

Abstract
Objectives
The purpose of this study was to test the hypothesis that the late Na current blocker ranolazine suppresses re-entrant and multifocal ventricular fibrillation (VF).
Background
VF can be caused by either re-entrant or focal mechanism.
Methods
Simultaneous voltage and intracellular Ca+2 optical mapping of the left ventricular epicardial surface along with microelectrode recordings was performed in 24 isolated-perfused aged rat hearts. Re-entrant VF was induced by rapid pacing and multifocal VF by exposure to oxidative stress with 0.1 mM hydrogen peroxide (H2O2).
Results
Rapid pacing induced sustained VF in 7 of 8 aged rat hearts, characterized by 2 to 4 broad propagating wavefronts. Ranolazine significantly (p < 0.05) reduced the maximum slope of action potential duration restitution curve and converted sustained to nonsustained VF lasting 24 ± 8 s in all 7 hearts. Exposure to H2O2 initiated early afterdepolarization (EAD)-mediated triggered activity that led to sustained VF in 8 out of 8 aged hearts. VF was characterized by multiple foci, appearing at an average of 6.8 ± 3.2 every 100 ms, which remained confined to a small area averaging 2.8 ± 0.85 mm2 and became extinct after a mean of 43 ± 16 ms. Ranolazine prevented (when given before H2O2) and suppressed H2O2-mediated EADs by reducing the number of foci, causing VF to terminate in 8 out of 8 hearts. Simulations in 2-dimensional tissue with EAD-mediated multifocal VF showed progressive reduction in the number of foci and VF termination by blocking the late Na current.
Conclusions
Late Na current blockade with ranolazine is effective at suppressing both pacing-induced re-entrant VF and EAD-mediated multifocal VF.
Keywords: early afterdepolarization, focal activity, optical mapping, oxidative stress, ranolazine, re-entry, triggered activity, ventricular fibrillation
Pharmacologic and genetic approaches to treatment of ventricular fibrillation (VF) must take into account its mechanistic complexity. VF can be caused by both reentrant and focal mechanisms, as well as mixtures of the 2. Rapid pacing-induced VF generally is attributed to reentrant mechanisms, resulting from either multiple wavelets (1) or a mother rotor (2), depending on experimental conditions (3). However, drugs and genetic defects that reduce repolarization reserve (prolong repolarization) and alter intracellular Ca+2 (Cai2+) cycling promote polymorphic ventricular arrhythmias degenerating to VF, in which focal mechanisms are involved in VF initiation and maintenance. For example, local application of aconitine, which impairs Na current inactivation, induces a form of VF that is maintained by focal activity originating from the site of aconitine application (4,5). Multifocal VF also can be induced by oxidative stress with hydrogen peroxide (H2O2), which is driven by early afterdepolarization (EAD)-mediated triggered activity generating multiple short-lived foci continuously shifting in location (6,7).
Ranolazine, which preferentially blocks the late Na current (INa-L) (8,9), is a clinically useful antianginal drug that has been shown to exert antiarrhythmic actions as well (10). In this respect, ranolazine is shown to reduce the dispersion of ventricular action potential duration (APD) (11–13), an effect that may account for the drug’s demonstrated efficacy to increase VF threshold and decrease ventricular defibrillation threshold of re-entrant VF. (14) Ranolazine also has been shown to suppress EADs and triggered activity in isolated cardiac myocytes exposed to H2O2 (8,15); however, its effects on the initiation and maintenance of EAD-mediated multifocal oxidative VF in intact hearts have not been evaluated. H2O2 has pleiotropic effects, including direct effects on ion channel and transporter proteins and indirect effects mediated by activating Ca-calmodulin kinase II signaling pathways (16–18), which enhance the late Na current, promoting EADs and triggered activity (8,15,19). The aim of this study was to compare the effects of ranolazine on pacing induced re-entrant VF and on H2O2-induced multifocal VF.
Methods
The research protocol was approved by the Institutional Animal Care and Use Committee and followed the guidelines of American Heart Association.
Langendorff setting
We used male Fisher344 rats age 24 to 26 months (n = 24) purchased from the National Institute on Ageing, Bethesda, Maryland. The hearts of the anesthetized rats were removed and the ascending aorta was cannulated for retrograde perfusion with warm (36.5 ± 0.50°C) oxygenated Tyrode’s solution, as described previously (6,20).
Optical mapping
The hearts were stained with RH237 and Rhod-2 AM (Invitrogen Molecular Probes, Carlsbad, California) for simultaneous dual voltage and Cai2+ fluorescent optical imaging, respectively, as described previously (6). Cai2+ transient decay rate constant, τ, was determined by a monoexponential fit during the relaxation before and after ranolazine (6). Cytochalasin D (5 μM) was added to the perfusate to inhibit motion (6). Single-cell action potentials were recorded with glass microelectrodes from the base of the left ventricle (LV) epicardium, the site of the focal activity during the onset of VT as determined by optical mapping (6). An epicardial wavefront was considered to be focal when it arose from within the mapped region (i.e., did not propagate into the mapped region from the outside) and was surrounded with nonexcited tissue. We previously showed that after extensive endomyocardial and midmyocardial cryoablation, the epicardial surface cells generate focal activity independent of breakthrough excitation from deeper cells (6).
Dynamic APD restitution
Epicardial APD was measured at 90% and 50% repolarization from multiple epicardial cells (3 to 5 cells) recorded with microelectrodes from base mid and apical regions in 8 hearts before and after ranolazine. APD restitution curves were determined using a dynamic pacing protocol, as described previously (6,20).
Pharmacological interventions
The isolated hearts first were perfused with normal Tyrode’s solution, and baseline pacing-induced arrhythmias were recorded and imaged. Then, the effect of ranolazine (10 μM) on inducible VF was evaluated 20 min after perfusion (n = 8). When the VF lasted longer than 3 min, it was defibrillated by electrical shock. In a separate series of experiments, the effect of ranolazine on H2O2 (0.1 mM)-initiated VF was evaluated. This involved 2 protocols. The purpose of the first protocol was to determine the efficacy of ranolazine to suppress the VF after it is initiated in the continuous presence of H2O2 (n = 8). The purpose of the second protocol was to test the efficacy of ranolazine to prevent the emergence of VF when administered 20 min before H2O2 (0.1 mM, n = 8).
