

Abstract
Enhanced cell survival and resistance to apoptosis during thermotolerance correlates with an increased expression of heat shock proteins (Hsps). Here we present additional evidence in support of the hypothesis that the induction of Hsp27 and Hsp72 during acquired thermotolerance in Jurkat T-lymphocytes prevents apoptosis. In thermotolerant cells, Hsp27 was shown to associate with the mitochondrial fraction, and inhibition of Hsp27 induction during thermotolerance in cells transfected with hsp27 antisense potentiated mitochondrial cytochrome c release after exposure to various apoptotic stimuli, despite the presence of elevated levels of Hsp72. Caspase activation and apoptosis were inhibited under these conditions. In vitro studies revealed that recombinant Hsp72 more efficiently blocked cytochrome c–mediated caspase activation than did recombinant Hsp27. A model is presented for the inhibition of apoptosis during thermotolerance in which Hsp27 preferentially blocks mitochondrial cytochrome c release, whereas Hsp72 interferes with apoptosomal caspase activation.
INTRODUCTION
Exposure of cells to transient, nonlethal elevations in temperature results in the synthesis and accumulation of Hsps, which induce a state of thermotolerance and render cells resistant to subsequent lethal insults (Li and Werb 1982; Parsell and Lindquist 1993). The major Hsps of mammalian cells include proteins with molecular masses of 110, 90, 70, 60, 40, and 27 kDa (for review, see Lindquist and Craig 1988; Moseley 1997). Several groups have shown that thermotolerant cells are less sensitive to cytotoxicity induced by hyperthermia, growth factor withdrawal, heavy metals, or anticancer drugs (Landry et al 1989; Jäättelä et al 1992; Mailhos et al 1993; Samali and Cotter 1996; Mosser et al 1997). Apart from the ability of mild heat stress to induce thermotolerance, more severe bouts of heat shock can cause a loss of cell viability by apoptosis or necrosis, if cellular defense mechanisms are incapable of coping with the stress.
Apoptosis is a highly regulated process characterized by condensation of nuclear chromatin, cytoplasmic shrinkage, membrane blebbing, nuclear fragmentation and, finally, the formation of apoptotic bodies (Kerr et al 1972; Wyllie et al 1980). This form of cell death is also associated with the activation of an evolutionarily conserved family of cysteine-aspartate proteases (for review, see Thornberry and Lazebnik 1998), referred to as caspases (Alnemri et al 1996). Caspase activation was recently proposed as the universal biochemical hallmark of apoptosis (Samali et al 1999c). At least 14 caspases have been hitherto identified in mammalian cells. They are synthesized as inactive precursor molecules, procaspases, and are activated by proteolytic cleavage (Thornberry and Lazebnik 1998).
Emerging evidence suggests that mitochondria are critical in the activation and/or amplification of the caspase cascade, via the release of cytochrome c and possibly other factors (Kluck et al 1997; Yang et al 1997). It has been demonstrated that after cytochrome c release from the mitochondrial intermembrane space, this molecule participates in apoptosome formation with Apaf-1 and procaspase-9, leading to the cleavage and activation of other procaspases, including procaspase-3 (Liu et al 1996; Li et al 1997; Zhou et al 1997). The ability of several antiapoptotic proteins, such as Bcl-2 and Bcl-XL, to inhibit apoptosis involves the prevention of cytochrome c release from mitochondria (Kluck et al 1997; Yang et al 1997) or interference with the function of the apoptosome complex (Kim et al 1997).
In recent years, Hsps have also emerged as regulators of apoptosis (for review, see Arrigo 1998; Samali and Orrenius 1998; Jäättelä 1999). These proteins fall within 2 categories: those that accelerate apoptosis—for example, Hsp60 (Samali et al 1999a; Xanthoudakis et al 1999), and those that inhibit the process, such as Hsp27 and Hsp72 (Mehlen et al 1996b; Samali and Cotter 1996; Gabai et al 1997; Mosser et al 1997; Jäättelä et al 1998; Gorman et al 1999; Robertson et al 1999). Despite an increasing number of reports on the modulation of apoptosis by Hsps, relatively little is known about the mechanism by which these proteins can render cells resistant to apoptosis. Here we present evidence suggesting that during thermotolerance, Hsp27 exerts its antiapoptotic effect at the level of the mitochondrion, whereas Hsp72 imparts its effect downstream of mitochondrial cytochrome c release by preventing caspase activation.
MATERIALS AND METHODS
Cell culture and heat shock conditions
Jurkat cells were grown in RPMI 1640 medium supplemented with 10% heat-inactivated fetal calf serum, 2 mM glutamine, 100 U/mL penicillin, and 100 mg/mL streptomycin in a humidified atmosphere of 5% CO2 in air at 37°C. For heat shock, cell numbers were determined with a Neubauer hemocytometer, and the density was adjusted to 106 cells per milliliter. The required numbers of cells were placed in culture flasks, which were sealed by wrapping parafilm around their lids. The flask was immersed in a water bath at the indicated temperatures (±0.5°C) for 1 hour. After the incubation period, cells were resuspended in fresh medium and incubated at 37°C for various times. Cell viability was assessed by the ability of cells to exclude trypan blue dye. Cell morphology was evaluated by staining cytospin preparations, and apoptotic or necrotic cells were scored as described previously (Samali and Cotter 1996).
Determination of cytochrome c release
Cells were washed in phosphate-buffered saline (PBS), and cytosolic fractions were separated from the pellet (mitochondrial fraction) as previously described (Samali et al 1999a). After determination of the protein concentration, the samples were frozen at −70°C until further analysis by Western blotting.
Western blotting
Protein samples were subjected to sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis, and the proteins were transferred onto a nitrocellulose membrane. Western blotting was performed by using mouse monoclonal antibodies to Hsp72 and Hsp27 (StressGen Biotechnologies Corp, Victoria, BC, Canada), PARP (BIOMOL, Plymouth Meeting, PA, USA), Bcl-2 (DAKO Corp, Glostrup, Denmark), cytochrome c (a gift from Dr Ronald Jemmerson), rat monoclonal antibody against Hsc70 and Grp94 (StressGen Biotechnologies Corp), and rabbit polyclonal antibodies to caspase-3 and DFF45 (gifts from Drs Donald Nicholson and Xiaodong Wang, respectively). The appropriate goat secondary antibodies conjugated to horseradish peroxidase were obtained from Pierce. Protein bands were then visualized by using the ECL Western Blot Detection Kit from Amersham Corp, Buckinghamshire, UK.
