NHS-SS-biotin (Thermo Scientific, Pittsburg, PA) for 30 min at 4 °C and quenched in PBS containing 200 mM glycine PBS for 25 min. Cells were harvested by scraping, washed twice in PBS and then solubilized in 1% Triton-X-100 for 60 min, and insoluble material was removed by centrifugation at 14,000 × g for 10 min. Protein concentrations were determined by detergent-compatible protein assay kit, DC™ Protein Assay (Bio-Rad, Hercules, CA) and equal amount of proteins were incubated overnight at 4 °C with 100 μL streptavadin-coated beads (Thermo Scientific, Pittsburg, PA). The beads were collected and washed, after which the proteins bound to the beads were cleaved with 5% β-mercaptoethanol in 2 × SDS electrophoresis sample buffer and subjected to Western blot analysis using an antibody to the myc tag or to CTR1 [33] and an antibody to the Na/K ATPase that was used as a lane loading control. Western blot analysis was performed as previously described [31]. Protein expression on the Western blots was quantified using an Odyssey Imaging System (Li-Cor Biosciences, Lincoln, NE) utilizing software version 3.0. Expression of CTR1 in the experimental cells was expressed as a percent of that in the control cells after normalization to the level of Na/K ATPase.
2.4. Reversible cross-link immunoprecipitation procedure
Cells were washed twice with PBS at room temperature to remove all traces of media and cell pellets were resuspended in PBS and incubated with freshly prepared dithiobis[succinimidyl propionate] (DSP) (Thermo Scientific, Pittsburg, PA) at a final concentration of 2.5 mM for 30 min at room temperature. The cross-linking reaction was quenched with 25 mM Tris–HCl pH 7.4 for 15 min at room temperature. The pelleted cells were lysed with RIPA buffer (50 mM Tris, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100) plus protease and phosphatase inhibitors (Thermo Scientific, Pittsburg, PA). Equal amount of proteins were incubated with either anti-FLAG or anti-myc antibody and protein A/G beads (Thermo Scientific, Pittsburg, PA) overnight at 4 °C with end-over-end rotation. The beads were then washed 5 times with RIPA buffer and proteins were eluted by incubating the beads with Laemmli’s 2× sample buffer (125 mM Tris–HCl, pH 6.8, 2 mM EDTA, 6% SDS, 20% glycerol, 5% β-mercaptoethanol and 0.25% bromophenol blue) at 95 °C for 5 min and subjected to Western blot analysis.
2.5. Proteomic analysis
Proteomic analysis was performed in UC San Diego Biomolecular/Proteomics Mass Spectrometry Facility. The beads containing the immunoprecipitated protein were diluted in TNE (50 mM Tris pH 8.0, 100 mM NaCl, 1 mM EDTA) buffer. RapiGest SF reagent (Waters Corp., Milford, MA) was added to the mixture to a final concentration of 0.1% and samples were boiled for 5 min. TCEP (Tris (2-carboxyethyl) phosphine) was added to a final concentration of 1 mM and the samples were incubated at 37 °C for 30 min. Subsequently, the samples were carboxymethylated with 0.5 mg/mL of iodoacetamide for 30 min at 37 °C followed by neutralization with TCEP at a final concentration of 2 mM. Proteins samples prepared as above were digested with trypsin (trypsin:protein ratio—1:50) overnight at 37 °C. RapiGest was degraded and removed by treating the samples with 250 mM HCl at 37 °C for 1 h followed by centrifugation at 14,000 rpm for 30 min at 4 °C. The soluble fraction was then added to a new tube and the peptides were extracted and desalted using C18 desalting columns (Thermo Scientific, Pittsburg, PA). LC-MS/MS analysis of the trypsin-digested peptides was carried out using ultra high pressure liquid chromatography coupled with tandem mass spectroscopy combined with nano-spray ionization. The nano-spray ionization experiments were performed using a Triple T of 5600 hybrid mass spectrometer (ABSCIEX, Framingham, MA) interfaced with nano-scale reverse-phase ultra high pressure liquid chromatography (Waters Corporation nano ACQUITY) using a 20 cm-75 micron ID glass capillary packed with 2.5-μm C18 (130) CSH™ beads (Waters Corp., Milford, MA). Peptides were eluted from the C18 column into the mass spectrometer using a linear gradient (5–80%) of acetonitrile at a flow rate of 250 μL/min for 1 h. The buffers used to create the acetonitrile gradient were: Buffer A (98% H2O, 2% acetonitrile, 0.1% formic acid, and 0.005% trifluoroacetic acid) and Buffer B (100% acetonitrile, 0.1% formic acid, and 0.005% trifluoroacetic acid). MS/MS data were acquired in a data-dependent manner in which the MS1 data was acquired for 250 ms at m/z of 400 to1250 Da and the MS/MS data was acquired from m/z of 50 to 2000 Da. The independent data acquisition parameters were as follows: MS1-TOF acquisition time of 250 ms, followed by 50 MS2 events of 48 ms acquisition time for each event. The threshold to trigger MS2 event was set to 150 counts when the ion had the charge state +2, +3 and +4. The ion exclusion time was set to 4 s. Finally, the collected data were analyzed using Protein Pilot 4.5 (ABSCIEX, Framingham, MA) for peptide identifications.
2.6. Measurement of cellular drug accumulation
Drug accumulation was measured by inductively-coupled plasma mass spectroscopy using sulfur as a surrogate for total protein as described previously [6]; the only change made was the utilization of 1 mL volume of medium and the use of DMEM-RS in place of OptiMEM media.
2.7. Statistical analysis
All two-group comparisons utilized Student’s t-test with the assumption of unequal variance. Data are presented as mean ± SEM of a minimum of 3 independent experiments.
