

Abstract
The discovery of new classes of antibacterial agents, particularly those with unique biological targets, is essential to keep pace with emerging drug resistance in pathogenic bacteria. We identified the minimal structural component of the cyclic acyldepsipeptide (ADEP) antibiotics that exhibits antibacterial activity. We found that N-acyldifluorophenylalanine fragments function via the same mechanism of action as ADEPs, as evidenced by the requirement of ClpP for the fragments’ antibacterial activity, the ability of fragments to activate B. subtilis ClpP in vitro, and the capacity of an N-acyldifluorophenylalanine affinity matrix to capture ClpP from B. subtilis cell lysates. N-acyldifluorophenylalanine fragments are much simpler in structure than the full ADEPs and are also highly amenable to structural diversification. Thus, the stage has been set for the development of non-peptide activators of ClpP that can be used as antibacterial agents.
Keywords: Antibacterials, ClpP, Peptides, Fragments, Proteolysis
Recently, the cyclic acyldepsipeptide (ADEP) antibiotics have garnered considerable attention because of their potent antibacterial activity against a broad range of Gram-positive bacterial pathogens and their efficacy in animal models of bacterial infection.[1] The cellular target of the ADEPs is ClpP, a component of the highly conserved Clp proteolytic complexes in bacteria.[1c, 2] ClpP is a self-compartmentalized, tetradecameric serine peptidase that functions in conjunction with accessory AAA+ partners (ATPases associated with diverse cellular activities), which themselves play important roles in substrate recognition and regulation of ClpP proteolytic activity.[2d, 3] Clp proteolytic complexes degrade a variety of misfolded and native target proteins, including those involved regulating stress responses and virulence-factor production.[4] ADEPs bind ClpP and dysregulate its activity by opening its axial pores, yielding a peptidase that indiscriminately degrades large peptides and unstructured proteins without the involvement of AAA+ partners, ultimately leading to inhibition of cell division and cell death.[1c, 2a]
In crystal structures of the B. subtilis ClpP tetradecamer in complex with ADEPs, the small molecules bind with exquisite specificity at the intra-subunit sites to which AAA+ partners, like ClpX and ClpA, also bind.[2b, 2c, 5] Intriguingly, it has been proposed that the N-acylphenylalanine side chain appended to the ADEP peptidolactone mimics the highly conserved IGF or LGF tripeptide motifs of ClpX and ClpA that mediate interactions with ClpP.[2b, 5] We find this proposal to be thought-provoking in light of our recent finding that rigidification of the peptidolactone via incorporation of conformationally constrained amino acids can improve ClpP activation in vitro by as much as 7-fold.[6] These observations motivated us to carry out experiments to define the relative contributions of the ADEP peptidolactone and its side chain to ClpP binding and activation.
For this analysis, we prepared fragments of the ADEP structure (1) including an ADEP peptidolactone with an N-acetylated serine residue (2), an N-acetyldifluorophenylalanyl peptidolactone (3), a serine-prolyl ester coupled to N -E-2-heptenoyldifluoro-phenylalanine (4), and an N-E-2-heptenoyldifluorophenylalanine methyl ester (5) (Figure 1). The bioactivities of the fragments were assessed in growth inhibition assays using wild-type B. subtilis AG174, a highly ADEP-susceptible bacterium. Only, fragments 4 and 5 exhibited antibacterial activity, albeit with much lower potency than intact ADEP (Figure 1). Clearly, the N-acyldifluorophenylalanine moiety is necessary and sufficient for antibacterial activity. Interestingly, fragment 4, which has an acyclic portion of the ADEP peptidolactone, is less active than fragment 5, which is composed only of the N-acyldifluorophenylalanine moiety.
Figure 1.
