

Abstract
Chronic inflammatory and autoimmune diseases have been the focus of many genome-wide association studies (GWAS) because they represent a significant cause of illness and morbidity, and many are heritable. Almost a decade of GWAS studies suggests that the pathological inflammation associated with these diseases is controlled by a limited number of networked immune system genes. Chronic inflammatory and autoimmune diseases are enigmatic from an evolutionary perspective because they exert a negative affect on reproductive fitness. The persistence of these conditions may be partially explained by the important roles the implicated immune genes play in pathogen defense and other functions thought to be under strong natural selection in humans. The evolutionary reasons for chronic inflammatory and autoimmune disease persistence and uneven distribution across populations are the focus of this review.
Introduction
Like all other organisms, humans are the transient outcome of eons of ancestral creatures affected by evolutionary forces. It has long been considered that chief amongst the factors that influence human physiological composition is natural selection exerted on the immune system [1,2]. As our primary interface with the environment, our immune system is thought to have been under severe selective pressure mediated by pathogens [3–7,8••,9–14,15••]. Indeed, studies examining human genomes for signs of positive selection, or ‘selection for’ particular traits, repeatedly find an overrepresentation of immune system genes associated with these signatures [16–21]. While we would expect the individuals of our young species to be phenotypically very similar, humans considerably vary in their capacity to manifest chronic diseases characterized by long-term, overt and pathological inflammation such as chronic inflammatory and many autoimmune diseases (Table 1). The persistence and increasing incidence of conditions characterized by pathological inflammation is a particularly enigmatic aspect of the diversification of human immunity, as many manifest in pre- and peri-reproductive individuals and negatively affect reproductive fitness. The factors contributing to disparate chronic inflammatory and autoimmune disease incidence are myriad and include both genetic as well as current environmental factors. Here, we consider how past human immune system adaptation may have contributed to current disease disparities between individuals and human populations.
Table 1.
Prevalence of select chronic Inflammatory and autoimmune diseases per 1000 individuals, by United States population. Data represents the combined sex prevalence rate normalized to 1000 Individuals unless otherwise noted. All data here was collected on populations within the United States after 1994, with the exception of some data for Native North Americans, which pre-dates 1994 and Includes Canadian groups. With the understanding that the defined population names used here encompass heterogeneous populations that may overlap, population group names were chosen based on the majority use In publications, with few exceptions: ‘African American’ and ‘Black’ are aggregated with ‘non-Hispanic Black’, Puerto Rican Hispanics and ‘Latino American’ are aggregated with ‘Hispanic’, ‘White’ and ‘Caucasian’ are aggregated with ‘Non-Hispanic White’, ‘Alaskan Eskimo’ is lumped under ‘Native North American and Inuit’
| Per 1000 individuals | Non-hispanic white Mean (range) |
Non-hispanic black Mean (range) |
Hispanic American Mean (range) |
Asian American and Pacific Islander Mean (range) |
Native North American/Inuit Mean (range) |
|---|---|---|---|---|---|
| Type 1 diabetes | 2.13a (1.86–2.55) [106,107] | 1.65a (1.29–2.04) [106,108] | 1.12a (0.96–1.29) [106] | 0.62a (0.50–0.77) [106,109] | 0.32a (0.35–0.30) [106] |
| Psoriasis | 30.5b (25–36) [110,111] | 16.0b (13–19) [110,111] | 12b (8–16) [111,112] | – | – |
| Multiple sclerosis | 0.83c (0.56–0.99) [113] | 0.56c (0.22–0.90) [113] | 0.35c (0.11–0.56) [113,114] | – | – |
| Celiac disease | 0.1 [115] | – | – | – | – |
| Ulcerative colitis (UC) | 1.94 [116] | 0.78 (0.07–1.5) [116,117] | 0.56 (0.12–1.0) [116,13] | 1.00 [116] | 1.15 [116] |
| Crohn’s disease (CD) | 1.3 [116] | 0.51 (0.12–0.89) [116,117] | 0.26 (0.058–0.47) [116,118] | 0.62 [116] | 1.09 [116] |
| Inflammatory bowel disease (UC and CD combined) |
3.24 [116] | 2.39 [116] | 0.53 (0.06–1.47) [116,118] | 1.62 [116] | 2.24 [116] |
| Systemic lupus erythematosus (SLE) |
0.72 (0.34–1.11) [119,120] 1.19d (0.62–2.03) [119–121] |
1.69 (1.16–2.23) [119,120] 3.78d (1.97–6.94) [119–122] |
1.29 (1.03–1.59) [120,123] 1.81d(1.38–2.44) [120,121] |
1.75 [120] 1.5d (0.92–2.55) [120,121] |
1.71 (1.65–1.78) [120,124] 2.13d [120] |
Age–adjusted rate 15–19 years of age, or as close to 19 years of age as possible.
Age–adjusted to ≥20.
Average of non-contiguous U.S. regions.
Female only. Combined sex rates listed immediately above.
The genetic basis of susceptibility to autoimmune and inflammatory disorders
With the advent of whole-genome genotyping arrays, examinations of the entire genome for associations with complex phenotypes have become common practice. In less than a decade, such genome-wide association studies (GWAS) have found hundreds of loci associated with chronic inflammatory and autoimmune diseases. The hundreds of genes implicated in the progression of these diseases by GWAS have revealed two major patterns that make the persistence and uneven distribution of chronic conditions characterized by pathological inflammation particularly intriguing. First, genes implicated in infectious disease susceptibility overlap considerably those associated with chronic inflammatory and autoimmune diseases [22•,23•,24,25••,26]. Such observations have forced a shift from disease models that emphasize individual inflammatory disease pathways, to a model of pathological inflammation regulated by a tightly regulated network of genes that are implicated in multiple diseases [24,27,28]. A recent assessment of risk allele sharing across seven chronic inflammatory and autoimmune diseases (celiac disease, multiple sclerosis, rheumatoid arthritis, Crohn’s disease, psoriasis and systemic lupus erythematosus) found that over 40% of the associated single nucleotide polymorphisms (SNPs) were shared across multiple, though not by all seven, conditions [22•]. Via a large meta-analysis of multiple GWAS for inflammatory bowel disease (IBD), Jostins et al. found that 66 of 154 of loci associated with IBD were also associated with other ‘immune-mediated diseases’, including 8 loci associated with ankylosing spondylitis, and 14 loci associated with psoriasis [23•].
The overlap between chronic inflammatory and autoimmune implicated loci makes sense in the context of disease pathogenesis because such diseases tend to manifest in pairs and share expression of minor pathologies. For example, approximately 50% of patients with the axial skeleton arthritis ankylosing spondylitis (AS) acquire small gut lesions that, in 10% of patients, develop into Crohn’s disease (CD) [29,30]. Other such diseases that co-manifest include Crohn’s disease with psoriasis/arthritis, as well as IBD with multiple sclerosis/optic neuritis/rheumatoid arthritis/asthma [31•] (reviewed in Ref. [32]). A model of similar or shared mechanisms regulating the pathological inflammation of these conditions is further supported by the observation that anti-TNFa (infliximab) treatment for Crohn’s disease significantly influences the risk of developing psoriasis [33–36]. Disease-associated gene overlap, disease co-occurrence and the influence that neutralizing a single but ubiquitous proinflammatory cytokine can have on the manifestation of multiple and seemingly diverse pathologies suggest that the essential mechanism of pathological inflammation across conditions is conserved and that such disease is the outcome of regulatory perturbations of a tightly regulated network of genes [22•,25••]. Importantly, the effect size of most GWAS identified loci appear to be rather small, suggesting that the manifestation of most chronic inflammatory and autoimmune diseases occurs via a combination of genetic risk loci and environmental triggers (e.g., gluten consumption in celiac disease development) that lead to various small shifts in the expression of these gene networks in an individual [37••].
Natural selection as a contributing factor to disease susceptibility
The second major pattern noted for GWAS data is that many GWAS ‘hits’ occur proximal to immune genes in regions with signatures of positive natural selection [15••,38–41]. For this review we gathered a measure of recent positive selection, Integrated Haplotype Scores (iHS), from Hapmap phase II data for the most recent catalogue of all GWAS implicated loci (Figure 1) [21]. When we examined GWAS loci with an absolute iHS value greater than the 99th percentile of the genomewide distribution we found the European sample to have >2-fold increase (P = 0.048) in the number of positively selected alleles among GWAS SNPs associated with autoimmune diseases, while the African sample’s strongest signals of positive selection occurred at loci associated with infectious disease (P = 0.001) (Figure 1a). This observation suggests that at least some of the present-day autoimmune risk loci have been adaptive and conferred some sort of functional benefit to Europeans in the past.
Figure 1.
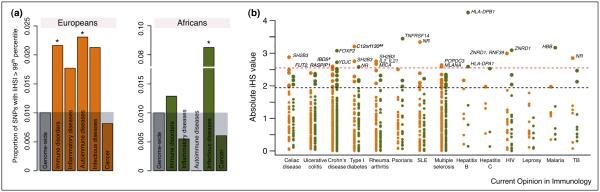
Recent positive selection targeting SNPs associated with susceptibility to chronic inflammatory, autoimmune and infectious diseases. (a) The proportion of GWAS SNPs that present evidence for recent positive selection, as attested by the Integrated Haplotype Score (iHS) statistic [21]. The complete list of GWAS-associated SNPs was obtained from the NIH Catalog of Published Genome-Wide Association Studies [105], which reports all SNP trait associations with a p-values <1.0 × 10−5. For the purpose of the current analyses we used all GWAS SNPs with an association at a significance threshold <1.0 × 10−6. We note, however, that the results are qualitatively similar if using more stringent cutoffs for the GWAS data. The y-axis represents the proportion of SNPs that present an absolute iHS values above the 99th percentile of the HapMap phase II genome-wide distribution for Europeans (left side — orange bars) and Africans (right side — green bars). Diseases were grouped into broader groups: the set of inflammatory disorders includes Crohn’s disease, ulcerative colitis and celiac disease. The set of autoimmune diseases includes rheumatoid arthritis, systemic lupus erythematosus, type 1 diabetes, multiple sclerosis, and psoriasis. The set of ‘Immune disorders’ encompasses both inflammatory and autoimmune diseases. The set of infectious diseases includes HIV/AIDS, Hepatitis B and C, Leprosy, Malaria and Tuberculosis. As a control we also looked at enrichment for signatures of selection among all cancer-related diseases. (b) iHS values for Europeans (orange) and Africans (green) for SNPs associated with several immune-related diseases by different GWAS studies. The genes reported by the GWAS as the most likely associated with the SNPs with absolute iHS values above the ninety-fifth percentile (orange dashed line) are reported. ‘NR’ stands for non-reported. # intergenic region. ## This is the same locus of SH2B3.
