

Abstract
Hypothermia can occur during fasting when thermoregulatory mechanisms, involving fatty acid (FA) utilization, are disturbed. CD36/FA translocase is a membrane protein which facilitates membrane transport of long-chain FA in the FA consuming heart, skeletal muscle (SkM) and adipose tissues. It also accelerates uptake of triglyceride-rich lipoprotein by brown adipose tissue (BAT) in a cold environment. In mice deficient for CD36 (CD36−/− mice), FA uptake is markedly reduced with a compensatory increase in glucose uptake in the heart and SkM, resulting in lower levels of blood glucose especially during fasting. However, the role of CD36 in thermogenic activity during fasting remains to be determined. In fasted CD36−/− mice, body temperature drastically decreased shortly after cold exposure. The hypothermia was accompanied by a marked reduction in blood glucose and in stores of triacylglycerols in BAT and of glycogen in glycolytic SkM. Biodistribution analysis using the FA analogue 125I-BMIPP and the glucose analogue 18F-FDG revealed that uptake of FA and glucose was severely impaired in BAT and glycolytic SkM in cold-exposed CD36−/− mice. Further, induction of the genes of thermogenesis in BAT was blunted in fasted CD36−/− mice after cold exposure. These findings strongly suggest that CD36−/−mice exhibit pronounced hypothermia after fasting due to depletion of energy storage in BAT and glycolytic SkM and to reduced supply of energy substrates to these tissues. Our study underscores the importance of CD36 for nutrient homeostasis to survive potentially life-threatening challenges, such as cold and starvation.
Keywords: CD36, Hypothermia, Fatty acid, Brown adipose tissue, Skeletal muscle
1. Introduction
In a cold environment, thermoregulatory heat production is increased by either non-shivering thermogenesis (NST), shivering thermogenesis, or physical activity. Among these, NST in brown adipose tissue (BAT) is the most important thermoregulatory mechanism in small mammals and human neonates [1,2]. BAT dissipates chemical energy and generates heat to help protect animals from cold temperatures. Upon stimulation by sympathetic nervous input, cellular triglyceride (TG) stores undergo lipolysis and in BAT the thermogenic regulator peroxisome proliferator-activated receptor gamma co-activator lα (PGClα) is induced [1—3]. For sustaining heat production in BAT, efficient uptake and subsequent utilization of fuels such as glucose and FA are required [4]. Thermogenesis in BAT is mediated through the BAT-specific mitochondrial protein, uncoupling protein 1 (UCP1) [1,2]. Skeletal muscle (SkM) is another crucial tissue for generating heat by shivering and physical activity. Shivering muscles are largely fueled by carbohydrates and lipids [5]. Although the contribution of circulating glucose to total heat generation remains minor (<15% of total heat produced), muscle glycogen becomes the dominant fuel, providing 30–40% of the total heat produced or 75% of the total carbohydrate oxidized [6].
CD36 plays an important role in membrane transport of long-chain FA in the heart, SkM and adipose tissue [7]. The expression of CD36 is increased by cold exposure, which enhances BAT uptake of TG-rich lipoprotein (TRL) and of albumin bound FA [8,9]. Increased activity of local lipoprotein lipase (LPL) accelerates hydrolysis of TG within TRL, followed by efficient engulfment of lipoprotein particles by BAT. Previous studied showed that CD36−/−mice have reduced uptake of FA with a remarkable increase in glucose utilization in the heart and oxidative SkM, lower levels of blood glucose and higher levels of serum NEFAs [10,11] (and our unpublished observation). Compared with oxidative SkM, uptake of FA in glycolytic SkM is less affected while uptake of glucose is marginally enhanced.
In this study, we examined the role of CD36 in thermogenesis in response to cold exposure during fasting. Using CD36−/− and wild-type (WT) mice, we document the indispensable role of CD36 for tolerance of cold temperature when nutrient supply is limited. Most energy substrates for thermogenesis, such as TG in BAT and glycogen in glycolytic SkM are rapidly consumed in the fasted state and mechanisms replenishing these energy substrates fail in CD36−/− mice, resulting in rapid occurrence of fatal hypothermia upon cold exposure.
