

SUMMARY
The recognition of modified histones by “reader” proteins constitutes a key mechanism regulating gene expression in the chromatin context. Compared with the great variety of readers for histone methylation, few protein modules that recognize histone acetylation are known. Here we show that the AF9 YEATS domain binds strongly to histone H3K9 acetylation and, to a lesser extent, H3K27 and H3K18 acetylation. Crystal structural studies revealed that AF9 YEATS adopts an eight-stranded immunoglobin fold and utilizes a serine-lined aromatic “sandwiching” cage for acetyllysine readout, representing a novel recognition mechanism that is distinct from that of known acetyllysine readers. ChIP-seq experiments revealed a strong co-localization of AF9 and H3K9 acetylation genome-wide, which is important for the chromatin recruitment of the H3K79 methyltransferase DOT1L. Together, our studies identified the evolutionarily conserved YEATS domain as a novel acetyllysine-binding module and established a direct link between histone acetylation and DOT1L-mediated H3K79 methylation in transcription control.
INTRODUCTION
The histone proteins are subjected to a number of post-translational modifications (PTMs) that play a critical role in regulating chromatin dynamics and the accessibility of the underlying DNA in eukaryotes (Strahl and Allis, 2000). Acetylation of lysine residues, one of the most frequent PTMs that occur on histones, has been well characterized as a mark of active transcription that is controlled by two families of counteracting enzymes: histone acetyltransferases (HATs) and histone deacetylases (HDACs) (Kouzarides, 2007). The addition of an acetyl moiety to the ε-amino group of histone lysine residues neutralizes their positive charge, thereby reducing the electrostatic interaction between histones and DNA and diminishing nucleosome stability. Furthermore, the bulky acetyl groups on lysine residues can also serve as docking sites for reader proteins, which recognize this specific modification and transduce the molecular signals downstream to elicit various biological outcomes (Jenuwein and Allis, 2001).
Bromodomain (BRD), a protein module evolutionarily conserved from yeast to human, has long been thought to be the sole protein module that specifically recognizes acetyllysine motifs (Dhalluin et al., 1999). There are 46 BRD-containing proteins in humans; despite sequence variations, all BRD modules share a conserved fold of four α helices. The acetyl group on the lysine substrate is recognized by a central deep hydrophobic pocket and is anchored by a hydrogen bond to an invariable asparagine residue in most acetyllysine-recognizing BRDs (Filippakopoulos et al., 2012). Recently, some tandem plant homeodomain (PHD) zinc fingers have been shown to bind histone H3 in an acetylation-sensitive manner (Ali et al., 2012; Qiu et al., 2012; Zeng et al., 2010). In addition, the tandem pleckstrin-homology (PH) domain of the yeast chaperone protein Rtt106 binds the H3K56-containing region in an acetylation-sensitive manner (Su et al., 2012). However, unlike the BRDs, these tandem domains also bind to the unmodified histone H3 relatively well; therefore, the acetylation on vicinal lysine residues is likely to promote existing interactions between the unmodified histones and the reader modules. Nevertheless, compared with our understanding of the protein modules known to recognize histone methylation, our knowledge of the protein modules that can recognize histone acetylation is very limited.
Here, we report our discovery of the YEATS domains as a novel family of histone acetylation readers. The YEATS domain, named for its five founding domain-containing proteins (Yaf9, ENL, AF9, Taf14 and Sas5), is evolutionarily conserved from yeast to human (Le Masson et al., 2003). There are four YEATS domain-containing proteins in humans and three in S. cerevisiae, all of which are associated with HAT complexes, chromatin-remodeling complexes or transcription-regulating complexes (Schulze et al., 2009). We found that the YEATS domains of AF9 from diverse species all bind to acetylated histone H3, with a strong preference for H3K9 acetylation (H3K9ac). The co-crystal structure of human AF9 YEATS with the H3K9ac peptide revealed a novel acetyl recognition mechanism that is distinct from those of BRDs and tandem PHD fingers. AF9 YEATS adopts an immunoglobin fold and utilizes a serine-lined aromatic “sandwiching” cage for the specific readout of acetyllysine within the “RK” consensus sequence. AF9 is part of the super elongation complex and also associates with the histone H3K79 methyltransferase DOT1L (Smith et al., 2011). ChIP-seq experiments revealed a strong co-localization of AF9 and H3K9ac genome-wide. We found that H3K9ac recognition by AF9 is essential for the chromatin recruitment of DOT1L and the subsequent deposition of H3K79 methylation on target genes. Together, our results identified the YEATS domain as a novel acetyllysine-binding module and established direct link between histone acetylation and DOT1L-mediated H3K79 methylation in transcription control.
RESULTS
The YEATS Domain Is a Novel Histone Acetylation-Recognizing Module
To identify novel protein modules that can recognize histone acetylation, we utilized a modified histone peptide array that contains peptides bearing most known acetylated lysines on histones (Table S1) to screen the domains with unknown functions. We found that the YEATS domain of the human AF9 protein (Figure 1A) bound strongly to H3K9ac and, to a lesser extent, the H3K27ac and H3K18ac peptides (Figure 1B). Pulldown assays using biotinylated histone peptides or acetylated full-length histones showed similar results (Figures 1C and S1A), suggesting that the binding of the AF9 YEATS domain to the histone H3 tail is acetylation-dependent. Quantitative isothermal titration calorimetry (ITC) analysis revealed a dissociation constant (KD) of 3.7 µM for AF9 YEATS domain to the H3K9ac peptide (Figure 1D), an affinity that is much higher than that of many BRDs to acetyllysines (Filippakopoulos et al., 2012). The binding KD dropped to 7.0 and 11.0 µM for H3K27ac and H3K18ac peptides, respectively; and no bindings were observed for the H3K14ac and other acetylated histone peptides (Figures 1C, 1D and S1B).
Figure 1. The AF9 YEATS Domain Is a Novel Histone Acetylation-Recognizing Module.

(A) Schematic representation of AF9 protein structure.
(B) A histone peptide microarray probed with GST-AF9 YEATS domain.
(C) Western blot analysis of histone peptide pulldowns with GST-AF9 YEATS domain and the indicated biotinylated peptides.
(D) The AF9 YEATS domain binds with highest affinity to the H3K9ac peptide. ITC curves of the indicated histone peptides titrated into the AF9 YEATS domain. ND: not detectable.
(E) Recognition of histone acetylation is a common property of the AF9/Yaf9 YEATS domains from diverse species. Western blot analysis of histone peptide pulldowns with YEATS domains from the indicated species and acetylated peptides.
AF9 is an evolutionarily conserved protein in virtually all eukaryotes (Schulze et al., 2009). Peptide pulldowns of the YEATS domains of AF9/Yaf9 proteins from human, mouse, Drosophila, S. pombe and S. cerevisiae revealed that all these YEATS domains bound to acetylated histone H3, albeit with slightly different specificities. The AF9 homologs in human, mouse and Drosophila shared similar substrate specificity, whereas the yeast Yaf9 protein exhibited a higher binding affinity towards H3K27ac than H3K9ac (Figures 1E and S1B). Together, these results suggest that histone acetylation recognition is an evolutionarily conserved function of the YEATS domains.
Crystal Structure of AF9 YEATS Bound to H3K9ac
To gain molecular insights into the reader function of the AF9 YEATS domain, we solved the crystal structure of human AF9 YEATS (1–138) bound to the H31–10K9ac peptide at 2.3 Å (Table S2). We found that the AF9 YEATS domain has two monomers in one asymmetric unit that are related by twofold noncrystallographic symmetry (Figure S2A). Similar to what is observed in the crystal of Yaf9 YEATS (Figure S2D) (Wang et al., 2009), the very N-terminal segment of each monomer takes on an extended β-strand conformation that stabilizes an AF9 YEATS dimer in the crystal through anti-parallel β-sheet formation (β1-β1’) (Figure S2B). Such a dimer is likely induced by crystal packing, as size exclusion chromatography followed by multiangle light scattering analysis revealed that AF9 YEATS primarily existed as monomer in solution (Figure S2C).
