


Abstract
The general structural fold of the LAGLIDADG endonuclease family consists of two similar α/β domains (αββαββα) that assemble either as homodimers or monomers with the domains related by pseudo-two-fold symmetry. At the center of this symmetry is the closely packed LAGLIDADG two-helix bundle that forms the main inter- or intra-molecular contact region between the domains of single- or double-motif proteins, respectively. In this work, we further examine the role of the LAGLIDADG residues involved in the helix–helix interaction. The interchangeability of the LAGLIDADG helix interaction was explored by grafting interfacial residues from the homodimeric I-CreI into the corresponding positions in the monomeric I-DmoI. The resulting LAGLIDADG exchange mutant is partially active, preferring to nick dsDNA rather than making the customary double-strand break. A series of partial revertants within the mutated LAGLIDADG region are shown to restore cleavage activity to varying degrees resulting in one I-DmoI mutant that is more active than wild-type I-DmoI. The phenotype of some of these mutants was reconciled on the basis of similarity to the GxxxG helix interaction found in transmembrane proteins. Additionally, a split variant of I-DmoI was created, demonstrating that the LAGLIDADG helices of I-DmoI are capable of forming and maintaining the protein–protein interface in trans to create an active heterodimer.
INTRODUCTION
The LAGLIDADG family of proteins, which includes more than 200 members, is defined by the presence of one or two copies of the consensus LAGLIDADG sequence (1,2). Originally characterized by primary sequence alignments, each conserved motif (Fig. 1, termed P1 and P2) is followed by ∼100 amino acids that share little or no sequence similarity (3,4). Some members of this family have only a single LAGLIDADG (P1) motif. Structural studies elucidated the role of the conserved residues in the context of both the structure and function of endonuclease members of this family [Fig. 2A; reviewed in (5)].
Figure 1.
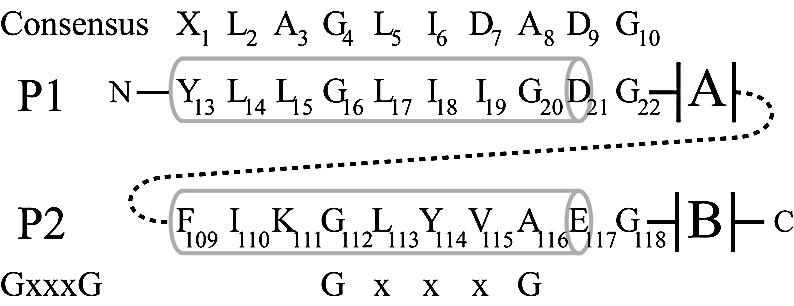
Schematic alignment of the LAGLIDADG residues of I-DmoI. Pertinent residues in P1 and P2 are identified below the consensus LAGLIDADG sequence. The alignment of the GxxxG motif is shown on the bottom. Numbers in the consensus sequences represent generic positions within the LAGLIDADG motif. Numbers in P1/P2 represent positions of residues in I-DmoI. Gray barrels indicate helical structure. A and B designate the two α/β domains that form the bulk of the protein. Dashed line represents the short (six to eight amino acids) linker between the subdomains A and B in I-DmoI.
Figure 2.

Structure of I-DmoI and the LAGLIDADG motif. (A) Overall fold of I-DmoI. Left, top view down the two-helix bundle axis. Right, side view perpendicular to bundle axis. White spheres represent the van der Waals radii of the interfacial LAGLIDADG residues highlighting the majority of subdomain contacts between helices α1 and α4. Linker residues 98–105 are shown in red. (B) The α1/α4 helices (P1 and P2 regions) of I-DmoI with the XLAGLIDADG residues labeled. Black type, residues above the plane of the page; gray type, residues below the plane of the page. A dashed horizontal line demarcates the conserved sequence. At the right is the superposition of the LAGLIDADG helices of four known structures (I-DmoI, I-CreI, PI-SceI and PI-PfuI) demonstrating the extent of structural similarity in naturally occurring proteins. Although E-DreI, I-MsoI and I-AniI display a similar overlap, for clarity they were omitted from the superposition. (C) Creating I-DmoICL. The consensus sequence and generic numbering are shown on the left. The interfacial P1/P2 regions of I-DmoI were replaced by the P1/P1′ of I-CreI to generate I-DmoICL; bold type, residues changed in the LAGLIDADG of I-DmoI; gray type, residues facing the protein core and thus not changed to their I-CreI counterparts; italics, the GxxxG motif residues within the LAGLIDADG helices. Revertants are indicated to the left and right of the helices. See Figure 4 and the text for details.
For single-motif LAGLIDADG family members, such as I-CreI, an α/β domain represents a single subunit of a homodimeric enzyme (6,7). In contrast, for double-motif members such as I-DmoI, two such domains within a single polypeptide form a pseudo-symmetric enzyme monomer (8–12). The domain exhibits a characteristic αββαββα fold that contains residues necessary to bind DNA and constitutes a unit of interaction for one half of the DNA target site. The LAGLIDADG sequence occurs within the N-terminal α-helix in each α/β domain and provides essential residues involved in both helix packing at the domain or subdomain interface and formation of the catalytic center. The LAGLIDADG two-helix bundle is reminiscent of the tightly packed GxxxG motif helix–helix interaction (where alanine may substitute for glycine as GxxxA or AxxxA) found in the transmembrane (TM) region of integral membrane proteins (13,14). The two LAGLIDADG α-helices at the interface juxtapose the C-terminal acidic residues (in bold type) in each helix within 3–6 Å to create the active site at the center of the two-fold or pseudo-two-fold symmetry (15–17). In the case of the single-motif proteins, this interaction assembles a homodimer capable of recognizing pseudo-palindromic homing sites. For double-motif proteins, this intramolecular pseudo-dimer allows for the recognition of more asymmetric DNA target sites.
Extensive studies involving sequence alignments (1,3), biochemical analyses (18,19) and structure determination (6,8) led to the idea that the double-motif LAGLIDADG proteins arose from a gene duplication event [reviewed in (20,21)]. The conserved nature of the LAGLIDADG motif is likely related to its direct involvement in both the active site and the protein–protein interface. As such, two questions arose: (i) if indeed the LAGLIDADG motif, being highly conserved at the amino acid level, forms a generalized dimerization interface, can two independent α/β domains from disparate proteins be fused together to form a functional endonuclease, and (ii) given the tethered nature of the double-motif proteins, have LAGLIDADG regions in the α/β domains co-evolved so that they can still function independently of one another? That is, could the double-motif proteins be split to form either functional heterodimers or novel homodimers without altering the existing LAGLIDADG motif?
The concept that the α/β domains of LAGLIDADG proteins are interchangeable was explored by Chevalier et al. (22) using computational analysis and by Epinat et al. (23) using structure modeling. The goal was to create an artificial, highly sequence-specific LAGLIDADG endonuclease by fusing the N-terminal domain of I-DmoI to the I-CreI monomer. In both cases, the result was a functional hybrid endonuclease capable of specifically cleaving a hybrid DNA sequence derived from one half of each of the respective native DNA targets.
This study focuses on further exploring the interactions of the LAGLIDADG two-helix bundle. The flexibility of the LAGLIDADG helix interface of I-DmoI was probed through directed mutagenesis guided by protein structure. I-DmoI mutants that contain the I-CreI LAGLIDADG helix interface are shown to be active to various extents. Computational analysis of the LAGLIDADG motif in the context of a GxxxG two-helix bundle was used to rationalize some of the mutant phenotypes. I-DmoI was also used to address the possibility of separating the functional domains of a double-motif LAGLIDADG protein. Generating active split variants of I-DmoI allowed us to investigate the functional importance of this conserved helix–helix interaction and gain new insight into the engineering and evolution of this family of proteins.
MATERIALS AND METHODS
Structure modeling and analysis
The structure coordinates of native I-DmoI and a high-resolution co-crystal of I-CreI were used to model the altered LAGLIDADG interface (PDB entries 1b24 and 1g9z, respectively). Key interfacial residues were mutated using the structurally determined rotamer positions where possible. The resulting model was analyzed for steric clashes, hydrogen bonding and van der Waals interactions at the preformed helix interface. Model manipulation and determination of bond angles and distances was carried out using the program O (24).
LAGLIDADG mutagenesis
Primers incorporating the desired residue changes were designed to amplify the separate LAGLIDADG regions of I-DmoI and subsequently combined by gene SOEing [splicing by overlap extension (25)]. The mutant construct was assembled from three separate PCRs that create cassettes of coding region that could be spliced together. The primers used for each cassette were as follows: (i) W1434, 5′-GCTCACTCATTAGGCACCC-3′, W1635, 5′-CACCATCTATGAATC-CAGCCAGGTAAGCAGATATTCCAC-3′; (ii) W1636, 5′-CTGGCTGGATTCATAGATGGTGATGGAGGACTTTACAAG-3′, W1637, 5′-CCATCATAGAACCCCGCTATGTACGCTATTTGCTCACGC-3′; and (iii) W1638, 5′-ACATA GCGGGGTTCTATGATGGTGAAGGAGATAAAACCC-3′, W1430, 5′-GCGATTAAGTTGGGTAACGC-3′. This initial construct, termed I-DmoICL (I-DmoI with the I-CreI LAGLIDADG interface), was then used as the target for further mutagenesis by similar methods to create the remaining constructs. The final products were confirmed by automated DNA sequencing over the entire coding region.
