


Abstract
DNA polymerase δ (Pol δ) plays a central role in eukaryotic chromosomal DNA replication, repair and recombination. In fission yeast, Pol δ is a tetrameric enzyme, comprising the catalytic subunit Pol3 and three smaller subunits, Cdc1, Cdc27 and Cdm1. Previous studies have demonstrated a direct interaction between Pol3 and Cdc1, the B-subunit of the complex. Here it is shown that removal of the tandem zinc finger modules located at the C-terminus of Pol3 by targeted proteolysis renders the Pol3 protein non-functional in vivo, and that the C-terminal zinc finger module ZnF2 is both necessary and sufficient for binding to the B-subunit in vivo and in vitro. Extensive mutagenesis of the ZnF2 module identifies important residues for B-subunit binding. In particular, disruption of the ZnF2 module by substitution of the putative metal-coordinating cysteines with alanine abolishes B-subunit binding and in vivo function. Finally, evidence is presented suggesting that the ZnF region is post-translationally modified in fission yeast cells.
INTRODUCTION
DNA polymerase δ (Pol δ) plays vital roles in the processes of DNA replication, repair and recombination in eukaryotic cells (1,2). In the yeasts Saccharomyces cerevisiae and Schizosaccharomyces pombe, Pol δ is required for complete chromosomal DNA replication (3). In these model eukaryotes, it seems likely that Pol δ functions to replicate the lagging strand although, in the absence of Pol ε, Pol δ also appears capable of leading strand synthesis (4–6). In the SV40 in vitro replication system, Pol δ is responsible for continuous synthesis of the leading strand and for the discontinuous synthesis of the lagging strand (7,8). However, the precise cellular roles of Pol δ and Pol ε, and their relationship with one another, is still unclear. Pol δ has also been implicated in various DNA repair processes, including nucleotide excision repair and base excision repair, and in V(D)J recombination (1).
Pol δ has been purified from a number of sources. In the fission yeast S.pombe, Pol δ is a heterotetrameric enzyme complex comprising the Pol3, Cdc1, Cdc27 and Cdm1 proteins (9–12). The catalytic subunit Pol3 is a family B DNA polymerase related in sequence, and presumably structure, to Escherichia coli Pol B (1). Sp Pol3 possesses both polymerase and proofreading exonuclease activities and interacts directly with the B-subunit of the complex, Cdc1. Cdc1 in turn interacts directly with the C-subunit Cdc27 (9). In S.pombe, each of these three proteins is encoded by an essential gene. In contrast, the D-subunit, Cdm1, is encoded by a non-essential gene (11); the function of this subunit is not known, nor is it known how it interacts with the rest of the complex.
In mammals, Pol δ also appears to exist as a heterotetramer, comprising orthologues of Pol3, Cdc1, Cdc27 and Cdm1 (13). Indeed, database searching identifies sequence homologues of Pol3, Cdc1, Cdc27 and Cdm1 in many eukaryotic species (14). One notable exception to this is the budding yeast S.cerevisiae, where no Cdm1 homologue has been identified. Consistent with this, budding yeast Pol δ purifies as a heterotrimer of Sc Pol3, Pol31 and Pol32, the latter two proteins being orthologues of Cdc1 and Cdc27, respectively (3).
On the basis of comparative protein sequence analysis, the Sp and Sc Pol3 proteins are likely to be composed of multiple structural domains. Central to the function of the enzyme are its polymerase and 3′–5′ exonuclease activities. The polymerase domain, which spans amino acids 534–990 in the 1084 residue Sp Pol3 protein, most probably adopts the classical palm–fingers–thumb structure of other family B polymerase enzymes. N-terminal to this is the 3′–5′ exonuclease domain, spanning amino acids 121–461, which performs a proofreading function during DNA synthesis. In budding yeast, mutation of conserved amino acids within the exonuclease domain causes a large increase in spontaneous mutation rates (15), while mice homozygous for the equivalent mutations display significantly increased rates of tumour formation and reduced lifespan (16,17).
Located near the C-terminus of the catalytic subunit are two putative C4 zinc finger (ZnF) modules (see Fig. 1). Previous work from Faye and co-workers (18–20) has demonstrated an important role for the ZnF modules in budding yeast. In addition to being found in the catalytic subunit of Pol δ, tandem zinc finger modules are also a feature of the catalytic subunits of Pol α, ε and ζ, consistent with the genes encoding these proteins being descended from a common ancestor. In the case of mouse Pol α, an important role for the ZnF modules in binding to the B-subunit p68 been demonstrated (21) and the structure of one of the two ZnF modules recently solved (22). In Pol ε, the extended C-terminal domain of the protein, which includes the ZnF modules, has also been implicated in B-subunit binding, as well as in Pol ε dimerization and in checkpoint signalling (4).
Figure 1.

Removal of the C-terminal zinc finger domain of Sp Pol3 by targeted proteolysis abolishes protein function in vivo. (A) Schematic of the Sp Pol3 protein showing the relative locations of the exonuclease and polymerase motifs in the catalytic domain and the ZnF modules (labelled 1 and 2). Also shown is the location of the TEV site insertion in the Sp Pol3-T2 protein. (B) Induction of TEV in haploid pol3-T2 cells prevents colony formation. Approximately 1000 pol3-T2 cells carrying either pREP3X (indicated as –TEV) or pREP3X-TEVNLS (+TEV) were plated on supplemented EMM plates either with or without thiamine to either repress or depress the nmt1 promoter (labelled OFF or ON). Colony formation was not possible when TEVNLS was induced (top right panel). (C) Induction of TEV in haploid pol3-T2 cells halts cell number increase in liquid culture. TEVNLS expression was induced by transferring pol3-T2 (pREP3X-TEVNLS) cells from thiamine-containing to thiamine-free medium at 32°C at time zero. Cell number increase was monitored from 9 to 18 h using an electronic particle counter (filled circles). As a control, pol3-T2 (pREP3X) cells were analysed in parallel (open circles). (D) Induction of TEVNLS expression. Samples for total protein extraction and immunoblotting were taken from the pol3-T2 (pREP3X-TEVNLS) culture at hourly intervals from 9 to 18 h. TEVNLS induction was apparent from 11 h. (E) Flow cytometric analysis of propidium iodide-stained cells from 10 to 18 h post-induction. Left panel: pol3-T2 (pREP3X) cells. Right panel: pol3-T2 (pREP3X-TEVNLS) cells.