AF9 YEATS adopts an immunoglobin (Ig) fold that consists of a two-layer β-sandwich between eight antiparallel β-strands arranged in a Greek key topology (Figures 2A and 2C). Two α-helices (α1, αC) cap the β-sandwich from the N-proximal end and stabilize a compact Ig fold through hydrophobic contacts (Figure S3A). On the basis of electron density, we could model all 138 residues of AF9 YEATS and trace the “T3-K4-Q5-T6-A7-R8-K9ac-S10” residues of the H31–10K9ac peptide (Figure 2A). The histone peptide is docked onto a surface formed by loops L4, L6 and L8 on top of the β(4,3,6,7)-sheets in an orientation perpendicular to the β-stands (Figures 2A and 2B). Notably, recognition of K9ac is achieved by a serine-lined aromatic cage in the cleft of loops L4 and L6 (Figures 2A). The long side chain of H3K9ac snugly inserts into the acetyllysine-binding pocket at the N-distal end of the elongated AF9 YEATS domain (Figure 2B). The H3 binding surface is negatively charged, thereby electrostatically facilitating the recognition of the basic H3 peptide (Figure 2B).
Figure 2. Crystal Structure of the AF9 YEATS–H3K9ac Complex.

(A) Overall structure of AF9 YEATS domain bound to the H3K9ac peptide. AF9 YEATS is shown as green ribbon with key residues of Kac pocket depicted as salmon stick. H3K9ac peptide is shown as yellow sticks covered by the simulated annealing Fo-Fc omit map countered at 2.5 σ level.
(B) Electrostatic surface view of the AF9 YEATS-H3K9ac complex structure. Electrostatic potential is expressed as a spectrum ranging from −6 kT/e (red) to +6 kT/e (blue). The H3K9ac peptide is depicted as space-filling sphere with yellow for carbon, blue for nitrogen and red for oxygen atoms.
(C) Topology diagram of AF9 YEATS domain. β-strands in green are numbered sequentially from N- to C-terminus and helices are shown in purple. The H3K9ac peptide is depicted as thick yellow line with the acetyl group highlighted as magenta star.
(D) Conservation mapping around the H3-binding surface among AF9 homologues listed in panel E. White and green colors indicate low (≤0.25) and high (1.0) sequence conservation, respectively. The H3K9ac peptide is shown in yellow stick.
(E) Sequence alignment of YEATS domain homologues from yeast to human. Conserved residues are shaded in yellow; identical residues are shaded in blue and cyan. Red dots, residues forming K9ac pocket; green star, the H3R8-binding residue; blue asters, the H3T6-binding residues; purple pound sign, H3K4 pocket residues.
See also Figure S2.
Residue conservation analysis among YEATS paralogs revealed strict conservation of the amino acids that compose the H3K9ac-binding pocket (Figures 2D and 2E), suggesting that acetyllysine readout is likely a common feature of all YEATS domains. Structural alignment of the YEATS domain of AF9 with those of yeast Yaf9 and Taf14 revealed a high degree of conservation of the core β-strands and the loops L4, L5 and L6, with some conformational variations at loops L1, L8 and N-proximal loops and α-helices (Figure S3B). We calculated a Cα root-mean-square deviation (RSMD) of 0.767 Å between AF9-Yaf9 and of 1.681 Å between AF9-Taf14. Since L1 and L8 loops participate in the formation of histone-binding surfaces, the conformational and sequence variations of L1 and L8 (Figures 2E and S3B) indicate that different YEATS domains may recognize acetyllysine in distinct sequence contexts.
A Novel Acetyllysine Recognition Mechanism of the AF9 YEATS Domain
In the AF9 YEATS-H3K9ac complex, the YEATS domain uses loops L1, L4, L6 and L8 and strands β2 and β7 to form extensive interactions with the H3 segment of T3-S10 (Figure 3A, stereo view). As highlighted in the LigPlot diagram (Laskowski and Swindells, 2011), these interactions include nine direct hydrogen bonds, four sets of water-mediated hydrogen bonds and a number of hydrophobic contacts (Figure 3B). Upon complex formation, we calculated 764 Å2 of the buried solvent accessible surface (SAS) area that accounts for 43% of the SAS area of the H3 peptide.
Figure 3. Molecular Details for Type- and Site-Specific Recognition of H3K9ac by the AF9 YEATS Domain.

(A) Stereo view of hydrogen bonding network involving H3 side chains (yellow sticks) and residues in AF9 YEATS (green sticks). Magenta dashes, hydrogen bonds; Small cyan balls, waters.
(B) LIGPLOT diagram listing critical contacts between the H3K9ac peptide and the AF9 YEATS domain. H3 segment (orange) and key residues of AF9 YEATS (green) are depicted in ball-and-stick mode. Grey ball, carbon; Blue ball, nitrogen; Red ball, oxygen; Big cyan ball, water molecule.
(C) Close-up view of the K9ac-binding pocket of the AF9 YEATS domain. The pocket is displayed as semi-transparent surface with key residues shown as green sticks. Kac is depicted in both yellow stick and space-filling sphere modes.
(D) Hydrogen bonding networks involving H3 main chain and the AF9 YEATS domain. For clarity, H3 side chains are omitted from stick representation except for K9ac.
AF9 YEATS organizes the highly conserved residues H56, S58 and F59 of loop L4, Y78 and F81 of loop L6, and F28 of loop L1 to generate a serine-lined aromatic cage for the acetyllysine readout (Figure 3A). The addition of an acetyl moiety causes the lysine residue to lose a positive charge while gaining hydrophobicity, extra hydrogen bonding capability, and an extended side-chain dimension. AF9 YEATS utilizes the side-chain hydroxyl (OH) of S58 and the backbone amide (NH) of Y78 to form relayed hydrogen bonding interactions with the amide NεH and carbonyl oxygen of the acetyllysine side chain, respectively (Figure 3A). The alignment of the acetyl group is further strengthened by water-mediated hydrogen bonding that involves the acetyl carbonyl oxygen and residues W32 and A79 of AF9 YEATS (Figure 3A). The long acetyllysine side chain is sandwiched by bulky aromatic residues H56, F59 and F81 from the bottom and F28 and Y78 from the top, with the flat acetyl group snugly clamped in position (Figure 3C). Notably, in addition to hydrophobic effects, the positioning of acetyllysine hydrocarbon chain over the π-electrons of a cluster of aromatic rings (H56, Y78, F81, F59 and F28) (Figure 3C) introduces multiple sets of CH-π interactions, which collectively contributes to stable acetyllysine binding as has been reported in many other cases of chemical or biological recognition (Nishio et al., 1998). All these structural features suggest that the aromatic cage in the AF9 YEATS fold is optimal for acetyllysine readout, but not for the unmodified or K9-methylated H3 tails (Figures 1C and S1). The loss of hydrogen bond forming potential and steric hindrance originated from a branched feature of methyllysine render both unmodified and methylated lysine inappropriate for AF9 YEATS recognition.
Site-Specific Recognition of Histone Acetylation by AF9 YEATS
In addition to acetylation recognition by the serine-lined aromatic cage, the H3K9 site-specific readout by AF9 YEATS is contributed by K9ac-flanking residues of histone H3. The H3 backbone interacts across loops L4, L6 and L8 of AF9 YEATS through five direct and one water-mediated hydrogen bonds (Figure 3D). In respect of side-chain mediated interactions, H3R8 forms charge-stabilized hydrogen bonds with D103; H3T6 inserts into a shallow pocket formed by A79, L106 and L108, and is stabilized by a hydrogen bonding network organized by a buried water; H3Q5 participates in direct and water-mediated hydrogen bonds with H3R8 and YEATS D103, respectively. Additionally, H3K4 contributes to H3 binding by hydrophobic contacts with the imidazole rings of H107 and H111 of AF9 YEATS (Figures 3A and 3B).