Interestingly, the African sample exhibits less of a signal for positive selection than the European sample overall, which may indicate disparate types of selection acting on these populations. Indeed, no signatures of selection on GWAS-associated SNPs appear to be shared between Europeans and Africans. Moreover, fifty-three percent of GWAS-associated SNPs showing evidence of recent selection in the European sample are either completely absent or found at very low frequency (minimum allele frequency <5%) within the African sample. We note, however, that virtually all GWAS studies to date have focused exclusively on individuals of European ancestry [42]. It is therefore possible that the genetic determinants of susceptibility to chronic inflammatory and autoimmune diseases in individuals of African descent are distinct from those found among Europeans and that, if we were to identify those variants, they would also show evidence of selection in Africans. Fortunately, an increasing number of cohorts of individuals of diverse ancestries are being assembled. Hopefully these data will soon allow less biased evaluations of the relative contribution of past selection to susceptibility to chronic inflammatory and autoimmune diseases in large array of human populations.
Asymmetrical interbreeding between archaic human and modern human populations is an interesting possibility that may have contributed to differences in the repertoires of disease-causing loci of African and European descended individuals. The temporal overlap between modern humans and Neanderthals in Europe has been estimated as 2600–5400 years, between ~35 000 and 40 000 years ago [43]. Interestingly, recent sequence studies of ancient DNA from Neanderthals suggests that some of the alleles presently associated with susceptibility to Crohn’s disease, systemic lupus erythematosus, IL-18 levels and type-II diabetes in Europeans have been introduced in non-African populations via interbreeding of modern humans with archaic Neanderthal species at the time of the Out-of Africa exodus [44]. Similarly, an analysis of human leukocyte antigen (HLA) class I sequences from Denisovan, Vindija Neanderthal and modern human genomes revealed that several of the most common and functionally distinct HLA haplotypes found in Eurasian populations (e.g., HLA-C12:02, HLA-C15 and HLA-A11) were also present in the archaic genomes, which suggests that these alleles came into modern Eurasian populations through admixture with archaic humans [45•]. The shared presence of these alleles in modern and archaic populations opens up the possibility that adaptive introgression of archaic alleles into modern human genetic diversity is amongst the factors contributing to the diversification of the immune system between populations.
To more formally assess if European and African populations have significantly differentiated disease-associated loci, we collected the levels of population differentiation (Fst) at GWAS-SNPs between European and African populations using the allele frequencies reported by the 1000 Genomes Project [46]. We found many GWAS-SNPs highly differentiated between the two populations (Fst values >0.4). SNPs associated with important inflammatory pathways appear to have very rapidly differentiated in humans. The most extremely differentiated SNPs (Fst >0.6) in the European and African samples occur proximal to genes that are the regulators of NFKB activation or are known to be expressed after NFKB regulation (i.e., PRKCH, TNIP1, TRAF3IP2) or occur in the chemokine cluster on chromosome 4q13.3 (Figure 2). Generally, SNPs associated with the NFKB and JAK-STAT (i.e., REL, STAT3, STAT4) pathway are well represented amongst polymorphisms highly differentiated between European- and African-descent individuals.
Figure 2.

Levels of population differentiation at GWAS SNPs for immune-related diseases. (a) Boxplots of Fst values between European- (CEU) and African-descent individuals (YRI) based on the data from the 1000 Genomes Project. The Fst statistic examines variation in SNP allele frequencies between populations. Under neutrality, Fst is determined by genetic drift, which affects all loci across the genome similarly. Conversely, positive selection will cause an increase in Fst values in the population where selection occurs. Orange dots highlight highly differentiated GWAS SNPs with an Fst value above the 95th percentile of the genome-wide distribution. (b) Worldwide frequency distribution of two highly differentiated SNPs. The top panel shows the worldwide distribution of allele frequencies for rs2472649, an SNP associated with IBD located in a chemokine cluster on chromosome 4 (e.g., CXCL5,CXCL1,CXCL3,IL8,CXCL6,PF4,CXCL2,PF4V1) and the bottom panel shows the worldwide distribution of allele frequencies for rs653178 an SNP associated with celiac disease linking the SH2B3/ATXN2 locus with susceptibility to celiac disease.
Within the European sample, multiple GWAS identified loci associated with a signature of positive selection are implicated repeatedly across several auto-immune conditions (i.e., HLA-DRB1, SH2B3), including celiac disease, type 1 diabetes, rheumatoid arthritis (Figure 1b). Previous authors have noted signatures of positive selection at these GWAS loci and suggested pathogens as the potential selective factor [6,15••,47]. The possible link between infectious and chronic inflammatory diseases is further supported by reports that some pathogens may be a contributing and possibly causal factor in chronic inflammatory and autoimmune disease (e.g., Epstein–Barr virus and SLE, RA and MS; Mycobacterium avium and Crohn’s disease, Yersinia enterocolica and IBD) [48–55]. However, the relationship between infection with a present day pathogen and the co-occurrence of a chronic disease is very difficult to interpret in terms of past selective events, as the original mitigating factors of allele fixation can only be vaguely reconstructed. Mycobacterium species are, perhaps, the most well supported candidates for pathogen-mediated selection altering inflammatory pathways and increasing the frequency of chronic disease alleles in humans [2,23•,56]. That virtually all identified Mycobacterium leprae risk loci are also associated with genes implicated in IBD progression (e.g., NOD2, LRRK2, TNFSF15) [57••] hints at a possible evolutionary trade off between the probability of reproductive success after acquiring a pathogen with epitopes or tropism similar to M. leprae, and the likelihood of long-term pathological inflammation that may affect reproduction. Caution must be taken with such an interpretation; however, because it assumes that any other role an implicated gene might play in any other biological process is less important to ensure reproductive success than the process of surviving an infection. While pathogens are likely a very important factor driving an increase in chronic inflammatory and autoimmune disease risk loci, the relationship between the two is far from straight forward.
Discerning the influential factors in an evolutionary trade off
Most of our evidence of an association between chronic inflammatory/autoimmune risk alleles and adaptation to past infectious disease is diffuse, primarily because the etiological agent(s) at core of this question is/are fundamentally unknowable. Most of the support for specific pathogens exerting a past evolutionary effect leading to chronic disease consists of genes implicated in chronic disease progression also being implicated in host responses to present day pathogens [23•,57••,58–60]. For all of the attempts to connect historical pathogen exposure to present day human immune characteristics, it’s worth noting the physiological promiscuity of the immune system. Many immune genes are cross-referenced and fulfill functions in other bodily systems thought to be under strong selective pressure, including reproduction, lung maintenance and embryonic and brain development [61–64]. An examination of the 20 GWAS implicated genes associated with the strongest signatures of positive selection reveals that along with host responses to infection, these genes are also involved other activities extremely important for reproductive success, such as embryo implantation into the uterine lining, embryonic morphogenesis and hematopoiesis (Table 2). Chronic inflammatory and autoimmune disease risk alleles associated with signatures of positive selection could indicate such alleles conferred greater reproductive success via a broader range of beneficial phenotypes than simply infection survival.
Table 2.
Non-immune functions of genes associated with 20 SNPs with the strongest signatures of positive selection (IHS scores) and implicated in chronic inflammatory and autoimmune disease
| GWAS gene | Physiological system/role | Process |
|---|---|---|
| NAA25 | Universal | Cell cycle progression [125] |
| SH2B3 | Embryonic development | Embryonic hematopoiesisa [61,126] |
| Wound healing/haematological | Platelet architecture [61] | |
| PTPN11 | Reproduction | Genitalia development [127] |
| Embryonic development | Genitalia development [127], heart development [128], growth plate architecture [129], brain development [130,62], face morphogenesis [130] |
|
| Growth and development | Growth plate architecture [129], brain development [130], face morphogenesis [7] |
|
| Digestive/metabolism | Insulin reception [128], energy metabolism [128,131] | |
| ZNRD1 | Reproduction | Testis (expressed in) [132,133] |
| IL2 | Locomotion/cognitive | Sensorimotor gating [134] |
| IRF1 | Reproduction | Uterine remodeling [135,63] |
| Skeletal | Bone remodeling [136] | |
| DNA maintenance/repair | Telomere maintenancea [137] | |
| IL13 | Respiration | Regulator of lung cilia cell and goblet cell differentiation [64] |
| CSF2 | Reproduction | Trophoblast differentiation [138], placenta development [138] |
| SLC22A4 | Haematological/respiration | Haeme biosynthesis [139] |
| IL4 | Reproduction | Regulates decidua [140], downregulates placental inflammation [141], contributes to normal maternal blood pressure [141], prevents reproductive failurea [142–144] |
| IL3 | Brain development | Nervous system development [145], determinant of brain volume [145] |
| PDLIM4 | Skeletal | Osteoblast development/function [146] |
| SLC22A5 | Digestive | Maintenance of gut epithelial barriera [147] |
| ACSL6 | Brain development/cognitive | Neuronal cell proliferation [148] |
| Growth and maintenance | Lipid synthesis/degradation [149,150] | |
| GNA12 | Reproduction | Sperm development [151] |
| TNXB | Growth and maintenance | Dermal collagen fibril development and organization [152] |
| POPDC3 | Embryonic development | Heart development [153] |
| Growth and maintenance | Skeletal muscle development [153] | |
| RASIP1 | Embryonic development | Vasculogenesisa [154], angiogenesisa [154] |
| MLANA | Skin colour | Melanogensis [155] |
| CPEB4 | Digestive | Glucose metabolism/proinsulin production [156] |
| ETS1 | Embryonic development | Angiogenesis [157], fetal membrane remodeling [158] |
| Reproduction | Uterine decidualization [159] | |
| Respiration/cardiac/growth and maintenance |
Response to hypoxia [160] |
Process with dual role in immunity.