In this report, direct interaction between the catalytic subunit of yeast Pol δ and the B-subunit (either Cdc1 in S.pombe or Pol31 in S.cerevisiae) is demonstrated to occur via the most C-terminal ZnF module (designated ZnF2). This region of the protein is shown to be both necessary and sufficient for B-subunit binding in vivo and in vitro. Mutational analysis demonstrates the importance for binding of the four conserved cysteine residues implicated in metal ion coordination as well as additional conserved residues both N- and C-terminal to the cysteine pairs. In addition, replacement of any of the four cysteines in the ZnF2 of Sp Pol3 abolishes Pol δ function in vivo, consistent with previous studies in budding yeast. Finally, evidence is presented suggesting that the ZnF region of Sp Pol3 is post-translationally modified in vivo, although the precise nature of the modification is unknown.
MATERIALS AND METHODS
Yeast strains and media
The following fission yeast diploid strain was used for mutation construction in this study: leu1-32/leu1-32 ura4-D18/ura4-D18 ade6-M210/ade6-M216 h–/h+. This was constructed by mating leu1-32 ura4-D18 ade6-M210 h– with leu1-32 ura4-D18 ade6-M216 h+ and selecting for ade+ diploids following 24 h incubation at 25°C on malt extract medium. For spore germination experiments, the following strain was used as the wild-type control: leu1-32/leu1-32 ura4+/ura4-D18 ade6-M210/ade6-M216 h–/h+. The S.cerevisiae strains used were: L40 [MATa ade2 trp1-901 leu2-3-112 his3Δ200 lys2-801am URA3::(lexAop)8-LacZ LYS2::(lexAop)4-HIS3] (23); Y187 (MATα gal4-542 gal80–538 ade2-101 his3Δ200 trp1-901 leu2-3-112 ura3-52 URA3::GAL1-lacZ) (Clontech); and CTY105d (MATa ade2 met- trp1-901 leu2-3-112 his3-Δ200 gal4-gal80-URA3::lexA-LacZ) (9). Fission yeast media (YE, EMM and ME) were as previously described (24). For budding yeast, YE and EMM were substituted with YPDA and SD (YMM). Cells were grown at 30 or 32°C unless otherwise indicated.
Generation of TEV site insertion mutants
Fission yeast cells carrying mutant pol3-T alleles were constructed as follows. First, plasmid pTZ19RU-Pol3DraI was created by cloning the 2346 bp DraI genomic DNA fragment that includes the 3′ end of the pol3+ open reading frame from S.pombe cosmid SPBC336 into the unique SmaI site of plasmid pTZ19RU, a derivative of pTZ19R (Fermentas) containing the S.pombe ura4+ marker (1764 bp genomic HindIII fragment) inserted at the polylinker HindIII site. pTZ19RU-Pol3DraI was then used as a template for PCR overlap extension mutagenesis, which was carried out using Taq polymerase essentially as described elsewhere (25). Following the second round of PCR (with oligonucleotides POL3-CTW: 5′-AAAGGTAAAACTGCTTTATGTGAG-3′ and POL3-CTY, above), the product was digested with NheI and AflII, re-cloned into plasmid pTZ19RU-Pol3DraI from which the wild-type NheI–AflII fragment had been excised, and sequenced (oligonucleotide POL3-CTX: 5′-TTCATC ATTACGTTTGATATACAC-3′) to confirm the presence of the desired mutation and the absence of PCR-introduced errors. Additional oligonucleotide sequences can be obtained from the authors on request.
The resulting mutant pTZ19RU-Pol3DraI plasmids were then linearized by SalI digestion and used to transform the S.pombe diploid strain leu1-32/leu1-32 ura4-D18/ura4-D18 ade6-M210/ade6-M216 h–/h+ by electroporation (26). Transformants obtained were analysed to identify those in which the ura4+ marker was stably maintained, indicative of plasmid integration. Chromosomal DNA was prepared for 4–6 integrants and subjected to PCR to confirm correct integration at the pol3+ locus by a single copy of the pTZ19RU-Pol3DraI plasmid. Integrants in which the sequence encoding the tobacco etch virus (TEV) site were found in the full-length pol3+ were then identified by PCR, using an oligonucleotide corresponding to the TEV insert (POL3-TEVPCR: 5′-ACCCGACTGAAAATATAAATTCTC-3′) in combination with POL3-CTW. Note that the resulting mutant alleles encoded proteins that deviated from the wild-type Pol3 by the insertion of a seven amino acid TEV recognition site (ENLYFQS) flanked by 2–4 glycine residues as follows (Pol3 amino acids in upper case, TEV cleavage site underlined): Pol3-T1 (LNRggggenlyfqsggggSAE), Pol3-T2 (APSggenlyfqsggggVGG), Pol3-T3 (RQVggenlyfqs ggggAQV) and Pol3-T4 (NDLggenlyfqsggggEVR). Diploid pol3+/pol3-T strains were then sporulated at 25°C for 48–72 h on malt extract medium before asci were dissected on YE medium using a micromanipulator (Singer Instruments). In initial experiments, the dissected spores were incubated at 32°C. Later, the viability of pol3-T spores was examined at temperatures ranging from 18 to 36.5°C.
Induction of TEV protease
In order to test the effects of TEV induction, a pol3-T2 strain (pol3: pTZ19RU-Pol3-T3 DraI leu1-32 ura4-D18 ade6–) was transformed with either pREP3X or pREP3X-TEVNLS (X.Yang and S.E.Kearsey, in preparation). Transformants were obtained on EMM medium supplemented with adenine and 5 µM thiamine (the latter to repress the nmt1 promoter in pREP3X-TEV). The cells were then grown up to mid-exponential phase in EMM medium containing 5 µM thiamine, washed three times with EMM, and resuspended at 1 × 105 cells/ml in EMM. Samples were taken for cell number determination, analysis of DNA content by fluorescence-activated cell sorting (FACS) and immunoblotting every hour from 10 h post-induction onwards.
Two-hybrid screening
Two-hybrid screening was accomplished using the bait plasmid pBTM116-Pol31 (a gift of Dr K. Sugimoto, University of Nagoya) and the FRYL library (27) in S.cerevisiae L40 (a generous gift of Professor J. Beggs, University of Edinburgh, by permission of Dr P. Legrain, Institut Pasteur). Briefly, S.cerevisiae Y187 cells containing pBTM116-Pol31 were mated with L40 cells carrying the FRYL library and plated on YMM plates lacking histidine and containing 5 mM 3-amino-1,2,4-triazole (3-AT) to select for interacting clones. Six positives were identified and plasmids recovered for sequencing. The recovered plasmids were also re-tested by (i) individual transformation into Y187 and mating to L40 containing pBTM116-Pol31, with the resulting diploids being tested on plates lacking histidine and containing 5 mM 3-AT; and (ii) by β-galactosidase assay in CTY10-5d. Sequence analysis showed that the six clones contained two different plasmids, with the inserts in the plasmids encoding amino acids 981–1097 and 1032–1097 of Pol3 fused to the Gal4 activation domain.
