

Abstract
Exome sequencing has greatly impacted the speed at which new disease genes are identified. In the last year alone, six studies have used exome sequencing to identify new genes involved in intellectual disability, a genetically heterogeneous condition affecting 1–3% of the population. These studies encompass the full gamut of modes of inheritance and phenotypic presentation, including syndromic and non-syndromic conditions, sporadic and familial cases, and dominant and recessive inheritance patterns. Because different disease presentations require different approaches to gene discovery, studies of intellectual disability provide a nearly comprehensive showcase of strategies for exome-driven gene discovery. Despite these successes, the etiology of ~60% of cases of intellectual disability remains unknown. The application of exome sequencing to the clinical diagnosis of intellectual disability in the near future will ultimately reduce the number of idiopathic cases and provide a rich source of sequence variation for the identification of new intellectual disability genes.
Keywords: Cognitive Impairment, Diagnostics, Exome, Exome Sequencing, Intellectual Disability, Mental Retardation, Next-Generation Sequencing, Rare Disease
Introduction
Exome sequencing, the technique of capturing, sequencing and analyzing the 1% of the genome that contains protein coding information (reviewed in (1)), was first applied to gene discovery in 2009 (2), and has since radically increased the speed at which researchers have identified new disease genes. Exome sequencing has had a particularly major impact on the rate of discovery of genes for intellectual disability, and has proven exceptionally well suited to addressing some of the specific challenges of gene discovery in this category of diseases.
Intellectual disability, also sometimes referred to as cognitive impairment or mental retardation (3), is a broad diagnosis encompassing a wide variety of phenotypes and severities. Clinically, it is characterized by a substantially below-average score on tests of mental ability or intelligence, and limitations in the ability to function in areas of daily life (4–6). Intellectual disability can range from mild to profound, and can be associated with other clinical findings as part of a syndrome or can occur as an isolated phenotype. Taken together, syndromic and non-syndromic forms of intellectual disability affect over two million individuals in the U.S alone — approximately 1–3% of the population (7, 8). The cost of care over a lifetime can be extremely high; the Center for Disease Control (CDC) estimates over $1M per individual over the course of his or her lifetime (9). Beyond the financial challenges, caring for a dependent with intellectual disabilities can have substantial social and emotional effects on a family. Parents and siblings of individuals with intellectual disabilities can experience chronic stress due to stigmatized social interactions and an alienation from unaffected families, a prolonged burden of care that can last the dependent’s entire life, and/or the deep emotional challenges of adapting to a reality significantly different than the one they had imagined (10, 11). Knowing the genetic cause of intellectual disability in a relative can often bring comfort to families, allow for anticipation and treatment of associated clinical findings, and support the testing of additional family members for carrier or affected status. Unfortunately, the genetic cause of most cases of intellectual disability remains unknown.
Genetic Causes of Intellectual Disability
Although non-genetic factors (e.g., infection, trauma) can result in cognitive impairment, most severe forms of intellectual disability have a genetic basis, with underlying mutations occurring at scales of resolution ranging from large cytogenetic abnormalities to point mutations and even including epigenetic alterations. For example, microscopically visible cytogenetic aberrations (aneuploidies, gross deletions, inversions and rearrangements) cause ~15% of cases (12); smaller copy number variants (CNVs) visible by microarray explain another ~15% (13); point mutations and small insertions or deletions (indels) are found in over 90 X-linked forms, which account for an additional ~10% of cases (14). In addition, epigenetic defects in methylation have been identified in rare cases (15). Overall, there have been over 100 autosomal and X-linked genes implicated in non-syndromic forms of intellectual disability (14). Currently, the genetic etiology of 60% of intellectual disability remains unexplained (16).
There are many reasons why the genetics of intellectual disability have been difficult to unravel, but the most important are extensive genetic and phenotypic heterogeneity. Families with multiple affected children are rare, either because many sporadic cases of intellectual disability are caused by de novo mutations (17, 18) or because of the deliberate limitation of family sizes after the birth of an intellectually disabled child (19). As a result, linkage studies, which require multiple affected individuals per family, often yield insufficient resolution to identify the disease gene.
The history of genetic studies of intellectual disability highlights these challenges. Large pedigrees are more common in X-linked conditions, and linkage analysis has led to the discovery of over 60 X-linked genes for non-syndromic intellectual disability (20). However, only seven autosomal genes have been identified in non-syndromic intellectual disability (21–27), primarily by homozygosity mapping in consanguineous families. This approach has also been used for the identification of several of the genes implicated in severe intellectual disability associated with primary microcephaly (28–34). Cytogenetic abnormalities have also played an important role in the identification of intellectual disability genes and over 30 genes have been identified by examination of breakpoints of cytogenetic anomalies in patients with intellectual disability (20). Examples include the ATP7A gene in Menkes disease (35) and the CDKL5 gene in West syndrome/atypical Rett syndrome (36). Cytogenetic microdeletions, detected by FISH or array comparative genomic hybridization (CGH) technologies, have led to the localization and identification of intellectual disability genes such as LIS1 in lissencephaly (37) and STXBP1 in early infantile epileptic encephalopathy (38). Another successful approach has been the interrogation of additional genes in pathways (39) or within protein complexes (40, 41) that include known intellectual disability genes. As an extreme example of this approach, Tarpey et. al. (42) screened all genes on the X chromosome, due to the preponderance of X-linked intellectual disability conditions, and identified nine new intellectual disability genes. Finally, some intellectual disability genes have been identified through studies of mouse models with specific neurological phenotypes; such studies led to the identification of the ARX and TUBA1A genes in lissencephaly (43, 44).