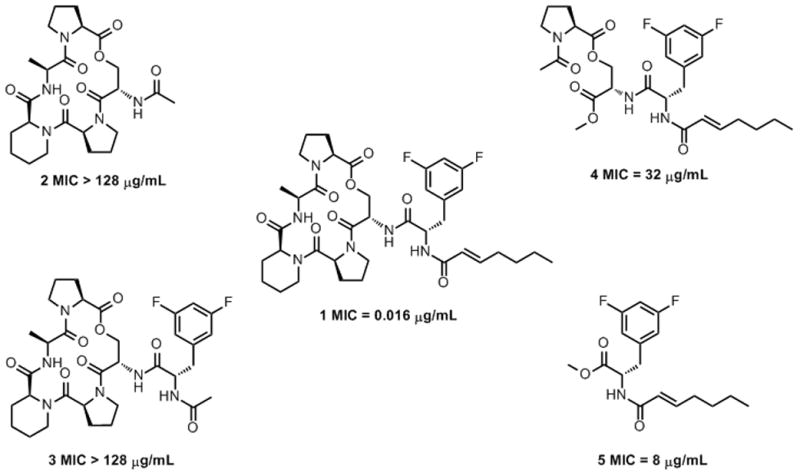
Fragments of a cyclic acyldepsipeptide have differential antibacterial activity against B. subtilis.
Based on our recent report that conformational flexibility of the ADEP peptidolactones strongly influences ClpP binding,[6] we predicted that substitution of the serine in 4 with either allo-threonine (6) or threonine (7) (Figure 2) would yield more rigid and bioactive fragments. However, both compounds exhibited the same antibacterial activity as fragment 4 (MIC = 32 μg/mL), suggesting that the allo-threonine and threonine residues do not significantly or favorably restrict the conformations of the acyclic fragments.
Figure 2.

Structures and antibacterial activities of N-acyldifluorophenylalanine fragment analogs against B. subtilis.
A positional scanning approach was used to define the minimal structural requirements for the bioactivity of fragment 5. First, we synthesized analogs with varied acyl chains (8-10; Figure 2). Fragment 8 bears an acyl chain that is common to previously reported synthetic ADEPs[1c] and fragment 9 bears the acyl chain of the A54556 ADEP natural products.[1a] Fragment 10 is an analog that we synthesized to test the effects of acyl group carbon-branching. Fragments 5, 8, and 10 were equally potent (MIC = 8 μg/mL), whereas fragment 9 exhibited attenuated activity (MIC = 32 μg/mL). This result is consistent with the literature, wherein analogs with polyunsaturated side chains are reportedly less active compared to analogs with α,β-unsaturation.[1d] Fragments 11 and 12 (Figure 2) with saturated acyl groups had reduced antibacterial activity like their ADEP counterparts.[1d] We next studied a pair of analogs in which the difluorophenylalanine was replaced by either phenylalanine (13) or leucine (14). The relatively modest activity of the fragment with the phenylalanine residue (13, MIC = 64 μg/mL) is consistent with the proposal that the fluorine atoms of difluorophenylalnine are engaged in hydrogen bonds with backbone amides of ClpP.[1d] In contrast, the analog containing leucine (14) was completely inactive. Collectively, the results are consistent with published studies of ADEP structure activity relationships.[1d]
Next, we explored the effects of several C-terminal manipulations on the antibacterial activity of fragment 5. Interestingly, the methyl ester (5) was more potent that than either the carboxylic acid or amide analogs thereof (15-18). The low potencies of the carboxamides were surprising, because the N-acyldifluorophenylalanine moiety in the ADEPs is linked to the peptidolactone via an amide bond. In any case, three additional ester analogs (19-21) were prepared. Only the propargyl ester 21 (MIC = 2 μg/mL) was more potent than 5. The potency of the intact ADEPs and the findings that modification of the carboxy-terminus of 5 can improve potency suggest that the structural context in which the bioactive N-acyldifluorophenylalanine moiety is presented is important and can be optimized.