2. Material and methods
2.1. Mice and sample collection
Mice with a homozygous null mutation in CD36 were generated as previously described [12]. Control male wild-type C57BL6j mice were purchased from Japan SLC, Inc. The ages (10—12 weeks) and body weights (22—27 g) of WT and CD36−/− mice were comparable for all experiments. All study protocols were approved by The Institutional Animal Care and Use Committee (Gunma University Graduate School of Medicine). The mice were housed in a temperature-controlled room (22 C) in a 12-h light/12-h dark cycle and had unrestricted access to water and standard chow (CE-2, Clea Japan, Inc.). For the fasting experiments, the mice were individually housed and food was withdrawn for 20 h; water was provided ad libitum. One sample of BAT and one sample of SkM were snapped frozen in liquid nitrogen and conserved at −80 °C until further use and another sample of BAT was maintained for histological examination.
2.2. Cold tolerance test
Cold tolerance was tested by exposing individually housed fasted (20 h) or non-fasted mice to 4 °C for a maximum of 4 h or until body temperature dropped below 25 °C. It has been shown that mice with temperatures below 25 °C do not recover, and thus this body temperature can be considered terminal without using death as an end-point [13]. Body temperature was measured from the shaved mid dorsal body surface using a ThermoScan thermometer (PRO 4000, Braun, Kronberg, Germany) as described previously [13]. Blood was collected from retro-orbital plexus before and after cold exposure to measure biochemical parameters and centrifuged at 1500 × g for 15 min at 4 °C to separate the serum.
2.3. Measurement of blood parameters
Blood glucose was measured by glutest sensor (Sanwa Kagaku, Aichi, Japan). Serum levels of triglyceride (Triglyceride E-test, Wako Chemical, Osaka), non-esterified fatty acid (NEFA C-test Wako Chemical, Osaka) and ketone body (EnzyChrom Ketone Body Assay Kit,BioAssaySystems, California) were measured according to the manufacturer's protocols.
2.4. Triglyceride measurements in the liver and BAT [4]
BAT was homogenized in RIPA buffer (50 mMTris-HC1, pH 7.4, 1% NP40, 0.25% Na-deoxycholate, 150 mM NaCl and 1 mM EDTA) and centrifuged at 18,000 × g for 10 min at 4 °C. Lipids in the supernatant were extracted with methanol/chloroform (1:2), evaporated with NO2 and dissolved in isopropanol. Triglyceride content was measured by the Triglyceride E-test Wako (Wako Chemical, Osaka).
2.5. Glycogen measurement in SkM [4]
The SkM was powder-pulverized in liquid nitrogen, and a 10 mg sample was homogenized in distilled water and boiled at 99 °C. After centrifugation, the supernatants were collected to measure the glycogen concentration (BioVision).
2.6. Biodistribution of 125I-BMIPP (15-(p-iodophenyl)-3-(R,S)-methyl pentadecanoic acid) and 18F-FDG (2-fluorodeoxyglucose)
Biodistribution of 125I-BMIPP and 18F-FDG was determined as described previously [14]. Mice received intravenous injection of 125I-BMIPP (5 kBq) and 18F-FDG (100 kBq) via the lateral tail vein in a volume of 100 μl. 125I-BMIPP was a gift from Nihon Medi-Physics Co. Ltd. 18F-FDG was obtained from batches prepared for clinical PET imaging in Gunma University. The animals were sacrificed at 2 h after injection. The isolated tissues were weighed and counted in a well-type gamma counter (ARC-7001, ALOKA).
2.7. RNA isolation and quantitative real-time polymerase chain reaction analysis
Total RNA was isolated from BAT using the RNAiso Plus reagent (Takara, Japan). Semi-quantitative RT-PCR was performed with RT-PCR kit (Takara, Japan) according to manufacturer's protocol. RNA was prepared by reverse transcription using oligo-dTand dNTP, and each sample was processed with the RT-PCR kit (TAKARA, Japan). Quantitative real time-PCR was performed using the SYBR Green PCR Master Mix (Applied Biosystems, CA, USA) according to the manufacturer's instructions, and then evaluated using the Light-Cycler 480 Real-Time PCR system (Roche, CA, USA). The expression level of the target gene was normalized to the TATA binding protein (TBP) mRNA level. The sequences of primers for quantitative realtime PCR used in this study are described previously [4].