Measurement of mitochondrial transmembrane potential
Changes in mitochondrial transmembrane potential were detected by using tetramethylrhodamine ethyl ester perchlorate (TMRE, Molecular Probes, Leiden, The Netherlands). TMRE (25 nM) was added to the culture medium 30 minutes before analysis. Samples were then washed and resuspended in buffer (10 mM N-2-hydroxyethylpiperazine-N′2-ethane-sulfonic acid [HEPES] and NaOH, pH 7.4; 150 mM NaCl; 5 mM KCl; 1 mM MgCl2; 1.8 mM CaCl2) containing 25 nM TMRE. Changes in fluorescence were detected by using a FACScan flow cytometer (Becton Dickinson).
Subcellular fractionation
For isolation of the nuclear fraction, cells (2.5 × 108 per milliliter) were resuspended in buffer A (10 mM Tris-HCl [pH 7.5], 2.5 mM KCl, 2.5 mM MgCl2, and 0.5 M sucrose) and kept on ice. Cells were lysed in 0.5 vol of buffer B (10 mM HEPES [pH 7.9], 10 mM KCl, 1.5 MgCl2, and 0.05% NP-40), followed by homogenization (20 strokes) in a Dounce homogenizer. The resulting lysate was immediately diluted with 3 vol of buffer A and transferred to a centrifuge tube. Before centrifugation, 1 vol of buffer C (50 mM Tris-HCl [pH 7.5], 5 mM MgCl2, and 2.1 M sucrose) was added as a cushion to the bottom of the tube and the suspension centrifuged at 1000 × g for 4 minutes (4°C). Supernatants were discarded, and the resultant pellet was resuspended in the same volume of buffer A, along with a buffer C cushion, and centrifuged at 1000 × g for 4 minutes (4°C). Again, supernatants were discarded and the sample layer was resuspended in the bottom layer of sucrose; and 7 mL of buffer C was added as a cushion. After centrifugation at 90 000 × g for 30 minutes (4°C), the resulting nuclei-rich pellet was resuspended in buffer A. For isolation of mitochondrial, cytosolic, and microsomal subfractions, cells were washed in buffer D (100 mM sucrose, 1 mM ethyleneglycotetraacetic acid [EGTA], 20 mM 3-[N-morpholino]propanesulfonic acid (MOPS), pH 7.4) and resuspended in buffer E (buffer D plus 5% Percoll, 0.01% digitonin and a cocktail of protease inhibitors: 10 μM aprotinin, 10 μM pepstatin A, 10 μM leupeptin, 25 μM calpain inhibitor I, and 1 mM pluripotent myeloid stem cell [PMSF]). After 15 minutes of incubation on ice, unbroken cells and nuclei were pelleted by centrifugation at 2500 × g for 10 minutes at 4°C. The supernatant was centrifuged at 15 000 × g for 15 minutes to pellet mitochondria, which were resuspended in buffer F (300 mM sucrose, 1 mM EGTA, 20 mM MOPS, pH 7.4, and the above cocktail of protease inhibitors). The supernatant was further centrifuged at 100 000 × g for 1 hour. The resultant supernatant and the pellet were designated as the cytosolic and microsomal fractions, respectively. The protein concentration of each subfraction was determined by using the Pierce protein assay.
Transfection procedures
Transfection was performed according to the protocol for DOTAP liposomal transfection reagent provided by Boehringer Mannheim, Mannheim, Germany. Cells were transfected with either pCIneohsp27 antisense vector, containing the entire coding sequence of hsp27 placed in reverse orientation under the control of cytomegalovirus (CMV) promoter, or a corresponding control vector. pCIneohsp27 antisense was constructed by using an EcoRI-EcoRI DNA fragment of plasmid psvhsp27, which was inserted in the EcoRI site of pCIneo vector (Promega Corp, Madison, WI, USA). Stable single-cell clones were selected with 250 μg/mL G418.
Preparation of the cytosolic fraction and in vitro caspase activation with cytochrome c and dATP
Control or thermotolerant cells were washed in PBS and resuspended in S-100 buffer (100 mM sucrose, 1 mM EGTA, 20 mM MOPS, pH 7.4, 10 μM aprotinin, 10 μM pepstatin A, 10 μM leupeptin, calpain inhibitor I, and 1 mM PMSF). After 10 minutes of incubation on ice, cells were centrifuged at 900 × g for 5 minutes, followed by a further centrifugation of the supernatant at 100 000 × g for 45 minutes to obtain the cytosolic fraction. For in vitro caspase activation, 30 μL of the cytosolic fraction (2 mg/ mL) was incubated with cytochrome c and deoxyadenosine triphosphate (dATP) for 60 minutes at 30°C.
Caspase assay
The activity of group II caspases (DEVDases) was determined fluorometrically by using a modified version of the method developed by Nicholson et al (1995). Briefly, lysate from 1 × 106 cells and substrate (DEVD-AMC, 50μM) were combined in reaction buffer. A total of 100 mM HEPES, 10% sucrose, 5 mM dithiothreitol, 0.0001% NP-40, and 0.1% 3-[(3-cholamidopropyl) dimethylammonio] propane-1-sulphonic acid, pH 7.25, was added in triplicate to a microtiter plate. The cleavage of the florigenic peptide substrate DEVD-AMC was monitored using a Fluoroscan II plate reader (Labsystems, Stockholm, Sweden) with 355-nm excitation and 460-nm emission wavelengths. Fluorescence was measured at 70-second intervals over a 30-minute period, and fluorescence units were converted to picomoles of AMC by using a standard curve generated with free AMC. Data from triplicate samples were then analyzed by linear regression.