Recent technological advances that have made the ability to sequence entire human coding regions (exomes) both affordable and practical have turned this field on its head, making it theoretically possible to now identify causal mutations in most individuals with intellectual impairment regardless of frequency, heterogeneity, and inheritance.
Exome Sequencing
In the last year alone, six studies have used exome sequencing to identify new genes involved in intellectual disability. These cases, discussed below, encompass the full gamut of modes of inheritance and phenotypic presentation, including syndromic and non-syndromic conditions, sporadic and familial cases, and dominant and recessive inheritance patterns. Different disease presentations have required different analytic approaches, making intellectual disability a nearly comprehensive showcase of strategies for exome-driven gene discovery.
Exome Sequencing in Sporadic, Syndromic Conditions Involving Intellectual Disability
Many syndromic conditions with consistent and clearly identifiable phenotypes may have monogenic etiologies. Astute clinicians can therefore aggregate patients with classic manifestations of the syndrome, and expect that many of them will have mutations in the same gene. The general exome strategy for identifying causal genes is to scan for genes harboring potentially disruptive mutations in all or most of the patients’ exomes assayed, a process that has been termed “intersection-filtering” (45) (Figure 1a). This is the general approach that led to the recent discoveries the genes for Schinzel-Giedion syndrome (46) and Kabuki syndrome (47).
Figure 1.
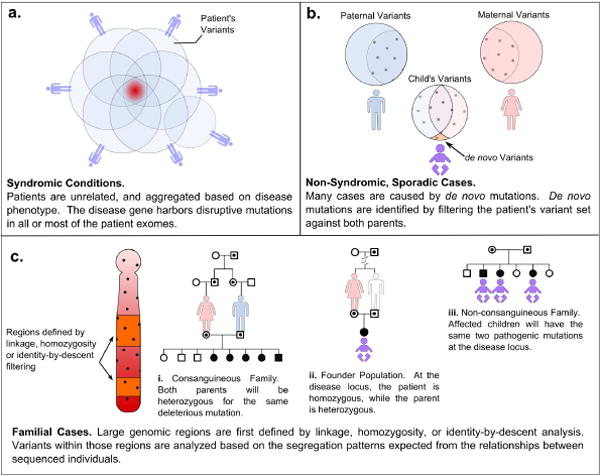
Analysis Strategies for Identifying Disease Genes Using Exome Sequencing Data. a | Syndromic Conditions. For syndromic conditions, it is possible to group unrelated patients based on a shared collection of phenotypes. The causative gene will harbor disruptive mutations in all or most of the patient exomes. In this example, one patient does not have a mutation in the gene responsible for the syndrome in the other patients. This is the strategy used to identify the causative genes for Schinzel-Giedeon syndrome and Kabuki syndrome. b | Non-Syndromic, Sporadic Cases. Many sporadic cases of intellectual disability are caused by small de novo mutations, acting in a dominant mode. De novo mutations are identified by sequencing the exomes of the patient and both parents, and filtering the patient’s variants against the parents’. c | Familial Cases. In recurrent, familial cases of intellectual disablity, genotyping multiple individuals in an extended pedigree can be used to localize the disease gene to genomic regions based on linkage, homozygosity mapping, or identity-by-descent filtering. The exomes of a few key individuals can then be sequenced, and variants within the implicated regions can be further filtered based on family structure and the specific relationships between the sequenced individuals.
Schinzel-Giedion Syndrome
Schinzel-Giedion syndrome is a recognizable syndrome characterized by distinctive facial features, severe intellectual disability, multiple congenital abnormalities, and a higher-than-normal prevalence of tumors (46). Schinzel-Giedion syndrome occurs sporadically and is, therefore, not amenable to linkage mapping. Instead, Hoischen et. al. sequenced the exomes of four individuals with Schinzel-Giedion syndrome, identifying over 21,000 variants per patient (46). Because it was assumed that the disease was dominant, the investigators hypothesized that the syndrome was caused by de novo heterozygous mutations in each individual. They filtered the variants present in the four individuals by first excluding variants in dbSNP and those observed frequently in this and other exome sequencing projects in their own laboratory. They then focused on the non-synonymous changes in the remaining set. Only two genes harbored unique missense changes in all four patients. The sequence data for one of those genes was unreliable because many of the variants fell in regions displaying high homology to other genomic loci, suggesting that many variant calls were likely artifacts of sequence misalignment. The remaining candidate gene was SETBP1, on chromosome 18q12.3.
All four patients had different missense changes in SETBP1, and targeted Sanger sequence analysis in the parents indicated that the variants were indeed de novo mutations. Targeted Sanger sequencing of nine additional children with Schinzel-Giedion syndrome identified SETPB1 mutations in eight of them, whereas no mutations in this gene were found in 188 control chromosomes. The missense changes clustered in a highly conserved 11 base pair region, affecting three of four conserved, consecutive amino acids. Little is known about the function of the SETBP1 protein, except that it is ubiquitously expressed, and contains SET domains. The mutations cluster around a predicted glycosaminoglycan attachment that has homology to the oncogene SKI. Because all mutations in Schinzel-Giedion syndrome patients were missense, and because rare individuals with 18q deletions that include the SETBP1 gene have a different phenotype than that associated with Schinzel-Giedion syndrome (48), the authors suggested that Schinzel-Giedion syndrome is caused by gain of function mutations in SETBP1.