Further two-hybrid analysis
Routine two-hybrid analysis was performed in S.cerevisiae CTY10-5d, as described previously (9). Plasmids constructed for two-hybrid analysis were as follows: pACT-ScPol3 (full-length POL3 gene PCR amplified from S.cerevisiae genomic DNA and cloned into pACT2), pACT-ScPol3ΔZnF2 (POL3 gene truncated for sequences encoding amino acids 1045–1097, cloned into pACT2), pACT-SpPol3-ZnF1F2 (sequences encoding amino acids 954–1084 of S.pombe Pol3, cloned into pACT2) and pACT-SpPol3-ZnF2 (sequences encoding amino acids 1016–1084 of S.pombe Pol3, cloned into pACT2). For analysis of interactions with the Pol ζ ZnF modules, sequences encompassing the ZnF-encoding regions of the S.cerevisiae REV3 and S.pombe rev3+ (SPAC688.10) genes were PCR amplified from genomic DNA and cloned into pACT2, to generate pACT-ScRev3ZnF and pACT-SpRev3ZnF, respectively. The cloned regions encoded amino acids 1368–1504 of S.cerevisiae Rev3 (oligonucleotides SCZETA5, 5′-GTT GTGTGTTGGATCCTGAACTGGGCTCAGGAAATAG-3′ and SCZETA3, 5′-GTTTGTTTGGGGATCCTTTGCGAGA CATATCTGTGTC-3′) and amino acids 1344–1480 of S.pombe Rev3 (oligonucleotides SPZETA5, 5′-GTGTT GTGTTGGATCCAGTCATGGTATCATGAAATGC-3′ and SPZETA3, 5′-GTTTGTTTGGGGATCCTTCAAGTCCTT CACACCAATA-3′). These regions are equivalent to the ZnF1F2 region of the Pol3 protein.
Construction of mutant Pol3 and Pol31 proteins for two-hybrid analysis
Mutants were introduced into pACT-ScPol3-ZnF2 by the PCR overlap extension method as described elsewhere (25) with the final PCR product being digested with BamHI and EcoRI, recloned into pACT2 and sequenced prior to use. The same method was used to make pBTM116-Pol31-P15. The mutation introduced sees Lys358 in Pol31 being replaced with glutamate. Fission yeast Pol3 mutants were generated in pACT-SpPol3-ZnF1F2 by overlap extension PCR, cloned and sequenced as above. Full oligonucleotide sequences are available from the authors on request.
Generation of fission yeast ZnF2 mutants
Fission yeast cells carrying mutant pol3 alleles were constructed exactly as described above for pol3-T1 through pol3-T4, i.e. by integrating mutated pTZ19RU-Pol3DraI plasmids (constructed by overlap extension mutagenesis) into diploid S.pombe cells, confirming the location of the mutation by PCR and sequencing of genomic DNA, sporulating the diploids and performing tetrad dissection. For analysis of pol3 mutant behaviour following spore germination in liquid culture, previously published protocols were followed (9).
Purification of recombinant ZnF modules
For expression in E.coli as N-terminally His6-tagged fusion proteins, BamHI fragments encoding either both ZnF modules at the C-terminus of Sp Pol3 (designated ZnF1F2) or only ZnF2 were cloned separately into pQE32. The resulting plasmids were transformed into E.coli M15 [pREP4] (Qiagen) and fusion protein expression induced by addition of isopropyl-β-d-thiogalactopyranoside (IPTG) to 1 l cultures growing at 30°C with shaking at an OD600 nm of 0.6–0.8. Following overnight growth, cells were harvested by centrifugation, washed once in phosphate-buffered saline (PBS), and the pellet frozen at –80°C until required. To prepare the protein, the pellets were resuspended in buffer A (100 mM Na2HPO4, 10 mM Tris–HCl, 6 M guanidine HCl pH 8.0) at 5 g/ml wet weight. The resuspended material was then stirred at room temperature for 1 h, before being centrifuged at 12 000 r.p.m. for 30 min at room temperature. The cleared cell lysate was then added to 2 ml of Ni-agarose (Qiagen) and incubated on a rotating wheel for 3 h at room temperature, before being poured into a 15 ml polypropylene chromatography column and allowed to settle. The column was then washed twice with eight column volumes (16 ml) of buffer B (100 mM Na2HPO4, 10 mM Tris–HCl, 8 M urea HCl pH 6.3). Proteins were then eluted from the column by sequential addition of four 1 ml volumes of buffer C (100 mM Na2HPO4, 10 mM Tris–HCl, 8 M urea HCl pH 5.9) followed by four 1 ml volumes of buffer D (100 mM Na2HPO4, 10 mM Tris–HCl, 8 M urea HCl pH 4.5). Eluted fractions were analysed by SDS–PAGE, and peak fractions were pooled and stored at –80°C until required.
For refolding, the proteins were diluted using buffer E (10 mM Tris–HCl, 8 M urea pH 8.0) to 50 µg/ml, and dialysed overnight against >125 vols of buffer F (50 mM borate buffer pH 8.0, 100 mM KCl, 0.1% Triton X-100, 100 µM ZnSO4, 10% glycerol). Prior to use in binding assays (see below), the sample was spun at maximum speed for 30 min at 4°C in a microcentrifuge and the pellet discarded.
In vitro Pol3–Cdc1 interactions
A 500 µl aliquot of refolded protein (∼25 µg/ml) was mixed with 3 µl of [35S]methionine-labelled Cdc1 that had been produced in vitro using the TNT Coupled T7 Transcription–Translation kit (Promega) as described previously (9). Following 30 min incubation on a rotating wheel, 80 µl of 50% (v/v) Ni-agarose slurry (Qiagen) was added and incubation continued for an additional 90 min. The samples were then spun down, and the supernatant was trichloroacetic acid (TCA)-precipitated. The pellet was washed repeatedly with buffer F, then heated to 65°C before being subjected to SDS–PAGE. Alternatively, the His6-tagged ZnF modules were immunoprecipitated using anti-MRGSH6 antibodies (Qiagen).