2.8. Statistical analysis
Statistical analysis was performed using unpaired Student's t-test or One-way ANOVA with Bonferroni's post-hoc multiple comparison. A p-value < 0.05 was considered statistically significant. The data are presented as the means ± S.E. Statistical analysis of the data was performed with IBM SPSS (version 20.0 for Windows, IBM, NY, USA).
3. Results
3.1. Effects of cold stress on body temperature in fed and fasted mice
To determine the effects of cold stress, wild-type (WT) and CD36−/− mice were exposed to a cold environment (4 °C) for 2 h with or without prior fasting for 20 h (Fig. 1). The body surface temperature was comparable between WT and CD36−/− mice without prior fasting (Fig. 1A). However, the body temperature of all CD36−/− mice that were fasted for 20 h rapidly declined and reached below 25 °C within 2 h of cold exposure (Fig. 1B). In contrast, the body temperature of WT mice was higher than 30 °C 2 h after fasting. The mice in both the fed and fasted states showed similar shivering behavior and similarly reduced physical activity. Thus, fasted CD36−/− mice exhibited severe cold intolerance compared with the WT mice when acutely exposed to a cold environment.
Fig. 1.
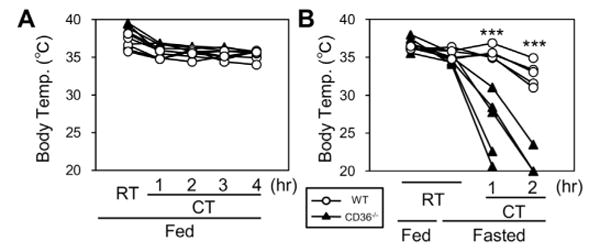
Cold intolerance in CD36−/− mice with prior fasting. Cold tolerance was tested for a maximum of 4 h or until body temperature dropped below 25 °C as described in Methods. Body temperature was measured from the shaved mid-dorsal body surface. Fasting was for 20 h n = 5/group. *p < 0.05. RT, room temperature (22 °C); CT, cold temperature (4 °C).
3.2. Effects of cold stress on blood glucose, lipids, and ketone bodies in fasted mice
We then examined biochemical parameters before and after brief cold exposure of 20 h fasted mice. Serum levels of glucose in CD36−/−mice were lower than those in WT before and after cold exposure (Fig. 2A). The levels of serum TG in CD36−/− mice were also lower than those in WT and were more reduced after cold exposure (Fig. 2C). Levels of NEFAs and ketone bodies (β-hydroxybutyrate, BHB) were comparable between CD36−/− and WT mice before and after cold exposure (Fig. 2B and D). Given that glucose and TG are crucial energy substrates for thermogenesis under a cold environment, our data suggest that energy supply into thermogenic tissues such as BAT and SkM is markedly limited in fasted CD36−/− mice.
Fig. 2.