Kabuki Syndrome
Kabuki syndrome is characterized by mild to moderate intellectual disability, a distinctive facial gestalt including highly arched, disrupted eyebrows, and cardiac and skeletal abnormalities (47). Ng et. al. focused their initial analysis on the exomes of 10 unrelated individuals with Kabuki syndrome. They also used a variation of intersection filtering to identify the causative gene, although a number of interesting complications necessitated a more complex analytic approach. These complications include a more variable disease phenotype, false positives due to the incompleteness of current databases of genetic variation, and false negatives related to incompleteness of sequence coverage.
Ng, et. al. also first filtered out variants present in dbSNP, the 1000 genomes project, and in-house databases. Filtering for genes containing novel variants in every one of the 10 patients identified a single gene, MUC16. However, the authors argued that this was a false lead because MUC16 is one of the largest genes in the human genome (190 kb) and would, therefore, be expected to harbor more variation solely on the basis of its size. Moreover, the function of MUC16 and its expression pattern made it an unlikely candidate for a neurological phenotype. The investigators then relaxed their criteria and allowed for the possibility of misdiagnoses, genetic heterogeneity, and unequal sequence coverage at the causal locus between patients. In this re-analysis, they ranked patients by disease severity, and gave more weight to truncating mutations (nonsense mutations and frame shifts). This led to the identification of MLL2 as a strong candidate for the causative gene for Kabuki Syndrome, with truncating mutations present in seven of 10 patients. MLL2 is a histone 3 lysine 4 methyl-transferase involved in chromatin remodeling and regulation of gene expression (49). In mice, heterozygous deletions are embryonic lethal (50), but no previous evidence linked the gene directly to neurological processes or phenotypes.
Because the identification of indels remains a major challenge in next generation sequencing, the investigators further examined samples from the remaining three patient using comparative genomic hybridization (CGH) arrays and Sanger sequencing of the full MLL2 gene. In two of the three patients, they found truncating indel mutations that had been missed in the exome sequence data. Indel detection generally requires more sequence reads mapping to the chromosomal position (i.e., greater read depth), especially for short read sequence data such as that generated by earlier iterations of Illumina instruments. Indel detection is growing more robust as newer sequencers generate longer reads, and as algorithms for identifying this class of variants improve (51, 52).
Subsequent confirmatory studies using Sanger sequencing identified MLL2 mutations in 26 of 43 additional Kabuki patients. Among 12 of the 26 patients for which parental samples were available, all causative mutations were de novo. Furthermore, no truncating mutations in MLL2 were found in 190 control chromosomes from individuals of matched ancestry. Together, these data indicated that mutations in MLL2 cause Kabuki syndrome. Previously, more than 10 different chromosomal anomalies had been identified in Kabuki patients (53), but none were in the region including the MLL2 gene, or, for that matter, even involved the chromosome that harbors MLL2 (chr12). This suggests that additional genes may be causative for Kabuki syndrome, or that some of the patients with chromosome abnormalities may have been misdiagnosed.
Exome Sequencing in Sporadic, Non-syndromic Cases of Intellectual Disability
Many individuals with intellectual disability present as isolated cases in an otherwise unaffected extended family, and without associated phenotypes (i.e., nonsyndromic). Discovering the genetic cause of these cases is exceptionally challenging because it is not possible to assume a priori that any particular group of patients with non-syndromic intellectual disability have mutations in the same gene, or to use SNP or haplotype information in family members to limit the genomic search space for a causative gene. It is likely that de novo mutations are responsible for many of these cases, as humans have a relatively high per-generation mutation rate (54, 55) and de novo CNVs haven been shown to be causal in ~15% of cases of intellectual disability (56, 57).
To test the hypothesis that small de novo mutations are responsible for many sporadic cases of non-syndromic intellectual disability, Vissiers et. al. (58) sequenced the exomes of 10 affected children and their unaffected parents to identify exonic or splice-site variants present in the child but not in either parent (Figure 1b). Their a priori criteria for a trio’s inclusion in this study were no previous family history, no clear syndromic indications, no evidence of Fragile X syndrome, and no de novo chromosomal abnormalities detected by comparative genomic hybridization microarray studies.
On average, they found less than two de novo exonic variants per individual. Further filtering of those variants based on evolutionary constraint and predicted effect on protein function resulted in the identification of likely causative mutations in six patients, in six different genes. Two of those genes (RAB39B and SYNGAP1) had been previously implicated in intellectual disability (23, 59), and evidence from network studies or mouse models provided evidence that the other four genes (DYNC1H1, YY1, DEAF1 and CIC) may be essential for proper development of the nervous system. Although these data provide strong support for the hypothesis that de novo small mutations cause many isolated cases of intellectual disability, functional studies are necessary to validate the causative role of the newly identified mutations. Nonetheless, this approach promises to be quite powerful for identifying de novo mutations in the large number of sporadic cases of intellectual impairment in the population, and may even be useful as a diagnostic approach.
Exome Sequencing in Familial Cases of Intellectual Disability
Although many cases of intellectual disability are sporadic, there are families with multiple affected children and pedigrees consistent with both autosomal recessive and X-linked inheritance patterns. In these cases, linkage studies and haplotype analysis can be used to localize causal genes to genomic regions. This targeting can provide a useful filtering tool in the analysis of the extensive genetic variation uncovered in exome data (Figure 1c). The remaining three examples of successful exome sequencing studies utilized pedigree data in combination with exome sequences to discover mutations in autosomal recessive forms of intellectual disability.