3. Results
3.1. Expression of CTR1 variants in CTR1−/− mouse embryo fibroblasts and HEK293T cells
Prior studies of the importance of various components of hCTR1 for Cu homeostasis have sometimes been confounded by the presence of endogenous CTR1. To avoid this problem, wild type and variant forms of hCTR1 were re-expressed in mouse embryo fibroblasts in which both alleles of CTR1 had been knocked out (CTR1−/− cells), and in human HEK293T cells that express low levels of endogenous CTR1. Lentiviral vectors containing a blastocidin resistance marker were constructed to express either an N-terminal myc-tagged form of wild type hCTR1, a variant in which the Y103 was converted to alanine, a variant in which C189 was converted to serine, or one in which the dilysine motif K178/K179 was replaced with alanines. CTR1−/− cells were infected and selected with blastocidin to generate the CTR1−/−/WT, CTR1−/−/Y103A, CTR1−/−/C189S and CTR1−/−/K178/K179A sublines that were then characterized with respect to the expression of each form of exogenous CTR1. HEK293T cells were infected with the same vectors to produce the HEK293/myc-CTR1WT, HEK293/myc-CTR1Y103A, HEK293/myc-CTR1C189S and HEK293/myc-CTR1K178A/K179A cell lines. To assess the level of CTR1 protein expression at the plasma membrane, the cell surface proteins were biotinylated by exposure to sulfo-NHS-SS-biotin before lysis, then recovered on streptavadin-coated beads and subjected to Western blot analysis using an antibody to the myc tag or to CTR1. The wild type and both variant forms of hCTR1 were present in the plasma membrane at nearly equivalent levels as determined by quantitative imaging and normalized for Na/K ATPase content. The CTR1−/−/Y103A cells expressed plasma membrane CTR1 at a mean of 96 ± 4% of that in the CTR1−/−/WT cells (n = 3), the CTR1−/−/C189S cells at 91 ± 5% (n = 3) and the CTR1−/−/K178A/K179A at 87 ± 8% (n = 3) of that in the CTR1−/−/WT cells. Western blot analyses of the expression of each of the variants in the plasma membrane of the HEK293T cells showed nearly equivalent expression of the wild type and Y103A and C189S variants but lower expression of the K178A/K179A variant. To document that the sulfo-NHS-SS-biotin reacted with only cell surface proteins, biotinylated proteins from each of the cell lines were analyzed by Western blot for the presence of the cytosolic protein glyceraldehyde 3-phosphate dehydrogenase and none was found (data not shown).
3.2. Effect of mutations on Cu and cDDP influx
To determine the effect of the Y103A, C189S and K178A/K179A mutations on the steady-state levels of cellular Cu we used the mouse embryo cells since they do not express any endogenous CTR1. The CTR1−/−/WT, CTR1−/−/Y103A, CTR1−/−/C189S and CTR1−/−/K178/K179A cells were grown in complete DMEM medium containing ~0.3 μM Cu. Fig. 2A shows that basal Cu was not significantly altered by re-expression of wild type CTR1 in the CTR1−/−/WT cells or by re-expression of any of the variant forms of CTR1.
Fig. 2.
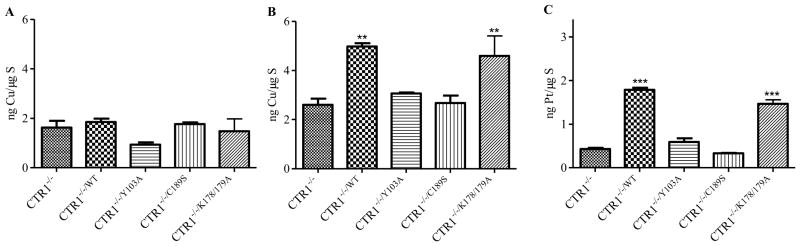
Effect of modifying Y103, C189 and K178/K179 on Cu and cDDP accumulation. (A) basal whole cell Cu content; (B) total Cu following 1 h exposure to 100 μM Cu; (C) net accumulation of Pt following 5 min exposure to 30 μM cDDP. All values represent means of at least 3 independent experiments each containing sextuplet replicates. Vertical bars, ±SEM. * p < 0.05; *** p < 0.001.
The effect of the CTR1 variants on the accumulation of Cu was analyzed by exposing the cells to media containing 100 μM Cu for 1 h. As shown in Fig. 2B, re-expression of wild type hCTR1 resulted in a 2-fold increase in the amount of Cu accumulation when compared to uptake in the CTR1−/− cells (p = 0.003). While the K178A/K179A variant was nearly as effective in increasing Cu uptake as the wild type, neither of the Y103A or the C189S variant was able to significantly increase Cu uptake. Thus, the integrity of both Y103 and C189 was required for Cu transport to occur in this system.
To analyze the effect of the Y103A and C189S substitutions on the initial influx of cDDP, the 4 types of cells were exposed to 30 μM cDDP for 5 min, washed thoroughly and the Pt content measured by ICP-MS. A short duration of drug exposure was used because prior studies have shown that the greatest effect of CTR1 is on initial cDDP influx. As shown in Fig. 2C, re-expression of wild type CTR1 resulted in a 4.2-fold increase in cDDP accumulation (p = 9 × 10−5). Expression of the Y103A variant resulted in only a 1.4-fold increase in cDDP accumulation, which was only 33% of the increase produced by re-expression of wild type CTR1 (p = 0.0009). Expression of the C189S variant failed to increase cDDP uptake at all, and in fact reduced it by a small amount to 78% of that observed for the CTR1−/− cells. In contrast, the K178A/K179A variant increased cDDP uptake to 83% of that produced by the wild type CTR1. Thus, while the K178A/K179A variant retained almost normal transport activity, mutation of Y103 severely disabled cDDP transport and the C189S variant appeared to be transport dead.