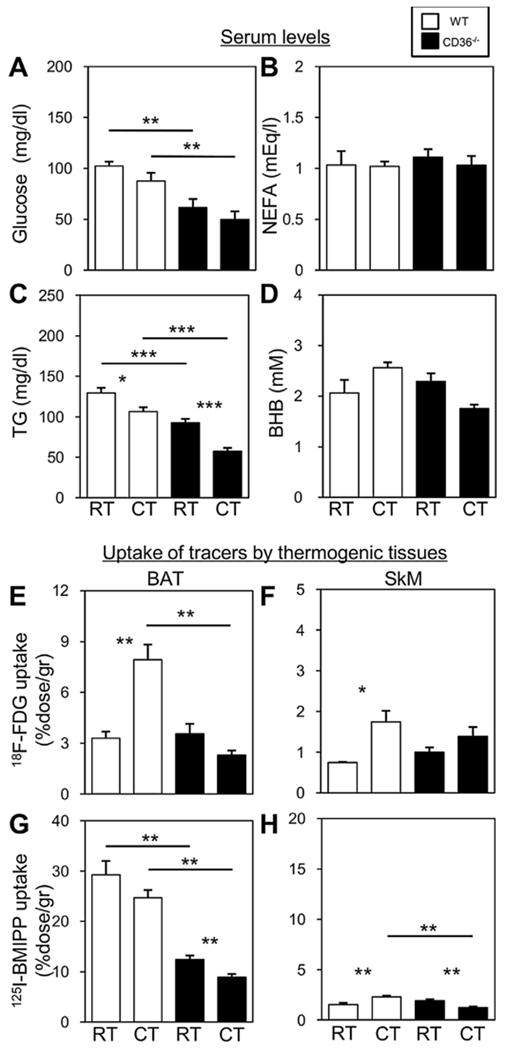
Blood energy substrates and uptake of glucose and NEFAs by BAT and SkM of fasted WT and CD36−/− mice before and after a 2 h cold exposure. (A to D) Blood was collected from the retro-orbital plexus before and 2 h after cold exposure to measure serum levels of glucose (A), NEFAs (B), TG (C), and ketone bodies (D) in the fasted states. n = 5—6/group. (E to H) The mice received intravenous injections of 18F-FDG (100 kBq) and 125I-BMIPP (5 kBq) via the lateral tail vein after a 20 h fast. The mice were maintained at room temperature or in cold rooms (4 °C) for 2 h and then sacrificed to isolate tissues and determine uptake of 18F-FDG (E and F) and 125I-BMIPP (G and H) by BAT (E and G) and SkM (F and H) (n = 4—6/group). *p < 0.05; **p < 0.01; ***p < 0.001. RT, room temperature; CT, cold temperature.
3.3. Effects of cold stress on glucose and FA uptake in BAT and SkM of fasted mice
We then estimated the uptake of glucose and NEFAs using the glucose analogue 18F-FDG and the FA analogue 125I-BMIPP, respectively. In the fasted state, uptake of 18F-FDG was enhanced in BAT of WT but not in BAT of CD36−/− mice (Fig. 2E). Uptake of 125I-BMIPP by BAT of CD36−/− mice was lower than that of WT mice and declined further after cold exposure (Fig. 2G). Uptake of 18F-FDG was not enhanced in SkM of CD36−/− mice after cold exposure while it doubled in WT mice (Fig. 2F). Uptake of 125I-BMIPP by SkM was low as compared to that of BAT in both WT and CD36−/− mice and while it was enhanced in WT by cold exposure, it was reduced in CD36−/− mice (Fig. 2H). These findings, together with the lower serum levels of glucose and TG, suggest that uptake of energy Substrates, such as glucose, NEFAs and TG, into BAT and SkM of CD36−/− mice was severely decreased supporting the hypothesis that a reduced supply of energy substrates to BAT and SkM could result in impaired thermogenesis.
3.4. Cold stress on TG storage in BAT and on glycogen storage in SkM
We next examined TG storage in interscapular BAT. In the fasted state, BAT weight relative to body weight was lower in CD36−/− as compared to WT mice (Fig. 4A). BAT TG content was also significantly lower (Fig. 4B). Cold exposure reduced BAT TG in both WT and CD36−/− mice and this reduction was much more pronounced in CD36−/− mice. The available TG storage, already low before cold exposure in CD36−/− mice was, nearly depleted after a short cold exposure (Fig. 4A and B). Consistent with the drop in BAT TG levels, the color of BAT was darker, and the multilocular lipid droplets became smaller (data not shown).
Fig. 4.