We tested our operating assumption that ADEPs and N-acyldifluorophenylalanine fragments have the same mechanism of killing bacteria in a series of genetic and biochemical experiments. In initial genetic experiments, an ADEP (1) and fragments 4 and 5 were tested for antibacterial activity against two engineered strains of B. subtilis- a spx null strain (AG 1927 spx::neo) and a strain lacking both spx and clpP (AG 1246 spx::neo and clpP::cat).[7] The clpP-spx null strain was selected because the spx null mutation suppresses the slow growth defect exhibited by a B. subtilis strain lacking clpP.[7] The spx null and wild-type strains of B. subtilis, both of which contain a functional ClpP, were equally susceptible to each of the three compounds. Importantly, neither intact ADEP nor fragments 4 and 5 were toxic to the spx-clpP null strain (MIC > 128 μg/mL). The essentiality of a functional clpP gene for the toxicity of both compounds indicates that the fragments share the same mechanism as the ADEPs.
We also tested for cross-resistance by selecting for spontaneously resistant mutants to either 1 or 5 in the spx null strain. Mutants with resistance to the intact ADEP and fragment 5 were observed at frequencies of 3 × 10−6 colony forming units (cfu) and 7 × 10−5 cfu, respectively. As expected, all mutants resistant to 1 were resistant to 5 and vice-versa (MICs >300 μg/mL). By sequencing the clpP locus in the mutants, we determined that resistance was highly correlated with mutations in the promoter of the clpP gene or with mis-sense or frameshift mutations in the clpP open-reading frame (see supporting information).
To biochemically validate the proposal that ADEP fragments activate ClpP peptidase activity, they were tested for activation of B. subtilis ClpP in vitro (Figure 3; Figure S2). Fragments were incubated with B. subtilis ClpP and a fluorogenic decapeptide and initial rates of ClpP mediated decapeptide hydrolysis were measured. All fragments exhibited concentration-dependent activation of ClpP decapeptidase activity and exhibited apparent activation constants (Kapp) ranging from 3.9 – 7.9 μM. Since the binding affinities fall into a narrow range, the large differences in bioactvities of the compounds can be primarily atributed to their in vivo stability and/or cell-permeability. Nevertheless, the fragment with the most potent antibacterial activity (21) was also the tightest ClpP binder. In any case, fragment binding to and activation of ClpP were much weaker than those of ADEP (1) (Kapp = 12 nM, Hill coefficient 2.02±0.08). However, the ADEP (1) and all fragments tested exhibited modest positive cooperativity in ClpP binding (i.e., Hill coefficients >1), suggesting that the ADEP and fragments bind in the same general fashion.
Figure 3.
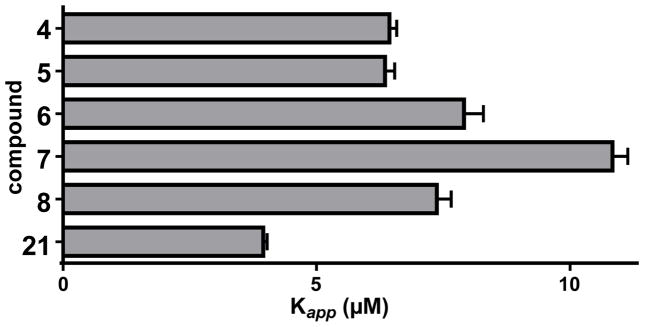
ClpP binding of key N-acyldifluorophenylalanine fragment analogs. Hydrolysis of a fluorogenic decapeptide substrate (15 μM) by B. subtilis ClpP (25 nM) was assayed in the presence of increasing concentrations of ADEP fragments, and activity was fit to a cooperative binding model (Figure S2) to determine apparent binding constants (Kapp) and Hill Coefficients.