Consanguineous Family
In a consanguineous family, affected children are assumed to be homozygous for an identical-by-descent allele derived from a single, common ancestor, for which both parents are heterozygous (Figure 1.c.i). Caliskan et. al. (27) studied a large consanguineous family in which five of 13 children had a non-syndromic form of intellectual disability encompassing developmental delay and poor language and fine motor skills. The group first conducted linkage studies that narrowed their search to a gene-rich region on chromosome 19p13. They then used high resolution SNP genotyping to identify a two megabase (Mb) region that was homozygous in all affected individuals, and which contained over 30 genes. Only the affected individuals shared the homozygous stretch, suggesting that this is indeed a fully penetrant recessive condition.
Because the parents were assumed to be obligate carriers for the causative mutation, the investigators chose to sequence the parent’s exomes and to limit their analysis to a search for novel, disruptive, heterozygous variants in the 2Mb region that were not present in dbSNP. These filters quickly led to the identification of a single variant that was a missense mutation in a highly conserved residue in the TECR gene. TECR is a synaptic glycoprotein involved in the biosynthesis of long chain fatty acids, and highly expressed in the brain. Follow-up Sanger sequencing and genotyping studies confirmed that the variant segregated with the disease phenotype in the family. Furthermore, homozygosity for the mutation was not found in >1000 unaffected members of this community. Although over 90 X-linked genes have been implicated in non-syndromic intellectual disability, TECR was only the seventh gene identified as causing an autosomal recessive form.
Founder Population
The frequency of particular alleles can be higher in some ethnic groups than in the larger population, due to founder effects and population bottlenecks. It therefore becomes reasonable to expect that a recessive condition may result from the homozygous presentation of a rare allele, even if the parents are not immediately related (Figure 1.c.ii). Edvardson et. al. (60) used this premise as the basis of a study of Joubert Syndrome, an autosomal recessive condition characterized by intellectual disability, a specific midhindbrain malformation, cerebellar ataxia, hypotonia, and frequently, oculomotor and renal phenotypes. The investigators sequenced the exomes of just two Ashkenazi Jewish individuals, an affected child and her unaffected mother, and searched for variants that were homozygous in the daughter and heterozygous in the mother. They identified seven candidate variants within a genomic region that had been previously defined by homozygosity mapping in a larger pedigree, similar to the example above. Five of those variants were present in dbSNP and only one of the two remaining variants was non-synonymous; an arginine to leucine change in the TMEM216 gene. This group performed exome sequencing in parallel with Sanger-based sequencing of candidate genes in the same locus, and both methods identified the same mutation. The exome sequencing approach provided the added benefit of demonstrating that no other disruptive mutations existed in the larger linked region. Homozygosity of the variant segregated with the disease phenotype in the larger pedigree. Currently, little is known about the function of TMEM216.
Non-Consanguineous Family
Approaches for identifying genetic causes of recessive conditions in non-consanguineous families that are not members of a founder population are necessarily different and more challenging than the studies described, above because the disease locus will not necessarily be homozygous in affected individuals. However, estimating the parental source of haplotypes can be used to narrow the search space (Figure 1.c.iii).
Krawitz et. al. (61) studied a non-consanguineous family with three children affected by Hyperphosphatasia Mental Retardation Syndrome (HMRS, also known as Marby syndrome), an autosomal recessive condition that manifests as intellectual disability, elevated alkaline phosphatase levels, a distinctive facial gestalt, and brachytelephalangy. Although they did not assume that the mother and father both carried the same mutant allele, the group reasoned that all affected children in this family would have inherited the same combination of maternal and paternal haplotypes (identity-by-descent (IBD) = 2) at the disease locus.
After acquiring exome sequence data for the three children with HMRS, they identified regions that were consistent with IBD=2. This reduced their search space to ~20% of the exome. They then searched within these regions for genes carrying rare variants in all three affected children, which further narrowed their candidates to two genes. The activity of one of those genes, PIGV, was highly consistent with the disease etiology. PIGV is a mannosyltransferase in the GPI anchor biosynthesis pathway, and similar clinical phenotypes arise from mutations in other interacting genes in this pathway. Follow up work identified homozygous or compound heterozygous mutations in three additional families.
Benefits and Challenges of Exome Sequencing
The benefits of exome sequencing studies are clear. In a single experiment, nearly all the coding content of the genome, the region likely to contain most of the disease-causing mutations, can be assayed. It is not necessary to limit the exploration to a pre-defined region, and it is possible to identify genes acting in unexpected pathways or cellular processes. Perhaps more importantly, the sample size required for a sufficiently powered study is dramatically reduced, often to a few phenotypically related individuals, a single extended family, or even a single parent and child. This efficiency could be the aspect of the exome sequencing approaches that will have the most profound impact on the field.
Despite the great potential of exome sequencing for identifying mutations for rare diseases, there remain many challenges. As mentioned previously, exome data generated by the current technologies is incomplete for several reasons. First, depth of coverage is not uniform across the exomes, and some regions are routinely missed. This can occur due to biases in the capture, sequencing and alignment processes. The capture process is biased by differences in GC content and secondary sequence structure. Exact matches to a probe sequence are also more likely to be captured, so heterozygous variants can represent substantially less than half of the captured DNA from a locus; this is especially true for genomic DNA fragments containing indels. Second, sequence alignment can introduce biases, and can perform poorly in disambiguating sequences among highly homologous sequences or in regions with repeated sequences. Lastly, mapping exome sequences to a single reference sequence can further bias variant calling because reads with variants are often assigned lower mapping quality scores, which can ultimately exclude those variants from subsequent analysis. Many of these problems are alleviated by longer capture probes and longer, paired-end sequence reads. Thus, as the technology improves many of these current limitations will be less problematic, although investigators should always be cognizant of these factors because any of them could potentially introduce biases, regardless of technology advancements.