AF9 displayed preference to H3K9ac with compromised affinities to H3K27ac and H3K18ac and weak or no detectable bindings to the H3K14ac or acetylated H4 peptides (Figures 1D and S1). Sequence context comparison revealed that H3K9ac (K4-Q5-T6-A7-R8-Kac9-S10), H3K27ac (T22-K23-A24-A25-R26-Kac27-S28) and H3K18ac (G13-K14-A15-P16-R17-Kac18-Q19) share a common “R-Kac” signature motif, which likely accounts for their relatively high binding affinities with AF9 YEATS, whereas sequence discrepancy at other positions among these peptides may explain the 2 to 3-fold binding differences.
Mutagenesis and Binding Studies of the AF9 YEATS-H3K9ac Interaction
Calorimetric titration assays using a variety of histone H3 peptides revealed that the KD values were 3.7 µM for H31–10K9ac, 5.7 µM for H31–15K9ac and 2.8 µM for H34–10K9ac, whereas 230 µM and no binding were observed for the H35–15K9ac and H37–11K9ac peptides, respectively (Figure 4A), demonstrating the requirement of the H3 segment K4-Q5-T6 for AF9 YEATS binding. Notably, substitution of H3R8 with alanine caused ~200-fold binding reduction, underscoring the importance of the arginine-mediated polar interactions at the -1 position (Figure 4A). By contrast, H3K4 trimethylation or H3S10 phosphorylation displayed minimal effects on H3K9ac binding (Figure 4A), suggesting that AF9 could serve as an effective reader accommodating active H3 modification patterns of “H3K4me3-K9ac” and “H3K9ac-S10ph”.
Figure 4. Analysis of the AF9 YEATS-H3K9ac interactions.

(A) ITC fitting curves of AF9 YEATS titrated with different frames of the H3K9ac peptides (left) and the H3 peptides containing different modifications (right).
(B) The AF9 YEATS-H3K9ac complex structure highlighting the residues used for the mutagenesis and binding studies.
(C) ITC fitting curves of H31–10K9ac peptide with point mutants clustered to H3 binding (left) or non-binding (right) surfaces.
(D) Western blot analysis of the peptide pulldown analysis using the WT AF9 YEATS domain and the indicated point mutants.
Similarly, point mutations of F28, S58, H56, F59, G77, Y78, and D103 around the H3K9ac binding surface of AF9 YEATS (Figure 4B) resulted in approximately 8 to 130-fold binding reduction (Figures 4C and 4D). Compared with a mild binding reduction of F28A (KD = 30.4 µM), the marked binding loss of F59A (KD = 464 µM) and Y78A (KD = 490 µM) underscored the importance of these two sandwiching residues in acetyllysine recognition. A small side chain of AF9 G77, highly conserved among YEATS domains (Figure 2E), is critical to provide an ample space for the acetyl group (Figure 3C). Mutations of G77 to alanine or serine with bigger side chains diminished or disrupted binding, respectively. Furthermore, mutation of AF9 D103, which directly interacts with H3R8, resulted in ~50-fold affinity drop, highlighting the importance of the “R-D” ion pair (Figure 4B). In comparison, mutations of the residues that do not participate in direct H3 binding, including Q8, N43, F47 on the N-proximal surface of AF9 YEATS, exhibited only minor defects in H3K9ac binding (Figure 4C).
Comparison of Distinct Acetyl-Binding Pockets
Compared with known histone acetyllysine readers such as the BRD4 BRD and the DPF3 tandem PHD fingers (Filippakopoulos et al., 2012; Zeng et al., 2010), AF9 YEATS represents a novel class of acetyllysine reader. The YEATS domain adopts an Ig-fold and makes use of two loops (L4 and L6) connecting four β-strands to generate an acetyllysine reader pocket at one corner of the YEATS domain; and acetyllysine is inserted into a deep pocket formed by a serine and several aromatic residues (Figures 5A and 5B). In contrast, a typical bromodomain adopts an α-helical bundle fold and makes use of two loops (LZA and LBC) connecting four α-helices to generate a central deep acetyllysine pocket lined by an indispensable asparagine and a few hydrophobic residues, as represented by the first BRD of BRD4 (Figures 5C and 5D). The DPF3b tandem PHD fingers utilize a β2-surface to generate a shallow pocket formed by an aspartate and three hydrophobic residues for acetyllysine readout (Figures 5E and 5F).
Figure 5. Structural Comparison of AF9 YEATS with Other Acetyllysine Readers.

(A) Side-insertion of H3K9ac into AF9 YEATS. Deep insertion of Kac is highlighted in a close-up cutaway view at the bottom. AF9 YEATS is shown in both ribbon and semi-transparent molecular surface view. The two Kac pocket-forming loops are labeled L4 and L6.
(B) Acetyllysine recognition by the AF9 YEATS reader pocket. Relayed hydrogen bonding is shown as magenta dashes.
(C) Top-insertion of H3K14ac into the first BRD of BRD4 (Bromo1). Coordinates are taken from PDB entry, 3JVK. LZA and LBC denote two loops used for Kac pocket formation. Close-up view illustrates the deep insertion of Kac into a half-open pocket.
(D) Acetyllysine recognition by the reader pocket of BRD4 Bromo1. Note the hydrogen bond between Asn side chain and the carbonyl oxygen of the acetyl group.
(E) Top-insertion of H3K14ac into the first PHD finger (PHD1) of the DPF3b tandem PHD fingers (PHD12). Coordinates are taken from PDB entry, 2KWJ. Note the shallow nature of the Kac pocket highlighted in a close-up cut-away view.
(F) Acetyllysine recognition details of the DPF3b PHD1 reader pocket.
See also Figure S4.