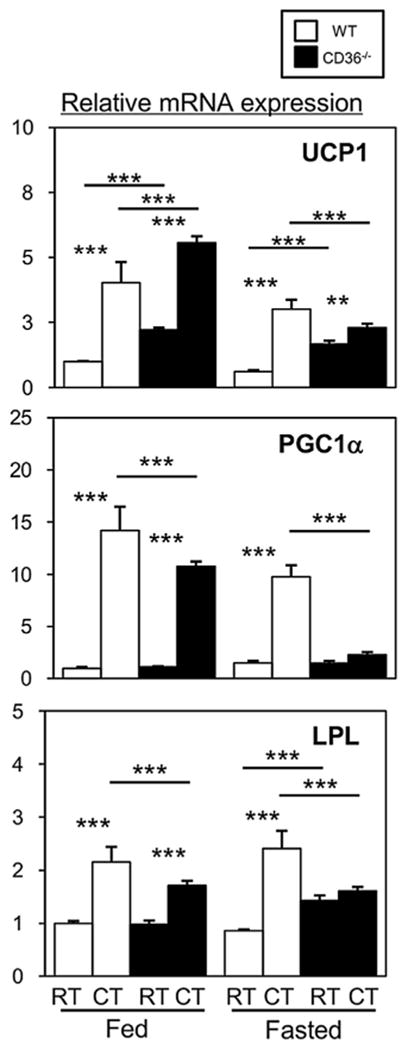
Induction of genes associated with thermogenesis in BAT of fasted WT and CD36−/− mice after a 2 h cold exposure. The mice were maintained at room temperature or in a cold room (4 °C) for 2 h with or without prior fasting. The total RNA from BAT was extracted for quantitative real-time PCR. n = 4—5/group. *p < 0.05; **p < 0.01; ***p < 0.001. RT, room temperature; CT, cold temperature.
We also examined the glycogen concentration in the quadriceps femoris muscle. In the fasted state, the level of glycogen was significantly lower before cold exposure and markedly reduced after cold exposure in CD36−/− mice (Fig. 3C). Thus, the energy storage in BAT and SkM was nearly depleted in fasted CD36−/− mice shortly after cold exposure, and this depletion would explain the reduced thermogenic activity observed during cold exposure.
Fig. 3.
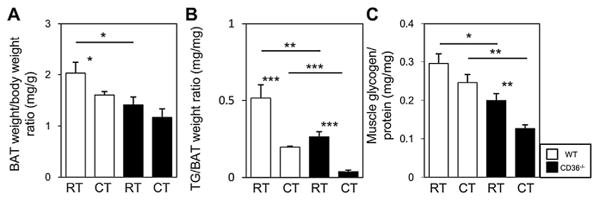
Storage of TG in BAT and glycogen in SkM in fasted WT and CD36−/− mice after a 2 h cold exposure. Interscapular BAT and quadriceps femoris muscle were isolated before and 2 h after cold exposure (4 °C) of 20 h fasted mice. (A) The ratio of the BAT weight to the body weight (B) TG concentration in the BAT. (C) Glycogen content in the muscle was normalized by protein concentration, n = 4—5/group. *p < 0.05; **p < 0.01; ***p < 0.001. RT, room temperature; CT, cold temperature.
3.5. Expression of the genes required for thermogenesis
We next evaluated the expression levels of the genes associated with thermogenesis in BAT in response to cold exposure (Fig. 4). In the fed state, cold exposure upregulatedmRNA expression of UCP1, PGC1α and LPL in both WT and CD36−/− mice. In the fasted state, however, while upregulation was still observed in WT mice it was markedly blunted in CD36−/− mice. Thus, fasting impairs thermogenesis at the gene expression level in BAT of CD36−/− mice.
4. Discussion
In this study, we demonstrate an indispensable role of CD36 in thermogenesis when cold exposure is combined with a prior fast. During fasting, glucose utilization continues to be enhanced in the heart and oxidative SkM of CD36−/− mice leading to accelerated hypoglycemia. Further, uptake of glucose in addition to the impaired FA uptake, were severely affected in thermogenic tissues such as BAT and glycolytic SkM of CD36−/− mice upon brief cold exposure. Consequently, CD36−/− mice displayed depletion of energy substrates such as TG in BAT and glycogen in glycolytic SkM. Thus, abnormal uptake and distribution of energy substrates cause impaired nutrient homeostasis in the fasted state, resulting in rapid occurrence of fatal hypothermia upon cold exposure.