Isolation of nuclei and incubation with cytosolic fraction
Rat thymic nuclei were isolated as described above. Cytosols were prepared and activated with cytochrome c and dATP for 30 minutes, as described above. A cell-free system was then set up to study nuclear apoptosis, consisting of 10 μL of cytosol (7 mg/mL), 2.5 μL nuclei (2 × 106), and 10 μL of adenosine triphosphate (ATP)-regeneration system, containing 2 mM ATP, 10 mM creatine phosphate, and 50 μg/mL creatine kinase. The volume was adjusted to 25 μL with S-100 buffer. Samples were incubated at 37°C for 2 hours and prepared for conventional DNA gel electrophoresis.
Conventional DNA gel electrophoresis
DNA fragmentation was assessed by 1-stage agarose gel electrophoresis. Briefly, 2 × 106 nuclei were pelleted and resuspended in buffer containing RNase (10 mg/mL) for 20 minutes. Samples were then loaded onto a 1.8% agarose gel with a digestion gel (0.8% Ultrapure agarose (Life Technologies), 2% SDS, and 0.6 mg/mL proteinase K) at the upper end. The gel was run at 20 V overnight, and electrophoresis was then continued for 3 hours at 90 V to separate the DNA fragments. The gel was subsequently stained with ethidium bromide, visualized under 305 nm ultraviolet illumination, and photographed with Polaroid 665 positive/negative film.
RESULTS
Acquisition of thermotolerance correlates with inhibition of apoptosis
We have demonstrated previously that a sublethal heat shock (42°C for 1 hour) in Jurkat cells results in an accumulation of Hsp27 and Hsp72 and an attenuation of apoptotic morphology induced by subsequent heat shock at 44°C (Samali et al 1999b). To determine whether inhibition of apoptosis during acquired thermotolerance is linked to an inhibition of caspase activity, cells were either maintained at 37°C or heat-shocked at 42°C for 1 hour, followed by a 12-hour recovery period at 37°C to allow the maximal accumulation of Hsps. Subsequently, cells were subjected to 1 hour of heat shock at the apoptosis-inducing temperature (44°C) and later processed after a 1- or 2-hour recovery at 37°C. Western blot analysis demonstrated that Hsp72 and Hsp27 were induced in cells preconditioned at 42°C (Fig 1a). The preconditioned cells resisted apoptosis induced by a subsequent exposure to heat shock at 44°C, as determined by the lack of procaspase-3 processing and cleavage of PARP and DFF45 (Fig 1b). Furthermore, the number of cells displaying apoptotic morphology was significantly lower in the thermotolerant cell population (Fig 1c). The inhibition of apoptosis was not limited to heat shock, because preconditioned cells were also resistant to induction of apoptosis by hydrogen peroxide as well as etoposide (data not shown). Overall, these results suggest that thermotolerance blocks apoptosis upstream of caspase-3 activation.
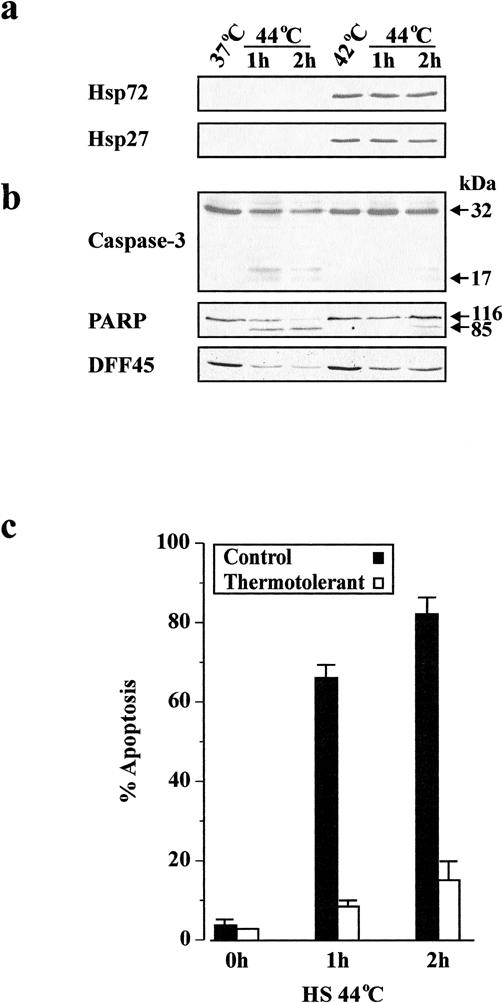
Fig 1. Inhibition of apoptosis in thermotolerant cells. Cells were incubated at either 37°C or 42°C for 1 hour and allowed to recover for 12 hours at 37°C. Cells were then subjected to heat shock at 44°C, followed by recovery for 1 and 2 hours at 37°C. (a) Equal amounts of protein (25 μg per lane) were subjected to SDS-PAGE, followed by Western blot analysis with specific antibodies against Hsp72 or Hsp27. (b) Determination of the extent of processing of caspase-3 and cleavage of PARP and DFF45 by Western blotting. (c) The percentage of apoptosis was assessed by morphological examination of stained cytospin preparations from 3 independent experiments. The results are presented as mean percentage ± SEM
Stress-induced loss of ΔΨm and cytochrome c release are blocked in thermotolerant cells
From the previous set of experiments, it was evident that thermotolerance prevented procaspase-3 processing and apoptosis in Jurkat T cells. However, whether the apoptotic process was blocked before, or downstream of, cytochrome c release from mitochondria was not clear. It has been reported previously that thermotolerance leads to the inhibition of hydrogen peroxide–induced loss of ΔΨm and that mitochondria are targets for the protective effects of thermotolerance against subsequent oxidative injury (Polla et al 1996). Furthermore, we recently demonstrated that heat-induced apoptosis is accompanied by a drop in ΔΨm and a release of cytochrome c from mitochondria (Samali et al 1999b). We therefore hypothesized that inhibition of apoptosis in thermotolerant cells occurs through stabilization of ΔΨm and prevention of cytochrome c release from mitochondria. In support of this hypothesis, monitoring ΔΨm with TMRE demonstrated a lack of dissipation of the ΔΨm after heat shock at 44°C (Fig 2a) as well as hydrogen peroxide treatment (data not shown) of thermotolerant cells. Western blot analysis of the cytosolic and mitochondria-enriched fractions from control and thermotolerant cells after exposure to heat shock (44°C) revealed that the release of cytochrome c was also blocked during thermotolerance (Fig 2b).