The larger challenge, of course, is interpretation. Exome sequencing generates a huge amount of data — millions of bases of sequence information and tens of thousands of variants per individual. As the technological aspects of rapid data generation become routine, the challenge for both research and diagnostic applications will be to implement streamlined analytic strategies for quickly classifying relevant pathogenic versus benign variants. This should be possible as more and more exome sequences in healthy and diseased individuals become available.
Application of Exome Sequencing to Diagnostics
Exome sequencing will soon be an effective tool for identifying the genetic basis of many diagnostically intractable cases of intellectual disability, as has already been accomplished for a few other diseases (62, 63). However, reliable integration of exome sequencing into the realm of routine genetic diagnostics requires that substantial attention be paid to the unique practical and ethical challenges of this application. It will be necessary that national advisory bodies such as the American College of Medical Genetics (ACMG), Centers for Disease Control (CDC), the College of American Pathologists (CAP) and others generate technical guidelines to enable the integration of exome sequencing for diagnostic use. For example, guidelines regarding sequence depth and sequence coverage requirements, quality metrics for alignment and variant calling algorithms, reporting of variants and “missed” regions, validation and proficiency testing processes, and data storage and availability will all be important.
From a technical standpoint, a diagnostic test requires complete coverage of the target region, and extremely low false positive and false negative rates. Practically speaking, at least for genes already implicated in a particular disease, this means substantially greater sequencing depth than is usually required for research purposes, to support higher accuracy of variant calls, generally 20–30× at each base. The susceptibility of low-depth sequence coverage to false variant calls was illustrated in the study of Vissiers et. al. (58), in which nearly 75% of their originally identified de novo variant calls (with a median coverage 5×), were proven to be false. Exome sequence data may routinely have to be supplemented by Sanger-based sequencing to cover regions missed due to limitations in exome capture, sequencing or alignment. Improvements to alignment algorithms, especially for identifying indels, are also necessary and are being made (51, 52). Interpretation of novel variants remains a challenge, but one that will gradually be made easier by the continued development of deeper catalogs of genetic variation in healthy individuals, and more comprehensive estimates of allele frequencies, especially for alleles at the low end of the frequency spectrum.
The ethical issues surrounding diagnostic exome sequencing are also complex. Exome data include extensive genetic information that is unrelated to the disorder being tested. The return of these “incidental” results, such as variants that influence the likelihood of acquiring other diseases, or variants that indicate carrier status for unrelated diseases, raises ethical concerns, as does the failure to return that information. What information should be returned, and how one consents patients to a test with such expansive boundaries, are active subjects of debate (64). Additional research will be necessary to understand the medical and psychological impact of returning these kinds of data, and guidelines from supervisory councils will be required to ensure that harm is not inadvertently done. Ownership, storage and access to data become major issues when generating a data set that may have many unanticipated uses over the course of the patient’s lifetime. There are also substantial practical challenges for physicians, who may receive what are likely to be long reports, heavy with variants of unknown significance.
In the near term, we suggest that many of these technical and ethical issues may be alleviated by a targeted analysis approach, in which the full exome sequence is generated in patients, but analysis is initially limited to those genes already known to play a role in the presenting disorder (Figure 2). This approach makes achieving complete coverage practical, substantially limits the number of un-interpretable sequence variants obtained, and sidesteps many ethical issues by limiting the number of returned results to those related to the disease at hand. A consensus among the medical genetics community regarding the amount and kinds of evidence that would be considered sufficient to link a gene to a disease in a clinical setting will be useful in this regard.
Figure 2.
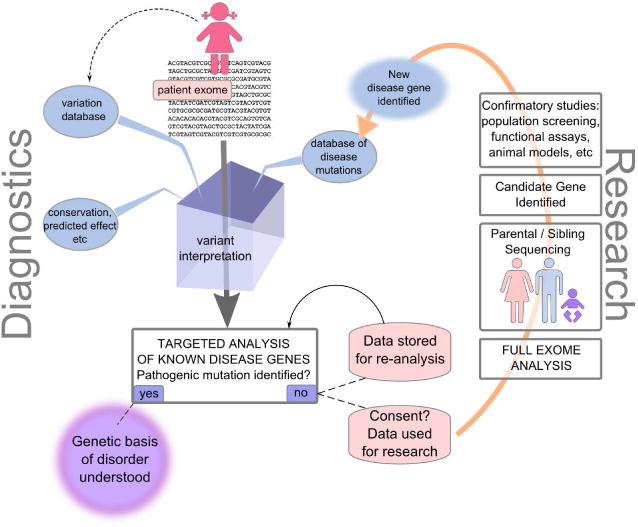
Integrated Diagnostic/Research Information Flow. A patient presents with idiopathic intellectual disability, and her exome is sequenced in a diagnostic laboratory. Variants are automatically annotated with respect to population frequency, evolutionary conservation, predicted effect on transcript expression or splicing, effect on protein function, and are checked against databases of proven disease-causing mutations. To make this process more robust, all variant data are also deposited into a local database of genetic variation. The laboratory then undertakes a targeted analysis of the set of genes known to be potentially causative of her condition, and information about mutations in those genes is reported back to the physician. In the case where no causative mutation is found, her sequence data are stored and periodically reanalyzed as additional genes are implicated in her disease. If research consent has been obtained, the exome data may be used for gene discovery. Full analysis of a patient’s exome, along with sequencing of parents’ or siblings’ exomes may result in the identification of new candidate genes. Confirmatory studies, such as analysis of other affected and unaffected individuals, in vitro functional assays of the normal and mutant sequences, and studies in animal models can confirm or refute the relevance of the gene and mutation for the disease in question. If confirmed, the result is added to databases of disease genes and informs future diagnostics and the re-interpretation of stored sequences from other patients.