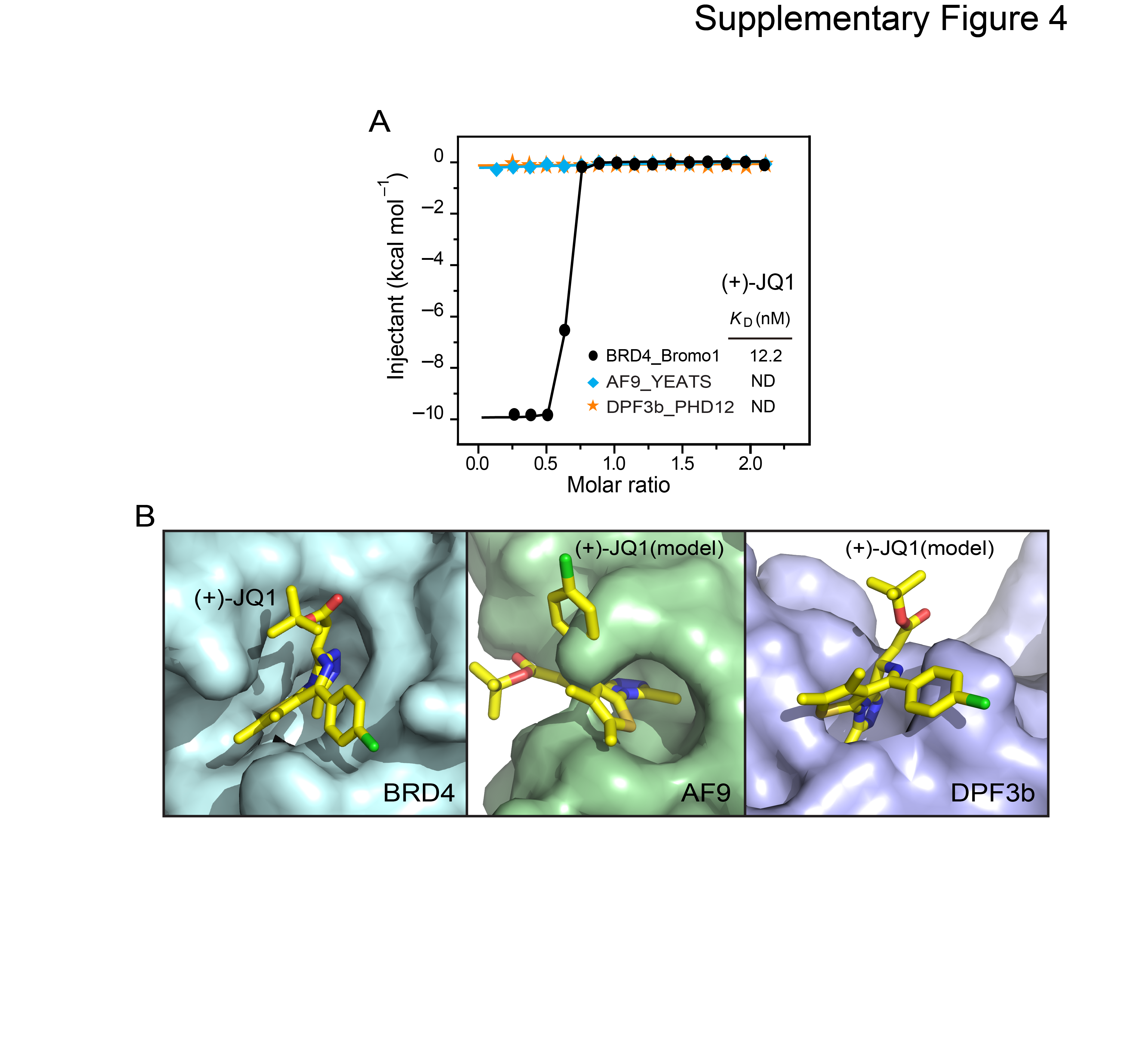
A close comparison of the acetyllysine pockets revealed that AF9 YEATS and BRD4 BRD have different pocket dimension and residue compositions. AF9 YEATS pocket possesses an intimate encapsulation of acetyllysine; specific recognition is achieved by relayed hydrogen bonding, CH-π interactions, and hydrophobic contacts associated with high degree of shape complementarity (Figures 3C). We calculated a shape correlation statistic (Sc) (Lawrence and Colman, 1993) of 0.85 between acetyllysine side chain and the AF9 YEATS reader pocket. In contrast, the reader pocket of the first BRD of BRD4 is relatively open in one dimension (Sc = 0.77) (Figure 5C), and acetyllysine is stabilized by one Asn-mediated hydrogen bond in addition to a few hydrophobic contacts (Figure 5D). In direct contrast to the site-specific recognition of a single acetyllysine by the YEATS domain, some BRDs exhibited much stronger interactions with multiply acetylated histone tails (Filippakopoulos et al., 2012; Moriniere et al., 2009), indicating that they may possess overlapping but also distinct roles in reading histone acetylation. The half-open pocket enables BRD4 an ideal target of the bulky inhibitor, (+)-JQ1 (Filippakopoulos et al., 2010). We observed a strong binding between BRD4 BRD1 and (+)-JQ1 by ITC (KD = 12.2 nM); whereas in sharp contrast, both AF9 YEATS and the DPF3b tandem PHD fingers had no interaction with (+)-JQ1 (Figure S4A), likely due to the unmatched dimensions between (+)-JQ1 and the reader pockets (Figure S4B).
AF9 Associates with H3K9ac-Enriched Chromatin in Cells
AF9 is a component of large protein complexes termed as super elongation complex (SEC) (Lin et al., 2010), elongation assisting proteins (EAP) (He et al., 2010) or AF4/ENL/P-TEFb complex (AEP) (Yokoyama et al., 2010) that contain overlapping subunits including AFF1/4, ELL, EAF1/2, ENL, P-TEFb and/or DOT1L (Sobhian et al., 2010). For simplicity, the term “SEC” is used in the current study. Within the SEC, the AFF1/4 scaffold proteins directly bind to the C-terminal region of AF9, whereas DOT1L can bind to the same region of AF9 independent of SEC (Biswas et al., 2011; He et al., 2011). Consistent with these published results, our co-immunoprecipitation (IP) experiments demonstrated that deletion of the C-terminal protein-protein interaction domain (ΔC) of AF9 abolished or reduced the interactions between AF9 and DOT1L, the SEC subunits AFF4 and ELL2, or the P-TEFb complex component CDK9, whereas deletion of the N-terminal YEATS domain (ΔN) had no effect (Figure 6A). In contrast, protein-ChIP experiments using crosslinked cell lysates revealed that the association with H3K9ac-enriched chromatin of the ΔN mutant, but not the ΔC mutant, was greatly impaired (Figure 6B). These results suggest that the C-terminal region of AF9 is responsible for protein-protein interactions between AF9 and DOT1L or SEC components, whereas the YEATS domain is indispensible for the chromatin recruitment of AF9.
Figure 6. AF9 Co-Localizes with H3K9ac Genome-Wide.

(A) Western blot analysis of co-IP using the M2 anti-Flag antibody in cells expressing Flag-AF9 and Myc-tagged DOT1L, AFF4, ELL2 or CDK9 proteins. FL: full-length; ∆N: deletion of aa1–112; ∆C: deletion of aa480–568 of AF9.
(B) Western blot analysis with the indicated antibodies of Flag ChIP in cells expressing the full-length or truncated Flag-AF9 proteins as in (A).
(C) Genomic distribution of AF9 ChIP-seq peaks in HeLa cells. The peaks are enriched in the promoter regions (Transcription Start Site [TSS] −/+3K). P < 1.8e-74 (binomial test).
(D) Venn diagram showing the overlap of AF9-, Flag-AF9- and H3K9ac-occupied genes. P< 3.6e-265 (3-way Fisher's exact test).
(E) Genome-browser view of the AF9- (blue), Flag-AF9- (green) and H3K9ac (red)-ChIP-seq peaks on the MYC, BMP2 and HOXA genes.
(F) Average genome-wide occupancies of AF9 (blue), Flag-AF9 (green) and H3K9ac (red) −/+5 kb around the TSS.
(G) qPCR analysis of the indicated ChIP in HeLa cells stably expressing Flag-tagged AF9 or DOT1L. Flag ChIP of cells stably transduced with empty vector was used as a negative control. Schematic of the genomic structure of the MYC gene and PCR primer–targeting regions are indicated in the top panel. The error bars represent the S.E.M. of three experiments.
See also Figure S5.
Next we sought to determine whether AF9 chromatin recruitment depends on histone H3K9ac levels. Although a number of HATs display acetylation activity on H3K9 in vitro, GCN5 and PCAF are the dominant enzymes that maintain H3K9ac levels in vivo (Jin et al., 2011). Compared with those in the mouse embryonic fibroblast (MEF) cells derived from PCAF single gene knockout (KO) mice, H3K9ac levels in the PCAF/GCN5 double knockout (DKO) MEF cells were greatly diminished or undetectable (Figure S5A). Immunofluorescence (IF) experiments of the stably expressed Flag-AF9 in these MEF cells revealed that AF9 proteins formed clear foci that overlapped with H3K9ac on chromatin in the PCAF KO MEF cells with normal H3K9ac levels; in contrast, in the PCAF/GCN5 DKO MEF cells with diminished H3K9ac levels, fewer AF9 chromatin foci were present, with the fluorescent signals evenly distributed in the nucleus (Figure S5B). Finally, treating HeLa cells with CPTH2, a selective GCN5/PCAF inhibitor, led to reduced histone H3 acetylation levels and AF9 occupancy on MYC (Figure S5C). Together, these results suggest that H3K9ac is required for AF9’s association with specific H3K9ac-enriched chromatin loci.