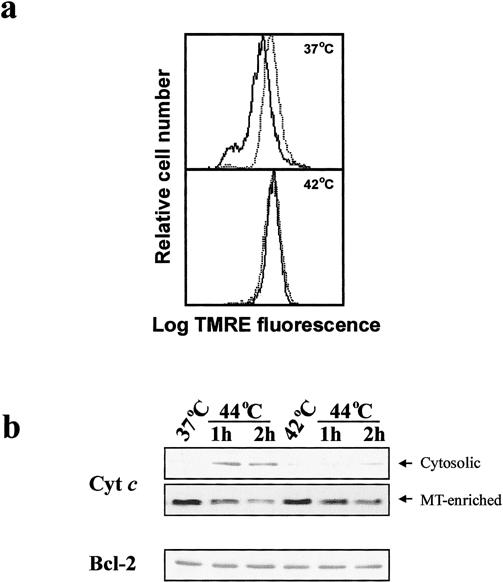
Fig 2. Inhibition of apoptosis occurs upstream of mitochondrial events. Cells were incubated at either 37°C or 42°C for 1 hour and allowed to recover for 12 hours at 37°C. Cells were then subjected to heat shock at 44°C, followed by recovery for 2 hours at 37°C. (a) Flow cytometric analysis of ΔΨm of cells incubated at the indicated temperatures using TMRE (25 nM), before (dotted line) or after (solid line) heat shock at 44°C. (b) Western blot analysis of cytochrome c in the cytosolic fractions and mitochondria-enriched pellets or Bcl-2 in whole cell extracts of control and thermotolerant cells subjected to heat shock at 44°C and recovery for 1 and 2 hours at 37°C
Resistance to apoptosis is often associated with increased expression of Bcl-2, an antiapoptotic protein associated with mitochondria. One study reported that heat shock may upregulate Bcl-2 expression (Polla et al 1996). However, in the present study, the thermotolerant cells did not show any detectable change in the Bcl-2 level, as compared with control cells (Fig 2b, lower panel). These data suggest that the inhibition of heat-induced apoptosis was not linked to altered Bcl-2 expression in preconditioned cells.
Hsp27 exerts its anti-apoptotic effect at the mitochondrial level
Because the resistance to apoptosis in thermotolerant cells was associated with the inhibition of cytochrome c release from mitochondria, we tested the possibility that Hsp27, Hsp72, or both could interact directly with mitochondria. To investigate this possibility, cytosolic, mitochondrial, nuclear, and microsomal subfractions were isolated from control and thermotolerant cells. In comparison to control cells, high levels of Hsp27 and Hsp72 (62% and 58% of the total, respectively) were present in the cytosolic fraction of thermotolerant cells (Fig 3). The mitochondrial fraction of thermotolerant cells also revealed the presence of a significant pool of Hsp27 (33%) and much less of Hsp72 (14%). The nuclear fraction, on the other hand, displayed a marked increase in the levels of Hsp72 (23%). There was no significant change in the Hsp content of the microsomal fraction of the thermotolerant cells (Fig 3).
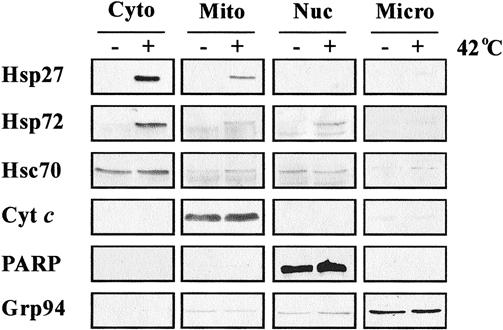
Fig 3. Subcellular fractionation of control and thermotolerant cells. Cytosols, mitochondria, nuclei, and microsomes were isolated from control (−) and heat-shocked (+) Jurkat cells, as described in Materials and Methods. The presence or absence of Hsp27 and Hsp72 in the different fractions was determined by Western blot analysis. Hsc70 (cytosolic/nuclear), cytochrome c (mitochondrial), PARP (nuclear), and Grp94 (microsomal) proteins were also analyzed to confirm the purity of the individual fractions
Induction of both Hsp27 and Hsp72 during thermotolerance makes it difficult to elucidate the respective antiapoptotic effects of these proteins. To determine at what levels in the apoptotic pathway Hsp27 and Hsp72 exert their respective effects, Jurkat cells were stably transfected with hsp27 antisense (Jurkat-AS), or the corresponding control vector (Jurkat-neo). Single-cell clones were selected by using G418. The expression of the antisense was verified by heat shocking the Jurkat-neo and Jurkat-AS cells at 42°C and monitoring the induction of Hsp27 by Western blot analysis. As demonstrated in Fig 4a, heat shock at 42°C resulted in the induction of Hsp27 and Hsp72 in both the wild-type and Jurkat-neo cells, whereas no significant increase in the level of Hsp27 in Jurkat- AS was detected, despite the induction of Hsp72. Moreover, the inhibition of Hsp27 induction in Jurkat-AS cells previously made thermotolerant (42°C for 1 hour) did not affect their viability (data not shown). Thermotolerant Jurkat-AS cells were subsequently heat shocked at 44°C. Suppression of Hsp27 induction in Jurkat-AS cells during thermotolerance enhanced the release of cytochrome c after heat shock at 44°C, despite the presence of Hsp72 (Fig 4b). The response of Jurkat-neo cells (both control and thermotolerant) to heat shock at 44°C was identical to that of wild-type cells (Fig 4b). Surprisingly, the release of cytochrome c in Jurkat-AS cells failed to induce any apparent caspase activation (data not shown) or apoptotic morphology (Fig 4c vs Fig 1c). These results suggest that Hsp27 induces a stabilizing effect on mitochondria and hence prevents the loss of cytochrome c in the presence of a proapoptotic stimulus, whereas some other component of thermotolerant cells, possibly Hsp72, exerts its effect downstream of mitochondria by preventing caspase activation.