Expression of V5-ZnF proteins
Sequences encoding amino acids 955–1084 of Sp Pol3 were amplified using oligonucleotide primers POL3-V5-5 (5′-GTGTGTGTCTCGAGATGGGAATACCCAATCCCC TTCTTGGACTTGATCTTGGTGAAAAAGCTAGCTCTTTAC-3′) and POL3-V5-3 (5′-GTTGGTTGGGATCCCA TCATTACGTTTGATATACAC-3′). The PCR product was digested with XhoI and BamHI (sites in oligos underlined), cloned into pREP4X and sequenced. The resulting plasmid (pREP4X-V5-ZnF) encodes a 141 amino acid protein comprising an N-terminal V5 epitope tag (MGIPNPLLGLD) followed by amino acids 955–1084 of Pol3 under the control of the thiamine-repressible nmt1 promoter. Plasmids expressing mutant forms of the V5-ZnF protein were constructed by repeating the initial amplification with POL3-V5-5 and POL3-V5-3 using mutant pTZ19RU-Pol3DraI templates (described above, for the cysteine to alanine substitutions), or by overlap extension PCR (for the K989R and ΔW1086 mutants). Plasmids were then transformed into S.pombe leu1-32 ura4-D18 h– cells and transformants obtained on EMM plates supplemented with leucine. Total protein extracts were then prepared by standard methods, subjected to SDS–PAGE and analysed by immunoblotting using anti-V5 (Serotec anti-Pk MCA1360), anti-ubiquitin (Chemicon or Sigma) or anti-Pmt3 antibodies (a generous gift of Dr F. Z. Watts, University of Sussex, UK).
RESULTS
The C-terminal zinc finger modules are essential for fission yeast Pol3 function
In common with its orthologues in other species, two tandem zinc finger modules are located at the C-terminus of the fission yeast Pol3 protein, Sp Pol3 (Fig. 1A). To test the importance of these modules for Sp Pol3 function, sequences encoding the recognition site of the TEV protease, flanked by 2–4 glycine residues, were introduced into the chromosomal pol3+ gene, so that the encoded protein would contain a cleavage site for the protease. Subsequent expression of a nuclear-targeted form of the TEV protease in the mutant cells was predicted to result in cleavage of the modified Sp Pol3 protein, resulting in the highly specific cleavage of one or both ZnF modules. In total, four mutant alleles were constructed to test this method (insertion points indicated by arrows in Fig. 2), but only one, pol3-T2, with the insertion N-terminal to the tandem zinc fingers, encoded a functional protein (see Discussion).
Figure 2.

Structure of the ZnF modules. (A) Schematic of the Sc Pol3 protein showing the extent of the protein encoded by the pACT-ScPol3-ZnF1F2 (top) and pACT-ScPol3-ZnF2 (bottom) plasmids recovered from the two-hybrid screen using Pol31 as bait. The locations of the cysteines are shown. (B) Protein sequence alignment of the C-terminal regions of Pol3 orthologues from various eukaryotic species. The two zinc finger modules ZnF1 and ZnF2 are indicated, with conserved cysteine residues boxed. The arrowheads labelled T1–T4 refer to the sites of insertion of TEV protease cleavage sites into the Sp Pol3 protein. Residues conserved in all species shown are indicated by the filled circles beneath the aligned sequences. Species and accession numbers: Sp (Schizosaccharomyces pombe, accession number P30316, length 1086 amino acids), Ca (Candida albicans, P46588, 1038), Sc (Saccharomyces cerevisiae, P15436, 1097), Nc (Neurospora crassa, CAD21389, 1104), Mg (Magnaporthe grisea, EAA57102, 1103), Rn (rat, CAA10946, 1103), Mm (mouse, BAC40275, 1105), Ma (golden hamster, AAB47255, 1102), Hs (human, P28340, 1107), Bt (cow, P28339, 1106), Dr (Danio rerio, EST, not known), Tb (Trypanosoma brucei, XP_340279, 1026), Dm (Drosophila, P54358, 1092), Ag (Anopheles gambiae, EAA00051, 1110), Ce (Caenorhabditis elegans, P90829, 1081), Pf (Plasmodium falciparum, P30315, 1094), Py (Plasmodium yoelii yoelii, EAA21409, 1097), Os (rice, Q9LRE6, 1105), Cp (Crytosporidium parvum, CAD98258, 1084) and Ec (Encephalitozoon cuniculi, CAD27015, 974).
Haploid pol3-T2 cells were phenotypically indistinguishable from wild type in all respects examined (data not shown). To examine the effects of TEVNLS expression in these cells, they were transformed with plasmid pREP3X-TEVNLS (X.Yang and S.E.Kearsey, in preparation), which expresses a nuclear-targeted form of TEV under the control of the thiamine-repressible nmt1 promoter. Expression of the TEVNLS protein from this plasmid has no discernible effects on wild-type cells (data not shown). pol3-T2 transformants were obtained on EMM plates containing 5 µM thiamine (EMM+T) and grown to mid-log phase in EMM+T medium, before 1000 cells were plated on EMM plates both with and without thiamine. Figure 1B shows the results of this experiment. In the presence of pREP3X-TEVNLS, colony formation was not possible on plates lacking thiamine, i.e. under conditions in which the nmt1 promoter was derepressed. When pREP3X-TEVNLS was substituted with pREP3X, colony formation was unaffected (Fig. 1B).
To examine this in greater detail, TEV expression was induced in pol3-T2 cells growing in liquid culture. Samples were taken over the course of the experiment to monitor cell number increase and DNA content (Fig. 1C and E), and for immunoblotting to detect the TEVNLS protease; Figure 1D shows that the TEV protease is undetectable at t = 0 but is detectable 11–12 h post-induction, i.e. following transfer of the cells to thiamine-free EMM medium. The appearance of the TEV protein coincides with the slowing of the rate of cell number increase, which all but ceases 14–16 h post-induction (Fig. 1C).
Flow cytometric analysis of propidium iodide-stained cells showed that the cells arrested 14–16 h post-induction with 2C DNA content (Fig. 1E), consistent with previous observation on the effects of inactivating Pol δ in exponentially growing cultures (9,28). At later time points (16–18 h), the slight increase in cell number observed (Fig. 1D) most probably reflects leaking through the cell cycle block. Significantly, cells with 1C DNA content also begin to accumulate at this time, suggesting that the newly divided daughter cells cannot perform bulk DNA replication in the absence of fully functional Pol δ (Fig. 1E). These experiments provide a graphic demonstration of the importance of the ZnF region for Sp Pol3 function, and also highlight the usefulness of the TEVNLS protease for studying protein structure–function relationships in fission yeast (see Discussion).