Computational analysis
A dataset of double-motif LAGLIDADG proteins was derived from Dalgaard et al. (1), Protein BLAST (NCBI) and Pfam (26). Protein BLAST searches were performed with the residue sequence XLAGLIDADG using the ‘Search for short nearly exact matches’ option. The extra N-terminal residue indicated as X (generally F, Y or W in most alignments) denotes the first structural consensus interfacial residue between the LAGLIDADG helices. References to generic amino acid positions within LAGLIDADG are based on this sequence, where X denotes position 1, L denotes position 2 and so forth (see Table 1 and Fig. 1 for details). The following criteria were applied to filter the data set: (i) the motif must fit the pattern XXX[AGST][FILWY]XX[ADGST][DE][AG] where X is any single residue and brackets enclose a subset of specific residues for a single position; and (ii) between 75 and 200 amino acids must be present following the consensus motif. The resulting set of 131 unique putative LAGLIDADG endonuclease sequences was analyzed with custom Perl scripts.
Table 1. Double-motif LAGLIDADG sequence analysisa.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| X | L | A | G | L | I | D | A | D | G |
| | | | | ||||||||
| G | x | x | x | G | |||||
| X | X | X | A | F | X | X | A | D | A |
| G | I | D | E | G | |||||
| S | L | G | |||||||
| T | W | S | |||||||
| Y | T |
aAlignment of the LAGLIDADG and GxxxG motifs with the query pattern (bottom) used to produce a dataset of 131 unique putative double-motif LAGLIDADG proteins representing a total of 262 helices.
Domain constructs
A total of eight single-domain constructs were created from the wild-type I-DmoI plasmid (27) using the GeneAmp XL PCR kit (Perkin-Elmer). Four different domain A constructs were designed, terminating at residue 98, 100, 102 or 104 (designated N098–N104, respectively) along with four domain B constructs, starting at residue numbers 99, 101, 103 or 105 (designated C099–C105, respectively). These individual domain constructs were then used as starting points for creating tandem expression clones. Each domain A plasmid (N098–N104) was digested with appropriate restriction enzymes for insertion of a domain B expression cassette, one per domain B construct (C099–C105). This produced a total of 16 tandem expression vectors termed I-DmoIA/B, where A/B signifies the domain construct numbers (98/99, 98/101, 98/103, 98/105, 100/99, 100/101, 100/103, 100/105, 102/99, 102/101, 102/103, 102/105, 104/99, 104/101, 104/103, 104/105). The final products were confirmed by automated DNA sequencing over the entire coding region.
Affinity tagged constructs
Constructs were N-terminally and/or C-terminally tagged with the FLAG (Sigma) and/or c-myc epitope, respectively, by direct PCR or the gene SOEing method. For FLAG-tagged constructs, a primer encoding the epitope (DYKDDDDK) was used to insert the tag following the first two residues (MH) of the protein. For c-myc-tagged constructs, a primer encoding the epitope (EQKLISEEDL) was used to insert the tag prior to the stop codon.
Overexpression and partial purification
All constructs were transformed into the expression strain B834(DE3)F′lacIq and protein overexpression was carried out as described previously (28). A modified protocol was used in which the final phenyl-Sepharose column was skipped and purification was carried out both with and without ethylene glycol in the buffer formulations. The final purity of the samples ranged from 30 to 70%, depending on the construct. Protein concentrations were determined using a modified Lowry assay (DC Protein Assay, Bio-Rad). For comparative activity assays, relative protein concentrations were derived by scanning and quantitating both Coomassie Blue- and SYPRO Ruby (Molecular Probes)-stained SDS–PAGE gels.
Activity assay and cleavage-site mapping
Cleavage activity was assayed by incubating 200 ng of pUD718 plasmid DNA (2.7 kb) with partially purified protein extracts (normalized to 10 ng of I-DmoI; a 4:1 molar ratio of protein:DNA) in cleavage buffer for 15 min at 65°C (28). Reactions were quenched with 5 µl stop-load buffer and products were analyzed by agarose gel electrophoresis, stained with SYBR Gold (Molecular Probes) then visualized and quantitated using a FluorImager and ImageQuant software (Molecular Dynamics). For cleavage-site mapping, the dsDNA target was a 171 bp PCR fragment amplified from pUD718 with primers W1461, 5′-TCCCGCGAAATTAATACGAC-3′, and W1430 (see LAGLIDADG mutagenesis) that were alternately 32P-end-labeled to visualize top and bottom strand cleavage, respectively. Products were separated on a 6% polyacrylamide–urea gel alongside their corresponding DNA sequencing reactions (USB Thermo Sequenase Cycle Sequencing kit) to map cleavage on the top (W1461) and bottom (W1430) strands.
DNA binding assay
A 38 bp dsDNA fragment was generated by annealing 32P-end-labeled homing-site oligonucleotides W68, 5′-AATTAAATGCCTTGCCGGGTAAGTTCCGGCGCGCATGA-3′, and W69, 5′-AATTTCATGCGCGCCGGAACTTACCCGGCAAGGCATTT-3′. Reactions were allowed to proceed for 5 min at 40°C in I-DmoI band-shift buffer (50 mM HEPES–KOH pH 8.0, 100 mM KCl, 10 mM MgCl2, 1 mM DTT, 0.05% Triton X-100, 5% glycerol). Samples were then loaded onto an 8% (w/v) polyacrylamide (29:1) non-denaturing gel and electrophoresed at 25 mA for 1 h at 4°C. Results were analyzed using a PhosphorImager and ImageQuant software (Molecular Dynamics).
Immunoprecipitation and western blots
Cells expressing tagged protein were resuspended in lysis buffer (50 mM Tris–HCl pH 7.6, 150 mM NaCl, 1 mM EDTA, 500 µM Pefabloc, 2 µg/ml leupeptin, 5 µg/ml aprotinin), disrupted by sonication then centrifuged for 15 min at 11 000 g. Cleared lysates were immunoprecipitated with appropriate antibody for 1 h at 4°C with gentle rocking, then supplemented with BioMag Protein G (Qiagen) magnetic beads (5 µg beads per 1 µg antibody) and incubated for an additional hour at 4°C. The magnetic beads were washed twice, resuspended in 40 µl of SDS loading buffer and heated for 5 min at 95°C. Equal amounts of the samples were loaded in duplicate on a single polyacrylamide gel to allow differential western blotting. Samples were separated using 15% PAGE, transferred to PVDF membranes and probed with anti-FLAG M2 (Sigma), anti-c-Myc (Sigma) or rabbit polyclonal antibodies against wild-type I-DmoI. Detection was performed using the ECL kit (Amersham) as per the manufacturer’s instructions.
RESULTS
Grafting the LAGLIDADG helix interface: I-DmoICL
The linear arrangement and detailed interaction of the LAGLIDADG helices (P1 and P2) of I-DmoI (corresponding to α1 and α4) are shown in Figures 1 and 2, respectively. The interface between the α/β subdomains is formed mainly by the α1/α4 LAGLIDADG two-helix bundle (Fig. 2A) (8). The residues involved in this interaction are shown in Figure 2B, with a least-squares fit of the P1/P2 helices for four known LAGLIDADG structures (I-DmoI, I-CreI, PI-SceI and PI-PfuI) (6,8–10) on the right. The region above the dashed horizontal line represents the conserved LAGLIDADG motif, which displays a near-perfect match for backbone and side-chain positioning. Below this line the N-terminal region, although helical in structure, diverges considerably in both residue composition and orientation.
We used the superposition of I-DmoI with I-CreI to assess the viability of grafting the LAGLIDADG helix interface from I-CreI onto I-DmoI. In order to retain the α/β domain core fold, only interfacial helix residues were analyzed (Fig. 2A and C). A construct was created in which the interfacial LAGLIDADG helix residues were mutated from those of I-DmoI to those of I-CreI. This LAGLIDADG exchange mutant, I-DmoICL, contains the following eight amino acid changes: L15A, L17F, I19D, F109Y, K111A, L113F, V115D and A116G (refer to Fig. 2C). The protein was soluble when overexpressed and displayed a similar elution profile to that of wild-type I-DmoI when partially purified on a heparin column. Figure 3A shows a representative DNA cleavage assay comparing the activity of wild-type I-DmoI with I-DmoICL. Because wild-type I-DmoI cleaves closed-circular more readily than linear substrate DNA by at least 2-fold (G. H. Silva, unpublished results), the closed-circular pUD718 plasmid was used as a substrate (28). For wild-type I-DmoI, a nicked, open-circle intermediate appeared immediately, corresponding to ∼20% of the products throughout the reaction (Fig. 3B, top). After ∼4 min, this intermediate began to disappear as more linear product was formed. A similar initial cleavage profile was seen for I-DmoICL. However, the conversion from nicked open-circular to linear DNA did not progress beyond 4 min (Fig. 3B, bottom). At the final time-point, wild-type I-DmoI had converted 76% of the substrate (63% linear/13% nicked) to products, while I-DmoICL had converted only 15% (10% linear/5% nicked).