To complement our finding that fragments activate ClpP in vitro, we used chemical proteomics experiments utilizing fragment-derived, affinity reagent.[8] The N-acyldifluorophenylalanine propargyl ester (21) was coupled to a biotin-azide conjugate via a Cu(I)-catalyzed Click reaction.[9] After the binding to avidin– agarose beads, the resulting affinity matrix was used to capture proteins from cell lysates of the wild-type and the clpP-spx null strain of B. subtilis. The specifically bound proteins were eluted using free fragment 5 and identified using MASCOT proteomic analysis. We were gratified to find ClpP among the thirty-nine unique and functionally annotated proteins that were positively identified (Table 2, supporting information). Interestingly, 20 of these proteins were also captured from the lysates of the clpP-spx null strain. The biological significance of these off-target binding events is not clear, as compound 14 and other fragments have no effect the growth of the clpP-spx null strain (i.e., the compounds’ bioactivity is dependent on ClpP). A speculative explanation for the off-target binding of the eighteen unique proteins in the lysates of the wild-type strain is that some of them are ClpP substrates (or degradation products thereof) that were trapped within the captured tetradecamer.[15]
Table 2.
Affinity Captured Proteins from B. subtilis Lystates Encoded by Functionally Annotated Genes
| 2-succinyl-6-hydroxy-2,4-cyclohexadiene-1-carboxylate synthase* | DnaB* | MinC* |
| 3-ketoacyl-ACP reductase* | DnaI | RapJ |
| Alpha-galactosidase | Fengycin synthetase* | Ribosomal protein B* |
| Aminoglycoside 6-adenylyltransferase | Flagellar switch protein* | RNA polymerase sigma-54 factor* |
| AnsB | Formamidopyrimidie-DNA glycosidase* | SHCHC synthase* |
| Aspartokinase II alpha subunit | Fructose-1-phosphate kinase | SinR |
| ATP-dependent nuclease | GerC3* | Spore coat protein |
| Bacillomycin synthetase A* | GltC* | SpoVS |
| Cell division ATP-binding protein* | Gluconate kinase* | SrfAA surfactin synthetase |
| Chemotaxis protein | Gramicidin S sythetase* | SubA |
| ClpP | Heptaprenyl diphosphate synthase* | Sucrase |
| Deoxyribodipyrimidine photolyase* | L-aspartase | Superoxide dismutase |
| DNA polymerase I* | Lysine decarboxylase* | TreP |
[a] Thirty-nine functionally annotated proteins positively identified by MASCOT Proteomic Analysis as specifically bound to N-E-2-heptenoyldifluoro-phenylalanine affinity matrix. See supporting information for a complete protein list. Proteins captured from both WT and the clpP null strain of B. subtillis are indicated by asterisks. ClpP is highlighted in bold text.
In conclusion, a truly remarkable example of perturbation of protein-protein interactions by a small molecule underlies the antibacterial activities of the ADEPs. Their binding to ClpP induces significant changes in the quaternary structure[2b, 2c] of the enzyme, which enhance off-target activity and precludes interaction with AAA+ partners.[2a] It has been proposed that binding and activation of ClpP are predicated on the mimicry of IGF and LGF motifs of the AAA+ partners by the ADEP side chain. Here, we report that only the N-acyldifluorophenylalanine moiety of the ADEPs is required for their bioactivities. These results are especially notable in light of reports that an IGF tripeptide alone does not activate ClpP or interfere with the binding of ClpP to ClpX.[11] We believe that the essentiality of the N-acyldifluorophenylalanine moiety for both ClpP activation and antibacterial activity can be reconciled with our recently reported finding that restriction of the peptidolactone dyanamics improves activity. Specifically, the strengthened trans-annular hydrogen bonding between the macrocycle and the side chain could enhance cell-permeability and lock the side chain in a conformation for optimized ClpP binding.