A good example of the complicated business of interpreting the evolutionary meaning of risk loci shared between chronic inflammatory and infectious diseases can be found in a celiac disease risk allele (exonic SNP rs3184504-A). This SNP is associated with SH2B3, a gene which encodes an adaptor important to T-cell activation and occurs in a region of 12q24 with a strong signature of positive selection. In 2010 Zhernakova et al. suggested that bacterial pathogens had acted as selective factors for rs3184504-A based on findings that peripheral blood mononuclear cells isolated from individuals homozygous for the selected allele under-expressed a mutated SH2B3 and exhibited increased pro-inflammatory cytokine production when challenged with ligands for the bacterial detecting receptor NOD2 [15••]. Although this observation is compatible with selection driven by bacterial pathogens the identity of what SH2B3 phenotype may be under selection is fogged by the pleiotropic nature of the gene. SH2B3 also acts as a regulator of two processes assumed to be under very high selective pressure — structural organization and development of platelets and endothelial cells (reviewed in Ref. [65]). Furthermore, the SNP rs3184504 is well known to co-segregate with intronic SNP (rs653178-C) in neighbouring RNA processing and amylosing lateral sclerosis (Lou Gherig’s) risk gene ATXN2 [66–69]. Both variants are found in high frequencies in Europeans, and are virtually non-existent in African populations (Figure 2b). The combination of both alleles is associated with multiple chronic diseases and, therefore, a wide range of pathologies (i.e., celiac disease, rheumatoid arthritis, psoriasis, cardiovascular disease, type 2 diabetes and thrombotic antiphospholipid syndrome) [24,70–73]. It seems very possible that signatures of positive selection in the region could be associated with other unknown beneficial variants that are being co-inherited within, at least, the 125Kb space between the SH2B3 and ATXN2 alleles, if not the entire >1 Megabase region of linkage disequilibrium that encompasses the two genes [66]. This might include ATXN2 variants that limit neurodegenerative disease and which have previously been proposed to be the target of positive selection [66]. Given the pleiotropy of most immune genes, and that natural selection is more easily detected in regions of high linkage disequilibrium, discerning the precise phenotypes in an evolutionary trade off can be a daunting task. Success could require extensive characterization of how putatively selected genetic variants might impact the multiple functions of the gene(s) located within the boundaries of the selected locus.
The rapid differentiation of chronic inflammatory risk alleles in humans
How pathogens, such as an ancestral Mycobacterium, may have exerted sufficient selective pressure to rapidly and significantly differentiate Eurasian and African populations at multiple chronic inflammatory and autoimmune disease risk loci is of significant interest. Our Fst analysis found 41 GWAS-implicated loci highly differentiated between European and Africans, with 8 loci exhibiting extreme Fst values (>0.6). It seems likely that in the ~60 000 years since these populations separated, major cultural shifts, such as the Mesolithic (~10 000–5000) transition from hunting-gathering to an agricultural lifestyle in Eurasia, to the initial exclusion of sub-Saharan Africa, would have profoundly affected human health and disease [74,75]. Pre-agricultural life after the end of Pleistocene was likely small group based, migratory or semi-sedentary, hunter-gatherer lifestyle. Most subarctic populations would have maintained a diverse diet and fairly egalitarian social structure (reviewed in Ref. [76•]). A shift to an agricultural economy would have led to significant changes in human ecology that altered the relationships between humans and pathogens. The advent of agriculture likely affected human immune phenotypes by encouraging many humans to live close together in large groups, with some suffering malnourishment due to eating an unevenly distributed and fairly homogenous diet of novel foods. With the emergence of moderate scale animal husbandry mixed and, potentially, stressed human and animal communities likely provided continuous stretches of human and nonhuman animal hosts for particular pathogens. The ease of pathogen transmission from one host to another also likely benefited from large and drastic alterations of the environment via mass waste accumulation, land clearance for farming, and diversion or development of local water supplies. All of these cultural and environmental changes would have contributed to the emergence of new infectious pathogens, nutritional deficiencies and ‘crowd diseases’ such as measles (reviewed in Ref. [77•]).
The dietary shifts associated with an agricultural economy likely transiently altered immune phenotypes on which selection could act. Efficient digestion of new foods could have conferred a selective advantage to agricultural populations via increased energy and indirectly affected the evolution of immunity. Novel foods likely also contributed to pathological immune phenotypes that reduce reproductive fitness. For example, with the emergence of cereal processing ~20 000 years ago, human populations began to consume considerable quantities gluten for, likely, the first time. It is the innovation of cereal processing that introduces many humans to high levels of the triggering antigen for celiac disease [78]. The negative effects of celiac disease, therefore, have only affected the human genome very recently. The likely ‘‘newness’’ of celiac disease may explain why the number of positively selected variants associated with the condition is high.
Similarly, the emergence of alcohol as a food stuff could have improved daily caloric intake, but might have also reduced reproductive fitness. Alcohol dehydrogenase (ADH) alleles that enhance alcohol metabolism are thought to have been under positive selection starting at the dawn of the Mesolithic [79–81]. Increased consumption or metabolism of alcohol, however, alters immune traits by changing innate immune modulation, triggering liver inflammation and promoting tumor development [82–88]. Similar arguments can be made about the indirect impact of other putative dietary adaptations such as the rise of multiple alleles upstream of the transcription start site for the gene that expresses Lactase/Lactase-Phlorizin Hydrolase (LCT) alleles in European populations approximately 2000–20 000 years ago [89,90]. These alleles confer lactase persistence and allow continued consumption of dairy products into old age. Milk, however, is iron poor, can cause gut microbleeding in human infants and toddlers (cow milk), and can disrupt iron absorption contributing to iron deficiency and potentially altering immune function [91–95]. After the Mesolithic–Neolithic transition, as agricultural practices disseminate, there is an increase in Eurasian skeletons with pathologies associated with nutritional disruptions such as increased carbohydrate and milk consumption including caries, cribia orbitalia and cranial pitting associated with anemia [96–100]. It is reasonable to expect that shifts in diet altered immune and, specifically, inflammatory phenotypes that later came under selective pressure via other factors.
Conclusion
The recent development of both high throughput sequencing and genotyping technologies, have led to a plethora of GWAS and genome-wide analyses of natural selection. These studies have revolutionized our understanding of how and why chronic inflammatory and autoimmune diseases manifest. Signatures of positive selection associated with risk alleles within a small network of, often, pleiotropic genes regulating these diseases, suggests that chronic inflammatory and autoimmune disease manifestation could be the outcome of an evolutionary trade off. While increased protection against pathogens seems a likely benefit, it is very possible that other traits such as anti-inflammatory conditions in utero, skin color and hypoxic responses associated with these genes could have been strong drivers of positive selection and contributed to increased frequencies of chronic disease risk alleles. The recent advent of new genome-editing technologies such as CRISPR together with induced pluripotent stem cells (known as iPS cells) open new exciting avenues to functionally test the impact of the selected alleles in different cell types and under different cellular conditions (e.g., in response to different pathogens, hypoxia, etc.) [101–104]. Determining which phenotypes might have been under selection in the past will allow us to delineate the immune functions that have been and still are essential for host survival, as well as help clarify the mechanisms contributing to pathological inflammation in contemporary human populations.
Acknowledgements
Thanks to Jeremy Sykes for his comments on the manuscript. This work was supported by the Canadian Institutes of Health Research (301538 and 231519 to LBB), The Human Frontiers Science Program (CDA-00025/2012 to LBB), Canada Research Chairs Program (950-228993 to LBB), Réseau de Médecine Génétique Appliquée (RMGA), the Fonds de Recherche du Québec – Santé(FRQS) and the Canadian Institutes of Health Research (CIHR, Grant # TGF-96109) issued as RGMA Postdoctoral fellowship (2013–2014, 2014–2015 to JFB); National Institutes of Health (1R01-GM102562) to Luis Barreiro (which supports JFB).