Figure 3.
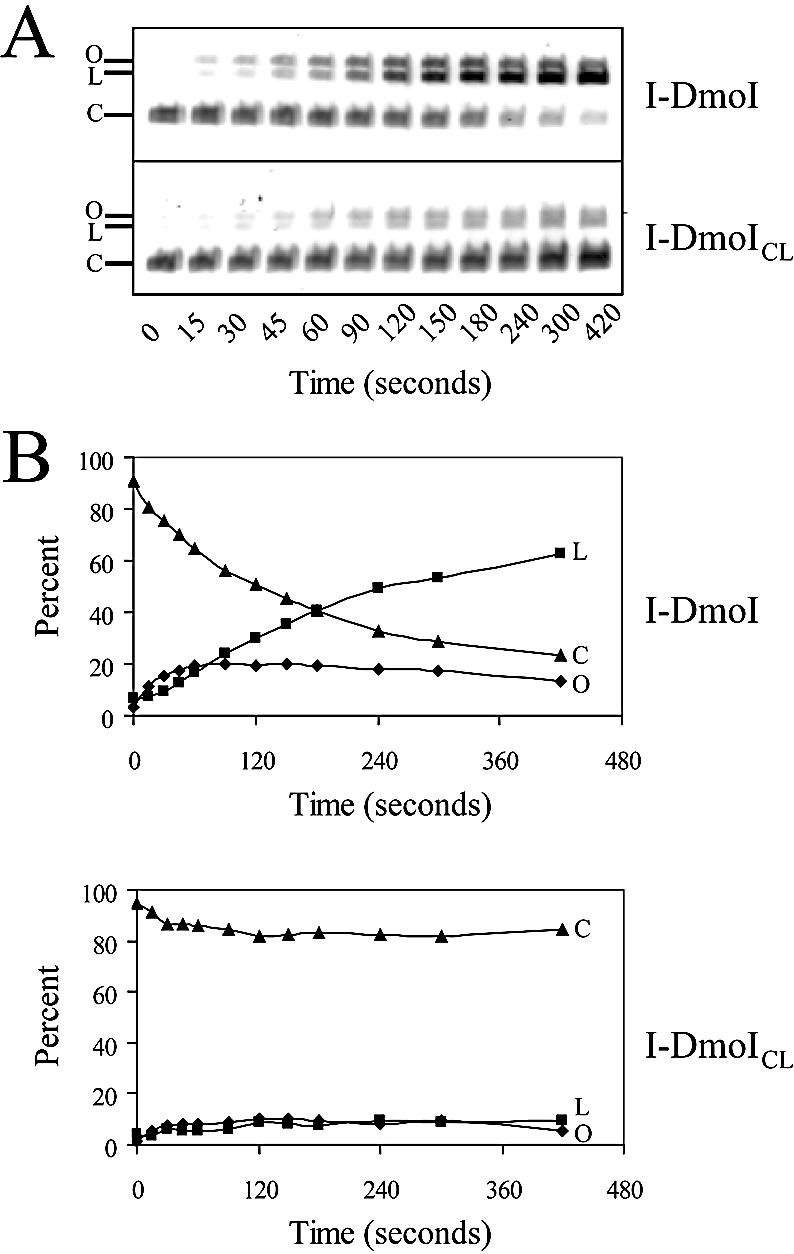
Cleavage activity of I-DmoI and I-DmoICL. (A) Typical time-course activity assay. The 7 min assay was performed with pUD718 plasmid dsDNA and I-DmoI (top) or I-DmoICL (bottom); 20 µl samples were removed at the times indicated. Closed-circular plasmid DNA (C) was cleaved forming a linear product (L); nicked, open circles (O) appeared as intermediates of the reaction. Residual nuclease activity was ruled out by incubating each construct with target plasmid lacking only the 14 bp recognition sequence. No nicking or cleavage activity was observed (G. H. Silva, unpublished results). (B) Plots of data in (A). Percent reaction products are shown over time. Diamonds, squares and triangles correspond to O, L and C, respectively, in (A).
Given that I-DmoICL has the LAGLIDADG helix interface of I-CreI, the decreased activity was originally attributed to improper active site residues (i.e. I-CreI has two Asp residues at position 20 and 20′ whereas I-DmoI has Asp21 and Glu117 at the catalytic center). To test this hypothesis, two additional constructs, I-DmoICLD and I-DmoICLE, were created from I-DmoICL by mutating either Glu117 to Asp or Asp21 to Glu, respectively. I-DmoICLD emulates the I-CreI active site by placing two Asp residues in the catalytic center, whereas I-DmoICLE serves as a control with two Glu residues in the active site. A third construct, I-DmoICLED, was also created wherein the identities of the catalytic residues in I-DmoICL are exchanged; i.e. having Glu21 and Asp117. In the standard cleavage assay, I-DmoICLD displayed only a slight (∼5%) increase in total DNA cleavage compared with I-DmoICL, whereas neither I-DmoICLE nor I-DmoICLED exhibited any detectable cleavage activity (data not shown). This was the case even when Mn2+ was used as a cofactor, which has been reported to enhance the activity of I-DmoI by ∼20% (29).
Restoring the activity of I-DmoICL
We next reasoned that the loss in I-DmoICL activity may involve mutations made in the helical scaffold near the active site that would perturb the catalytic center. The active site of LAGLIDADG endonucleases has been shown to require an ordered array of water molecules at the protein/DNA interface (16,17). This hydration shell appears to be critical for cleavage activity and is the basis for the apparent diversity in active site residues other than the LAGLIDADG consensus C-terminal acidic residues (bold). In I-DmoICL, disruption of the hydration shell can arise from changes in the surface potential proximal to the active site (e.g. I19D and V115D mutations, see Fig. 2) as well as decreased helical stability for placement of catalytic residues (e.g. A116G) (30,31). Using I-DmoICL as a base construct, partial revertants were created for the three suspected residues (D19, D115 and G116) either individually or in all possible combinations. Figure 2C highlights the modified residues of the I-DmoICL LAGLIDADG helix interface with amino acid changes indicated.
All constructs were overexpressed, partially purified and normalized for relative I-DmoI protein concentration. Assays were performed as in Figure 3 and the ratio of linear to closed-circular DNA was plotted (Fig. 4A) to determine relative cleavage activity. Figure 4B lists the results of the relative cleavage activity, as determined from the slope of the best fit line to the data in Figure 4A, for wild-type I-DmoI, I-DmoICL and all CL amino acid revertants. For comparison, cleavage activity is expressed relative to wild-type I-DmoI (100%), where I-DmoICL is 16% active. Taken individually both G116A and D115V improved relative cleavage to 51 and 58%, respectively, while the D19I mutation had a deleterious effect, with a relative cleavage of 11%. These effects are additive. Constructs with G116A and D115V together have a corresponding increase in activity to 118%, whereas those with D19I have a proportional decrease. This CLG116A, D115V mutant is hereafter termed I-DmoIH (hybrid-vigor). The deleterious effect caused by the D19I reversion is apparent in all combinations: down from 51 to 32% with CLG116A; from 58 to 22% with CLD115V; and from 118 to 74% with CLG116A, D115V.
Figure 4.

Relative cleavage activity of the I-DmoI constructs. (A) Time-course assays. Activity assays were performed in triplicate (average deviation <5%) as noted in Figure 3 for each construct listed below the plots. Percent cleavage reflects the ratio of linear to closed-circular dsDNA. (B) Relative cleavage activity. 1Cleavage activity on circular target, resulting in linear and nicked dsDNA; 2relative cleavage to wild-type I-DmoI based on the slope of the best fit line to each curve in (A); 3CL is I-DmoICL base construct with residue changes as indicated (see text for details); 4CLG116A, D115V is termed I-DmoIH in later experiments; 5I-DmoIS corresponds to split construct I-DmoI102/101
Computational analysis of the LAGLIDADG residue interaction
To better understand the phenotype of the LAGLIDADG mutants, a computational analysis was undertaken. Our original dataset, which included over 300 predicted LAGLIDADG proteins, was reduced to 131 unique helix–helix interactions by applying a filtering algorithm designed to retain only double-motif proteins with potential endonuclease activity. The stringency of the search precluded capturing outliers, e.g. I-SceI, a known endonuclease with non-canonical LAGLIDADG sequences. The criteria used to generate the filter were derived from what is currently known about the structure and function of LAGLIDADG endonucleases as well as data relating to helix packing (1,8,14,32). Specifically, the algorithm preserved the subset of residues known to be involved in helix–helix interactions as well as the key acidic residues that form the active site.