Our definition of the ADEPs’ simple pharmacophore has important implications. Firstly, the ease of fragment synthesis, relative to the ADEPs, makes ClpP activators more accessible to the growing number of groups interested in their mechanism of killing bacteria. In addition, the reported fragments are generally stable and can be stored at room temperature for weeks without any detectable degradation with two exceptions (9: light sensitive; 19: acid sensitive). Secondly, our findings provide a starting point for fragment-based design of non-peptide activators of ClpP. In this ligand-design strategy, weakly active fragments are structurally elaborated into higher affinity ligands. [12] Although fragment-based drug design is a relatively new, it has become widely appreciated as a powerful tool in drug discovery.[12d–f] The fragment-based design strategy is a viable alternative to screening libraries of compounds in the search for non-peptide ClpP activators.[13] Such efforts are motivated by concerns that the ADEP peptidolactone backbone could have pharmacological liabilities that are often associated with peptides, [14] despite the fact that structurally optimized ADEPs effectively cure bacterial infections in mice and rats.[1c, 1d, 1g] Conveniently, the N-acyldifluorophenylalanine moiety is highly amenable to structural elaboration as it possesses a reactive carboxylate functionality that can be easily coupled to a wide array of scaffolds and rapidly diversified. Work to develop more potent antibacterial agents using this strategy is underway in these laboratories.
Experimental Section
Experimental procedures for the preparation of chemical compounds, antibacterial assays, ClpP activation assays, ClpP mutant selection, and chemical proteomics are described in the supporting information
Supplementary Material
Table 1.
| Compound | ClpP Kapp (μM) | Hill Coefficient |
|---|---|---|
| 4 | 6.4±0.1 | 1.4±0.04 |
| 5 | 6.3±0.2 | 1.6±0.07 |
| 6 | 7.9±0.4 | 1.5±0.08 |
| 7 | 11±0.3 | 1.6±0.06 |
| 8 | 7.3±0.3 | 1.2±0.05 |
| 21 | 3.9±0.1 | 1.6±0.04 |
Acknowledgments
JKS and DCW gratefully acknowledge Brown University for funding. JKS is the recipient of a NSF CAREER Award. The work was supported in part by NIH grant GM-101988 to RTS. Expert technical support on NMR and mass spectrometry were provided by R. Hopson, T. Shen, and J. Clifton at Brown University. We thank Prof. A. Grossman of MIT for providing the B. subtilis strains.
Footnotes
Supporting information for this article is available
Contributor Information
Dr. Daniel W. Carney, Department of Chemistry, Brown University, 324 Brook Street, Providence, RI 02912
Corey L. Compton, Department of Chemistry, Brown University, 324 Brook Street, Providence, RI 02912
Dr. Karl R. Schmitz, Department of Biology, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139
Julia P. Stevens, Department of Chemistry, Brown University, 324 Brook Street, Providence, RI 02912
Prof. Dr. Robert T. Sauer, Department of Biology, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139
Prof. Dr. Jason K. Sello, Email: jason_sello@brown.edu, Department of Chemistry, Brown University, 324 Brook Street, Providence, RI 02912
References