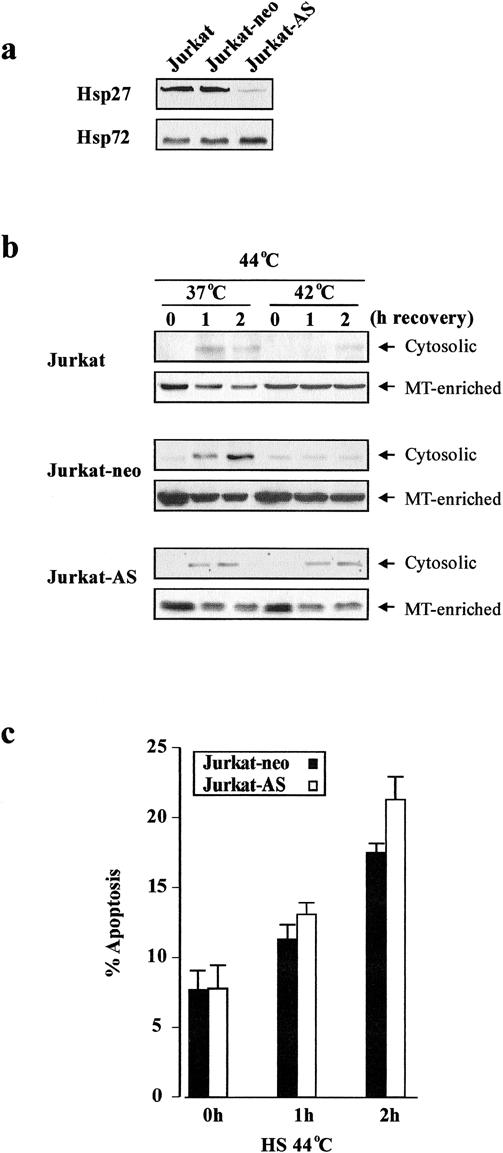
Fig 4. Effect of hsp27 antisense on cytochrome c release and apoptosis. Wild-type, Jurkat-neo, and Jurkat cells were transfected with hsp27 antisense (Jurkat-AS). Jurkat-AS were heat-shocked at 42°C for 1 hour, followed by a 12-hour recovery. (a) Changes in Hsp27 and Hsp72 were analyzed by Western blot analysis. (b) Cytosolic distribution of cytochrome c in control and thermotolerant wild-type, Jurkat- neo, and Jurkat-AS cells at 0, 1, and 2 h of recovery after heat-shock (44°C for 1 hour). (c) Thermotolerant Jurkat-neo and Jurkat-AS cells were heat-shocked (44°C for 1 hour), and the percentage of apoptotic morphology was determined at 0, 1, and 2 hours of recovery at 37°C. The degree of resistance is comparable to wild-type cells shown in Fig 1c.
Cytosolic fractions from thermotolerant cells are resistant to cytochrome c/dATP–stimulated caspase activation
A recent report indicated that apoptosis was inhibited downstream of caspase activation and before nuclear changes in Hsp72-overexpressing cells (Jäättelä et al 1998). We therefore set up an in vitro system to determine if accumulation of Hsps in thermotolerant cells could also inhibit apoptosis downstream of caspase activation. In this in vitro system, the cytosolic fraction from control or thermotolerant cells was incubated with exogenous cytochrome c and dATP to stimulate caspase activation. As demonstrated in Fig 5a, the extent of caspase activity after titration of cytochrome c against cytosols was lower in thermotolerant cells, as compared with control cells. However, with a high concentration of cytochrome c (400 nM), similar levels of caspase activity were detected in the cytosolic fraction from both control and thermotolerant cells. These results suggest that the inhibition of cytochrome c–mediated caspase activation depends on the level of the cytochrome c released from mitochondria. The activation of caspases in the cytosolic fraction led to the cleavage of DFF45 (Fig 5b, upper panel), the inhibitory counterpart of the endonuclease DFF40, even though inducible Hsps were present in this fraction from thermotolerant cells (Fig 5b, lower panel). Cleavage of DFF45 indicated that DFF40 could be activated in the cytosolic fraction from both nonthermotolerant and thermotolerant cells. Moreover, treatment of isolated nuclear fractions with active cytosols, ie, those where cytochrome c and dATP had been added, induced internucleosomal DNA fragmentation characteristic of apoptosis (Fig 5c).

Fig 5. Resistance of cytosolic fractions from thermotolerant cells to cytochrome c/dATP--stimulated caspase activation. Cells were incubated at either 37°C or 42°C for 1 hour and allowed to recover for 12 hours at 37°C. Cytosolic fractions were prepared and incubated with cytochrome c and dATP (1 mM) for 60 minutes at 30°C. (a) The caspase-3-like activity (DEVDase) induced by addition of various concentrations of cytochrome c and dATP in cytosolic fractions was assayed using DEVD-AMC substrate. Inset: the caspase-3-like activity within the cytosols was induced only when they were incubated with both exogenous cytochrome c (100 nM) and dATP. (b) Western blot analysis of DFF45 cleavage (top) and its disappearance in the cytosolic fractions after activation with 100 nM cytochrome c and 1 mM dATP, and (bottom) Hsp72 to demonstrate induction of Hsps in thermotolerant cells. (c) Cytosolic fractions treated with or without cytochrome c (100 nM) and dATP (1 mM) for 60 minutes at 30°C before co-incubation with isolated nuclei. The nuclear DNA was then subjected to agarose gel electrophoresis. The typical internucleosomal DNA fragmentation is detectable in isolated nuclei co-incubated with activated cytosolic fractions. (d) Effect of recombinant Hsp27 and Hsp72 on caspase-3-like activation stimulated by cytochrome c (100 nM) and dATP (1 mM) as determined by DEVD-AMC cleavage
The inhibition of cytochrome c–mediated caspase activation in the cytosolic fraction of thermotolerant cells could be caused by the effects of either one or both Hsps. In order to determine which of the two Hsps conferred a caspase inhibitory effect, cytosolic fractions from control cells were incubated with various concentrations of Hsp27, Hsp72, and bovine serum albumin (BSA) (as a control) before the addition of cytochrome c and dATP. The results showed that the same concentration of Hsp72 was more efficient than Hsp27 at inhibiting cytochrome c–mediated caspase activation (Fig 5d). The effect of BSA on caspase activation was negligible, ie, less than 10% even at the highest concentration (data not shown). An additive inhibitory effect on cytochrome c–mediated caspase activation was noticed when both Hsps were present together (data not shown).