Tables 1–3 summarize the results of the residue analysis. We examined the refined dataset of 131 unique sequences in terms of helix packing as GxxxG bundles [Table 1; (14,32,33)]. Gly/Ala (G/A) pairs represent backbone interactions characteristic of the GxxxG helix interface (Table 2). The G/A pairs are listed in P1↔P2 order and are divided into N- versus C-terminal residue positions 4 and 8, respectively, based on the XLAGLIDADG numbering scheme (Table 1). Allowed residues in the alignment are G, A, S and T at position 4 and G, A, S, T and D at position 8; thus creating 16 and 25 possible interacting pairs, respectively, for these positions in independent P1 and P2 helices. Table 2 lists statistics for the residue pairs that occurred as both P11↔P22 and P12↔P21, where subscript numbers denote the two different amino acids. The data indicate a clear difference for residue pairs observed in the two regions of the LAGLIDADG motif. The N-terminal region (position 4) is dominated by G–G interactions (80.9% observed, close to the 77.7% expected), which allow for tighter packing of the helix backbones.
Table 3. Relationship of intrahelical residues at XLAGLIDADG positions 8 and 9.
| Catalytic sequence (positions 8/9) | Counta | Exp (%) | Obs (%) |
|---|---|---|---|
| -AD- | 50 | 24.6 | 19.1 |
| -AE- | 48 | 12.8 | 18.3 |
| -GD- | 97 | 29.9 | 37.0 |
| -GE- | 22 | 15.6 | 8.4 |
| -SD- | 16 | 7.8 | 6.1 |
| -SE- | 15 | 4.1 | 5.7 |
| Otherb | 14 | 5.2 | 5.4 |
| χ2 = 25.3 (13.8, p = 0.001) | |||
aTotal of 262 independent LAGLIDADG helices (two per 131 unique double-motif proteins).
bCatalytic sequences in which position 8 is not A, G or S as in Table 2.
Table 2. Interhelical residue pairings at XLAGLIDADG positions 4 and 8.
| G/A pairsa | Position 4 (N-terminal) | Position 8 (C-terminal) | ||||
|---|---|---|---|---|---|---|
| P1↔P2 | Countb | Expc(%) | Obsd(%) | Count | Exp (%) | Obs (%) |
| A↔A | 1 | 0.6 | 0.8 | 7 | 12.7 | 5.3 |
| A↔G | 5 | 5.4 | 3.8 | 23 | 7.9 | 17.6 |
| G↔A | 9 | 8.1 | 6.9 | 52 | 29.5 | 39.7 |
| G↔G | 106 | 77.7 | 80.9 | 7 | 18.4 | 5.3 |
| G↔S | 1 | 0.7 | 0.8 | 15 | 7.8 | 11.5 |
| S↔G | 2 | 2.0 | 1.5 | 10 | 3.3 | 7.6 |
| Othere | 7 | 5.5 | 5.3 | 17 | 20.4 | 13.0 |
| χ2 = 0.59 (10.8, p = 0.001) | χ2 = 37.4 (10.8, p = 0.001) | |||||
aInterhelical residue pairings corresponding to the GxxxG motif; P1 and P2 data analyzed separately.
bTotal of 131 LAGLIDADG two-helix bundles.
cExpected frequency (%) derived from the product of the probability for each residue at the given position.
dObserved frequency (%).
eResidue pairing outliers that did not occur as both P11↔P22 and P12↔P21; for position 4, the following combinations were omitted: A with S or T, G with T, S with S or T, and T with T; for position 8, this included the position 4 omissions along with any combination having a D; see text for details.
Conversely, the C-terminal region (position 8) displays a significant (χ2 = 37.4) preference for pairing a G with a small, non-glycine residue, predominantly A and S (compare combined P11/2↔P22/1 totals for G with A and S of 76.4% observed versus 48.5% expected). The G–G interaction is 3.5-fold less prevalent than expected at this position (compare 18.4% observed with 5.3% expected). This suggests a preference for a somewhat less compact helix–helix interaction, most likely related to proper spacing of the ensuing acidic residues forming the active site.
Therefore we asked whether this discrepancy in G/A pairings at position 8 (Table 2, for residues A, G and S) could be related to the identity of the acidic residue. Table 3 shows that whereas both A and S precede D or E with approximately equal frequency (-A[D/E]- 19.1 versus 18.3% and -S[D/E]- 6.1 versus 5.7% observed), G is more than four times more likely to precede D (37.0%) than E (8.4%). Apart from the position 8/9 combination (χ2 = 25.3), no other residue pairs within the LAGLIDADG motif appear significant using chi-squared analysis (data not shown). Given the flexible nature of the glycine backbone, in particular within an α-helix (34,35), this nearest-neighbor effect can be viewed in terms of structural stability related to a crucial active site residue (30). This theoretical analysis helps explain the phenotype of some of the LAGLIDADG mutants (see Discussion).
Creating a functional heterodimer by tandem expression: I-DmoIS
Using the superposition of the four LAGLIDADG structures as a guide (Fig. 2B), cut sites were chosen in the subdomain linker of I-DmoI (Fig. 1, dashed line; Fig. 2A, marked in red) in order to create expression vectors for four independent A and four B subdomains (Fig. 5, path 1). After expression, cell lysates were systematically assayed either individually or in 16 combinations of all possible domain A (N098, N100, N102 and N104) and B (C099, C101, C103 and C105) pairs for both binding and cleavage of wild-type DNA substrate. In all cases, no activity was observed (data not shown).
Figure 5.

Creating a functional heterodimer. The wild-type I-DmoI plasmid was used as the template in efforts to create I-DmoIS via two pathways as shown. T7, promoter region; SD, Shine–Dalgarno sequence; ATG and TAA are the start and stop codons, respectively. Coding regions are indicated by arrows. (1) Domain constructs: individual A and B domain expression vectors were created. Protein expression was carried out separately for each domain and then lysates were combined and tested for heterodimer formation based on DNA cleavage activity. No active constructs were generated by this method. (2) Tandem constructs: individual A and B domain coding sequences were cloned into a single plasmid and co-expressed under the control of a single T7 promoter. Constructs were tested for heterodimer formation based on DNA cleavage activity as described in Table 4, Figure 6 and the text.
To overcome the potential problems in forming active I-DmoI heterodimers from the separate domains, a new expression system was developed (Fig. 5, path 2). The approach involved generating an operon on the plasmid, in which one promoter (T7) was used to express the two individual domains of the protein in tandem. Each coding region includes the upstream Shine–Dalgarno sequence as well as an initiation and termination codon. Additionally, to prevent read-through expression, the coding sequence of domain B was positioned out of frame with the coding sequence of domain A. A total of 16 tandem expression constructs were created from the initial four domain A (N098–N104) and four domain B (C099–C105) constructs and lysates expressed from clones were tested for DNA cleavage activity. The following constructs demonstrated the ability to specifically cleave the wild-type I-DmoI target DNA (Table 4): I-DmoI098/099, I-DmoI098/101, I-DmoI100/101, I-DmoI102/101, I-DmoI104/101 and I-DmoI102/103. Constructs that have domain B C101 displayed activity with all the A domain constructs, with the N102/C101 combination being most active. Therefore, I-DmoI102/101, renamed I-DmoIS (I-DmoI split), was selected for further analysis.
Table 4. Cleavage activity of I-DmoI split constructs.
| Domain B | Domain A | |||
|---|---|---|---|---|
| N098 | N100 | N102 | N104 | |
| C099 | + | – | – | – |
| C101 | ++ | +++ | ++++ | + |
| C103 | – | – | ++ | – |
| C105 | – | – | – | – |
The relative cleavage activity of the 16 tandem expression constructs was assessed in a standard cleavage assay (see Materials and Methods). Plus signs indicate the amount of dsDNA cleavage relative to other split constructs from – to ++++; minus signs indicate no detectable dsDNA cleavage and ++++ represents ∼5% of wild-type I-DmoI activity. The construct with the highest relative activity, I-DmoI102/101, was termed I-DmoIS and used in all subsequent experiments.
I-DmoIS was partially purified and the relative protein concentration was determined as before with additional corrections for the difference in molecular mass (A/B subunits are ∼11 kDa each versus full-length protein at 22 kDa). The relative cleavage activity, ∼5% of that of wild-type I-DmoI, was determined as for the I-DmoICL constructs (Fig. 4). The basis for the reduced activity of I-DmoIS was further explored in the dimerization experiments presented below.