Pol31 binds to the C-terminal ZnF of Sc Pol3
In order to identify proteins that interacted with Pol δ, a two-hybrid screen was performed. In order to take advantage of the availability of a two-hybrid library of particularly high quality (27), the budding Pol31 protein, rather than its fission yeast counterpart Cdc1, was used as the bait in this screen (see Materials and Methods). Screening was performed by mating L40 cells containing the bait plasmid pBTM116-Pol31 with Y187 cells containing the FRYL library (27), and diploids able to grow on selective medium containing 5 mM 3-AT (see Materials and Methods) were identified. In total, ∼1.4 × 107 diploids were screened and six independent Pol31-interacting clones identified. Sequence analysis of recovered prey plasmids revealed that the six represented two independent library clones. Both of these were derived from the POL3 gene (Table 1A). One of the clones, named pACT2-ScPol3-ZnF1F2, contained sequences encoding amino acids 981–1097 of Sc Pol3 fused to the Gal4 activation domain, while the other, pACT2-ScPol3-ZnF2, contained amino acids 1032–1097 fused to Gal4 only (Fig. 2A). These regions of Sc Pol3 correspond to the extreme C-terminus of the protein; the larger includes both zinc finger (ZnF) modules, whereas the smaller spans the most C-terminal ZnF only (ZnF2).
Table 1. Two-hybrid interactions.
| Prey | Bait | Co-expressed with | Interaction |
|---|---|---|---|
| A. Budding yeast proteins Sc Pol3 and Pol31 | |||
| – | Pol31 | – | – |
| Pol3 | – | – | – |
| Pol3 | Pol31 | – | ++ |
| Pol3-ΔZnF2 | – | – | – |
| Pol3-ΔZnF2 | Pol31 | – | – |
| Pol3-ZnF1F2 | – | – | – |
| Pol3-ZnF1F2 | Pol31 | – | +++ |
| Pol3-ZnF2 | – | – | – |
| Pol3-ZnF2 | Pol31 | – | +++ |
| B. Fission yeast proteins Sp Pol3 and Cdc1 with Cdc27 | |||
| – | Cdc1 | Cdc27 | – |
| Pol3-ZnF2 | Cdc1 | – | – |
| Pol3-ZnF2 | – | Cdc27 | – |
| Pol3-ZnF2 | Cdc1 | Cdc27 | – |
| Pol3-ZnF1F2 | Cdc1 | – | – |
| Pol3-ZnF1F2 | – | Cdc27 | – |
| Pol3-ZnF1F2 | Cdc1 | Cdc27 | +++ |
| Pol3-ZnF1F2-C1038A | Cdc1 | Cdc27 | – |
| Pol3-ZnF1F2-C1041A | Cdc1 | Cdc27 | – |
| Pol3-ZnF1F2-C1051A | Cdc1 | Cdc27 | – |
| Pol3-ZnF1F2-C1056A | Cdc1 | Cdc27 | – |
| Pol3-ZnF1F2-C1056S | Cdc1 | Cdc27 | – |
| C. Interactions with Pol ζ ZnF modules | |||
| Rev3-ZnF (Sc) | – | – | – |
| Rev3-ZnF (Sc) | Pol31 | – | – |
| Rev3-ZnF (Sp) | Cdc1 | – | – |
| Rev3-ZnF (Sp) | – | Cdc27 | – |
| Rev3-ZnF (Sp) | Cdc1 | Cdc27 | – |
Prey or bait proteins were expressed as Gal4 activation domain or LexA binding domain fusions from pACT2 or pBTM116, respectively. In A, Pol3-ZnF1F2 and Pol3-ZnF2 were identified by two-hybrid screening using a Pol31 bait (see text). Cdc27 was expressed from pAA-Cdc27. The positive interaction recorded with budding yeast Sc Pol3 and Pol31 corresponded to 15 U of β-galactosidase activity. The positive interaction recorded with fission yeast Sp Pol3 and Cdc1 corresponded to 110 U of β-galactosidase activity. All negative interactions were <2% of these values. See text for details.
In the two-hybrid system, therefore, ZnF2 is sufficient to bind to Pol31. To test whether ZnF2 was necessary for binding to Pol31, the ability of Pol31 to bind to Sc Pol3 from which ZnF2 had been deleted was investigated. It was found that Pol31 binds to full-length Sc Pol3 fused to the Gal4 activation domain but not to Pol3-ΔZnF2, a construct lacking the ZnF2 module. ZnF2 is therefore both necessary and sufficient for Sc Pol3 binding, at least in the two-hybrid system (Table 1A).
Mutational analysis of the ZnF2 module
To identify key residues within ZnF2 for Pol31 binding, mutational analysis of the Sc Pol3 ZnF2 module was performed. A series of 32 mutant Sc Pol3-ZnF2 proteins were generated by PCR overlap mutagenesis of the prey plasmid pACT2-ScPol3-ZnF2 (see Materials and Methods) and tested for their ability to bind to Pol31 in the two-hybrid system. The results are shown in Figure 3A. Mutation of any of the four cysteine residues to either alanine or serine reduced β-galactosidase activity to <2% of the value recorded with the wild-type ZnF2. Similar reductions were seen when residue E1046 was mutated to alanine or when sequences beyond amino acid 1084 were removed by truncation (CΔ1085 mutant). Other significant reductions in binding were seen throughout the ZnF2 region. For example, mutation of the highly conserved Q1057 and R1058 residues located between the first pair of cysteines in ZnF2 (see Fig. 2) reduces binding to ∼10% of wild-type levels, whereas mutation of His1064 to alanine reduced binding to ∼20% of wild-type levels. Only three mutations failed to significantly reduce ZnF2–Pol31 interaction and, in two of these cases, the level of interaction recorded was reproducibly higher than that seen with the wild-type ZnF protein. The mutated residues were E1066, located in the region between the cysteine pairs; K1072, located between the second pair of cysteines; and Arg1080, located towards the C-terminus of the Sc Pol3 protein, C-terminal to the second pair of cysteines (Fig. 3A). Note that for those mutants unable to bind to Pol31, the level of the Gal4AD-Pol3-ZnF protein was checked by immunoblotting using anti-Gal4 activation domain antibodies to eliminate the possibility that mutant proteins were present at a low level only; in each case, however, the level of the mutant ZnF proteins matched that of the wild-type ZnF protein (data not shown).