3.3. Effect of mutations on Cu-induced internalization of CTR1
Exposure to Cu causes the rapid internalization of CTR1, whereas in HEK293T and small cell lung cancer cells, exposure to cDDP has been noted to increase the relative amount of higher molecular weight forms interpreted as being dimers and trimers [7,34]. To determine how the mutations we made in CTR1 affected these responses we used the HEK293T cells since CTR1 levels were higher and could be quantified more precisely. Fig. 3A shows the effect of a 30 min exposure to 100 μM Cu on plasma membrane CTR1 in the wild type and the Y103A, C189S and K178A/K179A variants. Using a rabbit monoclonal antibody that we have previously extensively characterized [33], CTR1 was detected as bands of 33–35 and 62–64 kDa. The Cu exposure reduced CTR1 in the plasma membrane of the HEK293/myc-CTR1WT cells to 15.9 ± 2.2% of that in the untreated cells (p = 0.0007), whereas it reduced it to 67.0 ± 6.2% (p = 0.07) of that in the untreated HEK293/myc-CTR1Y103A cells, to 52.6 ± 3.6% (p = 0.004) of that in the untreated HEK293/myc-CTR1C189S cells and to 34.0 ± 9.0% (p = 0.051) of that in the untreated HEK293/myc-CTR1K178A/K179A cells as determined by quantitative imaging of 5 independent Western blot analyses normalized for Na/K ATPase content (Fig. 3B). Thus, both the Y103A and C189S mutations impaired the ability of Cu to trigger internalization of CTR1 while the K178A/K179A mutation had less effect. Exposure to cDDP did not cause loss of CTR1 from the plasma membrane in any of the 4 cell types indicating that it does not trigger the same internalization mechanism as Cu (data not shown).
Fig. 3.

Effect of modifying Y103, C189 and K178/K179 on the ability of Cu to trigger CTR1 internalization. HEK293/myc-CTRWT and variant cells were exposed to 100 μM Cu for 30 min prior to being biotinylated. (A) Western blots of biotinylated proteins were probed with anti-CTR1 antibody. Na/K ATPase served as a loading control. (B) Quantification of Western blots. Relative amount CTR1 in cells treated with Cu (black bars) is shown as a percentage of that in untreated control cells. Values are the mean of 5 independent experiments ± SEM. * p < 0.05; ** p < 0.01.
3.4. Analysis of CTR1 for phosphorylation
CTR1 contains 6 serines, 3 threonines but only a single tyrosine in the solvent exposed intracellular portion of the molecule. Given that phosphorylation of plasma membrane transporters is one mechanism by which internalization is triggered, and given that conversion of the one tyrosine (Y103) to alanine blocked internalization, we were curious as to whether Cu triggered the phosphorylation of CTR1. We first sought to determine whether CTR1 was phosphorylated under basal conditions using the HEK293 FLP-In T-Rex cells in which FLAG-tagged CTR1 was induced by exposure to tetracycline. Cells were pretreated with the phosphatase inhibitor orthovanadate or calf intestinal alkaline phosphatase, or the combination of both orthovanadate and the alkaline phosphatase. CTR1 was immunoprecipitated using an antibody to the FLAG tag and subjected to Western blot analysis with an antibody capable of detecting phosphorylation on serine, threonine or tyrosine, and with an antibody specific for phosphotyrosine. No change in the signal detected by Western blot analysis was observed in response to either the phosphatase inhibitor or alkaline phosphatase (Fig. 4A). CTR1 was also immunoprecipitated from HEK293 FLP-In T-Rex cell cells before and after a 30 min exposure to 100 μM Cu using the same technique. As shown in Fig. 4B, no phosphorylation of the tyrosine was detected. To refine the search further, CTR1 was immunoprecipitated before and 5 min after the start of exposure to 100 μM Cu and then digested with trypsin and examined by mass spectroscopy of the presence of phosphopeptides. Peptides containing the possible phosphorylation sites from the internal portion of CTR1 were detected using this approach (103YNSMPVPGPNGTILMETHK and 180AVVVDI-TEHCH); however, no phosphorylation of these was consistently demonstrated by this proteomic analysis. Based on these results, it appears that Cu triggers removal of CTR1 from the plasma membrane by some other mechanism.
Fig. 4.
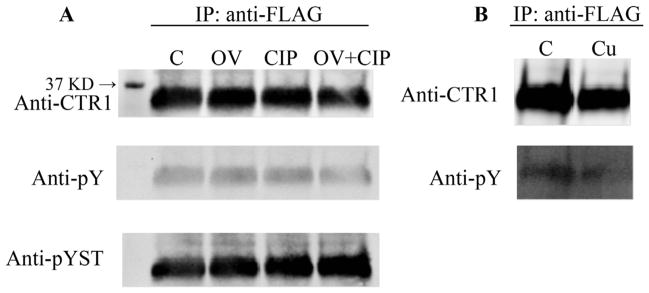
Analysis of CTR1 for phosphorylation following exposure to Cu. (A) FLAG-CTR1-expressing HEK293 FLP-In T-Rexcells were treated with 1 mM orthovanadate for 30 min and CTR1 was immunoprecipitated with anti-FLAG antibody and A/G beads. The beads were treated with calf intestinal alkaline phosphatase for 1 h at 37 °C; C, control; OV, orthovanadate; CIP, calf intestinal alkaline phosphatase. (B) CTR1 was immunoprecipitated from HEK293 FLP-In T-Rex cells before and after a 30 min exposure to 100 μM Cu.