DISCUSSION
Although the expression of Hsps in response to mild stress induces thermotolerance (Li and Werb 1982; Parsell and Lindquist 1993) and protection from apoptosis (Samali and Cotter 1996; Mehlen et al 1996b; Gabai et al 1997; Mosser et al 1997; Jäättelä et al 1998; Gorman et al 1999; Robertson et al 1999), it is still unclear whether the antiapoptotic influence of thermotolerance is caused by one particular Hsp acting at a single level of the process or to a cooperative action of several Hsps acting at different levels. In this study, we focused our attention on two of the stress-inducible Hsps—Hsp27 and Hsp72— with particular interest in their respective abilities to inhibit apoptosis. We have identified a significant pool of Hsp27 in association with the mitochondrial fraction from thermotolerant cells. This observation, combined with data demonstrating the ability of hsp27 antisense to facilitate mitochondrial cytochrome c release, clearly indicates a direct, stabilizing effect of Hsp27 on the mitochondrion. However, the release of cytochrome c did not result in caspase activation or in apoptosis. The latter observation, together with the results showing the inhibition of in vitro caspase activation by recombinant Hsp72, clearly suggests that Hsp72 interferes with apoptosomal procaspase processing. These results are in agreement with recent reports suggesting that inducible Hsp70 (Hsp72) interferes with the formation of the apoptosome (Beere et al 2000; Saleh et al 2000). In this study we also detected a weaker inhibitory effect of recombinant Hsp27 on cytochrome c--mediated caspase activation in an in vitro system. This seems to be a secondary effect and may be caused by the interaction of Hsp27 with apoptosome components (Garrido et al 1999; Pandey et al 2000). Taken together, these experiments suggest that Hsp27 and Hsp72 differentially and selectively inhibit mitochondrial and postmitochondrial events in apoptosis, respectively.
On the basis of our in vitro experiments, we suggest that Hsp27 inhibits the apoptotic process by blocking mitochondrial cytochrome c release (Fig 4). The mechanisms involved may depend on one or more functions traditionally ascribed to this protein. These include (1) the ability of the Hsp27 to function as a molecular chaperone (Jakob et al 1993; Ehrnsperger et al 1997; Rogalla et al 1999); (2) maintenance of the cellular redox state via sequential upregulation of pentose-phosphate metabolism and glutathione levels (Mehlen et al 1996a; Preville et al 1999), and (3) the stabilization or reorganization of cytoskeletal structure during times of stress (Lavoie et al 1993; Lavoie et al 1995). However, it is unclear which, if any, of these established functions of Hsp27 is responsible for the inhibition of mitochondrial cytochrome c release. Our data demonstrating the preferential association of Hsp27 with mitochondria favors the model in which Hsp27 acts as a molecular chaperone, interacting with protein targets on the mitochondrial outer-membrane and specifically preventing the release of cytochrome c. This effect might be achieved through a direct interaction of Hsp27 with one or more components of the permeability transition pore (PTP). Changes in ΔΨm accompany mitochondrial permeability transition, resulting in the opening of the PTP and the release of cytochrome c (Piotrowicz and Levin 1997; Kroemer et al 1998), although other models describing the release of cytochrome c, involving swelling and rupture, have been proposed (Vander Heiden et al 1997). Thus, it is tempting to speculate that the regulation of PTP by Hsp27 occurs in a manner similar to that elicited by Bcl-2 or Bcl-xL, which bind to the voltage-dependent anion channel, a central component of the PTP (Shimizu et al 1999). This is not a far-fetched possibility, because recent emerging evidence supports this hypothesis. A small heat shock protein, reactive with antibodies to Hsp25 (murine form of human Hsp27) was shown to associate with the mitochondria of mammalian cells after heat stress and protect the mitochondrial complex I activity against heat and oxidative stress (Downs et al 1999).
In contrast to Hsp27, this study shows that the antiapoptotic effect of Hsp72 in thermotolerant Jurkat cells is directed downstream of mitochondrial cytochrome c release. This is in agreement with a previous report by Jäättelä et al (1998), but unlike those previous findings, in our studies the blockade of apoptosis caused by Hsp72 occurred upstream of caspase activation. This observation supports recent findings that Hsp72 exerts its antiapoptotic effect by preventing apoptosomal caspase activation (Beere et al 2000; Saleh et al 2000). The lack of any detectable procaspase-3 processing during the inhibition of apoptosis in thermotolerant cells seen here is in agreement with other studies that examine the cytoprotective potential of Hsp72 in cells overexpressing this protein (Gabai et al 1997; Mosser et al 1997). Moreover, our conclusions are supported by observations of a lack of an effect of recombinant Hsp72 on the proteolytic capacities of preactivated caspases (Mosser et al 1997).
In conclusion, we propose a model for the inhibition of apoptosis during thermotolerance in which Hsp27 preferentially blocks mitochondrial cytochrome c release, whereas Hsp72 interferes with apoptosomal caspase activation (Fig 6). According to this model, the collective, antiapoptotic effects of these proteins exceed the ability of each one of them to protect against apoptosis independently, perhaps suggesting why both are indeed coordinately induced during acquired thermotolerance.