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Haldane JBS. The rate of mutation in human genes. Proceedings VIII International Congress Genetics Hereditas. 1949;35:267–273. [Google Scholar]
- 2.Martin RD. Earth history, disease, and the evolution of primates. In: Greenblatt CL, Spigelmann M, editors. Emerging Pathogens: Archaeology, Ecology and Evolution of Infectious Disease. Oxford University Press; 2003. pp. 13–24. [Google Scholar]
- 3.Barreiro LB, Quintana-Murci L. From evolutionary genetics to human immunology: how selection shapes host defence genes. Nat Rev Genet. 2010;11:17–30. doi: 10.1038/nrg2698. [DOI] [PubMed] [Google Scholar]
- 4.Ferrer-Admetlla A, Bosch E, Sikora M, Marques-Bonet T, Ramirez-Soriano A, Muntasell A, Navarro A, Lazarus R, Calafell F, Bertranpetit J, et al. Balancing selection is the main force shaping the evolution of innate immunity genes. J Immunol. 2008;181:1315–1322. doi: 10.4049/jimmunol.181.2.1315. [DOI] [PubMed] [Google Scholar]
- 5.Filip LC, Mundy NI. Rapid evolution by positive Darwinian selection in the extracellular domain of the abundant lymphocyte protein CD45 in primates. Mol Biol Evol. 2004;21:1504–1511. doi: 10.1093/molbev/msh111. [DOI] [PubMed] [Google Scholar]
- 6.Fumagalli M, Sironi M, Pozzoli U, Ferrer-Admetlla A, Pattini L, Nielsen R. Signatures of environmental genetic adaptation pinpoint pathogens as the main selective pressure through human evolution. PLoS Genet. 2011;7:e1002355. doi: 10.1371/journal.pgen.1002355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hamblin MT, Di Rienzo A. Detection of the signature of natural selection in humans: evidence from the Duffy blood group locus. Am J Hum Genet. 2000;66:1669–1679. doi: 10.1086/302879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karlsson EK, Harris JB, Tabrizi S, Rahman A, Shlyakhter I, Patterson N, O’Dushlaine C, Schaffner SF, Gupta S, Chowdhury F, et al. Natural selection in a Bangladeshi population from the cholera-endemic Ganges river delta. Sci Transl Med. 2013;5:192ra186. doi: 10.1126/scitranslmed.3006338. An important examination of how the human genome has been shaped by a historically important infectious disease. The authors examine the genomes of cholera patients and healthy family members and identify 300+ regions under selection, and identifies a number of genes under associated with cholera susceptibility. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Metzger KJ, Thomas MA. Evidence of positive selection at codon sites localized in extracellular domains of mammalian CC motif chemokine receptor proteins. BMC Evol Biol. 2010;10:139. doi: 10.1186/1471-2148-10-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakajima T, Ohtani H, Satta Y, Uno Y, Akari H, Ishida T, Kimura A. Natural selection in the TLR-related genes in the course of primate evolution. Immunogenetics. 2008;60:727–735. doi: 10.1007/s00251-008-0332-0. [DOI] [PubMed] [Google Scholar]
- 11.Novembre J, Han E. Human population structure and the adaptive response to pathogen-induced selection pressures. Philos Trans R Soc Lond B Biol Sci. 2012;367:878–886. doi: 10.1098/rstb.2011.0305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sawyer SL, Wu LI, Emerman M, Malik HS. Positive selection of primate TRIM5alpha identifies a critical species-specific retroviral restriction domain. Proc Natl Acad Sci U S A. 2005;102:2832–2837. doi: 10.1073/pnas.0409853102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilson JN, Rockett K, Keating B, Jallow M, Pinder M, Sisay-Joof F, Newport M, Kwiatkowski D. A hallmark of balancing selection is present at the promoter region of interleukin 10. Genes Immun. 2006;7:680–683. doi: 10.1038/sj.gene.6364336. [DOI] [PubMed] [Google Scholar]
- 14.Wlasiuk G, Khan S, Switzer WM, Nachman MW. A history of recurrent positive selection at the toll-like receptor 5 in primates. Mol Biol Evol. 2009;26:937–949. doi: 10.1093/molbev/msp018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhernakova A, Elbers CC, Ferwerda B, Romanos J, Trynka G, Dubois PC, de Kovel CG, Franke L, Oosting M, Barisani D, et al. Evolutionary and functional analysis of celiac risk loci reveals SH2B3 as a protective factor against bacterial infection. Am J Hum Genet. 2010;86:970–977. doi: 10.1016/j.ajhg.2010.05.004. An important paper that assesses signatures of selection for multiple GWA celiac disease loci in a large cohort, provides a confirmation of altered bacterial ligand sensing function in the PBMCs of individuals homozygous for a SH2B3 allele. This paper can be interpreted to provide evolutionary genomic and functional support for an evolutionary trade off between pathogen sensing and celiac disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pickrell JK, Coop G, Novembre J, Kudaravalli S, Li JZ, Absher D, Srinivasan BS, Barsh GS, Myers RM, Feldman MW, et al. Signals of recent positive selection in a worldwide sample of human populations. Genome Res. 2009;19:826–837. doi: 10.1101/gr.087577.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barreiro LB, Laval G, Quach H, Patin E, Quintana-Murci L. Natural selection has driven population differentiation in modern humans. Nat Genet. 2008;40:340–345. doi: 10.1038/ng.78. [DOI] [PubMed] [Google Scholar]
- 18.Andersen KG, Shylakhter I, Tabrizi S, Grossman SR, Happi CT, Sabeti PC. Genome-wide scans provide evidence for positive selection of genes implicated in Lassa fever. Philos Trans R Soc Lond B Biol Sci. 2012;367:868–877. doi: 10.1098/rstb.2011.0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sabeti PC, Schaffner SF, Fry B, Lohmueller J, Varilly P, Shamovsky O, Palma A, Mikkelsen TS, Altshuler D, Lander ES. Positive natural selection in the human lineage. Science. 2006;312:1614–1620. doi: 10.1126/science.1124309. [DOI] [PubMed] [Google Scholar]
- 20.Bustamante CD, Fledel-Alon A, Williamson S, Nielsen R, Hubisz MT, Glanowski S, Tanenbaum DM, White TJ, Sninsky JJ, Hernandez RD, et al. Natural selection on protein-coding genes in the human genome. Nature. 2005;437:1153–1157. doi: 10.1038/nature04240. [DOI] [PubMed] [Google Scholar]
- 21.Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cotsapas C, Voight BF, Rossin E, Lage K, Neale BM, Wallace C, Abecasis GR, Barrett JC, Behrens T, Cho J, et al. 2011;7:e1002254. doi: 10.1371/journal.pgen.1002254. A large GWAS meta-analysis of seven chronic inflammatory and autoimmune diseases that finds extensive sharing of SNPs across multiple diseases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al. Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. A large meta-analysis of IBD GWAS that identifies dozens of new risk loci and finds many are under directional or balancing selection, and are implicated in other chronic inflammatory/autoimmune and infectious diseases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhernakova A, Stahl EA, Trynka G, Raychaudhuri S, Festen EA, Franke L, Westra HJ, Fehrmann RS, Kurreeman FA, Thomson B, et al. Meta-analysis of genome-wide association studies in celiac disease and rheumatoid arthritis identifies fourteen non-HLA shared loci. PLoS Genet. 2011;7:e1002004. doi: 10.1371/journal.pgen.1002004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raj T, Kuchroo M, Replogle JM, Raychaudhuri S, Stranger BE, De Jager PL. Common risk alleles for inflammatory diseases are targets of recent positive selection. Am J Hum Genet. 2013;92:517–529. doi: 10.1016/j.ajhg.2013.03.001. An important study that uses a unique multi-disease, GWAS, quantitative and evolutionary genomic approach to identify chronic inflammatory and autoimmune risk alleles and finds a small network of implicated genes under positive selection, shared across diseases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cooper GS, Bynum ML, Somers EC. Recent insights in the epidemiology of autoimmune diseases: improved prevalence estimates and understanding of clustering of diseases. J Autoimmun. 2009;33:197–207. doi: 10.1016/j.jaut.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhernakova A, van Diemen CC, Wijmenga C. Detecting shared pathogenesis from the shared genetics of immune-related diseases. Nat Rev Genet. 2009;10:43–55. doi: 10.1038/nrg2489. [DOI] [PubMed] [Google Scholar]
- 28.Parkes M, Cortes A, van Heel DA, Brown MA. Genetic insights into common pathways and complex relationships among immune-mediated diseases. Nat Rev Genet. 2013;14:661–673. doi: 10.1038/nrg3502. [DOI] [PubMed] [Google Scholar]
- 29.Van Praet L, Jans L, Carron P, Jacques P, Glorieus E, Colman R, Cypers H, Mielants H, De Vos M, Cuvelier C, et al. Degree of bone marrow oedema in sacroiliac joints of patients with axial spondyloarthritis is linked to gut inflammation and male sex: results from the GIANT cohort. Ann Rheum Dis. 2014;73:1186–1189. doi: 10.1136/annrheumdis-2013-203854. [DOI] [PubMed] [Google Scholar]
- 30.Mielants H, Veys EM, Cuvelier C, De Vos M, Goemaere S, De Clercq L, Schatteman L, Elewaut D. The evolution of spondyloarthropathies in relation to gut histology. II. Histological aspects. J Rheumatol. 1995;22:2273–2278. [PubMed] [Google Scholar]
- 31.Bernstein CN, Wajda A, Blanchard JF. The clustering of other chronic inflammatory diseases in inflammatory bowel disease: a population-based study. Gastroenterology. 2005;129:827–836. doi: 10.1053/j.gastro.2005.06.021. A powerful study demonstrating chronic inflammatory and autoimmune disease overlap in a large cohort (University of Manitoba cohort). Specifically, this study shows a significant association between IBD and neurological autoimmune diseases, and provides support for a chronic disease model of shared central disease mechanism. [DOI] [PubMed] [Google Scholar]
- 32.Lees CW, Barrett JC, Parkes M, Satsangi J. New IBD genetics: common pathways with other diseases. Gut. 2011;60:1739–1753. doi: 10.1136/gut.2009.199679. [DOI] [PubMed] [Google Scholar]