3.5. Proteomic analysis of CTR1 interacting proteins
After successfully detecting CTR1 peptides using mass spectrometry, we carried out reversible cross-link immunoprecipitation and mass spectrometry experiments to identify potential CTR1 binding partners [35]. Labile interactions were stabilized prior to cell lysis by treating the cells with the cell-permeable, thiol-cleavable cross-linker DSP. After immunoprecipitation of CTR1 from the HEK293 FLP-In T-Rex cells with anti-FLAG antibody, crosslinked binding partners were selectively released by reduction with β-mercaptoethanol which resulted in an extremely efficient recovery with exceptionally low background. Proteomic analysis of the released trypsin-digested proteins identified insulin receptor substrate 4 (IRS-4) and this was considered of interest because IRS proteins mediate a variety of biological effects downstream of the insulin and insulin-like growth factor 1 (IGF-1) receptors, and we had previously demonstrated that CTR1 plays an important role in regulating signaling from receptor tyrosine kinases [36]. The IRS-4 peptides identified by mass spectrometry are presented in Table 1. To confirm the interaction between CTR1 and IRS-4, FLAG-CTR1 was immunoprecipitated from the HEK293 FLP-In T-Rex cells after tetracycline induction and subjected to Western blot analysis for the presence of IRS-4. As shown in Fig. 5, there was a 21.5 ± 4.0-fold increase in the association of CTR1 with IRS-4 after tetracycline induction (p < 0.05, n = 4). The interaction was further confirmed by showing a 4.8 ± 0.4-fold increase when the CTR1 immunoprecipitated from in HEK293/myc-CTR1WT cells as compared to that precipitated from the empty vector control cells (p < 0.001, n = 4). No interaction between CTR1 and IRS-1 or IRS-2 was detected indicating that the interaction is specific to IRS-4 (data not shown). To determine whether either Y103 or C189 is important to the interaction with IRS-4, the HEK293/myc-CTR1Y103A and HEK293/myc-CTR1C189S cells were analyzed in the same way. As shown in Fig. 6 there was a 2.7 ± 0.2-fold increase in the CTR1-bound IRS-4 level in HEK293/myc-CTR1Y103A cells compared to that in the HEK293/myc-CTR1WT cells (p = 0.041, n = 3), but no significant interaction in the HEK293/myc-CTR1C189S cells. This suggests that not only is Y103 important to the ability of Cu to induce CTR1 internalization but it also participates in the interaction between CTR1 and IRS-4.
Table 1.
IRS-4 fragments identified by mass spectrometry from immunoprecipitated FLAG-CTR1.
| AAAAAAAAAASGAAIPPLIPPR |
| AAVSAFPTDSLER |
| AIGDGEDEMLFTR |
| DAASKPSGEGSFSKPGDGGSPSKPSDHEPPK |
| DLSPSSAPAVASAAEPTLALSQVVAAASALAAAPGIGAAAAAAGFDSASAR |
| EVSYNWDPK |
| FVTPSEPVAHSR |
| GAQDVAGGSNPGAHNPSANLAR |
| GDNQAGGAAAAAAAPEPPPR |
| GYMMMFPR |
| HLGLVPLEPGGWLR |
| LNTEVASVVVQLLSIR |
| LSSEVSGSGSGNFGEEGNPQGK |
| LNTEVASVVVQLLSIRR |
| VSPPPAPSPPK |
| LSQVVAAASALAAAPGI |
| ESNTPAPSTQGLPDSWGIIAEPR |
| AIGDGEDEMLFTR |
| DAASKPSGEGSFSKPGDGGSPSKPSDHEPPK |
| GAQDVAGGSNPGAHNPSANLAR |
| GDNQAGGAAAAAAAPEPPPR |
Fig. 5.

CTR1 interacts with IRS-4 in HEK 293 cells. (A) FLAG-CTR1 was immunoprecipitated from DSP-treated HEK293 FLP-In T-Rex cells before and after induction with tetracycline and subjected to Western blot analysis for the presence of IRS-2 and IRS-4. (B) myc-CTR1 was immunoprecipitated from DSP-treated HEK293/myc-EV and HEK293/myc-CTR1WT cells and subjected to Western blot analysis for the presence of IRS-4. Histograms show mean ± SEM determined from 4 experiments. * p < 0.05; *** p < 0.001.
Fig. 6.
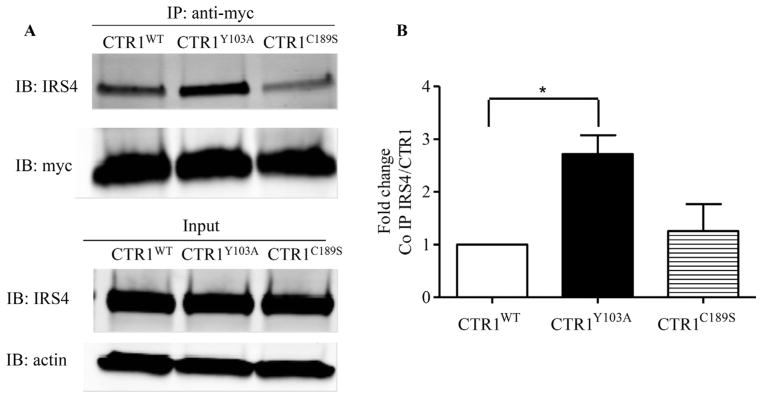
CTR1 mutants interact with IRS-4 in HEK 293 cells. (A) myc-CTR1 was immunoprecipitated from DSP-treated HEK293/myc-CTRWT, HEK293/myc-CTRY103A, HEK293/myc-CTRC189S and subjected to Western blot analysis for the presence of IRS-4. (B) Mean ± SEM determined from 3 independent experiments. * p < 0.05.
4. Discussion
The re-expression of wild type and variant forms of hCTR1 in cells in which both alleles of CTR1 have been knocked out provides a powerful way of identifying structural components essential for function. The results of this study provide further insight into the domains of CTR1 that are required for the transport of Cu and cDDP, and those required for the internalization of CTR1 in response to exposure to Cu. Perhaps the most important new finding to emerge from this study is that Y103, in addition to being required for the transport of Cu and cDDP, is important to the ability of Cu to trigger CTR1 internalization. Conversion of Y103 to alanine eliminated the increase in whole cell Cu produced by a 1 h exposure to even a very high concentration of Cu. The intracellular loop that spans transmembrane domains 1 and 2 has not previously been thought to play an important role in the accumulation of either Cu or cDDP. However, this domain contains several motifs that are of interest to a potential scaffold role of CTR1 that facilitates the binding of other proteins. In Xenopus CTR1 is important for signaling during embryonic development [37] and recent studies have shown that it is essential for signaling from multiple receptor tyrosine kinases in mouse cells [36,38]. Molecular modeling suggests that the intracellular loop is a likely site for the docking of proteins that participate in this pathway [12]. The phosphorylation of tyrosines in the cytosolic domains of transmembrane proteins is known to be involved in both clathrin and non-clathrin mediated endocytosis [24]. The Y103 site in hCTR1 is of particular interest because of its location within a YXXM motif that, once phosphorylated, is a candidate for binding to the p85 subunit of PI3K which mediates the endocytosis of PDGF and many other surface proteins [25,39]. PI3K is involved in protein sorting in a variety of trafficking pathways such as agonist-induced endocytosis [40] and multi-vesicular body formation [41]. However, despite the fact that Y103 is solvent exposed in the all atom model of CTR1 [12], we found no evidence that Y103 became phosphorylated during Cu-induced internalization. Using PhosphoSitePlus, NetPhos predictor, Phospho. ELM database and other major online phosphorylation site prediction programs, we also did not find suggestion that the tyrosine on 103YNSM of CTR1 can be phosphorylated. Nevertheless, we speculate that Y103 itself, or the conformation that it enforces on the cytoplasmic loop, is required for the engagement of CTR1 with the endosomal machinery and quite likely with proteins or peptides that can accept Cu from CTR1. Since Cu is not free inside the cell, engagement with such Cu acceptors is probably essential for transport to occur [42]. Whether such an acceptor is functionally linked to the endocytic machinery is an interesting area for future investigation.