2-dimensional computer simulation studies
Computer simulations were performed in a 2-dimensional (2D) tissue model with the membrane voltage described by the following partial differential equation:
| [1] |
where Cm is the membrane capacitance set at 1μF/cm, Iion is the total membrane ionic currents, and D is the diffusion coefficient and was set as 0.0005 cm2/ms with no-flux boundary conditions. Istim is the stimulation current (a square pulse with a strength 40 μF/cm and duration of 1 ms). We used the ventricular myocyte action potential model that we developed recently (21). The model was modified to generate EADs with detailed changes as described previously (7). To model the effects of H2O2, in addition to increasing the maximum conductance of ICaL by 2.6 and decreasing the maximum conductance of the rapid delayed rectified potassium current (IKr) by 20%, we also halved the rate of calcium uptake by the sarcoplasmic reticulum and added INa-L that was equivalent to 0.1% of the peak INa amplitude (7). The effects of ranolazine were simulated by decreasing the late INa. We first paced a single myocyte with a square pulse (Istim described above) to the steady-state and used it as the initial condition for the myocytes in the whole tissue to save computer time. The 2D tissue then was paced from the corner with 1 beat to allow EAD-mediated focal VF to develop. After 1 min, the dynamics of the focal VF-like state were analyzed in the presence and absence of INa-L to study ranolazine effect. Numerically, Equation 1 was discretized with Δx = Δy = 0.015 cm and was integrated with a forward Euler method with an adaptive time step varying from 0.01 to 0.1 ms.
Statistical analyses
Significant differences in the incidence of VF (dichotomous comparisons) were determined using the Fisher exact test or McNemar test whenever appropriate. A likelihood ratio test was used to determine the significance of site-specific origination of focal activity in the LV. Rate-dependent changes in the Cai2+ decline rate constant (monoexponential fit), APD, resting potential, maximum rate of phase-zero action potential depolarization (dV/Dtmax), and maximum slope of APD restitution curves were determined using 1-way repeated measures analyses of variance (ANOVA). Our 2-way repeated measures ANOVA between groups indicated linear trends between groups and did not show interaction effect. Differences among individual means were verified subsequently by Newman-Keuls post hoc tests. p ≤ 0.05 was considered significant. All data are presented as mean ± SD.
Results
Pacing-induced VF
When VF was induced by rapid pacing (Fig. 1A), activation mapping revealed 2 to 4 irregularly wandering wavelets that propagated freely over a relatively large area on the epicardium, often colliding with other wavefronts (Fig. 1B) and initiating transient re-entry (Online Video 1). VF persisted (for >3 min) unless electrically cardioverted (Fig. 1A). The activation patterns were comparable with those previously reported in pacing-induced multiple wavelet VF in isolated swine (22), canine (23), and human ventricles (24). The effects of ranolazine on the dynamics of pacing-induced VF were examined in 8 hearts. Ranolazine did not prevent the induction of VF by rapid pacing (7 of 8 at baseline vs. 6 of 8 after ranolazine, p = NS) (Fig. 1C). However, after ranolazine, VF terminated spontaneously in all 6 hearts after a mean of 12 ± 6 s (Fig. 1C). Activation maps showed that ranolazine caused progressive reduction in the number of wavelets until only a single wavefront remained, followed by quiescence and resumption of sinus rhythm (Fig. 1E). Ranolazine significantly reduced the maximum slope of epicardial APD restitution curve, from 1.3 ± 0.5 to 0.85 ± 0.3 (18 sites in 6 hearts) (p < 0.05).
Figure 1. Activation During Pacing-Induced Ventricular Fibrillation.
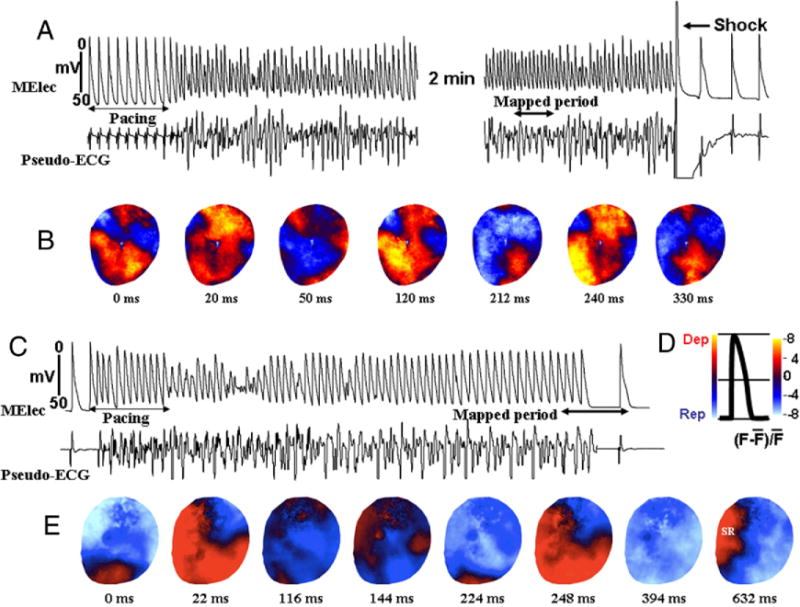
Snapshots of activation during pacing-induced ventricular fibrillation (A) at baseline and (B) after ranolazine (10 μM) perfusion. (A) Multiple (2 to 4) propagating and colliding wavefronts appear to maintain the induced ventricular fibrillation (Online Video 1), requiring electrical shock for termination. (B) However, after ranolazine perfusion and while the ventricular fibrillation could still be induced, it self-terminated. D is a schematic drawing of optical action potential showing the different phases of the action potential in color. E shows snap shots taken just prior to termination of the ventricular fibrillation after ranolazine as indicated by the double-headed arrow in C. SR is sinus rhythm. Dep = depolarization; ECG = electrocardiogram; F = fluorescence; F bar = mean fluorescence; MElec = microelectrode; Rep = repolarization; SR = sinus rhythm.