Figure 3.

Mutational analysis of the S.cerevisiae ZnF2 module. (A) Wild-type and mutant ZnF2 modules expressed from pACT2-ScPol3-ZnF2 were tested for their ability to bind to Pol31 in the two-hybrid system. The strength of interaction (wild-type = 100%) was tested by quantitative β-galactosidase assay of co-transformed CTY10-5d cells grown in liquid culture. The mutant labelled FY>AA corresponds to an F1077Y1078>AA double mutant. (B) A similar analysis but with Pol31 (light grey boxes) shown in comparison with Pol31-P15 protein (dark grey boxes). Note that the actual β-galactosidase values for the wild-type ZnF interacting with Pol31 and Pol31-P15 were ∼120 U of β-galactosidase activity for Pol31 versus ∼170 U for Pol31-P15.
Binding of ZnF2 by mutated Pol31
Previously, it was shown that budding yeast cells expressing the Sc Pol3-11 mutant protein (in which the third cysteine, C1069, in ZnF2 is replaced by a serine) were viable but temperature sensitive (18). The tempera ture sensitivity of the pol3-11 allele was suppressed by dominant mutations in an unlinked gene, originally desig nated SDP5 (18,20). Subsequent work showed that SDP5 and POL31 were isogenic, and that the sdp5-15 allele encoded a protein that differed from the wild-type protein at a single amino acid only (19). For convenience, in this report, this protein is referred to as Pol31-P15. The mutation in the Pol31-P15 protein sees Lys358 replaced with glutamic acid.
The ability of the mutant Pol31-P15 protein to bind to both wild-type and mutant ZnF2 modules was examined using the two-hybrid system. Binding of Pol31-P15 to the wild-type ZnF2 module was enhanced to 140% of the level seen with non-mutant Pol31 (from ∼120 to ∼170 U of β-galactosidase activity, see Materials and Methods), suggesting that the ability of sdp5-15 to suppress pol3 mutants in vivo may be due to the interaction between the Pol3 and Pol31 proteins being strengthened.
The ability of Pol31-P15 to bind to mutated Sc ZnF2 modules was also examined. In many cases, the ZnF2 mutations had a less marked effect on binding to Pol31-P15 compared with Pol31 (Fig. 3B). For example, binding of ZnF2-E1047A to Pol31 occurred at 12% of the level seen with non-mutant ZnF2; when tested against Pol31-P15, however, this value increased to 60% of the level seen with non-mutant ZnF2 and Pol31-P15. Other mutants clearly affected in this way include L1052A (from 39% with Pol31 to 83% with Pol31-P15), Q1057A (from 14 to 45%) and H1064A (from 20 to 63%). Increases were also seen with mutations at Cys1056 and Cys1059, irrespective of whether the cysteine is replaced by an alanine or a serine (Fig. 3B), but not with mutations at Cys1069 and Cys1074. The C1069S mutation is encoded by the pol3-11 allele, which is suppressed by sdp5-15 in vivo (20).
Sp Pol3–Cdc1 interaction in fission yeast
Next, the question of whether the interaction detected between the ZnF2 module and the B-subunit was conserved in fission yeast was addressed. Previously, a direct interaction between Sp Pol3 and Cdc1 was reported (9). However, in two-hybrid assays, it was not possible to detect a direct interaction between these proteins unless the C-subunit of the complex, Cdc27, was simultaneously co-expressed (data not shown). Interaction of the fission yeast ZnF modules and Cdc1 was therefore tested in the presence of Cdc27. Note that Cdc27 cannot itself bind to Sp Pol3, ruling out the possibility that this protein acts to bridge Sp Pol3 and Cdc1 in the two-hybrid assay. Instead, it is thought that by binding to Cdc1, Cdc27 stabilizes the latter, or alters its confirmation, in such a way that binding to Sp Pol3 is enhanced to detectable levels (9).
Table 1B shows the results of quantitative two-hybrid analysis with Sp Pol3, Cdc1 and Cdc27 proteins. The Pol3-ZnF1F2 protein interacted strongly with Cdc1 (110 U of β-galactosidase activity) but only when Cdc27 was present. All other pairwise combinations of bait, prey and bridging protein gave <2% of this value. Notably, in contrast to the situation in budding yeast, no interaction could be detected when only ZnF2 was present. However, the importance of ZnF2 for binding was demonstrated by mutational analysis of the cysteine residues. Substitution of any of the four cysteines with alanine abolished Cdc1 binding (Table 1B). Replacing Cys1056 with serine also abolished binding.
Note that while the interaction between the ZnF modules and the B-subunit is conserved in both yeasts, cross-species interactions (between the fission yeast Sp Pol3 ZnF and budding yeast Pol31 for example) were not detected using the available reagents (data not shown). This could reflect a role for non-conserved amino acids in Sp Pol3 in B-subunit binding. As an additional point (Table 1C), it was also not possible to detect an interaction, using the two-hybrid system, between Pol31 and the tandem ZnF modules at the C-terminus of the catalytic subunit of budding yeast Pol ζ, the Rev3 protein (29), or between Cdc1 and the ZnF modules at the C-terminus of the fission yeast Rev3 homologue SPAC688.10 (tested in the presence of Cdc27, as described above). Rev3 is the closest homologue of Pol3 in S.cerevisiae and S.pombe (closer than either Pol1 or Pol2, the catalytic subunits of Pol α or Pol ε, respectively) indicative of the relatively recent divergence of the genes encoding the Pol3 and Rev3 proteins.
ZnF2–Cdc1 binding in vitro
It is possible that the interactions observed in the two-hybrid system may be indirect ones, i.e. that endogenous budding yeast proteins may play a role in bridging bait and prey. To confirm that the ZnF–B-subunit interaction was direct, the Sp Pol3 ZnF region and Sp Pol3 ZnF2 alone were expressed in bacteria as His6-tagged fusion proteins, purified to near homogeneity and tested for their ability to bind to Cdc1 that had been produced in vitro using rabbit reticulocyte lysates (see Materials and Methods). The ZnF-containing proteins were found to be mostly insoluble, thereby necessitating their solubilization and subsequent refolding, which was carried out in the presence of 100 µM ZnSO4. Refolded protein was then mixed with radiolabelled Cdc1, and bound and unbound Cdc1 analysed by SDS–PAGE following pull-down and extensive washing. The results (Fig. 4) clearly show that the recombinant ZnF modules, and ZnF2 alone, are able to bind Cdc1 in vitro.