The results of this study confirm prior reports that C189 is required for the transport of Cu by human CTR1 [14,43] and indicate that it is also essential for the transport of cDDP. However, they are at odds with results obtained in a small cell lung cancer model in which Cu and cDDP transport where equivalent in cells forced to express either wild type CTR1 or a variant in which the last 3 amino acids 188HCH190 were deleted [4]. It is uncertain whether the difference in the two models is due to interaction of the variant with endogenous CTR1 in the small cell model or deletion of the terminal 3 amino acids rather than just substitution of C189 by serine. Electron crystallography studies of CTR1 indicate that it forms a pore with a narrow entrance at the extracellular end and a vestibule at the intracellular end [10,11]. The HCH motif in the C-terminal tail is one of the most highly conserved parts of CTR1, and the all atom molecular model of CTR1 suggests that the cysteines contributed by each CTR1 monomer participate in the formation of Cu binding sites at the exit of the vestibule [12]. In both human and yeast CTR1 the C-terminal domain appears to be involved in interactions between the monomers that affect the structural stability of the trimer [44], and this region may function as a Cu sensor that switches the pore between an open and closed configuration [17]. The HCH motif is also reported to be involved in interactions between CTR1 and ATOX1 [15,43,45]. In our study, conversion of C189 to serine reduced Cu and cDDP transport but did not ablate the ability of Cu to trigger internalization of CTR1 indicating that transport function is not essential to the internalization process. It is important to note it has been reported that the C189S variant does not assemble correctly as a trimer in the plasma membrane which may account for its failure to support normal Cu uptake [26]. It has also been reported that replacement of the entire HCH motif with AAA increases rather than decreases Cu transport [14] which suggests that the two histidines that flank C189 also serve a role in gating Cu influx, a concept consistent with their potential role in hosting a Cu binding site at the cytoplasmic end of the trimer [12].
Along with the 188HCH190 the 178K/K179 motif is conserved across species. Prior studies have demonstrated that yCTR1 and mCTR1 become ubiquitinated in response to Cu or cDDP exposure [23,27]. The 178K/K179 motif was selected for investigation because it is a potential site of ubiquitinylation. yCTR1 contains a typical ubiquitinylation signal in the form of a pair of lysines at 340K/K345 [23]. yCTR1 is ubiquitinylated by the ubiquitin ligase Rsp5p that is attracted to CTR1 by an adaptor protein in response to Cu exposure. This occurs prior to endocytosis, and mutation of the 340K/K345 motif in yCTR1 or deletion of the adaptor protein results in failure of trafficking of CTR1 to the vacuole and inhibition of degradation [23]. Similar to the 340K/K345 residues in yeast, the 178K/K179 motif in hCTR1 is also located in the C-terminal end of the protein. However, the results of the current study indicated that conversion of both K178 and K179 to alanines had no significant effect on the transport of Cu, produced only a 20% decrease in cDDP uptake, and only insignificantly impaired the ability of Cu to trigger internalization of CTR1 from the plasma membrane. The lack of effect on transport is consistent with the finding that the 340K/K345 in yCTR1 is not essential for Cu transport [23], and that truncation of hCTR1 at K178 still permitted some Cu transport when expressed in Sf9 cells[43]. We concluded that, despite the role of a similarly positioned KK motif in yCTR1, the 178K/K179 motif in hCTR1 either is either not a site of ubiquitinylation or ubiquitinylation is not essential for the Cu-induced internalization of CTR1.
The results of this study provide additional evidence that, despite the fact that Cu1+ and cDDP appear to be substrates for the CTR1 transporter, the molecular details of the mechanism by which CTR1 imports cDDP and Cu are different. This may be a reflection of the ability of Cu1+ to bind 3 or 4 sulfur atoms, whereas cDDP and its variants are expected to only form bis-adducts with just 2 sulfur atoms. Other evidence comes from studies in yeast where FRET analysis of the interaction between the C-terminal tails of yCTR1 monomers showed that Cu but not cDDP brought them closer together. A variant of yCTR1 defective in Cu transport nevertheless still enhanced cDDP accumulation [44]. Some N-terminal methionine-rich motifs that are dispensable for Cu transport are required for cDDP uptake [44]. Refinement of the understanding of how cDDP is transported by hCTR1 is important because cellular uptake is a direct determinant of cytotoxicity, and insights into how transport can be manipulated have the potential to enhance overall efficacy of chemotherapy with this agent.