- 33.Barthel C, Biedermann L, Frei P, Vavricka SR, Kundig T, Fried M, Rogler G, Scharl M. Induction or exacerbation of psoriasis in patients with Crohn’s disease under treatment with anti-TNF antibodies. Digestion. 2014;89:209–215. doi: 10.1159/000358288. [DOI] [PubMed] [Google Scholar]
- 34.Furst DE, Breedveld FC, Kalden JR, Smolen JS, Burmester GR, Sieper J, Emery P, Keystone EC, Schiff MH, Mease P, et al. Updated consensus statement on biological agents for the treatment of rheumatic diseases. Ann Rheum Dis. 2007;66(Suppl 3):iii2–iii22. doi: 10.1136/ard.2007.081430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Collamer AN, Guerrero KT, Henning JS, Battafarano DF. Psoriatic skin lesions induced by tumor necrosis factor antagonist therapy: a literature review and potential mechanisms of action. Arthritis Rheum. 2008;59:996–1001. doi: 10.1002/art.23835. [DOI] [PubMed] [Google Scholar]
- 36.Harrison MJ, Dixon WG, Watson KD, King Y, Groves R, Hyrich KL, Symmons DP, British Society for Rheumatology Biologics Register Control Centre C Bsrbr: rates of new-onset psoriasis in patients with rheumatoid arthritis receiving anti-tumour necrosis factor alpha therapy: results from the British Society for Rheumatology Biologics Register. Ann Rheum Dis. 2009;68:209–215. doi: 10.1136/ard.2007.087288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park JH, Wacholder S, Gail MH, Peters U, Jacobs KB, Chanock SJ, Chatterjee N. Estimation of effect size distribution from genome-wide association studies and implications for future discoveries. Nat Genet. 2010;42:570–575. doi: 10.1038/ng.610. An important paper that estimates the effect size of most GWAS variants is small, and provides support for a chronic inflammatory/autoimmune disease model based on cumulative effects of multiple SNPs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nielsen R, Williamson S, Kim Y, Hubisz MJ, Clark AG, Bustamante C. Genomic scans for selective sweeps using SNP data. Genome Res. 2005;15:1566–1575. doi: 10.1101/gr.4252305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fumagalli M, Pozzoli U, Cagliani R, Comi GP, Bresolin N, Clerici M, Sironi M. Genome-wide identification of susceptibility alleles for viral infections through a population genetics approach. PLoS Genet. 2010;6:e1000849. doi: 10.1371/journal.pgen.1000849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Casto AM, Feldman MW. Genome-wide association study SNPs in the human genome diversity project populations: does selection affect unlinked SNPs with shared trait associations? PLoS Genet. 2011;7:e1001266. doi: 10.1371/journal.pgen.1001266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prugnolle F, Manica A, Charpentier M, Guegan JF, Guernier V, Balloux F. Pathogen-driven selection and worldwide HLA class I diversity. Curr Biol. 2005;15:1022–1027. doi: 10.1016/j.cub.2005.04.050. [DOI] [PubMed] [Google Scholar]
- 42.Bustamante CD, Burchard EG, De la Vega FM. Genomics for the world. Nature. 2011;475:163–165. doi: 10.1038/475163a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Higham T, Douka K, Wood R, Ramsey CB, Brock F, Basell L, Camps M, Arrizabalaga A, Baena J, Barroso-Ruiz C, et al. The timing and spatiotemporal patterning of Neanderthal disappearance. Nature. 2014;512:306–309. doi: 10.1038/nature13621. [DOI] [PubMed] [Google Scholar]
- 44.Sankararaman S, Mallick S, Dannemann M, Prufer K, Kelso J, Paabo S, Patterson N, Reich D. The genomic landscape of Neanderthal ancestry in present-day humans. Nature. 2014;507:354–357. doi: 10.1038/nature12961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abi-Rached L, Jobin MJ, Kulkarni S, McWhinnie A, Dalva K, Gragert L, Babrzadeh F, Gharizadeh B, Luo M, Plummer FA, et al. The shaping of modern human immune systems by multiregional admixture with archaic humans. Science. 2011;334:89–94. doi: 10.1126/science.1209202. A comparison of archaic and modern human genomes that suggests some human immune alleles are the product of inter-species interbreeding. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Genomes Project C. Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abadie V, Sollid LM, Barreiro LB, Jabri B. Integration of genetic and immunological insights into a model of celiac disease pathogenesis. Annu Rev Immunol. 2011;29:493–525. doi: 10.1146/annurev-immunol-040210-092915. [DOI] [PubMed] [Google Scholar]
- 48.Yamazaki M, Kitamura R, Kusano S, Eda H, Sato S, Okawa-Takatsuji M, Aotsuka S, Yanagi K. Elevated immunoglobulin G antibodies to the proline-rich amino-terminal region of Epstein–Barr virus nuclear antigen-2 in sera from patients with systemic connective tissue diseases and from a subgroup of Sjögren’s syndrome patients with pulmonary involvements. Clin Exp Immunol. 2005;139:558–568. doi: 10.1111/j.1365-2249.2004.02704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harley JB, James JA. Epstein–Barr virus infection induces lupus autoimmunity. Bull NYU Hosp Jt Dis. 2006;64:45–50. [PubMed] [Google Scholar]
- 50.James JA, Neas BR, Moser KL, Hall T, Bruner GR, Sestak AL, Harley JB. Systemic lupus erythematosus in adults is associated with previous Epstein–Barr virus exposure. Arthritis Rheum. 2001;44:1122–1126. doi: 10.1002/1529-0131(200105)44:5<1122::AID-ANR193>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 51.Ramos-Casals M, Jara LJ, Medina F, Rosas J, Calvo-Alen J, Mana J, Anaya JM, Font J, Group HS. Systemic autoimmune diseases co-existing with chronic hepatitis C virus infection (the HISPAMEC Registry): patterns of clinical and immunological expression in 180 cases. J Intern Med. 2005;257:549–557. doi: 10.1111/j.1365-2796.2005.01490.x. [DOI] [PubMed] [Google Scholar]
- 52.Saebo A, Vik E, Lange OJ, Matuszkiewicz L. Inflammatory bowel disease associated with Yersinia enterocolitica O:3 infection. Eur J Intern Med. 2005;16:176–182. doi: 10.1016/j.ejim.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 53.Feller M, Huwiler K, Stephan R, Altpeter E, Shang A, Furrer H, Pfyffer GE, Jemmi T, Baumgartner A, Egger M. Mycobacterium avium subspecies paratuberculosis and Crohn’s disease: a systematic review and meta-analysis. Lancet Infect Dis. 2007;7:607–613. doi: 10.1016/S1473-3099(07)70211-6. [DOI] [PubMed] [Google Scholar]
- 54.Abubakar I, Myhill D, Aliyu SH, Hunter PR. Detection of Mycobacterium avium subspecies paratuberculosis from patients with Crohn’s disease using nucleic acid-based techniques: a systematic review and meta-analysis. Inflamm Bowel Dis. 2008;14:401–410. doi: 10.1002/ibd.20276. [DOI] [PubMed] [Google Scholar]
- 55.Olsen I, Tollefsen S, Aagaard C, Reitan LJ, Bannantine JP, Andersen P, Sollid LM, Lundin KE. Isolation of Mycobacterium avium subspecies paratuberculosis reactive CD4 T cells from intestinal biopsies of Crohn’s disease patients. PLoS ONE. 2009;4:e5641. doi: 10.1371/journal.pone.0005641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barreiro LB, Ben-Ali M, Quach H, Laval G, Patin E, Pickrell JK, Bouchier C, Tichit M, Neyrolles O, Gicquel B, et al. Evolutionary dynamics of human Toll-like receptors and their different contributions to host defense. PLoS Genet. 2009;5:e1000562. doi: 10.1371/journal.pgen.1000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang FR, Huang W, Chen SM, Sun LD, Liu H, Li Y, Cui Y, Yan XX, Yang HT, Yang RD, et al. Genomewide association study of leprosy. N Engl J Med. 2009;361:2609–2618. doi: 10.1056/NEJMoa0903753. An important GWAS that finds infectious disease progression alleles also known to be risk alleles for IBD. [DOI] [PubMed] [Google Scholar]
- 58.Cagliani R, Pozzoli U, Forni D, Cassinotti A, Fumagalli M, Giani M, Fichera M, Lombardini M, Ardizzone S, Asselta R, et al. Crohn’s disease loci are common targets of protozoa-driven selection. Mol Biol Evol. 2013;30:1077–1087. doi: 10.1093/molbev/mst020. [DOI] [PubMed] [Google Scholar]
- 59.Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fumagalli M, Pozzoli U, Cagliani R, Comi GP, Riva S, Clerici M, Bresolin N, Sironi M. Parasites represent a major selective force for interleukin genes and shape the genetic predisposition to autoimmune conditions. J Exp Med. 2009;206:1395–1408. doi: 10.1084/jem.20082779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Buza-Vidas N, Antonchuk J, Qian H, Mansson R, Luc S, Zandi S, Anderson K, Takaki S, Nygren JM, Jensen CT, et al. Cytokines regulate postnatal hematopoietic stem cell expansion: opposing roles of thrombopoietin and LNK. Genes Dev. 2006;20:2018–2023. doi: 10.1101/gad.385606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gauthier AS, Furstoss O, Araki T, Chan R, Neel BG, Kaplan DR, Miller FD. Control of CNS cell-fate decisions by SHP-2 and its dysregulation in Noonan syndrome. Neuron. 2007;54:245–262. doi: 10.1016/j.neuron.2007.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kitaya K, Yasuo T, Yamaguchi T, Fushiki S, Honjo H. Genes regulated by interferon-gamma in human uterine microvascular endothelial cells. Int J Mol Med. 2007;20:689–697. [PubMed] [Google Scholar]
- 64.Turner J, Roger J, Fitau J, Combe D, Giddings J, Heeke GV, Jones CE. Goblet cells are derived from a FOXJ1-expressing progenitor in a human airway epithelium. Am J Respir Cell Mol Biol. 2011;44:276–284. doi: 10.1165/rcmb.2009-0304OC. [DOI] [PubMed] [Google Scholar]
- 65.Devalliere J, Charreau B. The adaptor Lnk (SH2B3): an emerging regulator in vascular cells and a link between immune and inflammatory signaling. Biochem Pharmacol. 2011;82:1391–1402. doi: 10.1016/j.bcp.2011.06.023. [DOI] [PubMed] [Google Scholar]