I-DmoICL, I-DmoIH and I-DmoIS display wild-type cleavage specificity
A DNA mobility-shift assay was employed to determine whether the observed decrease in DNA cleavage of I-DmoICL and I-DmoIS or increase in DNA cleavage of I-DmoIH correlates with a decrease or increase, respectively, in DNA binding. As for PI-PfuI (36,37), it was determined that I-DmoI is only able to produce a DNA band-shift complex in the presence of Mg2+. All other divalent metal ions tested failed to produce a specific shifted complex (G. H. Silva, unpublished results). Even then, and as a consequence of partial cleavage activity in the presence of Mg2+, the maximum ratio of bound to unbound DNA never exceeded ∼35%. Nevertheless, the DNA mobility-shift analysis (see Supplementary Material) is in agreement with the observed DNA cleavage activity (Fig. 4B) for all constructs relative to wild type (taken as 100%): I-DmoICL, 5% bound, 16% cleaved; I-DmoIH, 110% bound, 118% cleaved; and I-DmoIS, 10% bound, 5% cleaved. DNA mobility-shift assays performed at 0, 40 and 65°C in the presence of Mg2+, and thus under cleavage conditions, did not indicate differences in the relative cleavage rates between wild-type and mutant constructs (data not shown). The correlation of DNA binding and cleavage activity suggests that the active sites of the test constructs are not compromised by the mutations, which exercise their effects primarily on DNA binding.
The cleavage sites on both top and bottom DNA strands were mapped for wild-type I-DmoI, I-DmoICL, I-DmoIH and I-DmoIS. As shown in Figure 6, all constructs cleave at the wild-type position, generating four-base, 3′-extended cohesive ends. Similar results were obtained from cleavage-site mapping performed with varying relative protein concentrations (0.05–0.5 µg) as well as in the presence of Mn2+ instead of Mg2+ (data not shown). The amount of strand cleavage from the mapping experiments, determined by taking the ratio of product to substrate DNA, was again consistent with the observed total DNA cleavage activity for all constructs, relative to wild-type: I-DmoICL, 20% cleaved; I-DmoIH, 125% cleaved; and I-DmoIS, 15% cleaved.
Figure 6.

Cleavage-site mapping of I-DmoICL, I-DmoIH and I-DmoIS. The cleavage positions (arrows) on the top (left) and bottom (right) strand were mapped for I-DmoI (lane 1), I-DmoICL (lane 2), I-DmoIH (lane 3) and I-DmoIS (lane 4). Lane 5, no-protein control. A, C, G and T indicate the base in the sequencing ladder; left and right, the sequence readout for the area of interest as marked; bottom, the I-DmoI homing site with the cleavage position indicated.
I-DmoIS is a heterodimer, not a read-through product
Before determining the extent of dimerization, it was important to confirm that the DNA cleavage activity of I-DmoIS was indeed the result of a functional heterodimer and not due to low-level read-through expression of a full-length protein. Two approaches were taken to address this issue. First, a new tandem expression construct was created in which the order of domain A (N102) and domain B (C101) expressed from the T7 promoter was switched. The new construct, named I-DmoISS (I-DmoI split, switched), was designed to test the influence of protein translation order since expression of the first protein can affect the expression and/or solubility of the second. Lysate prepared from cells expressing I-DmoISS was tested for activity in standard DNA cleavage assays. In comparison with I-DmoIS, a 2- to 5-fold decrease in overall protein expression was observed and the preparation was found to be proportionally 2- to 5-fold less active (data not shown). This corresponding decrease in activity indicated that the specific activity of I-DmoISS was similar to that of I-DmoIS. Together, these data suggest that the activity of the split constructs is unlikely to be attributable to translational read-through.
The second approach used to test for the separate I-DmoIS subdomains involved a western blot analysis of wild-type I-DmoI and I-DmoIS (Fig. 7A and B). Polyclonal anti-I-DmoI antibodies were produced in rabbits using purified wild-type I-DmoI (linear form) (28, and J. Z. Dalgaard and M. Belfort, unpublished results). Whereas the antibodies efficiently detected full-length I-DmoI as well as the individual subdomains (Fig. 7B, panel 1, middle two lanes), there was a complete absence of any 22 kDa band in I-DmoIS lysates despite the appearance of some cross-reactive proteins. These results, along with the data from the I-DmoISS construct, demonstrate that I-DmoIS is a split heterodimer.
Figure 7.
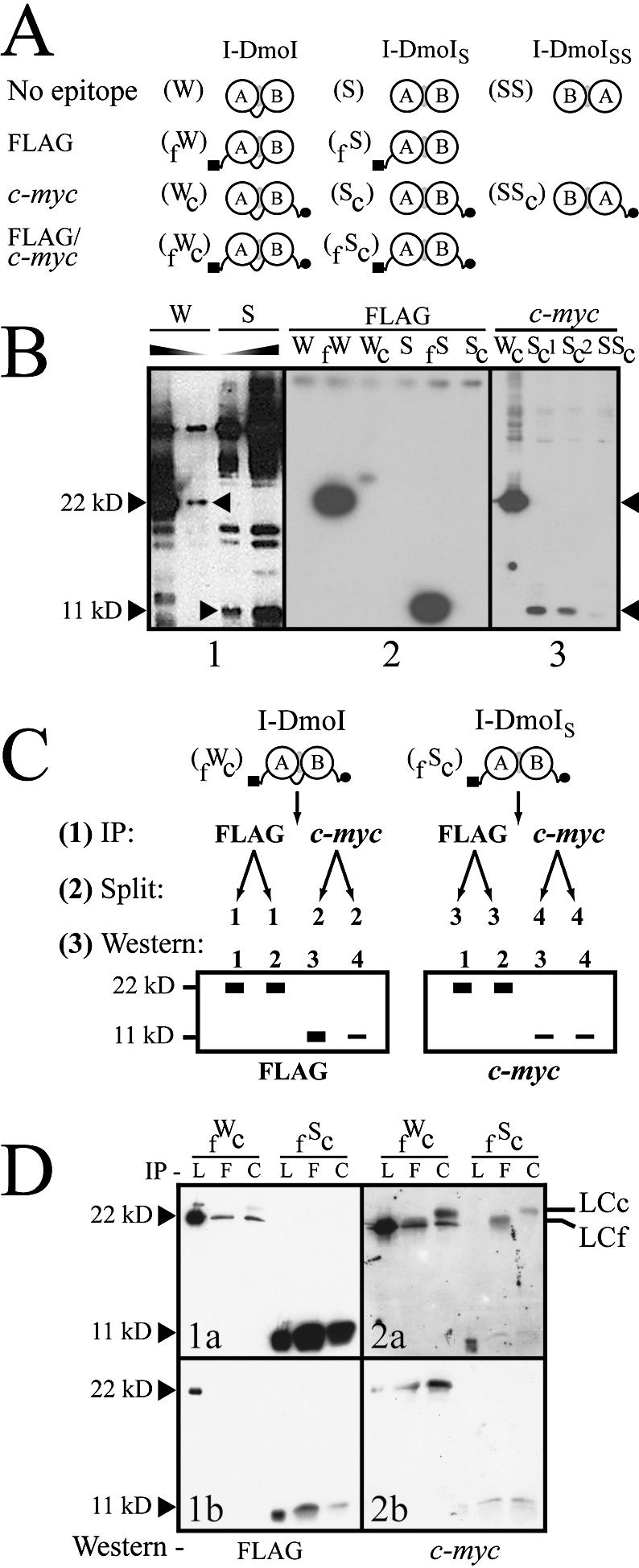
The extent of heterodimer formation in I-DmoIS. (A) Schematic of the I-DmoI constructs used to probe heterodimer formation. I-DmoI constructs were created with the N-terminal FLAG (black square) and/or C-terminal c-myc (black circle) epitopes. Abbreviations for construct names appear in parenthesis to the left of each diagram. (B) Western blots quantifying I-DmoI and I-DmoIS. For all panels, sizes indicate expected position of wild-type I-DmoI (22 kDa) and split constructs (11 kDa; I-DmoIS and I-DmoISS). Panel 1 shows that I-DmoIS does not contain detectable full-length protein using anti-I-DmoI polyclonal antibody. Shaded triangles indicate decreasing protein concentration. Panel 2 shows that no cross-reactivity is evident for untagged or c-myc-tagged I-DmoI and I-DmoIS using the anti-FLAG antibody (the shadow band between lanes fW and Wc is an exposure artifact). Panel 3 shows the dramatically reduced amount of domain B in protein lysates (total protein amounts loaded are as in panel 2) when c-myc-tagged I-DmoI, I-DmoIS and I-DmoISS proteins were probed with anti-c-Myc antibody. Sc1 and Sc2 represent crude and cleared lysates of Sc protein, respectively. (C) Schematic of IP/western blot protocol. This method was used to determine relative dimerization. See text for details. (D) Western blot of dimerization. Results from the protocol outlined in (C). Panels 1a and 1b show different exposures of the same experiment; Panels 2a and 2b show the results of two separate experiments. Size markers are as in (B). IP indicates antibody used for immunoprecipitation: L, lysate-only control (no IP); F, anti-FLAG antibody; and C, anti-c-Myc antibody. Western indicates antibody used for western blot. By-products of the IP step: LCc, light chain of the anti-c-Myc antibody; LCf, light chain of the anti-FLAG antibody.