The only prior evidence that CTR1 interacts with any other protein was provided in a Xenopus model in which the Src-related kinase Laloo was reported to physically interact with CTR1 and the FGFR docking protein FRS2α [37]. We and another group have reported that CTR1 plays an pivotal role in regulating receptor tyrosine kinase signaling [36,38] on the basis that knockout of CTR1 resulted in impaired insulin, EGF, FGF and PDGF-stimulated activation of Ras/MAPK signaling as evidenced by a reduction in ERK1/2 phosphorylation. We speculated that CTR1 is associated with other signaling proteins to mediate the Ras/MAPK signaling. In the current study reversible cross-link immunoprecipitation and mass spectrometric analysis identified IRS-4 as a potential CTR1 interacting protein. IRS-4 is the most recently identified member of the IRS family which contains IRS-1, IRS-2, IRS-3, and IRS-4 [46]. The IRS proteins are found associated with the insulin and IGF-1 receptors and upon receptor activation they become phosphorylated and provide docking sites for SH2 domain containing proteins such as PI3K and Grb2 [47,48]. The four IRS proteins have some redundant functions and some unique roles that are still being clarified. IRS-4 knockout mice showed almost no phenotype other than slightly reduced weight and slightly lower glucose levels [49]. IRS-4 was shown to compensate for IRS-1 in liver regeneration and brown adipocyte differentiation and for IRS-2 in preventing apoptosis of pancreatic β-cells [50,51]. Although IRS-4 has a very similar modular structure to other IRS members, it has only 27 and 29% sequence identity with IRS-1 and IRS-2, respectively. IRS-4 is also known to interact with Shp2 and WDR6 and all these proteins have been implicated in FGFR signaling [52,53]. No interaction was found between CTR1 and IRS-1 or IRS-2 indicating that CTR1 is specifically associated with IRS-4 only. We speculate that the interaction between CTR1 and IRS-4 is transient and labile because it was not detected in the absence of the cross-linker. The basis for the increased steady-state association of CTR1 and IRS-4 in the Y103A mutant is unknown but may be linked with the failure of the Y103A variant to internalize in response to Cu. How Cu exposure modulates the interaction of CTR1 and IRS-4 has yet to be investigated.
Acknowledgments
The authors would like to thank Dr. Dennis Thiele for providing the CTR1+/+ and CTR1−/− mouse embryo fibroblasts, Dr. Jack Kaplan for providing HEK293 FLP-In T-Rex cells and Preston Adams and Gerald Manorek for technical assistance. Valuable advice was provided by Drs. Brian Blair, Paolo Abada, Jacob Quail, Michael Petris and Xinjian Lin. This work was supported by grants from the National Institutes of Health (CA152185 and T32CA121938), and the U.S. Department of Defense (W81XWH-08-1-0135). Additional support for core laboratories was from the UCSD Cancer Center Support Grant P30 CA23100.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ information
Participated in research design: Tsai, Larson, Safaei, and Howell. Conducted experiments: Tsai, Larson, and Safaei. Performed data analysis: Tsai, Larson, Safaei and Howell. Wrote or contributed to the writing of the manuscript: Tsai, Larson and Howell.
Contributor Information
Cheng-Yu Tsai, Email: cht026@ucsd.edu.
Christopher A. Larson, Email: clarson@illumina.com.
Stephen B. Howell, Email: showell@ucsd.edu.
References
- 1.Lin X, Okuda T, Holzer A, Howell SB. The copper transporter CTR1 regulates cisplatin uptake in Saccharomyces cerevisiae. Mol Pharmacol. 2002;62:1154–9. doi: 10.1124/mol.62.5.1154. [DOI] [PubMed] [Google Scholar]
- 2.Holzer AK, Samimi G, Katano K, Naerdemann W, Lin X, Safaei R, et al. The copper influx transporter human copper transport protein 1 regulates the uptake of cisplatin in human ovarian carcinoma cells. Mol Pharmacol. 2004;66:817–23. doi: 10.1124/mol.104.001198. [DOI] [PubMed] [Google Scholar]
- 3.Safaei R, Holzer AK, Katano K, Samimi G, Howell SB. The role of copper transporters in the development of resistance to Pt drugs. J Inorg Biochem. 2004;98:1607–13. doi: 10.1016/j.jinorgbio.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 4.Song I, Savaraj N, Siddik Z, Liu P, Wei Y, Wu C, et al. Role of copper transporter Ctr1 in the transport of platinum-based antitumor agents in cisplatin-sensitive and resistant cells. Mol Cancer Ther. 2004;3:1543–9. [PubMed] [Google Scholar]
- 5.Holzer AK, Manorek GH, Howell SB. Contribution of the major copper influx transporter CTR1 to the cellular accumulation of cisplatin, carboplatin, and oxaliplatin. Mol Pharmacol. 2006;70:1390–4. doi: 10.1124/mol.106.022624. [DOI] [PubMed] [Google Scholar]
- 6.Larson CA, Blair BG, Safaei R, Howell SB. The role of the mammalian copper transporter 1 in the cellular accumulation of platinum-based drugs. Mol Pharmacol. 2009;75:324–30. doi: 10.1124/mol.108.052381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liang ZD, Stockton D, Savaraj N, Kuo MT. Mechanistic comparison of human copper transporter hCtr1-mediated transports between copper ion and cisplatin. Mol Pharmacol. 2009;76:843–53. doi: 10.1124/mol.109.056416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blair BG, Larson CA, Safaei R, Howell SB. Copper transporter 2 regulates the cellular accumulation and cytotoxicity of cisplatin and carboplatin. Clin Cancer Res. 2009;15:4312–21. doi: 10.1158/1078-0432.CCR-09-0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holzer AK, Katano K, Klomp LW, Howell SB. Cisplatin rapidly down-regulates its own influx transporter hCTR1 in cultured human ovarian carcinoma cells. Clin Cancer Res. 2004;10:6744–9. doi: 10.1158/1078-0432.CCR-04-0748. [DOI] [PubMed] [Google Scholar]