Retaining access to exome sequence data can provide a substantial benefit to patients without clinical diagnoses. For example, our understanding of the genetics of intellectual disability is extensive but far from complete, and new genes are identified regularly. In cases where a causative mutation cannot be found in the analysis of known disease genes, the exome data could be re-analyzed as new intellectual disability genes are identified. Likewise, as our understanding progresses regarding the roles that modifier genes play in phenotypic presentation or responsiveness to particular therapies, an available exome sequence can be used to ensure that individualized patient care takes advantage of new medical developments.
Also, with appropriate research consent, sequences from patients without mutations in known genes can be used to power gene discovery research. A patient’s exome sequence could be analyzed in its entirety or could support candidate gene analysis, leading to the discovery of mutations in novel genes that could be presented as candidates for further study. Close interactions between diagnostic and research laboratories will therefore become more and more important as previously uncharacterized mutations are found in patient exomes. Likewise, as more sequence data becomes available in the clinical laboratory, it may become possible to aggregate patients based on their disrupted genes, identify common phenotypic characteristics in these patients, or even accrue sufficient numbers of affected patients to enable a robust search for modifier genes. It is clear that exome sequencing has had, and will continue to have, a profound effect on the genetics and diagnosis of intellectual disability. With the further incorporation of exome sequencing into the diagnostic laboratory, the pace and yield of both diagnosis and gene discovery should continue to increase.
Acknowledgments
This work was supported in part by R01 HD21244 to C.O.
Footnotes
Conflicts of Interest:
There are no conflicts of interest for Scott Topper.
There are no conflicts of interest for Carole Ober.
There are no conflicts of interest for Soma Das
Contributor Information
Scott Topper, Email: stopper@bsd.uchicago.edu, Department of Human Genetics, University of Chicago.
Carole Ober, Email: c-ober@bsd.uchicago.edu, Department of Human Genetics, University of Chicago.
Soma Das, Email: sdas@bsd.uchicago.edu, Department of Human Genetics, University of Chicago.
References
- 1.Teer JK, Mullikin JC. Exome sequencing: the sweet spot before whole genomes. Hum Mol Genet. 2010;19:R145–151. doi: 10.1093/hmg/ddq333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ng SB, Turner EH, Robertson PD, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schalock RL, Luckasson RA, Shogren KA, et al. The renaming of mental retardation: understanding the change to the term intellectual disability. Intellect Dev Disabil. 2007;45:116–124. doi: 10.1352/1934-9556(2007)45[116:TROMRU]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 4.Luckasson R, Borthwick-Duffy S, Buntinx WHE, et al. Mental retardation: Definition, classification, and systems of supports. 10. Washington, DC: American Association on Mental Retardation. (BEST); 2002. [Google Scholar]
- 5.Moeschler JB. Medical genetics diagnostic evaluation of the child with global developmental delay or intellectual disability. Curr Opin Neurol. 2008;21:117–122. doi: 10.1097/WCO.0b013e3282f82c2d. [DOI] [PubMed] [Google Scholar]
- 6.CDC . About Intellectual Disability. Centers for Disease Control and Prevention; 2005. http://www.cdc.gov/ncbddd/dd/ddmr.htm. [Google Scholar]
- 7.CDC. State-specific rates of mental retardation—United States, 1993. MMWR Morb Mortal Wkly Rep. 1996;45:61–65. [PubMed] [Google Scholar]
- 8.Curry CJ, Stevenson RE, Aughton D, et al. Evaluation of mental retardation: recommendations of a Consensus Conference: American College of Medical Genetics. Am J Med Genet. 1997;72:468–477. doi: 10.1002/(sici)1096-8628(19971112)72:4<468::aid-ajmg18>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 9.CDC. Economic costs associated with mental retardation, cerebral palsy, hearing loss, and vision impairment—United States, 2003. MMWR Morb Mortal Wkly Rep. 2004;53:57–59. [PubMed] [Google Scholar]
- 10.Jamison JW. The impact of mental retardation on the family and some directions of help. Journal of the National Medical Association. 1965;57:136–138. [PMC free article] [PubMed] [Google Scholar]
- 11.Wikler L, Wasow M, Hatfield E. Chronic sorrow revisited: parent vs. professional depiction of the adjustment of parents of mentally retarded children. The American journal of orthopsychiatry. 1981;51:63–70. doi: 10.1111/j.1939-0025.1981.tb01348.x. [DOI] [PubMed] [Google Scholar]
- 12.Leonard H, Wen X. The epidemiology of mental retardation: challenges and opportunities in the new millennium. Ment Retard Dev Disabil Res Rev. 2002;8:117–134. doi: 10.1002/mrdd.10031. [DOI] [PubMed] [Google Scholar]
- 13.Zahir F, Friedman JM. The impact of array genomic hybridization on mental retardation research: a review of current technologies and their clinical utility. Clin Genet. 2007;72:271–287. doi: 10.1111/j.1399-0004.2007.00847.x. [DOI] [PubMed] [Google Scholar]
- 14.Ropers HH. Genetics of early onset cognitive impairment. Annual review of genomics and human genetics. 2010;11:161–187. doi: 10.1146/annurev-genom-082509-141640. [DOI] [PubMed] [Google Scholar]