Assessing the extent of heterodimer formation in I-DmoIS
Splitting I-DmoI into its subdomains places demands on the LAGLIDADG helical scaffold to maintain a novel heterodimer in solution. It was consequently pertinent to investigate the extent of heterodimer formation in I-DmoIS protein preparations. In our initial studies I-DmoIS appeared soluble and displayed a similar elution profile to that of wild-type I-DmoI when partially purified on a heparin column. Nevertheless, since both independent domains (N102 and C101) have a molecular mass of ∼11 kDa, we were not able to determine the relative concentration of each using SDS–PAGE analysis. Given this limitation, a caveat of the relative activity analysis was that relative protein concentrations were determined with the assumption of a 1:1 ratio of A:B domains in partially purified lysates. However, the above results with the I-DmoISS construct suggested that the expression of the domain B protein is a limiting step for heterodimer formation and hence for DNA cleavage activity. Determining the actual ratio of A:B domains would allow application of a correction factor for DNA cleavage activity.
We therefore developed a sensitive dimerization assay based on differential detection using two distinct affinity tags. Figure 7 shows the constructs created in the assay along with a schematic outline of the procedure and results for I-DmoIS. The epitopes used in the assay, the eight-residue FLAG and 10-amino-acid c-myc, were chosen not only for their high specificity but also for flexibility in placement within the coding sequence. The effects of introducing the extraneous amino acids to the termini of the proteins were assessed by first creating either N- or C-terminally tagged wild-type I-DmoI and I-DmoIS (Fig. 7A). These constructs displayed DNA cleavage activity similar to that of their untagged counterparts (data not shown).
Western blots were performed with the singly tagged constructs to evaluate both the specificity and the sensitivity of the particular antibody. Minimal cross-reactivity was seen either with the anti-FLAG antibody (Fig. 7B, panel 2) or with anti-c-Myc antibody (Fig. 7B, panel 3). From these initial trials we observed the first firm evidence for the reduced efficiency in expression of domain B. Whereas the FLAG-tagged wild-type and domain A proteins were present in equivalent amounts, this was not the case for the c-myc-tagged wild-type and domain B proteins (Fig. 7B, compare panel 2 with panel 3). Although an equivalent amount of lysate was used, the c-myc-tagged I-DmoIS lanes appear to contain much less protein than the c-myc-tagged wild-type control (Fig. 7B, panel 3; compare lane Wc with lanes Sc1 and Sc2). Moreover, the difference between lane Sc1 (crude lysate) and Sc2 (cleared lysate) indicates that some fraction of domain B is insoluble. It appears that the reduced expression and/or insolubility of domain B adversely affects the expression of the ensuing domain A protein. Although total I-DmoISS protein expression was only down 2- to 5-fold compared with I-DmoIS, the c-myc-tagged version of the I-DmoISS construct (lane SSc) contains significantly (>50-fold) less domain A.
The protocol used to evaluate relative heterodimer formation is outlined in Figure 7C. Doubly tagged wild-type I-DmoI (fWc) and I-DmoIS (fSc) were first immunoprecipitated with an excess amount of either anti-FLAG or anti-c-Myc antibody to capture all available protein (Step 1). Samples were then split (Step 2) and separate western blots (Step 3) were probed with each antibody used in the immunoprecipitation step. Theoretically, immunoprecipitation of the split construct will only pull down the sister domain if actively dimerized. Excess of either subdomain upon re-detection in western blots would indicate a skewed stoichiometry. Full-length protein allowed normalization of the relative reactivity of the given antibodies since immunoprecipitation with either antibody always produced equal amounts of both epitopes (Step 3, diagrams). This correction factor could then be applied to the quantities of subunits visualized to determine relative heterodimerization.
Typical results for the immunoprecipitation/western blot (IP/western) analysis in which nanogram quantities of heterodimerized I-DmoIS were visualized are shown in Figure 7D. Although the relative amount of full-length I-DmoI re-detected with anti-FLAG (specific for the N-terminus) was roughly equal when immunoprecipitated with either antibody (Fig. 7D, 1a, fWc lane F versus C, 22 kDa), the relative amount of I-DmoIS domain A differed depending on the antibody used for IP (Fig. 7D, 1a and 1b, fSc lane F versus C, 11 kDa). Yet when anti-c-Myc antibody (specific for the C-terminus) was used in the western blot, the relative quantity of I-DmoIS domain B appeared roughly equal (Fig. 7D, 2a and 2b); as did, again, the relative amount of full-length I-DmoI (Fig. 7D, 2a). These data demonstrate that the proportion of I-DmoIS heterodimer produced upon expression is less than anticipated owing to the absence of domain B. After standardizing for the antibody reactivity, it was determined that only 5–10% of domain A forms heterodimer in the protein preparations used for the DNA cleavage analysis. Extrapolating these results to the data in Figure 4B indicates that I-DmoIS has near wild-type levels of cleavage activity.
DISCUSSION
In grafting the LAGLIDADG interfacial residues of I-CreI into I-DmoI we demonstrate that the LAGLIDADG helix–helix interface is interchangeable to some extent between monomeric and dimeric proteins. This is not wholly unexpected given the conserved overall structure and sequence of this protein family (Figs 1 and 2). As demonstrated by the range of DNA cleavage activity for the partial-revertant CL constructs, exchanging residues within the LAGLIDADG interface is limited by the bifunctional nature of the LAGLIDADG two-helix bundle. Insight into the phenotype of the mutants is provided by the observation that the LAGLIDADG motif is related (bold residues) to the GxxxG helix–helix interaction found in TM proteins (13,14,32,33,38).
The LAGLIDADG helices as a subset of the GxxxG helices of TM proteins
Like LAGLIDADG helices, TM helices interact with a right-handed crossing angle (∼40°) and are stabilized by van der Waals interactions of side-chain and backbone atoms (39). In the majority of cases a pair of glycine (or alanine) residues on each helix, flanked by mostly hydrophobic, β-branched residues, comprise the core of the helix–helix interface (33). Using sequence data and structural alignments we determined that the LAGLIDADG motif is a specialized form of this GxxxG motif (Fig. 1 and Table 1). Whereas the N-terminal part of the LAGLIDADG helices adopts the classical helix–helix interaction of GxxxG two-helix bundles, the C-terminal part includes a novel functional component arising from the last two residues in the motif: the acidic residue (D) involved in the formation of an active site and the following glycine that acts to break the helical structure and leads into the first β-strand of the DNA binding region. Nonetheless this correlation of motifs allows us to apply the insights gained from GxxxG studies to our analysis of the LAGLIDADG helix–helix interface.
The LAGLIDADG helix can be modeled as two interacting regions. The N-terminal region, XLAGLIDADG residues 1, 3, 4 and 5 (Fig. 1), comprises a core, hydrophobic interaction and is mainly responsible for domain interactions or protein dimerization (Fig. 2A). Changes in this region appear to have a lesser effect on catalytic function as long as the GxxxG-type interaction is preserved. The very active I-DmoIH construct supports this theory as it retains better than wild-type activity yet incorporates five residue changes in the N-terminal region of the LAGLIDADG helices. Also consistent with this model are studies that mutate the key interfacial glycine residues in this region (XLAGLIDADG position 4) producing a disruptive effect on functionality (40,41). This agrees with the observation in Table 2 that a glycine is highly conserved at position 4 for both LAGLIDADG helices.
The C-terminal region, XLAGLIDADG residues 7–10 (Fig. 1), contributes to the endonuclease active site. This region is more sensitive to mutation, as supported by the differential activity of the I-DmoICL reversion mutants (Fig. 4B) and the analysis in Table 2 (position 8). For example, both I19D and V115D change the chemical environment in analogous areas on either side of the active site (Fig. 2A and B) but they display opposite effects (Fig. 4B). This would argue that although the helix–helix interface is maintained, as evidenced by the retained DNA cleavage activity, the chemical moiety introduced is detrimental on one side of the interaction (V115D) but favorable on the other (I19D). In the I-CreI structure these aspartic acids appear to reinforce the protein dimer interface by forming a hydrogen bond to a residue in the opposing subunit and a neighboring loop (6). Although this loop is absent in I-DmoI, in modeling the I-DmoICL structure we observed that the I19D and V115D mutants might also form alternative interactions to account for their phenotype (see Supplementary Material).
One of the more surprising findings in this work was the extreme effects on DNA cleavage activity of the A116G mutation. In the reversion analysis, the G116A change of a simple methyl group restores nearly 35% cleavage activity to I-DmoICL. However, A116 in I-DmoI, at XLAGLIDADG position 8, corresponds to the second G of the GxxxG motif interaction (Fig. 1 and Table 1) that is crucial for helical stability (14,32,42). It would appear that XLAGLIDADG position 8 may be involved not only in the interhelical interface but also in accurately positioning the ensuing active site residues. This proposal is supported by the analysis in Tables 2 and 3 for residues at XLAGLIDADG position 8 and the catalytic sequence residues 8/9. Not only is there a significant preference for a G–A interaction at position 8, consistent with the high activity of the G116A revertant, but also the identity of the residue at this position reflects a preference for the neighboring catalytic amino acid (D versus E). An A at position 8 occurs at equivalent frequency with D or E at position 9, whereas G at position 8 occurs >4-fold more frequently with D than E, likely accounting for the enhanced activity of the G116A mutant.