Figure 4.

Binding of purified recombinant fission yeast ZnF or ZnF2 modules to radiolabelled Cdc1 synthesized in vitro in reticulocyte lysates. N-terminally MRGS(His6)-tagged ZnF or ZnF2 fusion proteins were mixed with radiolabelled Cdc1 before being pulled-down using either Ni-NTA agarose (A) or anti-MRGS antibodies (B). Both the pulled-down (P, pellet) and supernatant (S) fractions are shown. Cdc1 was visualized by autoradiography following SDS–PAGE. (A) Lanes 1 and 2, MRGS(His6)-ZnF; lanes 3 and 4, control, no added fusion protein; lanes 5 and 6, MRGS(His6)-ZnF2; lanes 7 and 8, control, no added fusion protein. (B) Lanes 1 and 2, MRGS(His6)-ZnF; lanes 3 and 4, control, no added fusion protein.
In vivo function of the ZnF2 module in fission yeast
The results described above show that the most C-terminal ZnF module in Pol3 is necessary and sufficient for binding to the B-subunit of the Pol δ complex. Next the in vivo function of the ZnF2 module in S.pombe was examined, by creating strains expressing ZnF2 mutant Sp Pol3 proteins. The mutant alleles were constructed by modification of one copy of the gene in diploid yeast by plasmid integration by homologous recombination, as described above for the pol3-T1 through pol3-T4 alleles (see also Materials and Methods). In total, five diploid strains heterozygous at the pol3 locus were created, carrying the following mutations: pol3-C1038A, pol3-C1041A, pol3-C1051A, pol3-C1056A and pol3-C1056S. Following meiosis and sporulation, tetrad dissection showed that none of the haploid pol3 mutant strains were viable (data not shown). In each case, replacement of the cysteine rendered the Sp Pol3 protein non-functional over a wide temperature range (20–36.5°C).
To investigate the phenotype of the pol3 mutants further, two of the mutant alleles, pol3-C1038A and pol3-C1056A, were germinated in liquid culture, and cell number and DNA content monitored over a 24 h period. Spores of a pol3+ control strain were germinated in parallel (see Materials and Methods). The results are shown in Figure 5. In the wild-type pol3+ cells, DNA replication took place 8–12 h post-inoculation (Fig. 5B), with the first signs of cell number increase apparent at 12 h (Fig. 5A). Cell number increased exponentially thereafter. In contrast, no significant increases in cell number were seen with either of the pol3-C1038A or pol3-C1056A mutant alleles. Over the period 12–18 h, for example, cell number doubled in the pol3+ culture, but rose by <10% in the pol3 mutant cultures. DNA replication was possible in the pol3-C1038A and pol3-C1056A cells, as previously observed for pol3Δ, cdc1Δ or cdc27Δ cells (9). At later time points (18–24 h), the pol3 mutant cells became highly elongated, consistent with the loss of Pol δ function resulting in incomplete DNA replication and checkpoint activation.
Figure 5.

In vivo consequences of disrupting ZnF2. (A) Cell number (×106) per ml of culture (y-axis, log scale) versus time (x-axis, linear scale) following inoculation of pol3+/pol3+, pol3+/pol3-C1038A and pol3+/pol3-C1056A spores into supplemented minimal medium at time 0 and growth at 30°C. Values for the period 12–24 h are shown. (B) Flow cytometric analysis of propidium iodide-stained cells from (A). Samples from 6–14 h are shown. The positions of the 1C and 2C peaks are indicated. See text for details.
Next, the ability of Cdc1 overproduction to rescue each of the five pol3 mutants was tested by transforming plasmid pREP3X-Cdc1 into the pol3+/pol3– diploids, sporulating the transformants, and analysing the colonies that formed after re-growth over a wide range of temperatures (18–36.5°C). pREP3X-Cdc1 carries the cdc1+ gene under the control of the strong thiamine-repressible nmt1 promoter: under derepressed conditions, the levels of Cdc1 in cells are at least two orders of magnitude above endogenous Cdc1 levels (data not shown). However, no suppression by Cdc1 overproduction was observed at any temperature for any of the five mutant alleles. As would be expected, all five were rescued by near endogenous levels of expression of wild-type Sp Pol3 (from the reduced strength nmt81 promoter under repressed conditions). Overproduction of Cdc27 and Cdm1 also failed to rescue the mutants under the same conditions (expression from derepressed nmt1 promoter, 18–36.5°C).
Previously, a temperature-sensitive S.pombe pol3 allele encoding a protein with a mutation in ZnF2 has been identified (28); pol3-ts3 encodes a protein with an arginine to glutamine change at residue 1062, which is conserved across all known Pol3 orthologues (see Fig. 2). In the two-hybrid system, the corresponding mutation in S.cerevisiae ZnF2 did not affect Pol31 binding when assayed at 30°C (R1080A, see Fig. 3A). The ability of Cdc1 overproduction to rescue this allele was also examined; however, like the pol3 alleles described above, pol3-ts3 was not rescued by Cdc1 overproduction at a range of temperatures, from 32 to 36.5°C (data not shown).
Overproduction of the ZnF modules in fission yeast
The effects on fission yeast cells of overproducing the ZnF modules were tested. To allow detection of the overproduced ZnF modules, sequences encoding the ZnF module (amino acids 955–1084 of Sp Pol3) were fused to sequences encoding the Pk/V5 monoclonal antibody (mAb) epitope (30), such that the ZnF modules would be produced with an N-terminal Pk/V5 mAb tag (Fig. 6A). The expression construct was placed under the control of the strong thiamine-repressible nmt1 promoter in plasmid pREP4X. However, when transformed into wild-type cells, we failed to observe any phenotypic changes associated with overproduction of the V5-ZnF protein (data not shown).
Figure 6.