Initiation and maintenance of VF by H2O2-mediated EADs and triggered activity
INITIATION
Figure 2A illustrates the onset of VF in a heart exposed to 0.1 mM H2O2. Focal VT arose suddenly during normal sinus rhythm (mean CL of 395 ± 182 ms), and within 1 s degenerated to VF. The mean CL of VT was 78 ± 20 ms and that of VF was 60 ± 18 ms (n = 8). Focal VT arose preferentially (66%; p < 0.05, likelihood ratio test) from the base of the LV epicardium (Fig. 2B) in 12 of 18 episodes in 8 hearts, consistent with our previous study (6). In the remaining 6 episodes (34%), VT originated from the outside of the mapped region and swept across a mapped region from either the apex (4 episodes) or the left lateral LV (2 episodes) as a single wavefront. VF was sustained until cardioverted electrically. To obtain insight into the mechanism of VT preceding VF, after cardioversion of a VF episode, continuous action potential recordings were made with a roving glass microelectrode from the base of the LV before the next episode of spontaneous VT or VF. The LV base was selected because of frequent origination of the focal VT (p < 0.05) from this site. Figure 2D shows the onset of a spontaneous VT episode initiated by an EAD-mediated triggered activity that causes focal VT, which then degenerated to VF, consistent with our previous study (6). The EAD-mediated triggered activity coincided in time with the isoelectric interval of the simultaneously recorded pseudo-electrocardiography (Fig. 2D), suggesting the absence of electrical activity elsewhere in the LV during the emergence of the EAD. Furthermore the U-shaped smooth depolarization of the EAD preceding the QRS complex of the VT by a mean of 7 ± 3 ms further suggested that the triggered activity caused the VT.
Figure 2. Initiation of VF in an Aged Rat Heart Exposed to H2O2 (0.1 mM) by an EAD-Mediated Triggered Activity.
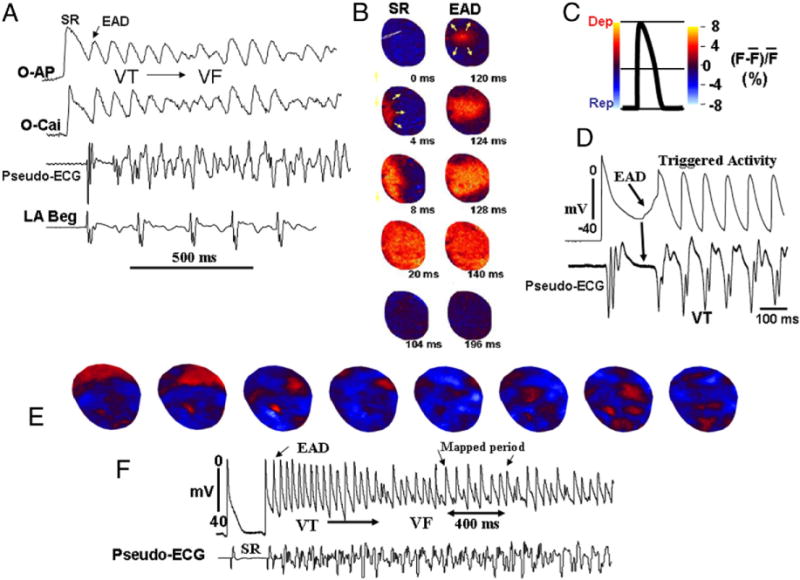
(A) Simultaneous optical action potential (O-AP), calcium transient (O-Cai) pseudo-ECG, and left atrial bipolar electrograms (LA Beg) at the onset of early afterdepolarization (EAD)-mediated ventricular tachycardia (VT)/ventricular fibrillation (VF). (B) Snapshots during the last sinus beat (SR) and the first EAD-mediated triggered beat that arose from the base of the LV. The arrows point to the direction of wave propagation and the numbers under each snapshot are the activation time (ms) with onset time arbitrarily set at 0. The color code is shown in C. (D) Glass microelectrode recording from the base of the heart in the same heart at the onset of another episode of EAD-mediated triggered activity causing the VT. Note the isoelectric interval during the initiation of EAD indicating the absence of electrical activity elsewhere in the heart. (E) Multifocal activation during an episode of VF in the same heart (Online Video 2). Abbreviations as in Figure 1.
MAINTENANCE
We mapped wavefront activation patterns between 2 to 30 s after the onset of VF. Wavefront activation patterns differed dramatically from those during pacing-induced VF in the same hearts before H2O2. As shown in Figure 2E, VF was maintained by multiple epicardial foci, appearing at a mean rate of 6.8 ± 3.2 new foci every 100 ms. These foci were caused by EAD-mediated triggered activity that arose from a mean membrane potential of 51 ± 8 mV. Unlike the multiple wandering wavelets VF (Fig. 1B), the multiple foci did not propagate freely across the epicardial surface. Instead, they typically remained confined to relatively small islands on the epicardium, with an average surface area of 2.8 ± 0.85 mm2 (Online Video 2). From a total of 1,945 epicardial foci analyzed in 6 hearts, the lifespan of a focus, from appearance to extinction, averaged 43 ± 16 ms (Fig. 2E). In 92% of the cases, extinction occurred because the depolarization failed to spread into adjacent, more repolarized, tissue. In only 8% of the cases, extinction of the foci was the result of collisions with other foci arising from adjoining sites <2 mm away.
Effects of ranolazine on H2O2-mediated multifocal VF
Ranolazine (10 μM) perfusion, beginning 5 s after the onset of H2O2-induced VF, terminated VF after a mean of 1.1 ± 0.4 min in 8 hearts (26 episodes) (Figs. 3A and 3B). No subsequent EADs or triggered activity emerged during ranolazine perfusion for up to 1 h. On washout of ranolazine, EADs and triggered beats progressively reemerged in 7 of 8 hearts, culminating in spontaneous VF after a mean washout period of 36 ± 10 min (Fig. 3B). In 8 additional hearts, ranolazine (10 μM) was perfused for 30 min before H2O2. Ranolazine prevented VF in all 8 hearts for up to 1 h of H2O2 exposure (Fig. 3C) (p < 0.01, compared with H2O2 with no ranolazine). On washout of ranolazine in the continued presence of H2O2, however, EADs progressively emerged in 7 of 8 hearts after a mean washout period of 25 ± 8 min leading to VF (Fig. 3D).
Figure 3. Suppression and Prevention of EAD-Mediated Triggered Activity and VF by Ranolazine in Hearts Exposed to 0.1 mM H2O2.