Functional independence of the α/β domains
Through protein engineering, two groups have recently demonstrated the functional independence of the α/β domains of LAGLIDADG proteins by fusing I-CreI with domain A of I-DmoI [E-DreI; (22); DmoCre; (23)]. Here we present additional evidence for the functional independence of the α/β domains by essentially reversing the fusion analysis. I-DmoI can therefore be viewed as a compact version of a naturally tethered I-CreI homodimer. Our aim was to separate the functional units of I-DmoI and test the tenacity of the LAGLIDADG two-helix bundle contacts. Whereas the aforementioned work involved modifying the LAGLIDADG interface (22,23), it was of interest to ascertain whether the naturally derived monomeric I-DmoI interface could support heterodimer formation.
Initial assays were designed to assess whether an active split I-DmoI heterodimer could be formed in solution from domains expressed in separate lysates (Fig. 5, path 1). Several possible explanations exist for the lack of activity in these single-domain, mixed construct experiments. First, although the expressed single domains appear soluble and intact on Coomassie-stained protein gels, there could be subtle degradation or the individual domains may not be folding properly in the absence of interactions with a pairing domain. Second, the formation of homodimers upon expression, either A/A or B/B, could conceivably preclude the formation of the proper wild-type heterodimer (A/B). In this case, a novel dimerized protein may be formed but in the absence of a proper target sequence would appear inactive. Initial attempts to assay potential homodimers for activity using symmetric homing sites failed to yield positive results (G. H. Silva, unpublished data).
In order to circumvent these potential expression and folding issues, we used a tandem expression system (Fig. 5, path 2) that allowed us to determine the viability of heterodimeric I-DmoI as well as to optimize the choice of domain pairings (Table 4). The final construct used in this study, I-DmoIS, consists of the first 102 (domain A) and the last 88 (domain B) residues of wild-type I-DmoI. An assay was developed to assess the extent of heterodimer formation in expressed lysates. Although based on principles similar to preparative work described in Wende et al. (43), our system differs in sensitivity and applicability. Detection of relatively small (nanogram) quantities of protein is limited only by the sensitivity of the antibodies. Also, this analytical assay can be used to assess dimerization in the context of expressed lysates. Using IP/western analysis it was determined that heterodimerized I-DmoIS accounts for a relatively small fraction of the expressed protein (5–10%). Thus, I-DmoIS is a highly specific (Fig. 6), heterodimeric LAGLIDADG endonuclease with near-wild-type levels of activity.
Implications for engineered endonucleases
As evidenced by numerous recent studies, LAGLIDADG proteins are ideal candidates for engineering gene-specific reagents (22,23,44–46). The work presented here provides clues to the dual structure/function properties of the LAGLIDADG helix–helix interaction. Although firm context rules could not be determined, the I-DmoICL (Figs 2–4) and informatics analyses (Tables 1–3) address the interchangeability of the LAGLIDADG interface. This information on LAGLIDADG flexibility will be useful for re-engineering non-native subunits to interact with each other. Additionally, the activity of a heterodimer formed by splitting I-DmoI holds promise for protein engineering. Whereas the heterodimerization experiment described here was limited by underexpression of one of the domains, such limitations can be overcome, with the goal of mixing and matching domains of different LAGLIDADG endonucleases to generate heterodimers with chimeric specificities. The sensitivity and applicability of the IP/western assay would then be useful for rapid identification of viable candidates in small-scale expression systems.
Evolution of LAGLIDADG endonucleases
The elucidation of the structures of diverse LAGLIDADG endonucleases (6,8,9) has added support for speculation that the monomeric, double-motif LAGLIDADG proteins arose from a gene duplication of their homodimeric, single-motif ancestors (18,20). From an evolutionary standpoint, a drawback of the single-motif proteins is the inherent need for palindromic or pseudo-palindromic target DNA sequences. The homing endonucleases circumvent this limitation to some extent by not making saturating contacts to all hydrogen bond donors and acceptors in any given base of the target DNA (5). However, this flexibility in DNA target recognition does not overcome the equally important restriction that a single mutation in one monomer of a homodimer is reflected in two halves of the protein. Thus there appears to be an intrinsic advantage to the tethering of functional units. Structurally, this allows either domain to evolve an independent specificity thereby broadening the array of available target sequences. This approach could then generate an increase in specificity as the generalized non-saturating hydrogen bonding pattern becomes tailored to specific base pairs [e.g. the highly specific I-SceI; see (11)]. Our ability to split a monomeric LAGLIDADG enzyme into two active domains that can dimerize lends support to the hypothesis that monomeric endonucleases were generated by fusion of genes encoding active single domains.
A recent study by Iyer et al. (47) provides evidence that the viral KilA–N and fungal APSES domains share a common fold with the LAGLIDADG DNA binding regions. The study suggests that lineage-specific gene expansion and domain shuffling was involved in the evolution of viral DNA binding regulatory proteins. Curiously, the alignments with the α/β domains do not include the LAGLIDADG helices. The LAGLIDADG helices bear the signature of the GxxxG motif which is considered a framework for TM helix–helix association and more recently for helix packing in general (14,32,33,42). It is possible that the LAGLIDADG unit of interaction arose as a result of modular evolution in which a preformed DNA binding domain (47) was linked to a natural helix–helix interaction domain (14). This would give rise to the ancestral single-motif LAGLIDADG proteins, which through gene duplication could form the double-motif members of this family. Herein would be the extended protein scaffold on which to build enhanced specificity.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Vicky Derbyshire for helpful discussions and technical advice, Carol Lyn Piazza for assistance with protein purifications and Maryellen Carl for expert manuscript preparation. We also thank Vicky Derbyshire, Lori Conlan, Arthur Beauregard and Alfred Pingoud for critical readings of the manuscript. We acknowledge the contribution of the Molecular Genetics Core Facility at the Wadsworth Center. National Institutes of Health Grants GM39422 and GM44844 funded this work.
REFERENCES
- 1.Dalgaard J.Z., Klar,A., Moser,M.J., Holley,W.R., Chatterjee,A. and Mian,I.S. (1997) Statistical modeling and analysis of the LAGLIDADG family of site-specific endonucleases and identification of an intein that encodes a site-specific endonuclease of the H-N-H family. Nucleic Acids Res., 25, 4626–4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lucas P., Otis,C., Mercier,J.P., Turmel,M. and Lemieux,C. (2001) Rapid evolution of the DNA-binding site in LAGLIDADG homing endonucleases. Nucleic Acids Res., 29, 960–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pietrokovski S. (1994) Conserved sequence features of inteins (protein introns) and their use in identifying new inteins and related proteins. Protein Sci., 3, 2340–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hensgens L.A.M., Bonen,L., de Haan,M., van der Horst,G. and Grivell,L.A. (1983) Two intron sequences in yeast mitochondrial COX1 gene: homology among URF-containing introns and strain-dependent variation in flanking exons. Cell, 32, 379–389. [DOI] [PubMed] [Google Scholar]
- 5.Chevalier B.S. and Stoddard,B.L. (2001) Homing endonucleases: structural and functional insight into the catalysis of intron/intein mobility. Nucleic Acids Res., 29, 3757–3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heath P.J., Stephens,K.M., Monnat,R.J.,Jr and Stoddard,B.L. (1997) The structure of I-CreI, a group I intron-encoded homing endonuclease. Nat. Struct. Biol., 4, 468–476. [DOI] [PubMed] [Google Scholar]
- 7.Chevalier B., Turmel,M., Lemieux,C., Monnat,R.J.,Jr and Stoddard,B.L. (2003) Flexible DNA target site recognition by divergent homing endonuclease isoschizomers I-CreI and I-MsoI. J. Mol. Biol., 329, 253–269. [DOI] [PubMed] [Google Scholar]
- 8.Silva G.H., Dalgaard,J.Z., Belfort,M. and Van Roey,P. (1999) Crystal structure of the thermostable archaeal intron-encoded endonuclease I-DmoI. J. Mol. Biol., 286, 1123–1136. [DOI] [PubMed] [Google Scholar]