- 15.Gropman AL, Batshaw ML. Epigenetics, copy number variation, and other molecular mechanisms underlying neurodevelopmental disabilities: new insights and diagnostic approaches. J Dev Behav Pediatr. 2010;31:582–591. doi: 10.1097/DBP.0b013e3181ee384e. [DOI] [PubMed] [Google Scholar]
- 16.Rauch A, Hoyer J, Guth S, et al. Diagnostic yield of various genetic approaches in patients with unexplained developmental delay or mental retardation. American journal of medical genetics Part A. 2006;140:2063–2074. doi: 10.1002/ajmg.a.31416. [DOI] [PubMed] [Google Scholar]
- 17.Uher R. The role of genetic variation in the causation of mental illness: an evolution-informed framework. Mol Psychiatry. 2009;14:1072–1082. doi: 10.1038/mp.2009.85. [DOI] [PubMed] [Google Scholar]
- 18.McClellan J, King MC. Genetic heterogeneity in human disease. Cell. 2010;141:210–217. doi: 10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
- 19.Collins J, Marvelle A, Stevenson R. Sibling recurrence in intellectual disability of unknown cause. Clin Genet. 2011;79:498–500. doi: 10.1111/j.1399-0004.2010.01601.x. [DOI] [PubMed] [Google Scholar]
- 20.Ropers HH. X-linked mental retardation: many genes for a complex disorder. Curr Opin Genet Dev. 2006;16:260–269. doi: 10.1016/j.gde.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 21.Basel-Vanagaite L, Attia R, Yahav M, et al. The CC2D1A, a member of a new gene family with C2 domains, is involved in autosomal recessive non-syndromic mental retardation. J Med Genet. 2006;43:203–210. doi: 10.1136/jmg.2005.035709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garshasbi M, Hadavi V, Habibi H, et al. A defect in the TUSC3 gene is associated with autosomal recessive mental retardation. Am J Hum Genet. 2008;82:1158–1164. doi: 10.1016/j.ajhg.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hamdan FF, Gauthier J, Spiegelman D, et al. Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. N Engl J Med. 2009;360:599–605. doi: 10.1056/NEJMoa0805392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Higgins JJ, Pucilowska J, Lombardi RQ, et al. A mutation in a novel ATP-dependent Lon protease gene in a kindred with mild mental retardation. Neurology. 2004;63:1927–1931. doi: 10.1212/01.wnl.0000146196.01316.a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Molinari F, Rio M, Meskenaite V, et al. Truncating neurotrypsin mutation in autosomal recessive nonsyndromic mental retardation. Science. 2002;298:1779–1781. doi: 10.1126/science.1076521. [DOI] [PubMed] [Google Scholar]
- 26.Motazacker MM, Rost BR, Hucho T, et al. A defect in the ionotropic glutamate receptor 6 gene (GRIK2) is associated with autosomal recessive mental retardation. Am J Hum Genet. 2007;81:792–798. doi: 10.1086/521275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caliskan M, Chong JX, Uricchio L, et al. Exome sequencing reveals a novel mutation for autosomal recessive non-syndromic mental retardation in the TECR gene on chromosome 19p13. Hum Mol Genet. 2011;20:1285–1289. doi: 10.1093/hmg/ddq569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alkuraya FS, Cai X, Emery C, et al. Human Mutations in NDE1 Cause Extreme Microcephaly with Lissencephaly. Am J Hum Genet. 2011 doi: 10.1016/j.ajhg.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bond J, Roberts E, Mochida GH, et al. ASPM is a major determinant of cerebral cortical size. Nat Genet. 2002;32:316–320. doi: 10.1038/ng995. [DOI] [PubMed] [Google Scholar]
- 30.Bond J, Roberts E, Springell K, et al. A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat Genet. 2005;37:353–355. doi: 10.1038/ng1539. [DOI] [PubMed] [Google Scholar]
- 31.Guernsey DL, Jiang H, Hussin J, et al. Mutations in centrosomal protein CEP152 in primary microcephaly families linked to MCPH4. Am J Hum Genet. 2010;87:40–51. doi: 10.1016/j.ajhg.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jackson AP, Eastwood H, Bell SM, et al. Identification of microcephalin, a protein implicated in determining the size of the human brain. Am J Hum Genet. 2002;71:136–142. doi: 10.1086/341283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumar A, Girimaji SC, Duvvari MR, et al. Mutations in STIL, encoding a pericentriolar and centrosomal protein, cause primary microcephaly. Am J Hum Genet. 2009;84:286–290. doi: 10.1016/j.ajhg.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nicholas AK, Khurshid M, Desir J, et al. WDR62 is associated with the spindle pole and is mutated in human microcephaly. Nat Genet. 2010;42:1010–1014. doi: 10.1038/ng.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vulpe C, Levinson B, Whitney S, et al. Isolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPase. Nat Genet. 1993;3:7–13. doi: 10.1038/ng0193-7. [DOI] [PubMed] [Google Scholar]
- 36.Kalscheuer VM, Tao J, Donnelly A, et al. Disruption of the serine/threonine kinase 9 gene causes severe X-linked infantile spasms and mental retardation. Am J Hum Genet. 2003;72:1401–1411. doi: 10.1086/375538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lo Nigro C, Chong CS, Smith AC, et al. Point mutations and an intragenic deletion in LIS1, the lissencephaly causative gene in isolated lissencephaly sequence and Miller-Dieker syndrome. Hum Mol Genet. 1997;6:157–164. doi: 10.1093/hmg/6.2.157. [DOI] [PubMed] [Google Scholar]