- 1.a) Michel KH, Kastner RE. 4492650 A. US Patent No. 1985; b) Osada H, Yano T, Koshino H, Isono K. J Antibiot. 1991;44:1463–1466. doi: 10.7164/antibiotics.44.1463. [DOI] [PubMed] [Google Scholar]; c) Brotz-Oesterhelt H, Beyer D, Kroll H, Endermann R, Ladel C, Schroeder W, Hinzen B, Raddatz S, Paulsen H, Henninger K, Bandow J, Sahl H, Labischinski H. Nat Med. 2005;11:1082–1087. doi: 10.1038/nm1306. [DOI] [PubMed] [Google Scholar]; d) Hinzen B, Raddatz S, Paulsen H, Lampe T, Schumacher A, Häbich D, Hellwig V, Benet-Buchholz J, Endermann R, Labischinski H, Brötz-Oesterhelt H. Chem Med Chem. 2006;1:689–693. doi: 10.1002/cmdc.200600055. [DOI] [PubMed] [Google Scholar]; e) Socha AM, Tan NY, LaPlante KL, Sello JK. Bioorg Med Chem. 2010;18:7193–7202. doi: 10.1016/j.bmc.2010.08.032. [DOI] [PubMed] [Google Scholar]; f) Ollinger J, O’Malley T, Kesicki EA, Odingo J, Parish T. J Bacteriol. 2012;194:663–668. doi: 10.1128/JB.06142-11. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Conlon B, Nakayasu E, Fleck L, LaFleur M, Isabella V, Coleman K, Leonard S, Smith R, Adkins J, Lewis K. Nature. 2013;503:365–370. doi: 10.1038/nature12790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Kirstein J, Hoffmann A, Lilie H, Schmidt R, Rübsamen-Waigmann H, Brötz-Oesterhelt H, Mogk A, Turgay K. EMBO Mol Med. 2009;1:37–49. doi: 10.1002/emmm.200900002. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li DHS, Chung YS, Gloyd M, Joseph E, Ghirlando R, Wright GD, Cheng Y, Maurizi MR, Guarne A, Ortega J. Chem Biol. 2010;17:959–969. doi: 10.1016/j.chembiol.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lee B, Park EY, Lee K, Jeon H, Sung KH, Paulsen H, Rubsamen-Schaeff H, Brotz-Oesterhelt H, Song HK. Nat Struct Mol Biol. 2010;17:471–478. doi: 10.1038/nsmb.1787. [DOI] [PubMed] [Google Scholar]; d) Alexopoulos JA, Guarné A, Ortega J. J Struct Biol. 2012;176:202–210. doi: 10.1016/j.jsb.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 3.a) Joshi SA, Hersch GL, Baker TA, Sauer RT. Nat Struct Mol Boil. 2004;11:404–411. doi: 10.1038/nsmb752. [DOI] [PubMed] [Google Scholar]; b) Yu AYH, Houry WA. FEBS Lett. 2007;581:3749–3757. doi: 10.1016/j.febslet.2007.04.076. [DOI] [PubMed] [Google Scholar]; c) Lee ME, Baker TA, Sauer RT. J Mol Biol. 2010;399:707–718. doi: 10.1016/j.jmb.2010.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Gottesman S, Wickner S, Maurizi MR, Beals CR, Clipstone NA, Ho SN, Crabtree GR, Zamir I, Zhang J, Lazar MA. Genes Dev. 1997 [Google Scholar]; b) Gottesman S, Roche E, Zhou Y, Sauer RT. Genes Dev. 1998;12:1338–1347. doi: 10.1101/gad.12.9.1338. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gaillot O, Pellegrini E, Bregenholt S, Nair S, Berche P. Mol Microbiol. 2000;35:1286–1294. doi: 10.1046/j.1365-2958.2000.01773.x. [DOI] [PubMed] [Google Scholar]; d) Robertson GT, Ng W, Foley J, Gilmour R, Winkler ME. J Bacteriol. 2002;184:3508–3520. doi: 10.1128/JB.184.13.3508-3520.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Frees D, Qazi SNA, Hill PJ, Ingmer H. Mol Microbiol. 2003;48:1565–1578. doi: 10.1046/j.1365-2958.2003.03524.x. [DOI] [PubMed] [Google Scholar]; f) Kwon H, Kim S, Choi M, Ogunniyi AD, Paton JC, Park S, Pyo S, Rhee D. Infect Immun. 2003;71:3757–3765. doi: 10.1128/IAI.71.7.3757-3765.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Gottesman S. Annu Rev Cell Dev Biol. 2003;19:565–587. doi: 10.1146/annurev.cellbio.19.110701.153228. [DOI] [PubMed] [Google Scholar]; h) Kwon H, Ogunniyi AD, Choi M, Pyo S, Rhee D, Paton JC. Infect Immun. 