4.1. Similarity and difference in nutrient homeostasis between FABP4/5- and CD36-mediated FA uptake
Our previous publication showed that thermogenesis is severely impaired in mice doubly deficient in FA-binding protein 4 (FABP4) and FABP5 (DKO mice) during cold exposure with a prior fast [4]. FABPs bind long-chain FA in the cytoplasm, and facilitate the transport of FAs to specific compartments in the cells [15]. We reported that in DKO mice the transport of FAs is disturbed in the heart and oxidative SkM at capillary endothelial cell level, where FABP4/5 are abundantly expressed [16]. As in the case with CD36−/−mice, the storage of both TG in BAT and glycogen in glycolytic SkM was nearly depleted in the fasted state after cold exposure in DKO mice [4]. In addition to accelerated hypoglycemia due to compensatory uptake of glucose by the heart and oxidative SkM, FAs are not efficiently taken up by BAT despite the robustly elevated levels of serum NEFAs [4]. In conjunction with these data, our present findings underscore the critical importance of cellular uptake of FA in FA consuming tissues such as the heart, oxidative SkM and BAT for maintaining nutrient homeostasis. When FA uptake is disrupted in these tissues, a compensatory increase in glucose uptake and utilization further impairs nutrient homeostasis in the fasted and cold exposure states.
We found that ketogenesis is enhanced in the fasted FABP4/5 DKO mice while it was not altered in the fasted CD36−/− mice [4,17]. Ketone bodies are possible substitutes for glucose even in the heart and SkM, which may save consumption of glucose in the heart and SkM for heat production by BAT. It is also unknown whether FABP4/5 play a role in uptake of TRLs under cold exposure like CD36. Further studies to explore if ketogenesis can contribute to ther-mogenesis and the potential involvement of FABP4/5 in TRLs uptake are warranted.
4.2. Clinical implications
Several reports documented the association of CD36 deficiency to metabolic diseases in humans. Patients with type I CD36 deficiency have insulin resistance and postprandial hyperlipidemia with the accumulation of small intestine-derived lipoproteins [18,19]. In contrast, subjects heterozygous for CD36 deficiency appear protected from insulin resistance [20,21]. Our findings have implications with respect to another potential consequence of CD36 deficiency, sudden infant death syndrome (SIDS), an FA utilization disorder. SIDS can be caused by mutation of the genes for mitochondrial FA oxidation enzymes, such as medium chain acyl-coenzyme A dehydrogenase, very long chain acyl-coenzyme A dehydrogenase, carnitine palmitoyl transferase and mitochondrial trifunctional protein [22,23]. Patients with this syndrome show hypothermia and hypoglycemia triggered by an increase in energy demand (e.g. fasting, infection, surgery and prolonged exercise). CD36 deficiency could have a similar clinical manifestation. This speculation is supported by the previous demonstration that fasting induces conduction anomalies and sudden death from bradycardia in CD36−/− mice [24] and by the fact that three patients with CD36 deficiency had a family history of sudden death in younger relatives [25].
In conclusion, our data provide the first experimental evidence to demonstrate that FA uptake through CD36 is crucial for ther-mogenesis during fasting, and imply that CD36 is indispensable for nutrient homeostasis when FA demand is increased and nutrient availability is limited.
Acknowledgments
We thank Miki Matsui, Yukiyo Tosaka, Keiko Matsukura and Takako Kobayashi for excellent technical help. This work was supported in part by a Grant-in-Aid for Scientific Research from the Japan Society for the promotion of Science, to T.I. (26461123), to M.K. (24390194). a grant from the Japan Cardiovascular Foundation (to MK) and a grant from the Vehicle Racing Commemorative Foundation and (to TI).
Footnotes
Conflict of interest: None.