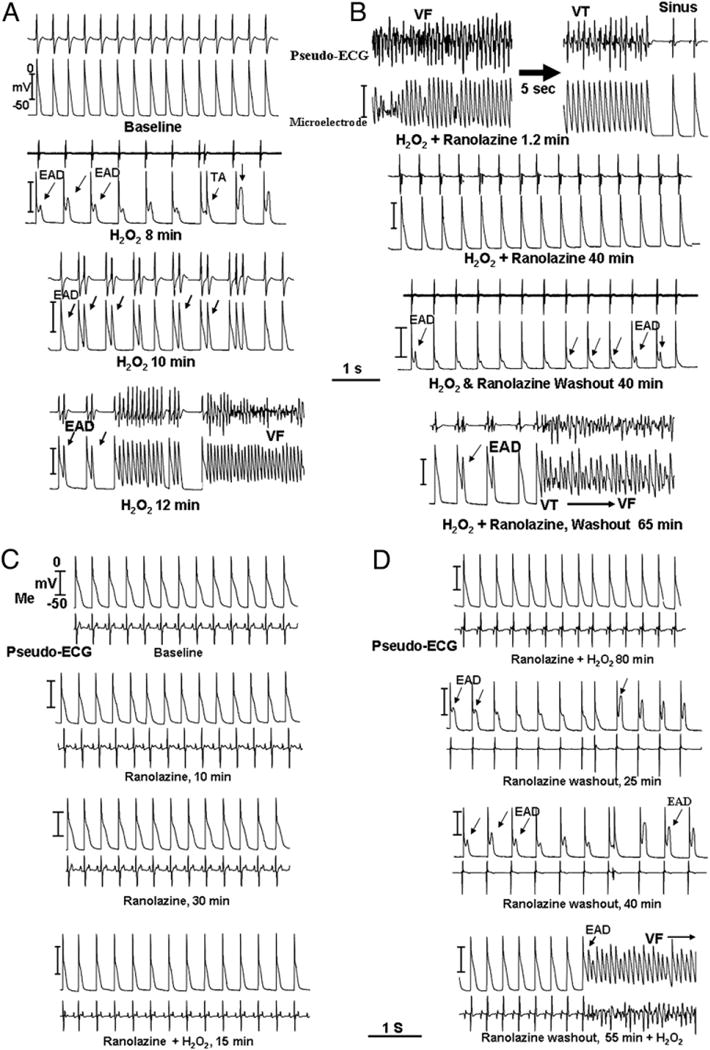
In all parts, the top recordings are pseudo-ECG and the bottom panels are glass microelectrode recordings. (A) Within 8 min of H2O2 perfusion, EADs emerge that then progressively degenerate to triggered beats, causing VT and VF 15 min after H2O2. (B) Thirteen min after ranolazine (10 μM) perfusion and in the continued presence of H2O2, the VF remains suppressed for the entire 40 min of ranolazine perfusion. However, after ranolazine washout and in the continued presence of H2O2, EADs emerged progressively, causing triggered beats and VF 65 min after washout of the drug. (C) Pre-treatment with ranolazine (10 μM) for 30 min before H2O2 perfusion in a different heart, preventing the formation of EADs and VF during the entire 60 min of ranolazine perfusion. However, on 40 min of ranolazine washout and in the continued presence of H2O2, the EADs emerged progressively, causing VT/VF 40 min after ranolazine washout as shown in D. Abbreviations as in Figures 1 and 2.
To gain insight into the mechanism of VF termination by ranolazine, we analyzed epicardial activation maps. As shown in Figures 4B and 4C, within 1 min, ranolazine caused a significant (p < 0.05) reduction in the number of epicardial foci, from 6.8 ± 3.2 foci to 3.5 ± 1.2 foci. Eventually, only a single focus remained, resulting in a brief (approximately 1 s) period of VT before the resumption of sinus rhythm (Fig. 4B).
Figure 4. Spontaneous VF 15 s After Its Onset and Its Termination With Ranolazine in a Heart Exposed to 0.1 mM H2O2.
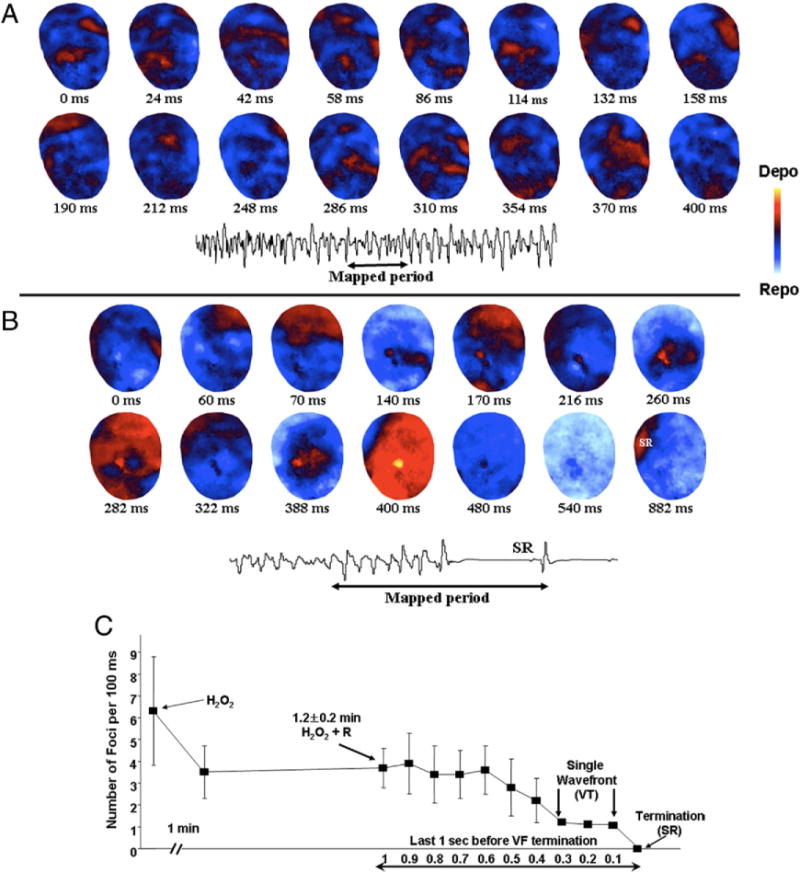
(A) Snapshots of spontaneous VF 15 s after its onset in a heart exposed to 0.1 mM H2O2 and (B) its termination with ranolazine (10 μM) in the continuous presence of H2O2. Multiple epicardial foci (red) during the VF separated by recovered tissue (blue) is evident. This pattern of VF persisted for more than 3 min as confirmed by periodic optical mapping of the VF. However, 1 min after ranolazine perfusion (B), the number of multiple foci progressively decreased, hastening the termination of VF and resumption of SR. (C) Time course of the reduction in the number of foci after ranolazine perfusion. Abbreviations as in Figures 1 and 2.