- 10.De Feo CJ, Aller SG, Unger VM. A structural perspective on copper uptake in eukaryotes. Biometals. 2007;20:705–16. doi: 10.1007/s10534-006-9054-7. [DOI] [PubMed] [Google Scholar]
- 11.De Feo CJ, Aller SG, Siluvai GS, Blackburn NJ, Unger VM. Three-dimensional structure of the human copper transporter hCTR1. Proc Nat Acad Sci USA. 2009;106:4237–42. doi: 10.1073/pnas.0810286106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsigelny IF, Sharikov Y, Greenberg JP, Miller MA, Kouznetsova VL, Larson CA, et al. An all-atom model of the structure of human copper transporter 1. Cell Biochem Biophys. 2012;63:223–34. doi: 10.1007/s12013-012-9358-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Howell SB, Safaei R, Larson CA, Sailor MJ. Copper transporters and the cellular pharmacology of the platinum-containing cancer drugs. Mol Pharmacol. 2010;77:887–94. doi: 10.1124/mol.109.063172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maryon EB, Molloy SA, Ivy K, Yu H, Kaplan JH. Rate and regulation of copper transport by human copper transporter 1 (hCTR1) J Biol Chem. 2013;288:18035–46. doi: 10.1074/jbc.M112.442426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiao Z, Loughlin F, George GN, Howlett GJ, Wedd AG. C-terminal domain of the membrane copper transporter Ctr1 from Saccharomyces cerevisiae binds four Cu(I) ions as a cuprous-thiolate polynuclear cluster: sub-femtomolar Cu(I) affinity of three proteins involved in copper trafficking. J Am Chem Soc. 2004;126:3081–90. doi: 10.1021/ja0390350. [DOI] [PubMed] [Google Scholar]
- 16.Xiao Z, Wedd AG. A C-terminal domain of the membrane copper pump Ctr1 exchanges copper(I) with the copper chaperone Atx1. Chem Commun (Cambridge England) 2002;6:588–9. doi: 10.1039/b111180a. [DOI] [PubMed] [Google Scholar]
- 17.Wu X, Sinani D, Kim H, Lee J. Copper transport activity of yeast Ctr1 is down regulated via its C-terminus in response to excess copper. J Biol Chem. 2009;284:4112–22. doi: 10.1074/jbc.M807909200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petris MJ, Smith K, Lee J, Thiele DJ. Copper-stimulated endocytosis and degradation of the human copper transporter, hCtr1. J Biol Chem. 2003;278:9639–46. doi: 10.1074/jbc.M209455200. [DOI] [PubMed] [Google Scholar]
- 19.Holzer AK, Howell SB. The internalization and degradation of human copper transporter 1 following cisplatin exposure. Cancer Res. 2006;66:10944–52. doi: 10.1158/0008-5472.CAN-06-1710. [DOI] [PubMed] [Google Scholar]
- 20.Jandial DD, Farshchi-Heydari S, Larson CA, Elliot GI, Wrasidlo WJ, Howell SB. Enhanced delivery of cisplatin to intraperitoneal ovarian carcinomas mediated by the effects of bortezomib on the human copper transporter 1. Clin Cancer Res. 2009;15:553–60. doi: 10.1158/1078-0432.CCR-08-2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Molloy SA, Kaplan JH. Copper-dependent recycling of hCTR1, the human high affinity copper transporter. J Biol Chem. 2009;284:29704–13. doi: 10.1074/jbc.M109.000166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo Y, Smith K, Lee J, Thiele DJ, Petris MJ. Identification of methionine-rich clusters that regulate copper-stimulated endocytosis of the human Ctr1 copper transporter. J Biol Chem. 2004;279:17428–33. doi: 10.1074/jbc.M401493200. [DOI] [PubMed] [Google Scholar]
- 23.Liu J, Sitaram A, Burd CG. Regulation of copper-dependent endocytosis and vacuolar degradation of the yeast copper transporter, ctr1p, by the rsp5 ubiquitin ligase. Traffic. 2007;8:1375–84. doi: 10.1111/j.1600-0854.2007.00616.x. [DOI] [PubMed] [Google Scholar]
- 24.Esposito DL, Li Y, Cama A, Quon MJ. Tyr(612) and Tyr(632) in human insulin receptor substrate-1 are important for full activation of insulin-stimulated phosphatidylinositol 3-kinase activity and translocation of GLUT4 in adipose cells. Endocrinology. 2001;142:2833–40. doi: 10.1210/endo.142.7.8283. [DOI] [PubMed] [Google Scholar]
- 25.Wu H, Windmiller DA, Wang L, Backer JM. YXXM motifs in the PDGF-beta receptor serve dual roles as phosphoinositide 3-kinase binding motifs and tyrosine-based endocytic sorting signals. J Biol Chem. 2003;278:40425–28. doi: 10.1074/jbc.C300225200. [DOI] [PubMed] [Google Scholar]
- 26.Lee S, Howell SB, Opella SJ. NMR and mutagenesis of human copper transporter 1 (hCtr1) show that Cys-189 is required for correct folding and dimerization. Biochim Biophys Acta. 2007;1768:3127–34. doi: 10.1016/j.bbamem.2007.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Safaei R, Maktabi MH, Blair BG, Larson CA, Howell SB. Effects of the loss of Atox1 on the cellular pharmacology of cisplatin. J Inorg Biochem. 2009;103:333–41. doi: 10.1016/j.jinorgbio.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee J, Petris MJ, Thiele DJ. Characterization of mouse embryonic cells deficient in the Ctr1 high affinity copper transporter. J Biol Chem. 2002;277:40253–59. doi: 10.1074/jbc.M208002200. [DOI] [PubMed] [Google Scholar]