- 38.Saitsu H, Kato M, Mizuguchi T, et al. De novo mutations in the gene encoding STXBP1 (MUNC18–1) cause early infantile epileptic encephalopathy. Nat Genet. 2008;40:782–788. doi: 10.1038/ng.150. [DOI] [PubMed] [Google Scholar]
- 39.Hamdan FF, Gauthier J, Araki Y, et al. Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am J Hum Genet. 2011;88:306–316. doi: 10.1016/j.ajhg.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deardorff MA, Kaur M, Yaeger D, et al. Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of cornelia de Lange syndrome with predominant mental retardation. Am J Hum Genet. 2007;80:485–494. doi: 10.1086/511888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jaglin XH, Poirier K, Saillour Y, et al. Mutations in the beta-tubulin gene TUBB2B result in asymmetrical polymicrogyria. Nat Genet. 2009;41:746–752. doi: 10.1038/ng.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tarpey PS, Smith R, Pleasance E, et al. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat Genet. 2009;41:535–543. doi: 10.1038/ng.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Keays DA, Tian G, Poirier K, et al. Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell. 2007;128:45–57. doi: 10.1016/j.cell.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kitamura K, Yanazawa M, Sugiyama N, et al. Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat Genet. 2002;32:359–369. doi: 10.1038/ng1009. [DOI] [PubMed] [Google Scholar]
- 45.Robinson PN. Whole-exome sequencing for finding de novo mutations in sporadic mental retardation. Genome Biol. 2010;11:144. doi: 10.1186/gb-2010-11-12-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoischen A, van Bon BW, Gilissen C, et al. De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat Genet. 2010;42:483–485. doi: 10.1038/ng.581. [DOI] [PubMed] [Google Scholar]
- 47.Ng SB, Bigham AW, Buckingham KJ, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010;42:790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Buysse K, Menten B, Oostra A, et al. Delineation of a critical region on chromosome 18 for the del(18)(q12.2q21.1) syndrome. American journal of medical genetics Part A. 2008;146A:1330–1334. doi: 10.1002/ajmg.a.32267. [DOI] [PubMed] [Google Scholar]
- 49.Issaeva I, Zonis Y, Rozovskaia T, et al. Knockdown of ALR (MLL2) reveals ALR target genes and leads to alterations in cell adhesion and growth. Mol Cell Biol. 2007;27:1889–1903. doi: 10.1128/MCB.01506-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Glaser S, Schaft J, Lubitz S, et al. Multiple epigenetic maintenance factors implicated by the loss of Mll2 in mouse development. Development. 2006;133:1423–1432. doi: 10.1242/dev.02302. [DOI] [PubMed] [Google Scholar]
- 51.Albers CA, Lunter G, Macarthur DG, et al. Dindel: Accurate indel calls from short-read data. Genome Res. 2010 doi: 10.1101/gr.112326.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Depristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adam MP, Hudgins L. Kabuki syndrome: a review. Clin Genet. 2005;67:209–219. doi: 10.1111/j.1399-0004.2004.00348.x. [DOI] [PubMed] [Google Scholar]
- 54.Roach JC, Glusman G, Smit AF, et al. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science. 2010;328:636–639. doi: 10.1126/science.1186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lynch M. Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci U S A. 2010;107:961–968. doi: 10.1073/pnas.0912629107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cook EH, Jr, Scherer SW. Copy-number variations associated with neuropsychiatric conditions. Nature. 2008;455:919–923. doi: 10.1038/nature07458. [DOI] [PubMed] [Google Scholar]
- 57.de Vries BB, Pfundt R, Leisink M, et al. Diagnostic genome profiling in mental retardation. Am J Hum Genet. 2005;77:606–616. doi: 10.1086/491719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vissers LE, de Ligt J, Gilissen C, et al. A de novo paradigm for mental retardation. Nat Genet. 2010;42:1109–1112. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- 59.Giannandrea M, Bianchi V, Mignogna ML, et al. Mutations in the small GTPase gene RAB39B are responsible for X-linked mental retardation associated with autism, epilepsy, and macrocephaly. Am J Hum Genet. 2010;86:185–195. doi: 10.1016/j.ajhg.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Edvardson S, Shaag A, Zenvirt S, et al. Joubert syndrome 2 (JBTS2) in Ashkenazi Jews is associated with a TMEM216 mutation. Am J Hum Genet. 2010;86:93–97. doi: 10.1016/j.ajhg.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Krawitz PM, Schweiger MR, Rodelsperger C, et al. Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat Genet. 2010;42:827–829. doi: 10.1038/ng.653. [DOI] [PubMed] [Google Scholar]
- 62.Choi M, Scholl UI, Ji W, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A. 2009;106:19096–19101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Worthey EA, Mayer AN, Syverson GD, et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genetics in medicine : official journal of the American College of Medical Genetics. 2011;13:255–262. doi: 10.1097/GIM.0b013e3182088158. [DOI] [PubMed] [Google Scholar]
- 64.Sharp RR. Downsizing genomic medicine: approaching the ethical complexity of whole-genome sequencing by starting small. Genetics in medicine : official journal of the American College of Medical Genetics. 2011;13:191–94. doi: 10.1097/GIM.0b013e31820f603f. [DOI] [PubMed] [Google Scholar]