AF9 Co-Localizes with H3K9ac Genome-Wide
To assess the chromatin occupancy of AF9 and its correlation with H3K9ac at a higher resolution, we performed ChIP experiments followed by high-throughput sequencing (ChIP-seq). Because no antibody was available for ChIP of endogenous AF9, we generated a HeLa cell line stably expressing Flag-AF9 (Figure S5D). ChIP-seq experiments using the anti-Flag M2 antibody and an anti-AF9 antibody revealed 2378 and 575 AF9-enriched peaks, respectively, that were widely distributed in the genome. Compared with the non-repetitive genome background, ChIP-seq peaks from both antibodies showed a strong enrichment in promoter, a region within −/+3 kb of the transcription starting site (TSS) (Figures 6C and S5E). Flag-AF9 and AF9 ChIP-seq uncovered 1520 and 338 AF9-occupied genes, respectively, with strong overlap on 311 genes that are enriched in the regulation of transcription and RNA metabolism (Table S3).
We also performed H3K9ac ChIP-seq, which revealed 25877 H3K9ac peaks over 11603 genes in HeLa cells. Strikingly, more than 95% of the AF9 and Flag-AF9-occupied genes were also enriched in H3K9ac occupancy (Figure 6D). The co-localization of AF9 with H3K9ac can be clearly visualized in a genome browser view of the ChIP-seq signals on MYC, BMP2 and the HOXA gene cluster (Figure 6E). Furthermore, the average distribution of all AF9- and Flag-AF9 ChIP-seq peaks exhibited a strong enrichment in the region immediate downstream of the TSS, which recapitulated the genomic distribution of H3K9ac extremely well (Figures 6F). Finally, we performed ChIP experiments followed by quantitative real-time PCR (qPCR) analysis using H3K9ac, H3K79me3 and the Flag M2 antibodies in the HeLa cells stably expressing Flag-tagged AF9 or DOT1L (Figure S5D). The results again demonstrated a co-localization of AF9 and H3K9ac around the TSSs and the immediate downstream regions, where DOT1L occupancy and H3K79me3 enrichment were also detected (Figures 6G and S5G).
AF9 Recruits DOT1L to Deposit H3K79 Methylation and Promote Active Transcription
DOT1L recruitment and the subsequent deposition of H3K79 methylation are ubiquitously coupled with active transcription (Steger et al., 2008). However, it still remains unknown how DOT1L is recruited to specific chromatin loci. Because AF9 binds to both DOT1L and the acetylated histone H3, we hypothesized that AF9 recruits DOT1L to target genes to deposit H3K79me3 and promote active transcription. To test this hypothesis, we first performed H3K79me3 ChIP-seq in HeLa cells. We identified 8021 H3K79me3-enriched genes that strongly overlapped with H3K9ac ChIP-seq peaks (Figures 7A and S6A). Importantly, approximately 90% of AF9-occupied genes and 80% of Flag-AF9-occupied genes overlapped with genes enriched in both H3K79me3 and H3K9ac. Notably, the H3K79me3 levels on the AF9- and the Flag-AF9-occupied genes were significantly higher than that on non-AF9-occupied genes (Figure 7B).
Figure 7. AF9 Is Required for DOT1L-Dependent H3K79me3 Deposition and Gene Activation.

(A) Venn diagram showing the overlap of AF9-, H3K9ac- and H3K79me3-occupied genes. P< 3.0e-82 (3-way Fisher's exact test).
(B) AF9-occupied genes have higher H3K79me3 levels. Box blots showing the average H3K79me3 levels in the AF9- (blue) or Flag-AF9- (green) occupied genes and the other genes (purple). *: P<2.4e-64, **: P<1.2e-163.
(C–E) AF9 depletion reduces the H3K79me3 levels on AF9-occupied genes.
(C) Average H3K79me3 occupancy along the transcription unit of the AF9-occupied genes in control (shControl, green) and AF9 KD (shAF9, blue) HeLa cells. H3K79me3 occupancy on the AF9-occupied genes in DOT1L KD cells (shDOT1L, red) is shown for comparison. The gene body length is aligned by percentage from the TSS to TTS. 5 kb upstream of TSS and 5kb downstream of TTS are also included.
(D) Genome-browser view of the H3K79me3-ChIP-seq peaks on the indicated genes or regions in cells as in (C). AF9 ChIP-seq peaks are shown on top.
(E) qPCR analysis of H3K79me3 and Flag-DOT1L ChIP on MYC in control and AF9 KD HeLa cells stably expressing Flag-DOT1L.
(F) qPCR analysis of Flag-AF9 ChIP in cells stably expressing WT or mutant Flag-AF9.
(G) AF9 YEATS–H3K9ac interaction is required for H3K79me3 deposition on MYC. qPCR analysis of H3K79me3 ChIP in control and AF9 knockdown cells expressing WT or the indicated mutant Flag-AF9.
(H) qPCR analysis of MYC expression in cells as in (G).
In panels E-H, the error bars represent the S.E.M. of three experiments. *: P<0.05 (two-way unpaired Student’s t-test).
(I) Working model of the recruitment of DOT1L and the SEC complex by AF9 via recognition of H3K9ac by the YEATS domain.
See also Figure S6.
Next, we sought to determine whether the H3K79me3 levels on the AF9-occupied genes are dependent on AF9. We knocked down AF9 in HeLa cells by shRNAs (Figure S6B and S6C) and performed H3K79me3 ChIP-seq experiments. As a control, we also performed the ChIP experiment in DOT1L knockdown cells. AF9 depletion led to a moderate reduction of H3K79me3 levels on the AF9- and Flag-AF9-occupied genes, whereas DOT1L knockdown resulted in a greater reduction of H3K79me3 levels on these genes (Figures 7C and S6D). A genome-browser view of the H3K79me3 ChIP-seq and ChIP-qPCR analysis in AF9 knockdown cells also demonstrated the reduction of H3K79me3 levels on individual AF9-occupied genes (Figures 7D, 7E and S6E), accompanied with diminished DOT1L occupancy (Figures 7E and S6E) and reduced gene expression (Figure S6F). Together, these results suggest that AF9 is required for DOT1L recruitment and H3K79me3 deposition to promote active transcription. Interestingly, knockdown of the SEC component AFF4 also slightly affected DOT1L recruitment and H3K79me3 deposition on MYC (Figure S6G).
The YEATS Domain Is Required for the Chromatin Recruitment of AF9 and DOT1L
Next, we asked whether the recognition of H3K9ac by the YEATS domain is required for AF9’s chromatin recruitment. We generated HeLa cells stably expressing Flag-tagged WT AF9 or the H3K9ac-binding deficient mutants (F59A and Y78A) (Figure S6H) and we performed IF experiments to assess their nuclear localization. WT AF9 proteins formed clear foci overlapping with H3K9ac distribution on chromatin in HeLa cells; in contrast, the H3K9ac-binding deficient mutants were evenly distributed in the nucleus without apparent foci (Figure S6I). Consistent with the IF data, ChIP experiments in these stable cells revealed that WT AF9 bound strongly on MYC and PABPC1, whereas the bindings of F59A and Y78A mutants on these genes were severely impaired (Figure 7F and Figure S6J). Taken together, these results suggest that the YEATS domain is essential for AF9 chromatin recruitment.
Finally, we sought to determine whether the recognition of H3K9ac by the AF9 YEATS domain is required for the chromatin recruitment of DOT1L and for target gene expression. We performed “rescue” experiments by ectopically expressing shRNA-resistant WT or mutant AF9 in AF9-depleted HeLa cells and assessed H3K79me3 and the expression levels of MYC and PABPC1. Depletion of endogenous AF9 reduced H3K79me3 and the expression levels of these genes. Ectopic expression of WT AF9, but not the F59A and Y78A mutants, in the AF9-depleted cells restored H3K79me3 on MYC and PABPC1 to a level similar to that in the control cells (Figures 7G and S6K). Consistently, WT AF9, but not the H3K9ac-binding deficient mutants, rescued target gene expression in AF9 knockdown cells (Figure 7H). Taken together, our findings support a model in which AF9, via the recognition of H3K9ac by its YEATS domain, recruits DOT1L (and possibly the SEC) to target genes, which in turn deposits methylation on histone H3K79 to promote active transcription (Figure 7I).