Fig 6. A proposed model of the antiapoptotic effects of Hsp27 and Hsp72. Some of the possible sites where Hsp27 and Hsp72 can exert their antiapoptotic effects. (1) Elevated levels of Hsp27 block ROS production and suppress changes in Δψm which can induce cytochrome c release from mitochondria. (2) Hsp27 may specifically interact with mitochondria and inhibit cytochrome c release or interfere with apoptosome formation. (3) However, Hsp72 exerts its effect primarily downstream of cytochrome c release by preventing the activation of one or more caspases
Acknowledgments
We thank Drs A. Gorman, J. Chandra, and B. Fadeel for critical discussions. We are grateful to Drs Donald Nicholson (Merck Frosst Center for Therapeutic Research, Pointe-Claire-Dorval, Quebec) and Ronald Jemmerson (University of Minnesota Medical School, Minneapolis, MN), for providing antibodies to p17 and cytochrome c, respectively. This work was supported by grants from Swedish Medical Research Council (03X-2471; SO and 32X-7140; IAC), the Association pour la Recherche sur le Cancer (5204; A-PA), Individual Marie Curie Fellowship from the European Commission (AS), The American- Scandinavian Foundation (JDR).
REFERENCES
- Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, Wong WW, Yuan J. Human ICE/CED-3 protease nomenclature. Cell. 1996;87:171. doi: 10.1016/s0092-8674(00)81334-3. [DOI] [PubMed] [Google Scholar]
- Arrigo AP. Small stress proteins: chaperones that act as regulators of intracellular redox state and programmed cell death. Biol Chem. 1998;379:19–26. [PubMed] [Google Scholar]
- Beere HM, Wolf BB, Cain K, et al. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to Apaf-1 apoptosome. Nat Cell Biol. 2000;2:469–475. doi: 10.1038/35019501. [DOI] [PubMed] [Google Scholar]
- Downs CA, Jones LR, Heckathorn SA. Evidence for a novel set of small heat-shock proteins that associates with the mitochondria of murine PC12 cells and protects NADH:ubiquinone oxidoreductase from heat and oxidative stress. Biochem Biophys Res Commun. 1999;365:344–350. doi: 10.1006/abbi.1999.1177. [DOI] [PubMed] [Google Scholar]
- Ehrnsperger M, Gräber S, Gaestel M, Buchner J. Binding of non-native protein to Hsp25 during heat shock creates a reservoir of folding intermediates for reactivation. EMBO J. 1997;16:221– 229. doi: 10.1093/emboj/16.2.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabai VL, Meriin AB, Mosser DD, Caron AW, Rits S, Shifrin VI, Sherman MY. Hsp72 prevents activation of stress kinases. A novel pathway of cellular thermotolerance. J Biol Chem. 1997;272:18033–18037. doi: 10.1074/jbc.272.29.18033. [DOI] [PubMed] [Google Scholar]
- Garrido C, Bruey JM, Fromentin A, Hammann A, Arrigo AP, Solary E. HSP27 inhibits cytochrome c-dependent activation of procaspase-9. FASEB J. 1999;13:2061–2070. doi: 10.1096/fasebj.13.14.2061. [DOI] [PubMed] [Google Scholar]
- Gorman AM, Heavey B, Creagh E, Cotter TG, Samali A. Antioxidant-mediated inhibition of the heat shock response leads to apoptosis. FEBS Lett. 1999;445:98–102. doi: 10.1016/s0014-5793(99)00094-0. [DOI] [PubMed] [Google Scholar]
- Jäättelä M. Heat shock proteins as cellular lifeguards. Ann Med. 1999;31:261–271. doi: 10.3109/07853899908995889. [DOI] [PubMed] [Google Scholar]
- Jäättelä M, Wissing D, Bauer P, Li GC. Major heat shock protein Hsp72 protects tumor cells from tumor necrosis factor cytotoxicity. EMBO J. 1992;11:3507–3512. doi: 10.1002/j.1460-2075.1992.tb05433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäättelä M, Wissing D, Kokholm K, Kallunki T, Egeblad M. Hsp72 exerts its anti-apoptotic function downstream of caspase-3-like proteases. EMBO J. 1998;17:6124–6134. doi: 10.1093/emboj/17.21.6124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakob U, Gaestel M, Engel K, Buchner J. Small heat shock proteins are molecular chaperones. J Biol Chem. 1993;268:1517–1520. [PubMed] [Google Scholar]
- Kerr JFR, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CN, Wang X, Huang Y, Ibrado AM, Liu L, Fang G, Bhalla K. Overexpression of Bcl-xL inhibits ara-C-induced mitochondrial loss of cytochrome c and other perturbations that activate the molecular cascade of apoptosis. Cancer Res. 1997;57:3115– 3120. [PubMed] [Google Scholar]
- Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Dallaporta B, Resche-Rigon M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu Rev Physiol. 1998;60:619–642. doi: 10.1146/annurev.physiol.60.1.619. [DOI] [PubMed] [Google Scholar]
- Landry J, Chretien P, Lambert H, Hickey E, Weber LA. Heat shock resistance conferred by expression of the human HSP27 gene in rodent cells. J Cell Biol. 1989;109:7–15. doi: 10.1083/jcb.109.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie JN, Hickey E, Weber LA, Landry J. Modulation of actin microfilament dynamics and fluid phase pinocytosis by phosphorylation of heat shock protein 27. J Biol Chem. 1993;268:24210– 24214. [PubMed] [Google Scholar]
- Lavoie JN, Lambert H, Hickey E, Weber LA, Landry J. Modulation of cellular thermoresistance and actin filament stability accompanies phosphorylation-induced changes in the oligomeric structure of heat shock protein 27. Mol Cell Biol. 1995;15:505– 516. doi: 10.1128/mcb.15.1.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GC, Werb Z. Correlation between synthesis of heat shock proteins and development of thermotolerance in Chinese hamster fibroblasts. Proc Natl Acad Sci USA. 1982;79:3218–3222. doi: 10.1073/pnas.79.10.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Lindquist S, Craig EA. The heat shock proteins. Annu Rev Genet. 1988;22:631–677. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- Liu X, Kim NC, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Mailhos C, Howard MK, Latchman DS. Heat shock protects neuronal cells from programmed cell death by apoptosis. Neuroscience. 1993;55:621–627. doi: 10.1016/0306-4522(93)90428-i. [DOI] [PubMed] [Google Scholar]