- 66.Yu F, Sabeti PC, Hardenbol P, Fu Q, Fry B, Lu X, Ghose S, Vega R, Perez A, Pasternak S, et al. Positive selection of a preexpansion CAG repeat of the human SCA2 gene. PLoS Genet. 2005;1:e41. doi: 10.1371/journal.pgen.0010041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Van Damme P, Veldink JH, van Blitterswijk M, Corveleyn A, van Vught PW, Thijs V, Dubois B, Matthijs G, van den Berg LH, Robberecht W. Expanded ATXN2 CAG repeat size in ALS identifies genetic overlap between ALS and SCA2. Neurology. 2011;76:2066–2072. doi: 10.1212/WNL.0b013e31821f445b. [DOI] [PubMed] [Google Scholar]
- 68.Scherer SE, Muzny DM, Buhay CJ, Chen R, Cree A, Ding Y, Dugan-Rocha S, Gill R, Gunaratne P, Harris RA, et al. The finished DNA sequence of human chromosome 12. Nature. 2006;440:346–351. doi: 10.1038/nature04569. [DOI] [PubMed] [Google Scholar]
- 69.Kottgen A, Pattaro C, Boger CA, Fuchsberger C, Olden M, Glazer NL, Parsa A, Gao X, Yang Q, Smith AV, et al. New loci associated with kidney function and chronic kidney disease. Nat Genet. 2010;42:376–384. doi: 10.1038/ng.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lu Y, Chen H, Nikamo P, Qi Low H, Helms C, Seielstad M, Liu J, Bowcock AM, Stahle M, Liao W. Association of cardiovascular and metabolic disease genes with psoriasis. J Invest Dermatol. 2013;133:836–839. doi: 10.1038/jid.2012.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ochoa E, Iriondo M, Bielsa A, Ruiz-Irastorza G, Estonba A, Zubiaga AM. Thrombotic antiphospholipid syndrome shows strong haplotypic association with SH2B3-ATXN2 locus. PLOS ONE. 2013;8:e67897. doi: 10.1371/journal.pone.0067897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hunt KA, Zhernakova A, Turner G, Heap GA, Franke L, Bruinenberg M, Romanos J, Dinesen LC, Ryan AW, Panesar D, et al. Newly identified genetic risk variants for celiac disease related to the immune response. Nat Genet. 2008;40:395–402. doi: 10.1038/ng.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Coenen MJ, Trynka G, Heskamp S, Franke B, van Diemen CC, Smolonska J, van Leeuwen M, Brouwer E, Boezen MH, Postma DS, et al. Common and different genetic background for rheumatoid arthritis and coeliac disease. Hum Mol Genet. 2009;18:4195–4203. doi: 10.1093/hmg/ddp365. [DOI] [PubMed] [Google Scholar]
- 74.Childe VG. The Dawn of European Civilization. K. Paul, Trench A.A. Knopf; London, New York: 1925. [Google Scholar]
- 75.Armitage SJ, Jasim SA, Marks AE, Parker AG, Usik VI, Uerpmann HP. The southern route “out of Africa”: evidence for an early expansion of modern humans into Arabia. Science. 2011;331:453–456. doi: 10.1126/science.1199113. [DOI] [PubMed] [Google Scholar]
- 76.Harper KN, Armelagos GJ. Genomics, the origins of agriculture, and our changing microbe-scape: time to revisit some old tales and tell some new ones. Am J Phys Anthropol. 2013;152(Suppl 57):135–152. doi: 10.1002/ajpa.22396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Harper K, Armelagos G. The changing disease-scape in the third epidemiological transition. Int J Environ Res Public Health. 2010;7:675–697. doi: 10.3390/ijerph7020675. Two very comprehensive reviews that discuss the impact of human cultural change on past disease epidemiology and the evolution and function of the human immune system. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Piperno DR, Weiss E, Holst I, Nadel D. Processing of wild cereal grains in the Upper Palaeolithic revealed by starch grain analysis. Nature. 2004;430:670–673. doi: 10.1038/nature02734. [DOI] [PubMed] [Google Scholar]
- 79.Peng Y, Shi H, Qi XB, Xiao CJ, Zhong H, Ma RL, Su B. The ADH1B Arg47His polymorphism in east Asian populations and expansion of rice domestication in history. BMC Evol Biol. 2010;10:15. doi: 10.1186/1471-2148-10-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li H, Gu S, Cai X, Speed WC, Pakstis AJ, Golub EI, Kidd JR, Kidd KK. Ethnic related selection for an ADH Class I variant within East Asia. PLoS ONE. 2008;3:e1881. doi: 10.1371/journal.pone.0001881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Han Y, Gu S, Oota H, Osier MV, Pakstis AJ, Speed WC, Kidd JR, Kidd KK. Evidence of positive selection on a class I ADH locus. Am J Hum Genet. 2007;80:441–456. doi: 10.1086/512485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lu Y, Ni F, Xu M, Yang J, Chen J, Chen Z, Wang X, Luo J, Wang S. Alcohol promotes mammary tumor growth through activation of VEGF-dependent tumor angiogenesis. Oncol Lett. 2014;8:673–678. doi: 10.3892/ol.2014.2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mercer KE, Hennings L, Sharma N, Lai K, Cleves MA, Wynne RA, Badger TM, Ronis MJ. Alcohol consumption promotes diethylnitrosamine-induced hepatocarcinogenesis in male mice through activation of the Wnt/beta-catenin signaling pathway. Cancer Prev Res (Phila) 2014;7:675–685. doi: 10.1158/1940-6207.CAPR-13-0444-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lieber CS, Jones DP, Decarli LM. Effects of prolonged ethanol intake: production of fatty liver despite adequate diets. J Clin Invest. 1965;44:1009–1021. doi: 10.1172/JCI105200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Boe DM, Nelson S, Zhang P, Quinton L, Bagby GJ. Alcoholinduced suppression of lung chemokine production and the host defense response to Streptococcus pneumoniae. Alcohol Clin Exp Res. 2003;27:1838–1845. doi: 10.1097/01.ALC.0000095634.82310.53. [DOI] [PubMed] [Google Scholar]
- 86.Pruett BS, Pruett SB. An explanation for the paradoxical induction and suppression of an acute phase response by ethanol. Alcohol. 2006;39:105–110. doi: 10.1016/j.alcohol.2006.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Goral J, Choudhry MA, Kovacs EJ. Acute ethanol exposure inhibits macrophage IL-6 production: role of p38 and ERK1/2 MAPK. J Leukoc Biol. 2004;75:553–559. doi: 10.1189/jlb.0703350. [DOI] [PubMed] [Google Scholar]
- 88.Saeed RW, Varma S, Peng T, Tracey KJ, Sherry B, Metz CN. Ethanol blocks leukocyte recruitment and endothelial cell activation in vivo and in vitro. J Immunol. 2004;173:6376–6383. doi: 10.4049/jimmunol.173.10.6376. [DOI] [PubMed] [Google Scholar]
- 89.Tishkoff SA, Reed FA, Ranciaro A, Voight BF, Babbitt CC, Silverman JS, Powell K, Mortensen HM, Hirbo JB, Osman M, et al. Convergent adaptation of human lactase persistence in Africa and Europe. Nat Genet. 2007;39:31–40. doi: 10.1038/ng1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bersaglieri T, Sabeti PC, Patterson N, Vanderploeg T, Schaffner SF, Drake JA, Rhodes M, Reich DE, Hirschhorn JN. Genetic signatures of strong recent positive selection at the lactase gene. Am J Hum Genet. 2004;74:1111–1120. doi: 10.1086/421051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ziegler EE. Adverse effects of cow’s milk in infants. Nestle Nutr Workshop Ser Pediatr Program. 2007;60:185–196. doi: 10.1159/000106369. [DOI] [PubMed] [Google Scholar]
- 92.Wilson JF, Lahey ME, Heiner DC. Studies on iron metabolism. V. Further observations on cow’s milk-induced gastrointestinal bleeding in infants with iron-deficiency anemia. J Pediatr. 1974;84:335–344. doi: 10.1016/s0022-3476(74)80713-4. [DOI] [PubMed] [Google Scholar]
- 93.Ziegler EE, Fomon SJ, Nelson SE, Rebouche CJ, Edwards BB, Rogers RR, Lehman LJ. Cow milk feeding in infancy: further observations on blood loss from the gastrointestinal tract. J Pediatr. 1990;116:11–18. doi: 10.1016/s0022-3476(05)90003-6. [DOI] [PubMed] [Google Scholar]
- 94.Ziegler EE. Consumption of cow’s milk as a cause of iron deficiency in infants and toddlers. Nutr Rev. 2011;69(Suppl 1):S37–S42. doi: 10.1111/j.1753-4887.2011.00431.x. [DOI] [PubMed] [Google Scholar]
- 95.Male C, Persson LA, Freeman V, Guerra A, van’t Hof MA, Haschke F, Euro-Growth Iron Study G Prevalence of iron deficiency in 12-mo-old infants from 11 European areas and influence of dietary factors on iron status (Euro-Growth study) Acta Paediatr. 2001;90:492–498. doi: 10.1080/080352501750197601. [DOI] [PubMed] [Google Scholar]
- 96.Silvana M, Borgognini T, Elena R. Dietary patterns in the mesolithic samples from Uzzo and Molara caves (Sicily): the evidence of teeth. J Hum Evol. 1985;14:241–254. [Google Scholar]
- 97.Temple DH, Larsen CS. Dental caries prevalence as evidence for agriculture and subsistence variation during the Yayoi period in prehistoric Japan: biocultural interpretations of an economy in transition. Am J Phys Anthropol. 2007;134:501–512. doi: 10.1002/ajpa.20694. [DOI] [PubMed] [Google Scholar]
- 98.Eshed V, Gopher A, Pinhasi R, Hershkovitz I. Paleopathology and the origin of agriculture in the Levant. Am J Phys Anthropol. 2010;143:121–133. doi: 10.1002/ajpa.21301. [DOI] [PubMed] [Google Scholar]
- 99.Angel JL. Health as a crucial factor in changes from hunting to developed farming in the eastern Mediterranean. In: Cohen MN, Armelagos G, editors. Paleopathology at the Origins of Agriculture. Academic Press; 1984. pp. 51–74. [Google Scholar]
- 100.Pechenkina EA, Vradenburg JA, Benfer RA, Franum JF. Skeletal biology of the central Peruvian coast: consequences of changing population density and progressive dependence on maize agriculture. In: Cohen M, Crane-Kramer GMM, editors. Ancient Health: Skeletal Indicators of Agricultural and Economic Intensification. Florida University Press; 2007. pp. 92–112. [Google Scholar]
- 101.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, Bogdanove AJ, Voytas DF. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics. 2010;186:757–761. doi: 10.1534/genetics.110.120717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, Baller JA, Somia NV, Bogdanove AJ, Voytas DF. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011;39:e82. doi: 10.1093/nar/gkr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hindorff LA, Macarthur JEBI, Morales JEBI, Junkins HA, Hall PN, Klemm AK, Manolio TA. A Catalog of Published Genome-Wide Association Studies. 2014 Available at: http://www.genome.gov/gwastudies.