2004;72:5646–5653. doi: 10.1128/IAI.72.10.5646-5653.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Frees D, Sorensen K, Ingmer H. Infect Immun. 2005;73:8100–8108. doi: 10.1128/IAI.73.12.8100-8108.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Sauer RT, Baker TA. Annu Rev Biochem. 2011;80:587–612. doi: 10.1146/annurev-biochem-060408-172623. [DOI] [PubMed] [Google Scholar]; k) Frees D, Gerth U, Ingmer H. Int J Med Microbiol. 2013;304:142–149. doi: 10.1016/j.ijmm.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 5.a) Kim Y, Levchenko I, Fraczkowska K, Woodruff RV, Sauer RT, Baker TA. Nat Struct Mol Biol. 2001;8:230–233. doi: 10.1038/84967. [DOI] [PubMed] [Google Scholar]; b) Martin A, Baker TA, Sauer RT. Mol Cell. 2007;27:41–52. doi: 10.1016/j.molcel.2007.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carney DW, Schmitz KR, Truong JV, Sauer RT, Sello JK. J Am Chem Soc. 2014;136:1922–1929. doi: 10.1021/ja410385c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakano MM, Hajarizadeh F, Zhu Y, Zuber P. Mol Microbiol. 2001;42:383–394. doi: 10.1046/j.1365-2958.2001.02639.x. [DOI] [PubMed] [Google Scholar]
- 8.Rix U, Superti-Furga G. Nat Chem Biol. 2009;5:616–624. doi: 10.1038/nchembio.216. [DOI] [PubMed] [Google Scholar]
- 9.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem Int Ed. 2002;114:2708–2711. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 10.Sass P, Josten M, Famulla K, Schiffer G, Sahl H, Hamoen L, Brötz-Oesterhelt H. Proc Natl Acad Sci USA. 2011;108:17474–17479. doi: 10.1073/pnas.1110385108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joshi SA, Hersch GL, Baker TA, Sauer RT. Nat Struct Mol Biol. 2004;11:404–411. doi: 10.1038/nsmb752. [DOI] [PubMed] [Google Scholar]
- 12.a) Erlanson DA, McDowell RS, O’Brien T. J Med Chem. 2004;47:3463–3482. doi: 10.1021/jm040031v. [DOI] [PubMed] [Google Scholar]; b) Hajduk PJ, Greer J. Nat Rev Drug Disc. 2007;6:211–219. doi: 10.1038/nrd2220. [DOI] [PubMed] [Google Scholar]; c) Congreve M, Chessari G, Tisi D, Woodhead AJ. J Med Chem. 2008;51:3661–3680. doi: 10.1021/jm8000373. [DOI] [PubMed] [Google Scholar]; d) Hung AW, Ramek A, Wang Y, Wilson JA, Clemons PA, Young DW. Proc Natl Acad Sci, USA. 2011;108:6799–6804. doi: 10.1073/pnas.1015271108. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Scott E, Coyne AG, Hudson SA, Abell C. Biochemistry. 2012;51:4990–5003. doi: 10.1021/bi3005126. [DOI] [PubMed] [Google Scholar]; f) McCarthy DJ, Campbell AJ, Kern G, Moustakas D. J Chem Inf Model. 2014;54:693–704. doi: 10.1021/ci400731w. [DOI] [PubMed] [Google Scholar]
- 13.Leung E, Datti A, Cossette M, Goodreid J, McCaw SE, Mah M, Nakhamchik A, Ogata K, El Bakkouri M, Cheng Y. Chem Biol. 2011;18:1167–1178. doi: 10.1016/j.chembiol.2011.07.023. [DOI] [PubMed] [Google Scholar]
- 14.a) Edwards CM, Cohen MA, Bloom SR. QJM. 1999;92:1–4. doi: 10.1093/qjmed/92.1.1. [DOI] [PubMed] [Google Scholar]; b) Loffet A. J Pept Sci. 2002;8:1–7. doi: 10.1002/psc.366. [DOI] [PubMed] [Google Scholar]; c) Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M. Drug Discov Today. 2010;15:40–56. doi: 10.1016/j.drudis.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 15.Kock H, Gerth U, Hecker M. J Bacteriol. 2004;186:5856–5864. doi: 10.1128/JB.186.17.5856-5864.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.