Other electrophysiological effects of ranolazine
Ranolazine had no significant effect on the resting membrane potential (measured with an intracellular microelectrode) or action potential duration to 50% and 90% repolarization, either before or after H2O2 (35 ± 7 ms vs. 32 ± 4 ms and 106 ± 5 ms vs. 102 ± 8 ms, respectively; pacing cycle length = 250 ms). Ranolazine also had no effect on the ventricular effective refractory period determined at a pacing cycle length of 250 ms (94 ± 10 ms vs. 97 ± 14 ms). However, ranolazine did cause significant rate-dependent reduction of dV/Dtmax when the pacing CL was reduced from 250 to 130 and 100 ms, both at baseline and after H2O2 consistent with ranolazine’s use-dependent block of the Na current (9,25) (Fig. 5A). Ranolazine had no significant effect on conduction velocity measured from the LV epicardial base to the LV apex at pacing cycle lengths longer than 250 ms (54 ± 8 cm/s vs. 56 ± 10 cm/s). However, at pacing cycle lengths of 250 ms and shorter, ranolazine caused a slight but significant rate-dependent decrease in conduction velocity, as shown in Figure 5B, consistent with its rate-dependent depressant of the fast INa reflected as reduction in dV/Dtmax. Ranolazine had no significant effect on the Cai2+ transient decline rate either before or after H2O2 (Fig. 5C), consistent with a previous study in rat hearts showing a minimal effect of ranolazine on the Cai2+ transient duration (26). However, H2O2 caused significant slowing of the rate constant of Cai2+ decline (84 ± 6 ms vs. 108 ± 10 ms, p < 0.05) (Fig. 5B), but had no significant effect on the maximum slope of the APD90 restitution curve (1.3 ± 0.5 vs. 1.2 ± 0.5), consistent with our previous report (6).
Figure 5. Effects of Ranolazine on Maximum Rate of Phase-Zero Action Potential Depolarization (dV/Dtmax) of Phase Zero Action Potential, Conduction Velocity, and Cai2+ Transient Decline Rate Constant (τ).
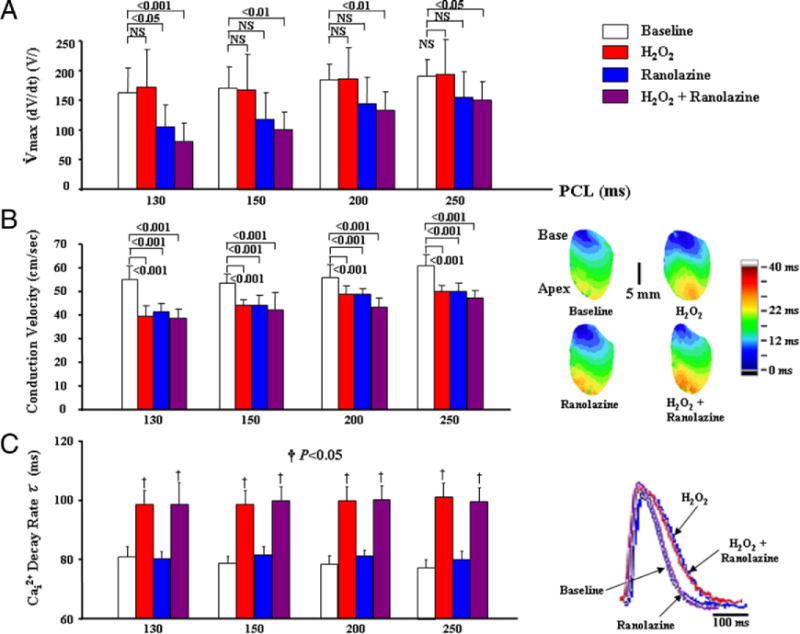
(A) Ranolazine causing rate-dependent significant decrease in dV/Dtmax at faster pacing cycle length (PCL) (<130 ms). (B, left) Isochronal activation maps with a color code (right) during pacing from the base at a PCL of 200 ms under different conditions. (C) H2O2 prolongs τ, whereas ranolazine (10 μM) has no effect on τ either before or after H2O2, as shown by the adjoining superimposed Cai2+ transients.
2D simulation of multifocal VF and the effect of ranolazine on VF maintenance
Based on the experimental findings described, we hypothesized that ranolazine suppressed multifocal VF by reducing the number of triggered foci below a critical number required to maintain VF. To test this hypothesis, we simulated 2D cardiac tissue exhibiting multifocal VF, using a realistic cardiac action potential model (21) modified to generate EADs by mimicking the effects of H2O2 on INa-L and other currents (27). INa-L then was blocked to simulate the effects of ranolazine. As shown in Figures 6A and 6C, the number of foci progressively decreased, such that wavefronts emanating from the foci spread over progressively wider regions, eventually leaving a single wavefront causing transient VT before the tissue became quiescent (Fig. 6A), in agreement with the experimental findings.
Figure 6. Simulation in 2-Dimensional Tissue of Multifocal VF Initiated With H2O2-Mediated Ionic Current Changes.
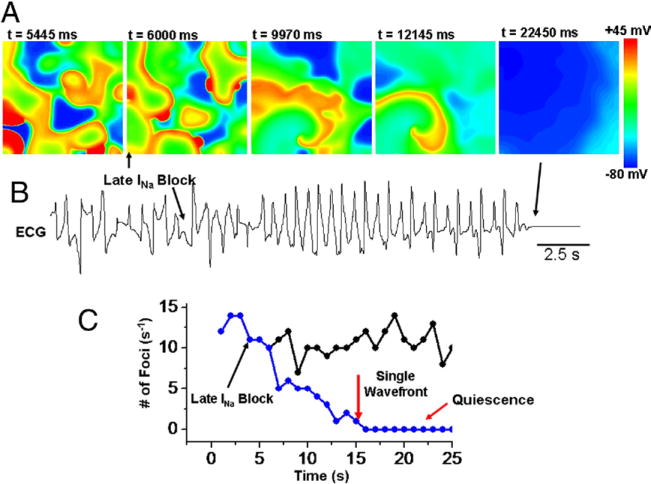
(A) Voltage snapshots in the presence of the late component of the fast sodium inward current (INa-L) (first frame) with its subsequent block (upward arrow) causing progressive decrease in the number of foci, eventually ending up with a single re-entrant wavefront causing transient VT, followed by termination. (B) Pseudo-ECG showing fibrillation-like state just before block of the INa-L with its subsequent block converting the VF to VT (first arrow), followed by tissue quiescence (second arrow). (C) Plot of the number of foci per second versus time for control (black circles) and with INa-L blocked (blue circles). Abbreviations as in Figures 1 and 2.