- Mehlen P, Preville X, Chareyron P, Briolay J, Klemenz R, Arrigo AP. Constitutive expression of human hsp27, Drosophila hsp27, or human alpha B-crystallin confers resistance to TNF- and oxidative stress-induced cytotoxicity in stably transfected murine L929 fibroblasts. J Immunol. 1995;154:363–374. [PubMed] [Google Scholar]
- Mehlen P, Kretz-Remy C, Preville X, Arrigo A-P. Human hsp27, Drosophila hsp27 and human αB-crystallin expression- mediated increase in glutathione is essential for the protective activity of these proteins against TNFα-induced cell death. EMBO J. 1996a;15:2695–2706. [PMC free article] [PubMed] [Google Scholar]
- Mehlen P, Schulze-Osthoff K, Arrigo AP. Small stress proteins as novel regulators of apoptosis. Heat shock protein 27 blocks Fas/APO-1- and staurosporine-induced cell death. J Biol Chem. 1996b;271:16510–16514. doi: 10.1074/jbc.271.28.16510. [DOI] [PubMed] [Google Scholar]
- Moseley PL. Heat shock proteins and heat adaptation of the whole organism. J Appl Physiol. 1997;83:1413–1417. doi: 10.1152/jappl.1997.83.5.1413. [DOI] [PubMed] [Google Scholar]
- Mosser DD, Caron AW, Bourget L, Denis-Larose C, Massie B. Role of the human heat shock protein Hsp72 in protection against stress-induced apoptosis. Mol Cell Biol. 1997;17:5317–5327. doi: 10.1128/mcb.17.9.5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson DW, Ali A, Thornberry NA, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- Pandey P, Farber R, Nakazawa A, Bharti A, Nalin C, Weichselbaum R, Kufe D, Kharbanda S. Hsp27 functions as a negative regulator of cytochrome c-dependent activation of procaspase- 3. Oncogene. 2000;19:1975–1981. doi: 10.1038/sj.onc.1203531. [DOI] [PubMed] [Google Scholar]
- Parsell DA, Lindquist S. The function of heat shock proteins in stress tolerance: degradation and reactivation of damaged proteins. Annu Rev Genet. 1993;27:437–496. doi: 10.1146/annurev.ge.27.120193.002253. [DOI] [PubMed] [Google Scholar]
- Piotrowicz RS, Levin G. Basolateral membrane-associated 27- kDa heat shock protein and microfilament polymerization. J Biol Chem. 1997;272:25920–25927. doi: 10.1074/jbc.272.41.25920. [DOI] [PubMed] [Google Scholar]
- Polla BS, Kantengwa S, Francois D, Salvioli S, Franceschi C, Marsac C, Cossarizza A. Mitochondria are selective targets for the protective effects of heat shock against oxidative injury. Proc Natl Acad Sci USA. 1996;93:6458–6463. doi: 10.1073/pnas.93.13.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preville X, Salvemini F, Giraud S, Chaufour S, Paul C, Stepien G, Ursini MV, Arrigo A-P. Mammalian small stress proteins protect against oxidative stress through their ability to increase glucose-6-phosphate dehydrogenase activity and by maintaining optimal cellular detoxifying machinery. Exp Cell Res. 1999;247:61–78. doi: 10.1006/excr.1998.4347. [DOI] [PubMed] [Google Scholar]
- Robertson JD, Datta K, Biswal SS, Kehrer JP. Heat-shock protein 70 antisense oligomers enhance proteasome inhibitor-induced apoptosis. Biochem J. 1999;344:477–485. [PMC free article] [PubMed] [Google Scholar]
- Rogalla T, Ehrnsperger M, Preville X, et al. Regulation of Hsp27 oligomerization, chaperone function, and protective activity against oxidative stress/tumor necrosis factor-α by phosphorylation. J Biol Chem. 1999;274:18947–18956. doi: 10.1074/jbc.274.27.18947. [DOI] [PubMed] [Google Scholar]
- Saleh A, Srinivasula SM, Balkiri L, Robbins PD, Alnemri E. Negative regulation of Apaf-1 apoptosome by Hsp72. Nature Cell Biol. 2000;2:476–483. doi: 10.1038/35019510. [DOI] [PubMed] [Google Scholar]
- Samali A, Cai J, Zhivotovsky B, Jones DP, Orrenius S. Presence of pre-apoptotic complex of pro-caspase-3 with Hsp60 and Hsp10 in mitochondrial fraction of Jurkat cells. EMBO J. 1999a;18:2040–2048. doi: 10.1093/emboj/18.8.2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samali A, Cotter TG. Heat shock proteins increase resistance to apoptosis. Exp Cell Res. 1996;223:163–170. doi: 10.1006/excr.1996.0070. [DOI] [PubMed] [Google Scholar]
- Samali A, Holmberg CI, Sistonen L, Orrenius S. Thermotolerance and cell death are distinct cellular responses to stress: dependence on heat shock proteins. FEBS Lett. 1999b;461:306–310. doi: 10.1016/s0014-5793(99)01486-6. [DOI] [PubMed] [Google Scholar]
- Samali A, Orrenius S. Heat shock proteins: regulators of stress response and apoptosis. Cell Stress Chap. 1998;3:228–236. doi: 10.1379/1466-1268(1998)003<0228:hspros>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samali A, Zhivotovsky B, Jones DP, Nagata S, Orrenius S. Apoptosis: cell death defined by caspase activation. Cell Death Differ. 1999c;6:495–496. doi: 10.1038/sj.cdd.4400520. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;28:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Chandel NS, Williamson EK, Schumacker PT, Thompson CB. Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997;91:627–637. doi: 10.1016/s0092-8674(00)80450-x. [DOI] [PubMed] [Google Scholar]
- Wyllie AH, Kerr JFR, Currie AR. Cell death: the significance of apoptosis. Int Dev Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- Xanthoudakis S, Roy S, Rasper D, et al. Hsp60 accelerates the maturation of pro-caspase-3 by upstream activator proteases during apoptosis. EMBO J. 1999;18:2049–2056. doi: 10.1093/emboj/18.8.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Liu X, Bhalla K, et al. Prevention of apoptosis by Bcl- 2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- Zhou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405– 413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]