- 106.Dabelea D, Mayer-Davis EJ, Saydah S, Imperatore G, Linder B, Divers J, Bell R, Badaru A, Talton JW, Crume T, et al. Prevalence of type 1 and type 2 diabetes among children and adolescents from 2001 to 2009. JAMA. 2014;311:1778–1786. doi: 10.1001/jama.2014.3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bell RA, Mayer-Davis EJ, Beyer JW, D’Agostino RB, Jr, Lawrence JM, Linder B, Liu LL, Marcovina SM, Rodriguez BL, Williams D, et al. Diabetes in non-Hispanic white youth: prevalence, incidence, and clinical characteristics: the SEARCH for Diabetes in Youth Study. Diabetes Care. 2009;32(Suppl 2):S102–S111. doi: 10.2337/dc09-S202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mayer-Davis EJ, Beyer J, Bell RA, Dabelea D, D’Agostino R, Jr, Imperatore G, Lawrence JM, Liese AD, Liu L, Marcovina S, et al. Diabetes in African American youth: prevalence, incidence, and clinical characteristics: the SEARCH for Diabetes in Youth Study. Diabetes Care. 2009;32(Suppl 2):S112–S122. doi: 10.2337/dc09-S203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liu LL, Yi JP, Beyer J, Mayer-Davis EJ, Dolan LM, Dabelea DM, Lawrence JM, Rodriguez BL, Marcovina SM, Waitzfelder BE, et al. Type 1 and Type 2 diabetes in Asian and Pacific Islander U.S. youth: the SEARCH for diabetes in youth study. Diabetes Care. 2009;32(Suppl 2):S133–S140. doi: 10.2337/dc09-S205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gelfand JM, Stern RS, Nijsten T, Feldman SR, Thomas J, Kist J, Rolstad T, Margolis DJ. The prevalence of psoriasis in African Americans: results from a population-based study. J Am Acad Dermatol. 2005;52:23–26. doi: 10.1016/j.jaad.2004.07.045. [DOI] [PubMed] [Google Scholar]
- 111.Rachakonda TD, Schupp CW, Armstrong AW. Psoriasis prevalence among adults in the United States. J Am Acad Dermatol. 2014;70:512–516. doi: 10.1016/j.jaad.2013.11.013. [DOI] [PubMed] [Google Scholar]
- 112.Sanchez MR. Cutaneous diseases in Latinos. Dermatol Clin. 2003;21:689–697. doi: 10.1016/s0733-8635(03)00087-1. [DOI] [PubMed] [Google Scholar]
- 113.Noonan CW, Williamson DM, Henry JP, Indian R, Lynch SG, Neuberger JS, Schiffer R, Trottier J, Wagner L, Marrie RA. The prevalence of multiple sclerosis in 3 US communities. Prev Chronic Dis. 2010;7:A12. [PMC free article] [PubMed] [Google Scholar]
- 114.Aguirre-Cruz L, Flores-Rivera J, De La Cruz-Aguilera DL, Rangel-Lopez E, Corona T. Multiple sclerosis in Caucasians and Latino Americans. Autoimmunity. 2011;44:571–575. doi: 10.3109/08916934.2011.592887. [DOI] [PubMed] [Google Scholar]
- 115.Rubio-Tapia A, Ludvigsson JF, Brantner TL, Murray JA, Everhart JE. The prevalence of celiac disease in the United States. Am J Gastroenterol. 2012;107:1538–1544. doi: 10.1038/ajg.2012.219. [DOI] [PubMed] [Google Scholar]
- 116.Betteridge JD, Armbruster SP, Maydonovitch C, Veerappan GR. Inflammatory bowel disease prevalence by age, gender, race, and geographic location in the U.S. military health care population. Inflamm Bowel Dis. 2013;19:1421–1427. doi: 10.1097/MIB.0b013e318281334d. [DOI] [PubMed] [Google Scholar]
- 117.Ogunbi SO, Ransom JA, Sullivan K, Schoen BT, Gold BD. Inflammatory bowel disease in African-American children living in Georgia. J Pediatr. 1998;133:103–107. doi: 10.1016/s0022-3476(98)70187-8. [DOI] [PubMed] [Google Scholar]
- 118.Appleyard CB, Hernandez G, Rios-Bedoya CF. Basic epidemiology of inflammatory bowel disease in Puerto Rico. Inflamm Bowel Dis. 2004;10:106–111. doi: 10.1097/00054725-200403000-00007. [DOI] [PubMed] [Google Scholar]
- 119.Lim SS, Bayakly AR, Helmick CG, Gordon C, Easley KA, Drenkard C. The incidence and prevalence of systemic lupus erythematosus, 2002–2004: the Georgia Lupus Registry. Arthritis Rheumatol. 2014;66:357–368. doi: 10.1002/art.38239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Feldman CH, Hiraki LT, Liu J, Fischer MA, Solomon DH, Alarcon GS, Winkelmayer WC, Costenbader KH. Epidemiology and sociodemographics of systemic lupus erythematosus and lupus nephritis among US adults with Medicaid coverage, 2000–2004. Arthritis Rheum. 2013;65:753–763. doi: 10.1002/art.37795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chakravarty EF, Bush TM, Manzi S, Clarke AE, Ward MM. Prevalence of adult systemic lupus erythematosus in California and Pennsylvania in 2000: estimates obtained using hospitalization data. Arthritis Rheum. 2007;56:2092–2094. doi: 10.1002/art.22641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Karlson EW, Watts J, Signorovitch J, Bonetti M, Wright E, Cooper GS, McAlindon TE, Costenbader KH, Massarotti EM, Fitzgerald LM, et al. Effect of glutathione S-transferase polymorphisms and proximity to hazardous waste sites on time to systemic lupus erythematosus diagnosis: results from the Roxbury lupus project. Arthritis Rheum. 2007;56:244–254. doi: 10.1002/art.22308. [DOI] [PubMed] [Google Scholar]
- 123.Molina MJ, Mayor AM, Franco AE, Morell CA, Lopez MA, Vila LM. Prevalence of systemic lupus erythematosus and associated comorbidities in Puerto Rico. J Clin Rheumatol. 2007;13:202–204. doi: 10.1097/RHU.0b013e318124a8af. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ferucci ED, Johnston JM, Gaddy JR, Sumner L, Posever JO, Choromanski TL, Gordon C, Lim SS, Helmick CG. Prevalence and incidence of systemic lupus erythematosus in a populationbased registry of American Indian and Alaska Native people, 2007–2009. Arthritis Rheumatol. 2014;66:2494–2502. doi: 10.1002/art.38720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Starheim KK, Arnesen T, Gromyko D, Ryningen A, Varhaug JE, Lillehaug JR. Identification of the human N(alpha)-acetyltransferase complex B (hNatB): a complex important for cell-cycle progression. Biochem J. 2008;415:325–331. doi: 10.1042/BJ20080658. [DOI] [PubMed] [Google Scholar]
- 126.Dravid G, Zhu Y, Scholes J, Evseenko D, Crooks GM. Dysregulated gene expression during hematopoietic differentiation from human embryonic stem cells. Mol Ther. 2011;19:768–781. doi: 10.1038/mt.2010.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Digilio MC, Conti E, Sarkozy A, Mingarelli R, Dottorini T, Marino B, Pizzuti A, Dallapiccola B. Grouping of multiplelentigines/LEOPARD and Noonan syndromes on the PTPN11 gene. Am J Hum Genet. 2002;71:389–394. doi: 10.1086/341528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Princen F, Bard E, Sheikh F, Zhang SS, Wang J, Zago WM, Wu D, Trelles RD, Bailly-Maitre B, Kahn CR, et al. Deletion of Shp2 tyrosine phosphatase in muscle leads to dilated cardiomyopathy, insulin resistance, and premature death. Mol Cell Biol. 2009;29:378–388. doi: 10.1128/MCB.01661-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bowen ME, Ayturk UM, Kurek KC, Yang W, Warman ML. SHP2 regulates chondrocyte terminal differentiation, growth plate architecture and skeletal cell fates. PLoS Genet. 2014;10:e1004364. doi: 10.1371/journal.pgen.1004364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, van der Burgt I, Crosby AH, Ion A, Jeffery S, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001;29:465–468. doi: 10.1038/ng772. [DOI] [PubMed] [Google Scholar]
- 131.Zhang EE, Chapeau E, Hagihara K, Feng GS. Neuronal Shp2 tyrosine phosphatase controls energy balance and metabolism. Proc Natl Acad Sci U S A. 2004;101:16064–16069. doi: 10.1073/pnas.0405041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bai X, Chan ED, Xu X. The protein of a new gene, Tctex4, interacts with protein kinase CK2beta subunit and is highly expressed in mouse testis. Biochem Biophys Res Commun. 2003;307:86–91. doi: 10.1016/s0006-291x(03)01118-5. [DOI] [PubMed] [Google Scholar]
- 133.Lepourcelet M, Coriton O, Hampe A, Galibert F, Mosser J. HTEX4, a new human gene in the MHC class I region, undergoes alternative splicing and polyadenylation processes in testis. Immunogenetics. 1998;47:491–496. doi: 10.1007/s002510050388. [DOI] [PubMed] [Google Scholar]