Discussion
The major conclusion of this study is that the late Na current blocking drug ranolazine demonstrates efficacy against both pacing-induced re-entrant VF and spontaneous oxidative multifocal VF. The suppression of multifocal VF by ranolazine is associated with a progressive reduction in the number of foci. This finding supports the hypothesis that multifocal VF requires the constant generation of new interacting foci to maintain themselves such that when the rate of production of new foci falls below a critical level, VF terminates. This is analogous to re-entrant VF, in which a critical mass is required to ensure that the rate of formation of new wavelets exceeds the rate of wavelet extinction (22).
Effects of ranolazine of pacing-induced re-entrant VF
The suppressant effect of ranolazine against re-entrant VF is compatible with previous studies demonstrating the drug’s efficacy to reducing dispersion of repolarization (28) and increasing the VF threshold (14). In addition, we found that ranolazine significantly reduced the maximum slope of APD restitution, which potentially contributes to its antifibrillatory effect by preventing APD restitution-driven wavebreak (29). Although the main target of ranolazine is the late Na current, other nonselective effects on IKr, IKs, and, to a lesser extent, ICaL (27) also are likely to be important in its overall antiarrhythmic effects. Ranolazine did not affect the Cai2+ decline rate and did not reverse H2O2-mediated slowing of the Cai2+ decline rate, consistent with a recent study showing a minimal effect of ranolazine on Cai2+ transient duration (26). However, consistent with previous studies (9,25) ranolazine caused a rate-dependent decrease in dV/Dtmax and conduction velocity, particularly at faster rates of activation.
Effects of ranolazine of spontaneous multifocal VF
Previous studies of isolated myocytes have shown that ranolazine has a potent suppressant effect on H2O2-mediated EADs and triggered activity (8). The present study extends these observations to EAD-mediated multifocal VF in intact hearts. Ranolazine progressively reduced the number EAD-mediated triggered foci, eventually terminating the multifocal VF. These findings suggest that the presence of a critical number of foci is necessary to maintain a multifocal VF, the rational being that EADs and triggered activity are afterpotentials, and therefore their formation is dependent on an obligatory prior excitation. This indicates that if no new foci seem to generate new foci, the VF can not be sustained, resulting in termination. The results of our 2D simulation corroborate this experimental dynamic scenario of multifocal VF termination. Reduction in the number of foci caused by the suppression of the late INa led to a progressive reduction of the number of foci resulting in VF termination. The role of wavebreak in the maintenance of H2O2-mediated VF seems to be minimal, because the foci remained confined to small regions (2 to 3 mm2) on the epicardium, with wavefront–wavefront collisions observed in only 4% of cases. There are several possible reasons why the foci remained spatially confined. The spread of foci may have been mediated by phase waves resulting from synchronization of EAD oscillations (7), rather than true propagation. Alternatively, outward propagation from the center of foci may have been decremental as a result of refractoriness of surrounding tissue. After ranolazine reduced the number of foci, regenerative outward propagation from foci was observed more frequently, consistent with the surrounding tissue having had more time to recover excitability.
Study limitations
In surface epicardial maps, an intramural wavefront breaking through the surface may have the appearance of a focus. However, we previously demonstrated that after endocardial and midmyocardial ablation, the epicardial cells have an intrinsic ability to generate H2O2-induced EADs, triggered activity, and VF (6). However, we can not dismiss the potential role of the endocardial Purkinje fiber network in the initiation and maintenance of focal VF (30,31). It could be argued that the observed epicardial focal mechanism may result from micro–re-entry, rather than triggered foci (32). Our single-cell microelectrode recordings from epicardial focal sites showing cellular EAD-mediated triggered activity and our 2D simulation showing EAD-mediated multifocal VF provide further suggestive evidence for a focal triggered activity, rather than a micro–re-entrant mechanism for VF. Although H2O2 is an artificial means of inducing oxidative stress, the 0.1-mM concentration used in this study is considered relevant to pathophysiological levels under conditions such as ischemia–reperfusion (33–35). Furthermore, it must be emphasized that the efficacy of ranolazine needs to be tested in different animal models of VF to substantiate its broad antifibrillatory efficacy. Finally, ranolazine’s IKr-blocking influence, which conceivably could account for its demonstrated ability to decrease ventricular defibrillation threshold (14) and perhaps re-entrant VF as well, was not tested in our computer model.
Conclusions
To our knowledge, this is the first study to demonstrate that ranolazine has a potent suppressant effect on both re-entrant and multifocal VF at concentrations considered therapeutic in humans (36,37). Our findings support previous studies demonstrating ranolazine’s efficacy against H2O2-mediated EADs and triggered activity in isolated cardiac myocytes (8,15), in isolated tissue (10), as well as ventricular arrhythmias in animal models (14,28,38) and in humans (39). Although H2O2 is an artificial means of inducing oxidative stress, there are many known triggers of oxidative stress in the heart, such as aging (40), heart failure (41), and ischemia–reperfusion (35), which are all conditions associated with increased risk of VF. The effect of ranolazine to reduce reperfusion arrhythmias significantly (42) indicates that the drug could exert a potent antiarrhythmic effect under conditions of oxidative stress (33).
Supplementary Material
Acknowledgments
The authors thank Pargol Samanianpour, MD, for her help with data analyses.
This study was supported by National Institutes of Health/National Heart, Lung, and Blood Institute grants P01 HL78931 and R01 HL103662. Dr. Morita received American Heart Association Western States Affiliate Fellowship Award 0725218Y. Dr. Lee is supported by Samsung Medical Center Research Award, Seoul, South Korea. Dr. Xie received 09POST2110079. Dr. Karagueuzian received Grant-in Aid 0555057Y. Drs. Karagueuzian and Weiss are supported by the Laubisch Endowment. Dr. Weiss is supported by the Kawata Endowment. Dr. Karagueuzian received a research grant from Gilead Sciences, Inc., Palo Alto, California.