- 9.Duan X., Gimble,F.S. and Quiocho,F.A. (1997) Crystal structure of PI-SceI, a homing endonuclease with protein splicing activity. Cell, 89, 555–564. [DOI] [PubMed] [Google Scholar]
- 10.Ichiyanagi K., Ishino,Y., Ariyoshi,M., Komori,K. and Morikawa,K. (2000) Crystal structure of an archaeal intein-encoded homing endonuclease PI-PfuI. J. Mol. Biol., 300, 889–901. [DOI] [PubMed] [Google Scholar]
- 11.Moure C.M., Gimble,F.S. and Quiocho,F.A. (2003) The crystal structure of the gene targeting homing endonuclease I-SceI reveals the origins of its target site specificity. J. Mol. Biol., 334, 685–695. [DOI] [PubMed] [Google Scholar]
- 12.Bolduc J.M., Spiegel,P.C., Chatterjee,P., Brady,K.L., Downing,M.E., Caprara,M.G., Waring,R.B. and Stoddard,B.L. (2003) Structural and biochemical analyses of DNA and RNA binding by a bifunctional homing endonuclease and group I intron splicing factor. Genes Dev., 17, 2875–2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brosig B. and Langosch,D. (1998) The dimerization motif of the glycophorin A transmembrane segment in membranes: importance of glycine residues. Protein Sci., 7, 1052–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Russ W.P. and Engelman,D.M. (2000) The GxxxG motif: a framework for transmembrane helix–helix association. J. Mol. Biol., 296, 911–919. [DOI] [PubMed] [Google Scholar]
- 15.Jurica M.S., Monnat,R.J.,Jr and Stoddard,B.L. (1998) DNA recognition and cleavage by the LAGLIDADG homing endonuclease I-CreI. Mol. Cell, 2, 469–476. [DOI] [PubMed] [Google Scholar]
- 16.Chevalier B.S., Monnat,R.J.,Jr and Stoddard,B.L. (2001) The homing endonuclease I-CreI uses three metals, one of which is shared between the two active sites. Nat. Struct. Biol., 8, 312–316. [DOI] [PubMed] [Google Scholar]
- 17.Moure C.M., Gimble,F.S. and Quiocho,F.A. (2002) Crystal structure of the intein homing endonuclease PI-SceI bound to its recognition sequence. Nat. Struct. Biol., 9, 764–770. [DOI] [PubMed] [Google Scholar]
- 18.Lykke-Andersen J., Garrett,R.A. and Kjems,J. (1996) Protein footprinting approach to mapping DNA binding sites of two archaeal homing enzymes: evidence for a two-domain protein structure. Nucleic Acids Res., 24, 3982–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gimble F.S. and Wang,J. (1996) Substrate recognition and induced DNA distortion by the PI-SceI endonuclease, an enzyme generated by protein splicing. J. Mol. Biol., 263, 163–180. [DOI] [PubMed] [Google Scholar]
- 20.Belfort M. and Roberts,R.J. (1997) Homing endonucleases: keeping the house in order. Nucleic Acids Res., 25, 3379–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jurica M.S. and Stoddard,B.L. (1999) Homing endonucleases: structure, function and evolution. Cell. Mol. Life Sci., 55, 1304–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chevalier B.S., Kortemme,T., Chadsey,M.S., Baker,D., Monnat,R.J. and Stoddard,B.L. (2002) Design, activity and structure of a highly specific artificial endonuclease. Mol. Cell, 10, 895–905. [DOI] [PubMed] [Google Scholar]
- 23.Epinat J.C., Arnould,S., Chames,P., Rochaix,P., Desfontaines,D., Puzin,C., Patin,A., Zanghellini,A., Paques,F. and Lacroix,E. (2003) A novel engineered meganuclease induces homologous recombination in yeast and mammalian cells. Nucleic Acids Res., 31, 2952–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jones T.A., Zou,J.Y., Cowan,S.W. and Kjeldgaard,M. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- 25.Horton R.M., Cai,Z., Ho,S.N. and Pease,L.R. (1990) Gene splicing by overlap extension: Tailor-made genes using the polymerase chain reaction. Biotechniques, 8, 528–536. [PubMed] [Google Scholar]
- 26.Bateman A., Birney,E., Cerruti,L., Durbin,R., Etwiller,L., Eddy,S.R., Griffiths-Jones,S., Howe,K.L., Marshall,M. and Sonnhammer,E.L. (2002) The Pfam protein families database. Nucleic Acids Res., 30, 276–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dalgaard J.Z., Garrett,R.A. and Belfort,M. (1993) A site-specific endonuclease encoded by a typical archaeal intron. Proc. Natl Acad. Sci. USA, 90, 5414–5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dalgaard J.Z., Garrett,R.A. and Belfort,M. (1994) Purification and characterization of two forms of I-DmoI, a thermophilic site-specific endonuclease encoded by an archaeal intron. J. Biol. Chem., 269, 28885–28892. [PubMed] [Google Scholar]
- 29.Aagaard C., Awayez,M.J. and Garrett,R.A. (1997) Profile of the DNA recognition site of the archaeal homing endonuclease I-DmoI. Nucleic Acids Res., 25, 1523–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matthews B.W., Nicholson,H. and Becktel,W.J. (1987) Enhanced protein thermostability from site-directed mutations that decrease the entropy of unfolding. Proc. Natl Acad. Sci. USA, 84, 6663–6667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rohl C.A., Fiori,W. and Baldwin,R.L. (1999) Alanine is helix-stabilizing in both template-nucleated and standard peptide helices. Proc. Natl Acad. Sci. USA, 96, 3682–3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kleiger G., Grothe,R., Mallick,P. and Eisenberg,D. (2002) GXXXG and AXXXA: common alpha-helical interaction motifs in proteins, particularly in extremophiles. Biochemistry, 41, 5990–5997. [DOI] [PubMed] [Google Scholar]
- 33.Senes A., Gerstein,M. and Engelman,D.M. (2000) Statistical analysis of amino acid patterns in transmembrane helices: the GxxxG motif occurs frequently and in association with beta-branched residues at neighboring positions. J. Mol. Biol., 296, 921–936. [DOI] [PubMed] [Google Scholar]
- 34.Chakrabartty A., Schellman,J.A. and Baldwin,R.L. (1991) Large differences in the helix propensities of alanine and glycine. Nature, 351, 586–588. [DOI] [PubMed] [Google Scholar]
- 35.Li S.C. and Deber,C.M. (1992) Influence of glycine residues on peptide conformation in membrane environments. Int. J. Pept. Protein Res., 40, 243–248. [DOI] [PubMed] [Google Scholar]
- 36.Komori K., Ichiyanagi,K., Morikawa,K. and Ishino,Y. (1999) PI-PfuI and PI-PfuII, intein-coded homing endonucleases from Pyrococcus furiosus. II. Characterization of the binding and cleavage abilities by site-directed mutagenesis. Nucleic Acids Res., 27, 4175–4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Komori K., Fujita,N., Ichiyanagi,K., Shinagawa,H., Morikawa,K. and Ishino,Y. (1999) PI-PfuI and PI-PfuII, intein-coded homing endonucleases from Pyrococcus furiosus. I. Purification and identification of the homing-type endonuclease activities. Nucleic Acids Res., 27, 4167–4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fleming K.G. and Engelman,D.M. (2001) Specificity in transmembrane helix–helix interactions can define a hierarchy of stability for sequence variants. Proc. Natl Acad. Sci. USA, 98, 14340–14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.MacKenzie K.R., Prestegard,J.H. and Engelman,D.M. (1997) A transmembrane helix dimer: structure and implications. Science, 276, 131–133. [DOI] [PubMed] [Google Scholar]
- 40.Henke R.M., Butow,R.A. and Perlman,P.S. (1995) Maturase and endonuclease functions depend on separate conserved domains of the bifunctional protein encoded by the group I intron aI4 alpha of yeast mitochondrial DNA. EMBO J., 14, 5094–5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Szczepanek T., Jamoussi,K. and Lazowska,J. (2000) Critical base substitutions that affect the splicing and/or homing activities of the group I intron bi2 of yeast mitochondria. Mol. Gen. Genet., 264, 137–144. [DOI] [PubMed] [Google Scholar]
- 42.Kleiger G. and Eisenberg,D. (2002) GXXXG and GXXXA motifs stabilize FAD and NAD(P)-binding Rossmann folds through Cα-H···O hydrogen bonds and van der Waals interactions. J. Mol. Biol., 323, 69–76. [DOI] [PubMed] [Google Scholar]
- 43.Wende W., Stahl,F. and Pingoud,A. (1996) The production and characterization of artificial heterodimers of the restriction endonuclease EcoRV. Biol. Chem., 377, 625–632. [DOI] [PubMed] [Google Scholar]
- 44.Posey K.L. and Gimble,F.S. (2002) Insertion of a reversible redox switch into a rare-cutting DNA endonuclease. Biochemistry, 41, 2184–2190. [DOI] [PubMed] [Google Scholar]
- 45.Seligman L.M., Chisholm,K.M., Chevalier,B.S., Chadsey,M.S., Edwards,S.T., Savage,J.H. and Veillet,A.L. (2002) Mutations altering the cleavage specificity of a homing endonuclease. Nucleic Acids Res., 30, 3870–3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fitzsimons Hall M., Noren,C.J., Perler,F.B. and Schildkraut,I. (2002) Creation of an artificial bifunctional intein by grafting a homing endonuclease into a mini-intein. J. Mol. Biol., 323, 173–179. [DOI] [PubMed] [Google Scholar]
- 47.Iyer L.M., Koonin,E.V. and Aravind,L. (2002) Extensive domain shuffling in transcription regulators of DNA viruses and implications for the origin of fungal APSES transcription factors. Genome Biol., 3, RESEARCH0012.0011–0012.0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.