- 29.Blair B, Larson C, Adams P, Abada P, Safaei R, Howell S. Regulation of copper transporter 2 expression by copper and cisplatin in human ovarian carcinoma cells. Mol Pharmacol. 2010;77:912–21. doi: 10.1124/mol.109.062836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blair BG, Larson CA, Adams PL, Abada PB, Pesce CE, Safaei R, et al. Copper transporter 2 regulates endocytosis and controls tumor growth and sensitivity to cisplatin in vivo. Mol Pharmacol. 2011;79:157–66. doi: 10.1124/mol.110.068411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Larson CA, Adams PL, Jandial DD, Blair BG, Safaei R, Howell SB. The role of the N-terminus of mammalian copper transporter 1 in the cellular accumulation of cisplatin. Biochem Pharmacol. 2010;80:448–54. doi: 10.1016/j.bcp.2010.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Larson CA, Adams PL, Blair BG, Safaei R, Howell S. The role of the methionines and histidines in the transmembrane domain of mammalian copper transporter 1 in the cellular accumulation of cisplatin. Mol Pharmacol. 2010;78:333–9. doi: 10.1124/mol.110.064766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quail JF, Tsai CY, Howell SB. Characterization of a monoclonal antibody capable of reliably quantifying expression of human copper transporter 1 (hCTR1) J Trace Elem Med Biol. 2014;28:151–9. doi: 10.1016/j.jtemb.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo Y, Smith K, Petris MJ. Cisplatin stabilizes a multimeric complex of the human Ctr1 copper transporter: requirement for the extracellular methionine-rich clusters. J Biol Chem. 2004;279:46393–99. doi: 10.1074/jbc.M407777200. [DOI] [PubMed] [Google Scholar]
- 35.Smith AL, Friedman DB, Yu H, Carnahan RH, Reynolds AB. ReCLIP (reversible cross-link immunoprecipitation): an efficient method for interrogation of labile protein complexes. PLoS One. 2011;6:e16206. doi: 10.1371/journal.pone.0016206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsai CY, Finley JC, Ali SS, Patel HH, Howell SB. Copper influx transporter 1 is required for FGF, PDGF and EGF-induced MAPK signaling. Biochem Pharmacol. 2012;84:1007. doi: 10.1016/j.bcp.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haremaki T, Fraser ST, Kuo YM, Baron MH, Weinstein DC. Vertebrate Ctr1 coordinates morphogenesis and progenitor cell fate and regulates embryonic stem cell differentiation. Proc Nat Acad Sci USA. 2007;104:12029–34. doi: 10.1073/pnas.0701413104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turski ML, Brady DC, Kim HJ, Kim BE, Nose Y, Counter CM, et al. A novel role for copper in Ras/mitogen-activated protein kinase signaling. Mol Cell Biol. 2012;32:1284–95. doi: 10.1128/MCB.05722-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tinti M, Kiemer L, Costa S, Miller ML, Sacco F, Olsen JV, et al. The SH2 domain interaction landscape. Cell Rep. 2013;3:1293–305. doi: 10.1016/j.celrep.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martin TF. Phosphoinositide lipids as signaling molecules: common themes for signal transduction, cytoskeletal regulation, and membrane trafficking. Annu Rev Cell Dev Biol. 1998;14:231–64. doi: 10.1146/annurev.cellbio.14.1.231. [DOI] [PubMed] [Google Scholar]
- 41.Futter CE, Collinson LM, Backer JM, Hopkins CR. Human VPS34 is required for internal vesicle formation within multivesicular endosomes. J Cell Biol. 2001;155:1251–64. doi: 10.1083/jcb.200108152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Banci L, Bertini I, Ciofi-Baffoni S, Kozyreva T, Zovo K, Palumaa P. Affinity gradients drive copper to cellular destinations. Nature. 2010;465:645–8. doi: 10.1038/nature09018. [DOI] [PubMed] [Google Scholar]
- 43.Eisses JF, Kaplan JH. The mechanism of copper uptake mediated by human CTR1: a mutational analysis. J Biol Chem. 2005;280:37159–68. doi: 10.1074/jbc.M508822200. [DOI] [PubMed] [Google Scholar]
- 44.Sinani D, Adle DJ, Kim H, Lee J. Distinct mechanisms for CTR1-mediated copper and cisplatin transport. J Biol Chem. 2007;282:26775–85. doi: 10.1074/jbc.M703973200. [DOI] [PubMed] [Google Scholar]
- 45.Eisses JF, Kaplan JH. Molecular characterization of hCTR1, the human copper uptake protein. J Biol Chem. 2002;277:29162–71. doi: 10.1074/jbc.M203652200. [DOI] [PubMed] [Google Scholar]
- 46.Qiu H, Zappacosta F, Su W, Annan RS, Miller WT. Interaction between Brk kinase and insulin receptor substrate-4. Oncogene. 2005;24:5656–64. doi: 10.1038/sj.onc.1208721. [DOI] [PubMed] [Google Scholar]
- 47.Sesti G. Insulin receptor substrate polymorphisms and type 2 diabetes mellitus. Pharmacogenomics. 2000;1:343–57. doi: 10.1517/14622416.1.3.343. [DOI] [PubMed] [Google Scholar]
- 48.Sesti G, Federici M, Hribal ML, Lauro D, Sbraccia P, Lauro R. Defects of the insulin receptor substrate (IRS) system in human metabolic disorders. FASEB J. 2001;15:2099–111. doi: 10.1096/fj.01-0009rev. [DOI] [PubMed] [Google Scholar]
- 49.Fantin VR, Sparling JD, Slot JW, Keller SR, Lienhard GE, Lavan BE. Characterization of insulin receptor substrate 4 in human embryonic kidney 293 cells. J Biol Chem. 1998;273:10726–32. doi: 10.1074/jbc.273.17.10726. [DOI] [PubMed] [Google Scholar]
- 50.Tseng YH, Kriauciunas KM, Kokkotou E, Kahn CR. Differential roles of insulin receptor substrates in brown adipocyte differentiation. Mol Cell Biol. 2004;24:1918–29. doi: 10.1128/MCB.24.5.1918-1929.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lingohr MK, Dickson LM, Wrede CE, Briaud I, McCuaig JF, Myers MG, Jr, et al. Decreasing IRS-2 expression in pancreatic beta-cells (INS-1) promotes apoptosis, which can be compensated for by introduction of IRS-4 expression. Mol Cell Endocrinol. 2003;209:17–31. doi: 10.1016/j.mce.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 52.Hinsby AM, Olsen JV, Mann M. Tyrosine phosphoproteomics of fibroblast growth factor signaling: a role for insulin receptor substrate-4. J Biol Chem. 2004;279:46438–47. doi: 10.1074/jbc.M404537200. [DOI] [PubMed] [Google Scholar]
- 53.Luo Y, Yang C, Jin C, Xie R, Wang F, McKeehan WL. Novel phosphotyrosine targets of FGFR2IIIb signaling. Cell Signal. 2009;21:1370–8. doi: 10.1016/j.cellsig.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]