- 134.Meola D, Huang Z, Petitto JM. Selective neuronal and brain regional expression of IL-2 in IL2P 8-GFP transgenic mice: relation to sensorimotor gating. J Alzheimers Dis Parkinsonism. 2013;3:1000127. doi: 10.4172/2161-0460.1000127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Johnson GA, Bazer FW, Burghardt RC, Spencer TE, Wu G, Bayless KJ. Conceptus–uterus interactions in pigs: endometrial gene expression in response to estrogens and interferons from conceptuses. Soc Reprod Fertil Suppl. 2009;66:321–332. [PubMed] [Google Scholar]
- 136.Salem S, Gao C, Li A, Wang H, Nguyen-Yamamoto L, Goltzman D, Henderson JE, Gros P. A novel role for interferon regulatory factor 1 (IRF1) in regulation of bone metabolism. J Cell Mol Med. 2014 doi: 10.1111/jcmm.12327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lee SH, Kim JW, Lee HW, Cho YS, Oh SH, Kim YJ, Jung CH, Zhang W, Lee JH. Interferon regulatory factor-1 (IRF-1) is a mediator for interferon-gamma induced attenuation of telomerase activity and human telomerase reverse transcriptase (hTERT) expression. Oncogene. 2003;22:381–391. doi: 10.1038/sj.onc.1206133. [DOI] [PubMed] [Google Scholar]
- 138.Sferruzzi-Perri AN, Macpherson AM, Roberts CT, Robertson SA. Csf2 null mutation alters placental gene expression and trophoblast glycogen cell and giant cell abundance in mice. Biol Reprod. 2009;81:207–221. doi: 10.1095/biolreprod.108.073312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Nilsson R, Schultz IJ, Pierce EL, Soltis KA, Naranuntarat A, Ward DM, Baughman JM, Paradkar PN, Kingsley PD, Culotta VC, et al. Discovery of genes essential for heme biosynthesis through large-scale gene expression analysis. Cell Metab. 2009;10:119–130. doi: 10.1016/j.cmet.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lin H, Mosmann TR, Guilbert L, Tuntipopipat S, Wegmann TG. Synthesis of T helper 2-type cytokines at the maternalfetal interface. J Immunol. 1993;151:4562–4573. [PubMed] [Google Scholar]
- 141.Chatterjee P, Kopriva SE, Chiasson VL, Young KJ, Tobin RP, Newell-Rogers K, Mitchell BM. Interleukin-4 deficiency induces mild preeclampsia in mice. J Hypertens. 2013;31:1414–1423. doi: 10.1097/HJH.0b013e328360ae6c. [DOI] [PubMed] [Google Scholar]
- 142.Piccinni MP, Beloni L, Livi C, Maggi E, Scarselli G, Romagnani S. Defective production of both leukemia inhibitory factor and type 2 T-helper cytokines by decidual T cells in unexplained recurrent abortions. Nat Med. 1998;4:1020–1024. doi: 10.1038/2006. [DOI] [PubMed] [Google Scholar]
- 143.Piccinni MP, Scaletti C, Vultaggio A, Maggi E, Romagnani S. Defective production of LIF, M-CSF and Th2-type cytokines by T cells at fetomaternal interface is associated with pregnancy loss. J Reprod Immunol. 2001;52:35–43. doi: 10.1016/s0165-0378(01)00111-5. [DOI] [PubMed] [Google Scholar]
- 144.Michimata T, Sakai M, Miyazaki S, Ogasawara MS, Suzumori K, Aoki K, Nagata K, Saito S. Decrease of T-helper 2 and Tcytotoxic 2 cells at implantation sites occurs in unexplained recurrent spontaneous abortion with normal chromosomal content. Hum Reprod. 2003;18:1523–1528. doi: 10.1093/humrep/deg280. [DOI] [PubMed] [Google Scholar]
- 145.Luo XJ, Li M, Huang L, Nho K, Deng M, Chen Q, Weinberger DR, Vasquez AA, Rijpkema M, Mattay VS, et al. The interleukin 3 gene (IL3) contributes to human brain volume variation by regulating proliferation and survival of neural progenitors. PLoS ONE. 2012;7:e50375. doi: 10.1371/journal.pone.0050375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Omasu F, Ezura Y, Kajita M, Ishida R, Kodaira M, Yoshida H, Suzuki T, Hosoi T, Inoue S, Shiraki M, et al. Association of genetic variation of the RIL gene, encoding a PDZ-LIM domain protein and localized in 5q31.1, with low bone mineral density in adult Japanese women. J Hum Genet. 2003;48:342–345. doi: 10.1007/s10038-003-0035-1. [DOI] [PubMed] [Google Scholar]
- 147.Shekhawat PS, Srinivas SR, Matern D, Bennett MJ, Boriack R, George V, Xu H, Prasad PD, Roon P, Ganapathy V. Spontaneous development of intestinal and colonic atrophy and inflammation in the carnitine-deficient jvs (OCTN2(S/S)) mice. Mol Genet Metab. 2007;92:315–324. doi: 10.1016/j.ymgme.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kee HJ, Koh JT, Yang SY, Lee ZH, Baik YH, Kim KK. A novel murine long-chain acyl-CoA synthetase expressed in brain participates in neuronal cell proliferation. Biochem Biophys Res Commun. 2003;305:925–933. doi: 10.1016/s0006-291x(03)00859-3. [DOI] [PubMed] [Google Scholar]
- 149.Soupene E, Kuypers FA. Mammalian long-chain acyl-CoA synthetases. Exp Biol Med (Maywood) 2008;233:507–521. doi: 10.3181/0710-MR-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tomoda H, Igarashi K, Cyong JC, Omura S. Evidence for an essential role of long chain acyl-CoA synthetase in animal cell proliferation. Inhibition of long chain acyl-CoA synthetase by triacsins caused inhibition of Raji cell proliferation. J Biol Chem. 1991;266:4214–4219. [PubMed] [Google Scholar]
- 151.Hu Y, Lu Y, Zhou Z, Du Y, Xing J, Wang L, Lin M, Sha J. Defective expression of Galpha12 in the testes of azoospermia patients and in the spermatozoa with low motility. J Mol Med (Berl) 2006;84:416–424. doi: 10.1007/s00109-005-0028-y. [DOI] [PubMed] [Google Scholar]
- 152.Zweers MC, van Vlijmen-Willems IM, van Kuppevelt TH, Mecham RP, Steijlen PM, Bristow J, Schalkwijk J. Deficiency of tenascin-X causes abnormalities in dermal elastic fiber morphology. J Invest Dermatol. 2004;122:885–891. doi: 10.1111/j.0022-202X.2004.22401.x. [DOI] [PubMed] [Google Scholar]
- 153.Andree B, Hillemann T, Kessler-Icekson G, Schmitt-John T, Jockusch H, Arnold HH, Brand T. Isolation and characterization of the novel popeye gene family expressed in skeletal muscle and heart. Dev Biol. 2000;223:371–382. doi: 10.1006/dbio.2000.9751. [DOI] [PubMed] [Google Scholar]
- 154.Xu K, Chong DC, Rankin SA, Zorn AM, Cleaver O. Rasip1 is required for endothelial cell motility, angiogenesis and vessel formation. Dev Biol. 2009;329:269–279. doi: 10.1016/j.ydbio.2009.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hoashi T, Watabe H, Muller J, Yamaguchi Y, Vieira WD, Hearing VJ. MART-1 is required for the function of the melanosomal matrix protein PMEL17/GP100 and the maturation of melanosomes. J Biol Chem. 2005;280:14006–14016. doi: 10.1074/jbc.M413692200. [DOI] [PubMed] [Google Scholar]
- 156.van Vliet-Ostaptchouk JV, den Hoed M, Luan J, Zhao JH, Ong KK, van der Most PJ, Wong A, Hardy R, Kuh D, van der Klauw MM, et al. Pleiotropic effects of obesity-susceptibility loci on metabolic traits: a meta-analysis of up to 37,874 individuals. Diabetologia. 2013;56:2134–2146. doi: 10.1007/s00125-013-2985-y. [DOI] [PubMed] [Google Scholar]
- 157.Hashiya N, Jo N, Aoki M, Matsumoto K, Nakamura T, Sato Y, Ogata N, Ogihara T, Kaneda Y, Morishita R. In vivo evidence of angiogenesis induced by transcription factor Ets-1: Ets-1 is located upstream of angiogenesis cascade. Circulation. 2004;109:3035–3041. doi: 10.1161/01.CIR.0000130643.41587.DB. [DOI] [PubMed] [Google Scholar]
- 158.Michaelis SA, Okuducu AF, Sarioglu NM, von Deimling A, Dudenhausen JW. The transcription factor Ets-1 is expressed in human amniochorionic membranes and is up-regulated in term and preterm premature rupture of membranes. J Perinat Med. 2005;33:314–319. doi: 10.1515/JPM.2005.056. [DOI] [PubMed] [Google Scholar]
- 159.Kessler CA, Schroeder JK, Brar AK, Handwerger S. Transcription factor ETS1 is critical for human uterine decidualization. Mol Hum Reprod. 2006;12:71–76. doi: 10.1093/molehr/gal008. [DOI] [PubMed] [Google Scholar]
- 160.Chan YC, Khanna S, Roy S, Sen CK. miR-200b targets Ets-1 and is down-regulated by hypoxia to induce angiogenic response of endothelial cells. J Biol Chem. 2011;286:2047–2056. doi: 10.1074/jbc.M110.158790. [DOI] [PMC free article] [PubMed] [Google Scholar]