Abbreviations and Acronyms
- 2D
2-dimensional
- APD
action potential duration
- Cai2+
intracellular calcium ion transient
- dV/Dtmax
maximum rate of phase-zero action potential depolarization
- EAD
early afterdepolarization
- H2O2
hydrogen peroxide
- INa-L
late component of the fast sodium inward current
- LV
left ventricle
- VF
ventricular fibrillation
- VT
ventricular tachycardia
APPENDIX
For supplementary videos and their legends, please see the online version of this article.
References
- 1.Moe GK, Rheinboldt WL, Abildskov JA. A computer model of atrial fibrillation. Am Heart J. 1964;64:200–20. doi: 10.1016/0002-8703(64)90371-0. [DOI] [PubMed] [Google Scholar]
- 2.Samie FH, Berenfeld O, Anumonwo J, et al. Rectification of the background potassium current: a determinant of rotor dynamics in ventricular fibrillation. Circ Res. 2001;89:1216–23. doi: 10.1161/hh2401.100818. [DOI] [PubMed] [Google Scholar]
- 3.Wu TJ, Lin SF, Weiss JN, Ting CT, Chen PS. Two types of ventricular fibrillation in isolated rabbit hearts: importance of excitability and action potential duration restitution. Circulation. 2002;106:1859–66. doi: 10.1161/01.cir.0000031334.49170.fb. [DOI] [PubMed] [Google Scholar]
- 4.Scherf D. Studies on auricular tachycardia caused by aconitine administration. Proc Exp Biol Med. 1947;64:233–9. doi: 10.3181/00379727-64-15754. [DOI] [PubMed] [Google Scholar]
- 5.Swissa M, Qu Z, Ohara T, et al. Action potential duration restitution and ventricular fibrillation due to rapid focal excitation. Am J Physiol Heart Circ Physiol. 2002;282:H1915–23. doi: 10.1152/ajpheart.00867.2001. [DOI] [PubMed] [Google Scholar]
- 6.Morita N, Sovari AA, Xie Y, et al. Increased susceptibility of aged hearts to ventricular fibrillation during oxidative stress. Am J Physiol Heart Circ Physiol. 2009;297:H1594–605. doi: 10.1152/ajpheart.00579.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sato D, Xie LH, Sovari AA, et al. Synchronization of chaotic early afterdepolarizations in the genesis of cardiac arrhythmias. Proc Natl Acad Sci U S A. 2009;106:2983–8. doi: 10.1073/pnas.0809148106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song Y, Shryock JC, Wagner S, Maier LS, Belardinelli L. Blocking late sodium current reduces hydrogen peroxide-induced arrhythmogenic activity and contractile dysfunction. J Pharmacol Exp Ther. 2006;318:214–22. doi: 10.1124/jpet.106.101832. [DOI] [PubMed] [Google Scholar]
- 9.Fredj S, Sampson KJ, Liu H, Kass RS. Molecular basis of ranolazine block of LQT-3 mutant sodium channels: evidence for site of action. Br J Pharmacol. 2006;148:16–24. doi: 10.1038/sj.bjp.0706709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Antzelevitch C, Belardinelli L, Zygmunt AC, et al. Electrophysiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation. 2004;110:904–10. doi: 10.1161/01.CIR.0000139333.83620.5D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Antzelevitch C, Belardinelli L, Wu L, et al. Electrophysiologic properties and antiarrhythmic actions of a novel antianginal agent. J Cardiovasc Pharmacol Ther. 2004;9(Suppl 1):S65–83. doi: 10.1177/107424840400900106. [DOI] [PubMed] [Google Scholar]
- 12.Sicouri S, Timothy KW, Zygmunt AC, et al. Cellular basis for the electrocardiographic and arrhythmic manifestations of Timothy syndrome: effects of ranolazine. Heart Rhythm. 2007;4:638–47. doi: 10.1016/j.hrthm.2006.12.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu L, Rajamani S, Li H, January CT, Shryock JC, Belardinelli L. Reduction of repolarization reserve unmasks the proarrhythmic role of endogenous late Na(+) current in the heart. Am J Physiol Heart Circ Physiol. 2009;297:H1048–57. doi: 10.1152/ajpheart.00467.2009. [DOI] [PubMed] [Google Scholar]
- 14.Kumar K, Nearing BD, Bartoli CR, Kwaku KF, Belardinelli L, Verrier RL. Effect of ranolazine on ventricular vulnerability and defibrillation threshold in the intact porcine heart. J Cardiovasc Electrophysiol. 2008;19:1073–9. doi: 10.1111/j.1540-8167.2008.01204.x. [DOI] [PubMed] [Google Scholar]
- 15.Xie LH, Chen F, Karagueuzian HS, Weiss JN. Oxidative stress-induced afterdepolarizations and calmodulin kinase II signaling. Circ Res. 2009;104:79–86. doi: 10.1161/CIRCRESAHA.108.183475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maier LS, Bers DM. Calcium, calmodulin, and calcium-calmodulin kinase II: heartbeat to heartbeat and beyond. J Mol Cell Cardiol. 2002;34:919–39. doi: 10.1006/jmcc.2002.2038. [DOI] [PubMed] [Google Scholar]
- 17.Anderson ME, Braun AP, Wu Y, et al. KN-93, an inhibitor of multifunctional Ca++/calmodulin-dependent protein kinase, decreases early afterdepolarizations in rabbit heart. J Pharmacol Exp Ther. 1998;287:996–1006. [PubMed] [Google Scholar]
- 18.Erickson JR, Joiner ML, Guan X, et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133:462–474. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ward CA, Giles WR. Ionic mechanism of the effects of hydrogen peroxide in rat ventricular myocytes. J Physiol. 1997;500(Pt 3):631–42. doi: 10.1113/jphysiol.1997.sp022048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ono N, Hayashi H, Kawase A, et al. Spontaneous atrial fibrillation initiated by triggered activity near the pulmonary veins in aged rats subjected to glycolytic inhibition. Am J Physiol Heart Circ Physiol. 2007;292:639–48. doi: 10.1152/ajpheart.00445.2006. [DOI] [PubMed] [Google Scholar]