References
- 1.Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004;84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- 2.Klingenspor M. Cold-induced recruitment of brown adipose tissue thermo-genesis. Exp Physiol. 2003;88:141–148. doi: 10.1113/eph8802508. [DOI] [PubMed] [Google Scholar]
- 3.Festuccia WT, Blanchard PC, Deshaies Y. Control of Brown Adipose Tissue Glucose and Lipid Metabolism by PPARgamma, Front Endocrinol. Lausanne. 2011:84–2. doi: 10.3389/fendo.2011.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Syamsunarno MR, Iso T, Yamaguchi A, Hanaoka H, Putri M, Obokata M, Sunaga H, Koitabashi N, Matsui H, Maeda K, Endo K, Tsushima Y, Yokoyama T, Kurabayashi M. Fatty acid binding protein 4 and 5 play a crucial role in thermogenesis under the conditions of fasting and cold stress. PLoS One. 2014;9:e90825. doi: 10.1371/journal.pone.0090825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haman F. Shivering in the cold: from mechanisms of fuel selection to survival. J Appl Physiol. 2006;100:1702–1708. doi: 10.1152/japplphysiol.01088.2005. [DOI] [PubMed] [Google Scholar]
- 6.Haman F, Peronnet F, Kenny GP, Massicotte D, Lavoie C, Weber JM. Partitioning oxidative fuels during cold exposure in humans: muscle glycogen becomes dominant as shivering intensifies. J Physiol. 2005;566:247–256. doi: 10.1113/jphysiol.2005.086272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glatz JF, Luiken JJ, Bonen A. Membrane fatty acid transporters as regulators of lipid metabolism: implications for metabolic disease. Physiol Rev. 2010;90:367–417. doi: 10.1152/physrev.00003.2009. [DOI] [PubMed] [Google Scholar]
- 8.Bartelt A, Bruns OT, Reimer R, Hohenberg H, Ittrich H, Peldschus K, Kaul MG, Tromsdorf UI, Weller H, Waurisch C, Eychmuller A, Cordts PL, Rinninger F, Bruegelmann K, Freund B, Nielsen P, Merkel M, Heeren J. Brown adipose tissue activity controls triglyceride clearance. Nat Med. 2011;17:200–205. doi: 10.1038/nm.2297. [DOI] [PubMed] [Google Scholar]
- 9.Bartelt A, Merkel M, Heeren J. A new, powerful player in lipoprotein metabolism: brown adipose tissue. J Mol Med (Berl) 2012;90:887–893. doi: 10.1007/s00109-012-0858-3. [DOI] [PubMed] [Google Scholar]
- 10.Coburn CT, Knapp FF, Jr, Febbraio M, Beets AL, Silverstein RL, Abumrad NA. Defective uptake and utilization of long chain fatty acids in muscle and adipose tissues of CD36 knockout mice. J Biol Chem. 2000;275:32523–32529. doi: 10.1074/jbc.M003826200. [DOI] [PubMed] [Google Scholar]
- 11.Hajri T, Han XX, Bonen A, Abumrad NA. Defective fatty acid uptake modulates insulin responsiveness and metabolic responses to diet in CD36-null mice. J Clin Invest. 2002;109:1381–1389. doi: 10.1172/JCI14596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Febbraio M, Abumrad NA, Hajjar DP, Sharma K, Cheng W, Pearce SF, Silverstein RL. A null mutation in murine CD36 reveals an important role in fatty acid and lipoprotein metabolism. J Biol Chem. 1999;274:19055–19062. doi: 10.1074/jbc.274.27.19055. [DOI] [PubMed] [Google Scholar]
- 13.Miinalainen IJ, Schmitz W, Huotari A, Autio KJ, Soininen R, Ver Loren van Themaat E, Baes M, Herzig KH, Conzelmann E, Hiltunen JK. Mitochondrial 2,4-dienoyl-CoA reductase deficiency in mice results in severe hypoglycemia with stress intolerance and unimpaired ketogenesis. PLoS Genet. 2009;5:el000543. doi: 10.1371/journal.pgen.1000543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goto K, Iso T, Hanaoka H, Yamaguchi A, Suga T, Hattori A, Irie Y, Shinagawa Y, Matsui H, Syamsunarno MR, Matsui M, Haque A, Arai M, Kunimoto F, Yokoyama T, Endo K, Gonzalez FJ, Kurabayashi M. Peroxisome proliferator-activated receptor-gamma in capillary endothelia promotes fatty acid uptake by heart during long-term fasting. J Am Heart Assoc. 