DISCUSSION
The YEATS Domain Possesses a Novel Mechanism for Histone Acetylation Recognition
Our studies identify YEATS domain as a novel class of histone acetyllysine reader that is distinct from other known readers in regarding to protein fold, pocket generation and acetyllysine recognition. The YEATS domains adopt an Ig-fold consisting of two-layer β-sandwich. Ig/Ig-like family members are one of the most abundant structural modules in human genome, and often involved in protein-protein and protein-ligand interactions at cell-surface or in cytoplasm (Bork et al., 1994). In addition, a few nuclear proteins, such as the histone chaperone Asf1, are also known to harbor an Ig/Ig-like module (Rudolph and Gergen, 2001). Structural alignment revealed that although AF9 YEATS and the Ig domain of the Fab antibody share a similar β-rich fold, but they utilize distinct functional surfaces for substrate recognition: AF9 YEATS binds to H3K9ac through the N-distal surface, whereas Fab Ig recognizes its antigen via an N-proximal surface (Figures 2C, S7A and S7B). Furthermore, Asf1 Ig-fold interacts with histone H3 core α-helices and an H4 C-terminal β-strand via a distinct β-sheet layer (Figures S7C and S7D) (Natsume et al., 2007). These structures suggest that many surfaces over an Ig/Ig-like fold can be exploited to achieve a particular function, and the Ig-fold of the YEATS domain represents a novel scaffold of histone acetyllysine readers.
The acetyllysine-binding pocket of the YEATS domain is characteristic of a Ser/Thr-lined sandwiching cage contributed by several aromatic residues conserved among all YEATS domains. Intimate encapsulation, hydrogen bonding relay and extensive CH-π interactions are unique features of the YEATS domain governing acetyllysine-specific readout. Furthermore, site-specific readout is contributed by additional interactions involving residues N-terminal to acetyllysine and a cross-loop surface of YEATS. In contrast, BRDs utilize a deep yet relatively open pocket for acetyllysine readout, with specific binding contributed by only one hydrogen bond and a few hydrophobic contacts. These differences underlying acetyllysine recognition between YEATS and BRD might partially explain the observed micromolar levels of binding affinity of H3K9ac for AF9 YEATS, as compared to much weaker affinities (usually submillimolar level) of Kac-peptide readout by many BRDs (Filippakopoulos et al., 2012). The unique features of the YEATS domains in histone acetylation recognition render them attractive targets of small molecule inhibitors.
The AF9 YEATS Domain Links DOT1L and SEC to Chromatin
Transcription is a multi-state process that coordinates the recruitment of the transcription apparatus, chromatin reorganization, and an orderly change of a series of histone PTMs. The SAGA HAT complex and the chromatin remodeling SWI/SNF complex are recruited to the proximal promoter regions at an early stage, and the resulting histone acetylation and nucleosome rearrangement are followed by pre-initiation complex assembly and histone H2B monoubiquitination (Sims et al., 2004). H2B ubiquitination, which is required for subsequent H3K4 methylation and H3K79 methylation, controls the binding of the H3K4 methyltransferase COMPASS complex to chromatin via interacting with the Cps35 subunit in yeast (Lee et al., 2007). However, it still remains unknown how H2B ubiquitination and other pre-existing histone PTMs, such as histone acetylation, influence DOT1L recruitment and H3K79me3 deposition. Our study demonstrates that DOT1L-mediated H3K79me3 requires the pre-deposition of acetylation on histone H3K9, and that AF9, through its recognition of H3K9ac by YEATS, plays a critical role in this process.
AF9 and its close homologous protein ENL are stoichiometric components of the SEC (He et al., 2010; Lin et al., 2010; Sobhian et al., 2010). The SEC is required for the proper induction of the HSP70 gene upon stress and is involved in HIV proviral transcription. However, it is unclear how the SEC is recruited to certain chromatin loci, besides interacting with sequence-specific transcription factors. Our findings suggest an appealing model in which AF9 (and possibly ENL) recruits or stabilizes the SEC to the proximal regions of active promoters that are enriched in histone acetylation levels. Interestingly, the AF9 YEATS domain has a strong specificity towards H3K9 acetylation, whereas the ENL YEATS domain exhibits a slightly higher affinity to H3K27 acetylation. AF9 and ENL exist in separate SECs with non-redundant functions (Biswas et al., 2011; He et al., 2011). Therefore, it is tempting to speculate that AF9 and ENL occupy promoter and enhancer regions, respectively, via interactions between their YEATS domains and the cognate acetylated histone substrates, and thus target DOT1L and the SECs to distinct chromatin loci. In support of this hypothesis, H3K79me3 and the Ell3-associated SEC were found to be enriched not only in the proximal transcribed regions but also on active enhancers (Bonn et al., 2012; Lin et al., 2013), the regions known to be marked with H3K27 acetylation (Rada-Iglesias et al., 2011).
A Common Role of the YEATS Domain in Chromatin Recruitment
The YEATS domain family of proteins has more than 100 members in more than 70 eukaryotic species. We propose that the recognition of histone acetylation is a general feature of the evolutionarily conserved YEATS domain. Previous studies have shown that the YEATS domains of human ENL (Zeisig et al., 2005) and yeast Yaf9 (Wang et al., 2009) can bind to histones, albeit no specific PTMs on the substrate histones were identified. Schulze et al proposed that Yaf9 YEATS domain might function as an acetylation reader (Schulze et al., 2010), though they lacked direct evidence. Our study demonstrates that the YEATS domains of AF9/Yaf9 proteins from human, mouse, fly, and yeast indeed bind to acetylated histone H3, albeit with slightly different specificities.
Yaf9 is a shared subunit of the SWR1 chromatin-remodeling complex and the NuA4 HAT complex and is required for both H2A.Z deposition and histone H4 acetylation in yeast (Krogan et al., 2003; Le Masson et al., 2003; Mizuguchi et al., 2004; Zhang et al., 2004). Sas5 is part of the SAS (something about silencing) complex that acetylates histone H4K16 and functions in silencing the heterochromatin-like mating type locus HMR in yeast (Meijsing and Ehrenhofer-Murray, 2001; Osada et al., 2001); and Taf14 is a member of several complexes, including the general transcription factor TAFIID complex, the NuA3 HAT complex, and the chromatin-remodeling SWI/SNF, INO80 and RSC complexes (Schulze et al., 2009). In addition to AF9/ENL, humans have two other YEATS domain proteins, GAS41 and YEATS2, which belong to the TIP60/SRCAP chromatin-remodeling complexes and the ATAC HAT complex, respectively. It will be interesting to determine in future studies whether the YEATS domains are required for the chromatin recruitment of all the YEATS domain proteins and the functions of their associated protein complexes.
Globally alterations in histone acetylation occur frequently in many types of human cancers. The identification of potent inhibitors targeting BRD4 and other BET family proteins proved epigenetic readers to be attractive therapeutic targets (Filippakopoulos P, 2010) Our findings indicate the importance of the YEATS domains in connecting histone acetylation to diverse processes during normal and neoplastic development, thus providing the YEATS domains as potential therapeutic targets for the treatment of human cancers.