- 21.Mahajan A, Sato D, Shiferaw Y, et al. Modifying L-type calcium current kinetics: consequences for cardiac excitation and arrhythmia dynamics. Biophys J. 2008;94:411–23. doi: 10.1529/biophysj.106.98590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim YH, Garfinkel A, Ikeda T, et al. Spatiotemporal complexity of ventricular fibrillation revealed by tissue mass reduction in isolated swine right ventricle. Further evidence for the quasiperiodic route to chaos hypothesis. J Clin Invest. 1997;100:2486–500. doi: 10.1172/JCI119791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garfinkel A, Chen PS, Walter DO, et al. Quasiperiodicity and chaos in cardiac fibrillation. J Clin Invest. 1997;99:305–14. doi: 10.1172/JCI119159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nash MP, Mourad A, Clayton RH, et al. Evidence for multiple mechanisms in human ventricular fibrillation. Circulation. 2006;114:536–42. doi: 10.1161/CIRCULATIONAHA.105.602870. [DOI] [PubMed] [Google Scholar]
- 25.Rajamani S, El-Bizri N, Shryock JC, Makielski JC, Belardinelli L. Use-dependent block of cardiac late Na(+) current by ranolazine. Heart Rhythm. 2009;6:1625–31. doi: 10.1016/j.hrthm.2009.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wasserstrom JA, Sharma R, O’Toole MJ, et al. Ranolazine antagonizes the effects of increased late sodium current on intracellular calcium cycling in rat isolated intact heart. J Pharmacol Exp Ther. 2009;331:382–91. doi: 10.1124/jpet.109.156471. [DOI] [PubMed] [Google Scholar]
- 27.Hale SL, Shryock JC, Belardinelli L, Sweeney M, Kloner RA. Late sodium current inhibition as a new cardioprotective approach. J Mol Cell Cardiol. 2008;44:954–67. doi: 10.1016/j.yjmcc.2008.03.019. [DOI] [PubMed] [Google Scholar]
- 28.Antoons G, Oros A, Beekman JD, et al. Late Na(+) current inhibition by ranolazine reduces torsades de pointes in the chronic atrioventricular block dog model. J Am Coll Cardiol. 2010;55:801–9. doi: 10.1016/j.jacc.2009.10.033. [DOI] [PubMed] [Google Scholar]
- 29.Weiss JN, Chen PS, Qu Z, Karagueuzian HS, Garfinkel A. Ventricular fibrillation: how do we stop the waves from breaking? Circ Res. 2000;87:1103–7. doi: 10.1161/01.res.87.12.1103. [DOI] [PubMed] [Google Scholar]
- 30.Tabereaux PB, Dosdall DJ, Ideker RE. Mechanisms of VF maintenance: wandering wavelets, mother rotors, or foci. Heart Rhythm. 2009;6:405–15. doi: 10.1016/j.hrthm.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu TJ, Lin SF, Hsieh YC, Chiu YT, Ting CT. Repetitive endocardial focal discharges during ventricular fibrillation with prolonged global ischemia in isolated rabbit hearts. Circ J. 2009;73:1803–11. doi: 10.1253/circj.cj-09-0260. [DOI] [PubMed] [Google Scholar]
- 32.Ideker RE, Rogers JM, Fast V, Li L, Kay GN, Pogwizd SM. Can mapping differentiate microreentry from a focus in the ventricle? Heart Rhythm. 2009;6:1666–9. doi: 10.1016/j.hrthm.2009.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zweier JL, Flaherty JT, Weisfeldt ML. Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proc Natl Acad Sci USA. 1987;84:1404–7. doi: 10.1073/pnas.84.5.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corretti MC, Koretsune Y, Kusuoka H, Chacko VP, Zweier JL, Marban E. Glycolytic inhibition and calcium overload as consequences of exogenously generated free radicals in rabbit hearts. J Clin Invest. 1991;88:1014–25. doi: 10.1172/JCI115361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bolli R, Patel BS, Jeroudi MO, Lei EK, McCay PB. Demonstration of free radical generation in “stunned” myocardium of intact dogs with the use of the spin trap alpha-phenyl N-tert-butyl nitrone. J Clin Invest. 1988;82:476–85. doi: 10.1172/JCI113621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sicouri S, Glass A, Bellardinelli L, Antzelevitch C. Antiarrhythmic effects of ranolazine in canine pulmonary vein sleeve preparations. Heart Rhythm. 2008;5:1019–26. doi: 10.1016/j.hrthm.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zaza A, Belardinelli L, Shryock JC. Pathophysiology and pharmacology of the cardiac “late sodium current”. Pharmacol Ther. 2008;119:326–39. doi: 10.1016/j.pharmthera.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 38.Burashnikov A, Di Diego JM, Zygmunt AC, Belardinelli L, Antzelevitch C. Atrium-selective sodium channel block as a strategy for suppression of atrial fibrillation: differences in sodium channel inactivation between atria and ventricles and the role of ranolazine. Circulation. 2007;116:1449–57. doi: 10.1161/CIRCULATIONAHA.107.704890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scirica BM, Morrow DA, Hod H, et al. Effect of ranolazine, an antianginal agent with novel electrophysiological properties, on the incidence of arrhythmias in patients with non ST-segment elevation acute coronary syndrome: results from the Metabolic Efficiency With Ranolazine for Less Ischemia in Non ST-Elevation Acute Coronary Syndrome Thrombolysis in Myocardial Infarction 36 (MERLIN-TIMI 36) randomized controlled trial. Circulation. 2007;116:1647–52. doi: 10.1161/CIRCULATIONAHA.107.724880. [DOI] [PubMed] [Google Scholar]
- 40.Jahangir A, Sagar S, Terzic A. Aging and cardioprotection. J Appl Physiol. 2007;103:2120–8. doi: 10.1152/japplphysiol.00647.2007. [DOI] [PubMed] [Google Scholar]
- 41.Ide T, Tsutsui H, Kinugawa S, et al. Direct evidence for increased hydroxyl radicals originating from superoxide in the failing myocardium. Circ Res. 2000;86:152–7. doi: 10.1161/01.res.86.2.152. [DOI] [PubMed] [Google Scholar]
- 42.Dhalla AK, Wang WQ, Dow J, et al. Ranolazine, an antianginal agent, markedly reduces ventricular arrhythmias induced by ischemia and ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2009;297:H1923–9. doi: 10.1152/ajpheart.00173.2009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.