2013;2:e004861. doi: 10.1161/JAHA.112.004861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Furuhashi M, Hotamisligil GS. Fatty acid-binding proteins: role in metabolic diseases and potential as drug targets. Nat Rev Drug Discov. 2008;7:489–503. doi: 10.1038/nrd2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iso T, Maeda K, Hanaoka H, Suga T, Goto K, Syamsunarno MR, Hishiki T, Nagahata Y, Matsui H, Arai M, Yamaguchi A, Abumrad NA, Sano M, Suematsu M, Endo K, Hotamisligil GS, Kurabayashi M. Capillary endothelial fatty acid binding proteins 4 and 5 play a critical role in fatty acid uptake in heart and skeletal muscle Arterioscler. Thromb Vase Biol. 2013;33:2549–2557. doi: 10.1161/ATVBAHA.113.301588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Syamsunarno MR, Iso T, Hanaoka H, Yamaguchi A, Obokata M, Koitabashi N, Goto K, Hishiki T, Nagahata Y, Matsui H, Sano M, Kobayashi M, Kikuchi O, Sasaki T, Maeda K, Murakami M, Kitamura T, Suematsu M, Yoshitotsushima, Endo K, Hotamisligil GS, Kurabayashi M. A critical role of fatty acid binding protein 4 and 5 (FABP4/5) in the systemic response to fasting. PLoS One. 2013;8:e79386. doi: 10.1371/journal.pone.0079386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuwasako T, Hirano K, Sakai N, Ishigami M, Hiraoka H, Yakub MJ, Yamauchi-Takihara K, Yamashita S, Matsuzawa Y. Lipoprotein abnormalities in human genetic CD36 deficiency associated with insulin resistance and abnormal fatty acid metabolism. Diabetes Care. 2003;26:1647–1648. doi: 10.2337/diacare.26.5.1647-a. [DOI] [PubMed] [Google Scholar]
- 19.Miyaoka K, Kuwasako T, Hirano K, Nozaki S, Yamashita S, Matsuzawa Y. CD36 deficiency associated with insulin resistance. Lancet. 2001;357:686–687. doi: 10.1016/s0140-6736(00)04138-6. [DOI] [PubMed] [Google Scholar]
- 20.Love-Gregory L, Sherva R, Sun L, Wasson J, Schappe T, Doria A, Rao DC, Hunt SC, Klein S, Neuman RJ, Permutt MA, Abumrad NA. Variants in the CD36 gene associate with the metabolic syndrome and high-density lipoprotein cholesterol. Hum Mol Genet. 2008;17:1695–1704. doi: 10.1093/hmg/ddn060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Su X, Abumrad NA. Cellular fatty acid uptake: a pathway under construction, Trends Endocrinol. Metab. 2009;20:72–77. doi: 10.1016/j.tem.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Houten SM, Wanders RJ. A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation. J Inherit Metab Dis. 2010;33:469–477. doi: 10.1007/s10545-010-9061-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spiekerkoetter U, Wood PA. Mitochondrial fatty acid oxidation disorders: pathophysiological studies in mouse models. J Inherit Metab Dis. 2010;33:539–546. doi: 10.1007/s10545-010-9121-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pietka TA, Sulkin MS, Kuda O, Wang W, Zhou D, Yamada KA, Yang K, Su X, Gross RW, Nerbonne JM, Efimov IR, Abumrad NA. CD36 protein influences myocardial Ca2+ homeostasis and phospholipid metabolism: conduction anomalies in CD36-deficient mice during fasting. J Biol Chem. 2012;287:38901–38912. doi: 10.1074/jbc.M112.413609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watanabe K, Ohta Y, Toba K, Ogawa Y, Hanawa H, Hirokawa Y, Kodama M, Tanabe N, Hirono S, Ohkura Y, Nakamura Y, Kato K, Aizawa Y, Fuse I, Miyajima S, Kusano Y, Nagamoto T, Hasegawa G, Naito M. Myocardial CD36 expression and fatty acid accumulation in patients with type I and II CD36 deficiency. Ann Nucl Med. 1998;12:261–266. doi: 10.1007/BF03164911. [DOI] [PubMed] [Google Scholar]