EXPERIMENTAL PROCEDURES
Crystallization, Data Collection, and Structure Determination
Crystallization was performed via the sitting drop vapor diffusion method under 18°C by mixing equal volumes (0.2–1.0 µl) of AF9 YEATS-H31–10K9ac (1:2 molar ratio, 6–8 mg/ml) and reservoir solution containing 20% (w/v) polyethylene glycol 4000, 5–15% 2-propanol, and 0.1 M sodium citrate tribasic dihydrate, pH 5.6. The complex crystals were briefly soaked in a cryoprotectant drop composed of the reservoir solution supplemented with 20% glycerol and then flash–frozen in liquid nitrogen for data collection. The diffraction data set was collected at the beamline BL17U of the Shanghai Synchrotron Radiation Facility at 0.9793 Å. All diffraction images were indexed, integrated, and merged using HKL2000 (Otwinowski and Minor, 1997). The structure was determined by molecular replacement using MOLREP (Vagin and Teplyakov, 2010) with the free Yaf9 structure (PDB ID: 3FK3) as the search model. Structural refinement was carried out using PHENIX (Adams et al., 2010), and iterative model building was performed with COOT (Emsley and Cowtan, 2004). Detailed data collection and refinement statistics are summarized in Supplemental Table 2. Structural figures were created using the PYMOL (http://www.pymol.org/) or Chimera (http://www.cgl.ucsf.edu/chimera) programs.
ChIP and ChIP-seq Analysis
ChIP analysis was performed essentially as described previously (Wen et al., 2014). Briefly, cells were crosslinked with 1% formaldehyde for 10 min and stopped with 125 mM glycine. The isolated nuclei were resuspended in nuclei lysis buffer and sonicated using a Bioruptor Sonicator (Diagenode). The samples were immunoprecipitated with 2–4 µg of the appropriate antibodies overnight at 4°C. Staph A cells were added and incubated for 15 min, and the immunoprecipitates were washed twice with dialysis buffer and four times with IP wash buffer. Eluted DNA was reverse-crosslinked, purified using PCR purification kit (Qiagene), and analyzed by quantitative real-time PCR on the ABI 7500-FAST System using the Power SYBR Green PCR Master Mix (Applied Biosystems). Statistic differences were calculated using a two-way unpaired Student’s t-test. The primers used for qPCR are listed in the Supplemental Table 4.
ChIP-seq was carried out essentially the same as described above, except using protein A/G beads instead of Staph A cells for IP and use low salt, high salt and LiCl buffer for washes. Samples were sequenced using the Illumina Solexa Hiseq 2000. The raw reads were mapped to human reference genome NCBI 36 (hg19) by Solexa data processing pipeline, allowing up to 2 mismatches. The genome ChIP-seq profiles were generated using MACS 1.3.6 (Zhang et al., 2008) with only unique mapped reads. Clonal reads were automatically removed by MACS. The ChIP-seq profiles were normalized to 10,000,000 total tag numbers, and peaks were called at p-values ≤1e-8. The ChIP-seq peaks distribution statistics were performed using the Cis-regulatory element annotation system (Eswaran et al., 2009).
The methods for protein production, peptide microarray, peptide pull-down, isothermal titration calorimetry, cell culture, viral transduction, RNA interference, RNA extraction, and real-time PCR analysis are described in the Extended Experimental Procedures.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
We thank M. Bedford, J. Tyler and B. Li for comments and reagents. We thank the staffs at beamline BL17U of the Shanghai Synchrotron Radiation Facility and Dr. S. Fan at Tsinghua Center for Structural Biology for their assistance in data collection and the China National Center for Protein Sciences Beijing for providing facility support. We thank the MD Anderson Sequencing and Microarray Facility and the Science Park Next-Generation Sequencing Facility for Solexa sequencing. We thank Joseph Munch for editing the manuscript. This work was supported in part by funds from Welch (G1719) and American Cancer Society (RSG-13-290-01-TBE) to X.S, The Major State Basic Research Development Program in China (2011CB965300) and Program for New Century Excellent Talents in University to H.L., NIH (R01HG007538 to W.L and R01GM067718 to S.Y.R.D), CPRIT (RP110471 to X.S., W.L. and S.Y.R.D), MDACC Institutional Research Grant and Center for Cancer Epigenetics pilot grant to H.W., China Postdoctoral Science Foundation (2014T70069 to Y.L.). Y.L. is a Tsinghua-Peking Center for Life Sciences postdoctoral fellow. W.L. is a recipient of a Duncan Scholar Award. X.S. is a Kimmel Scholar and a R. Lee Clark Fellow.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information includes Extended Experimental Procedures, seven figures and four tables.
ACCESSION NUMBERS
Structure data are deposited in the Protein Data Bank with the accession numbers of 4TMP. The ChIP-seq data are deposited in the GEO database with the accession number of GSE60366.
AUTHOR CONTRIBUTIONS
Y.L. and H.W. contributed equally to this work. X.S. and H.L. conceived the study. H.W. and K.T. performed the biochemical and cellular studies and discovered the acetylation binding activity; H.L. and Y.L. performed structural and calorimetric studies with assistance from H.B.W.; Y.X. performed bioinformatics analysis; D.P. and Y.R. provided technical assistance; Q.J. isolated PCAF/GCN5 DKO MEF cells; X.S. and H.L. wrote the paper with comments from Y.L., H.W., S.Y.D, and W.L.
The authors declare no competing financial interest.
REFERENCES
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali M, Yan KZ, Lalonde ME, Degerny C, Rothbart SB, Strahl BD, Cote J, Yang XJ, Kutateladze TG. Tandem PHD Fingers of MORF/MOZ Acetyltransferases Display Selectivity for Acetylated Histone H3 and Are Required for the Association with Chromatin. J Mol Biol. 2012;424:328–338. doi: 10.1016/j.jmb.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas D, Milne TA, Basrur V, Kim J, Elenitoba-Johnson KS, Allis CD, Roeder RG. Function of leukemogenic mixed lineage leukemia 1 (MLL) fusion proteins through distinct partner protein complexes. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:15751–15756. doi: 10.1073/pnas.1111498108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonn S, Zinzen RP, Girardot C, Gustafson EH, Perez-Gonzalez A, Delhomme N, Ghavi-Helm Y, Wilczynski B, Riddell A, Furlong EE. Tissue-specific analysis of chromatin state identifies temporal signatures of enhancer activity during embryonic development. Nature genetics. 2012;44:148–156. doi: 10.1038/ng.1064. [DOI] [PubMed] [Google Scholar]
- Bork P, Holm L, Sander C. The Immunoglobulin Fold - Structural Classification, Sequence Patterns and Common Core. J Mol Biol. 1994;242:309–320. doi: 10.1006/jmbi.1994.1582. [DOI] [PubMed] [Google Scholar]
- Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;399:491–496. doi: 10.1038/20974. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Eswaran J, Patnaik D, Filippakopoulos P, Wang F, Stein RL, Murray JW, Higgins JM, Knapp S. Structure and functional characterization of the atypical human kinase haspin. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:20198–20203. doi: 10.1073/pnas.0901989106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Muller S, Pawson T, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos PQJ, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, et al. Selective inhibition of BET bromodomains. Nature. 2010 doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He N, Chan CK, Sobhian B, Chou S, Xue Y, Liu M, Alber T, Benkirane M, Zhou Q. Human Polymerase-Associated Factor complex (PAFc) connects the Super Elongation Complex (SEC) to RNA polymerase II on chromatin. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:E636–E645. doi: 10.1073/pnas.1107107108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He NH, Liu M, Hsu J, Xue YH, Chou S, Burlingame A, Krogan NJ, Alber T, Zhou Q. HIV-1 Tat and Host AFF4 Recruit Two Transcription Elongation Factors into a Bifunctional Complex for Coordinated Activation of HIV-1 Transcription. Molecular cell. 2010;38:428–438. doi: 10.1016/j.molcel.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Jin Q, Yu LR, Wang L, Zhang Z, Kasper LH, Lee JE, Wang C, Brindle PK, Dent SY, Ge K. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. The EMBO journal. 2011;30:249–262. doi: 10.1038/emboj.2010.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Krogan NJ, Keogh MC, Datta N, Sawa C, Ryan OW, Ding HM, Haw RA, Pootoolal J, Tong A, Canadien V, et al. A Snf2 family ATPase complex required for recruitment of the histone H2A variant Htz1. Molecular cell. 2003;12:1565–1576. doi: 10.1016/s1097-2765(03)00497-0. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- Lawrence MC, Colman PM. Shape Complementarity at Protein-Protein Interfaces. J Mol Biol. 1993;234:946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- Le Masson I, Yu DY, Jensen K, Chevalier A, Courbeyrette R, Boulard Y, Smith MM, Mann C. Yaf9, a novel NuA4 histone acetyltransferase subunit, is required for the cellular response to spindle stress in yeast. Molecular and cellular biology. 2003;23:6086–6102. doi: 10.1128/MCB.23.17.6086-6102.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Shukla A, Schneider J, Swanson SK, Washburn MP, Florens L, Bhaumik SR, Shilatifard A. Histone crosstalk between H2B monoubiquitination and H3 methylation mediated by COMPASS. Cell. 2007;131:1084–1096. doi: 10.1016/j.cell.2007.09.046. [DOI] [PubMed] [Google Scholar]
- Lin C, Garruss AS, Luo Z, Guo F, Shilatifard A. The RNA Pol II elongation factor Ell3 marks enhancers in ES cells and primes future gene activation. Cell. 2013;152:144–156. doi: 10.1016/j.cell.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Smith ER, Takahashi H, Lai KC, Martin-Brown S, Florens L, Washburn MP, Conaway JW, Conaway RC, Shilatifard A. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Molecular cell. 2010;37:429–437. doi: 10.1016/j.molcel.2010.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijsing SH, Ehrenhofer-Murray AE. The silencing complex SAS-I links histone acetylation to the assembly of repressed chromatin by CAF-I and Asf1 in Saccharomyces cerevisiae. Genes & development. 2001;15:3169–3182. doi: 10.1101/gad.929001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuguchi G, Shen XT, Landry J, Wu WH, Sen S, Wu C. ATP-Driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex. Science. 2004;303:343–348. doi: 10.1126/science.1090701. [DOI] [PubMed] [Google Scholar]
- Moriniere J, Rousseaux S, Steuerwald U, Soler-Lopez M, Curtet S, Vitte AL, Govin J, Gaucher J, Sadoul K, Hart DJ, et al. Cooperative binding of two acetylation marks on a histone tail by a single bromodomain. Nature. 2009;461:U664–U112. doi: 10.1038/nature08397. [DOI] [PubMed] [Google Scholar]
- Natsume R, Eitoku M, Akai Y, Sano N, Horikoshi M, Senda T. Structure and function of the histone chaperone CIA/ASF1 complexed with histones H3 and H4. Nature. 2007;446:338–341. doi: 10.1038/nature05613. [DOI] [PubMed] [Google Scholar]
- Nishio M, Hirota M, Umezawa Y. The CH-[pi] interaction : evidence, nature, and consequences. New York: Wiley; 1998. [Google Scholar]
- Osada S, Sutton A, Muster N, Brown CE, Yates JR, Sternglanz R, Workman JL. The yeast SAS (something about silencing) protein complex contains a MYST-type putative acetyltransferase and functions with chromatin assembly factor ASF1. Genes & development. 2001;15:3155–3168. doi: 10.1101/gad.907201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods in Enzymology. 1997;276:19. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Qiu Y, Liu L, Zhao C, Han CC, Li FD, Zhang JH, Wang Y, Li GH, Mei YD, Wu MA, et al. Combinatorial readout of unmodified H3R2 and acetylated H3K14 by the tandem PHD finger of MOZ reveals a regulatory mechanism for HOXA9 transcription. Genes & development. 2012;26:1376–1391. doi: 10.1101/gad.188359.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279-+. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph MJ, Gergen JP. DNA-binding by Ig-fold proteins. Nature structural biology. 2001;8:384–386. doi: 10.1038/87531. [DOI] [PubMed] [Google Scholar]
- Schulze JM, Wang AY, Kobor MS. YEATS domain proteins: a diverse family with many links to chromatin modification and transcription. Biochemistry and Cell Biology-Biochimie Et Biologie Cellulaire. 2009;87:65–75. doi: 10.1139/O08-111. [DOI] [PubMed] [Google Scholar]
- Schulze JM, Wang AY, Kobor MS. Reading chromatin Insights from yeast into YEATS domain structure and function. Epigenetics : official journal of the DNA Methylation Society. 2010;5:573–577. doi: 10.4161/epi.5.7.12856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims RJ, 3rd, Belotserkovskaya R, Reinberg D. Elongation by RNA polymerase II: the short and long of it. Genes & development. 2004;18:2437–2468. doi: 10.1101/gad.1235904. [DOI] [PubMed] [Google Scholar]
- Smith E, Lin C, Shilatifard A. The super elongation complex (SEC) and MLL in development and disease. Genes & development. 2011;25:661–672. doi: 10.1101/gad.2015411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobhian B, Laguette N, Yatim A, Nakamura M, Levy Y, Kiernan R, Benkirane M. HIV-1 Tat Assembles a Multifunctional Transcription Elongation Complex and Stably Associates with the 7SK snRNP. Molecular cell. 2010;38:439–451. doi: 10.1016/j.molcel.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steger DJ, Lefterova MI, Ying L, Stonestrom AJ, Schupp M, Zhuo D, Vakoc AL, Kim JE, Chen JJ, Lazar MA, et al. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Molecular and cellular biology. 2008;28:2825–2839. doi: 10.1128/MCB.02076-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- Su D, Hu Q, Li Q, Thompson JR, Cui G, Fazly A, Davies BA, Botuyan MV, Zhang Z, Mer G. Structural basis for recognition of H3K56-acetylated histone H3–H4 by the chaperone Rtt106. Nature. 2012;483:104–107. doi: 10.1038/nature10861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr D Biol Crystallogr. 2010;66:22–25. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- Wang AY, Schulze JM, Skordalakes E, Gin JW, Berger JM, Rine J, Kobor MS. Asf1-like structure of the conserved Yaf9 YEATS domain and role in H2A.Z deposition and acetylation. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:21573–21578. doi: 10.1073/pnas.0906539106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen H, Li Y, Xi Y, Jiang S, Stratton S, Peng D, Tanaka K, Ren Y, Xia Z, Wu J, et al. ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature. 2014;508:263–268. doi: 10.1038/nature13045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A, Lin M, Naresh A, Kitabayashi I, Cleary ML. A Higher-Order Complex Containing AF4 and ENL Family Proteins with P-TEFb Facilitates Oncogenic and Physiologic MLL-Dependent Transcription. Cancer cell. 2010;17:198–212. doi: 10.1016/j.ccr.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisig DT, Bittner CB, Zeisig BB, Garcia-Cuellar MP, Hess JL, Slany RK. The eleven-nineteen-leukemia protein ENL connects nuclear MLL fusion partners with chromatin. Oncogene. 2005;24:5525–5532. doi: 10.1038/sj.onc.1208699. [DOI] [PubMed] [Google Scholar]
- Zeng L, Zhang Q, Li S, Plotnikov AN, Walsh MJ, Zhou MM. Mechanism and regulation of acetylated histone binding by the tandem PHD finger of DPF3b. Nature. 2010;466:258–262. doi: 10.1038/nature09139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HY, Richardson DO, Roberts DN, Utley R, Erdjument-Bromage H, Tempst P, Cote J, Cairns BR. The Yaf9 component of the SWR1 and NuA4 complexes is required for proper gene expression, histone H4 acetylation, and Htz1 replacement near telomeres. Molecular and cellular biology. 2004;24:9424–9436. doi: 10.1128/MCB.24.21.9424-9436.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.