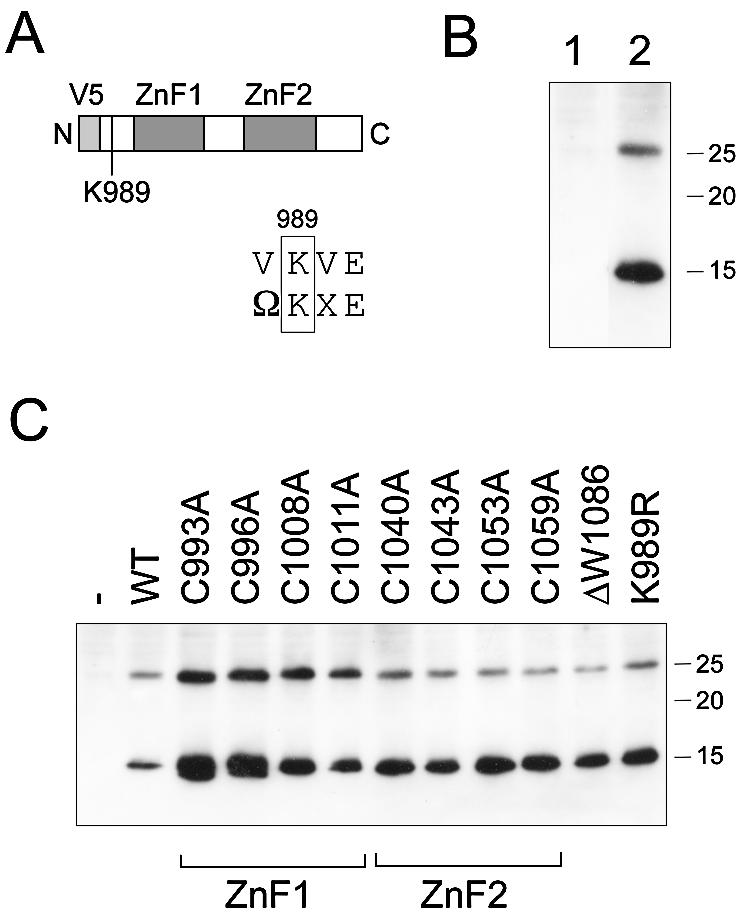
Putative post-translational modification of the fission yeast ZnF modules. (A) Schematic of the V5-ZnF fusion protein showing the relative location of the ZnF modules and the possible sumoylation site at Lys989 (inset: sequence surrounding K989 aligned with the consensus sumoylation site, where Ω indicates I, L or V). (B) Immunoblot using anti-V5 antibodies of total protein extracts prepared from cells carrying pREP4X (lane 1) or pREP4X-V5-ZnF (lane 2). Marker sizes are indicated (in kDa). (C) Immunoblot using anti-V5 antibodies of total protein extracts prepared from cells carrying pREP4X (lane 1), pREP4X-V5-ZnF (lane 2, labelled WT) or various ZnF1 or ZnF2 mutant pREP4X-V5-ZnF plasmids (lanes 3–12). Marker sizes are indicated (in kDa).
To confirm that the V5-Pol3 protein was being expressed, protein extracts were prepared from transformed cells, subjected to SDS–PAGE and immunoblotted using anti-V5 antisera. The ∼15 kDa V5-Pol3 protein was readily detectable by this method; however, a second immunoreactive species that migrated more slowly than the ∼15 kDa V5-Pol3 protein was also observed (Fig. 6B). This was only present in cells expressing V5-Pol3 (Fig. 6B) and was also seen, although at reduced intensity, in cells grown on thiamine, in which the nmt promoter is repressed (data not shown).
Next, the effect of mutating the cysteine residues in the V5-Pol3 protein on the appearance of the slower mobility form was examined. Each of the eight cysteines was mutated in turn to alanine and the mutant V5-Pol3 proteins expressed in S.pombe and visualized by immunoblotting following SDS–PAGE, as above. As shown in Figure 6C, none of the cysteine to alanine mutations affected the appearance of the slower mobility form. This suggests that the slower mobility form does not result from disulfide bond formation between adjacent cysteines affecting migration on SDS–polyacrylamide gels.
The possibility that the slower mobility form might be either mono-ubiquitinated or sumoylated was also examined, by immunoblotting using anti-ubiquitin and anti-fission yeast SUMO (Pmt3) antisera (31,32). However, no cross-reaction could be detected, suggesting that V5-Pol3 is not modified by either ubiquitin or SUMO. The latter was initially considered a possibility as the V5-Pol3 sequence contains a sequence similar to the SUMO consensus (VKVE where K989 represents a potential sumoylation site). Mutation of this sequence did not prevent the appearance of the slower mobility form (Fig. 6C). Further work aimed at characterizing this putative modified form of the Sp Pol3 ZnF region, and determining the biological role of the modification, is in progress. Note that it is unclear as yet whether the full-length Sp Pol3 protein is also present in two forms in wild-type cells, or whether the apparent modification is seen only when the ZnF modules are expressed on their own, in the absence of the catalytic domains. However, preliminary results indicate that the Pk/V5 epitope is not the target of the modification, as a protein in which the Pk/V5 epitope is replaced by the c-myc (9E10) mAb epitope is also detectable in low and high mobility forms, and the appearance of the modified form is not affected by treatment with λ protein phosphatase (F. Gray and S. A. MacNeill, unpublished results).
DISCUSSION
In eukaryotic cells, the processes of chromosomal DNA replication are dominated by three essential family B DNA polymerase enzymes: Pol α, Pol δ and Pol ε (3,33). Each of these enzymes is a multi-subunit entity, comprising an essential catalytic subunit and several additional subunits. The catalytic subunits of the three enzyme complexes are related to one another, and to the catalytic subunit of Pol ζ, consistent with their descent from a common ancestor. Phylogenetic studies have indicated that the catalytic subunit of Pol α was the first to diverge from the common lineage, then Pol ε. Finally, Pol δ and Pol ζ diverged. All four catalytic subunits possess two putative zinc finger modules close to their C-termini, and genetic analyses in budding yeast have shown that mutations within these modules abolish polymerase function in vivo, at least in the cases of Pol δ and Pol ε (4,18). Amongst the non-catalytic subunits, only the B-subunits of Pol α, Pol δ and Pol ε are related to one another (34). In budding yeast, each B-subunit (Pol12, Pol31 or Dpb2) is essential for complete chromosomal DNA replication and, therefore, for cell viability (3).
In this report, targeted proteolytic cleavage of the fission yeast Sp Pol3 protein at a position N-terminal to the ZnF region is shown to inactivate the protein (Fig. 1). This is presumably due to the separation of the N-terminal catalytic domains (amino acids 1–977, including both exonuclease and polymerase domains) from the ZnF modules (amino acids 978–1084), but it remains a formal possibility that removal of the ZnF modules actually results in significant destabilization of the catalytic domains. Due to technical limitations, it was not possible to test whether the catalytic domain remained intact following proteolytic cleavage of the Pol3-T2 protein. Cleavage was effected by a nuclear-localized form of the TEV protease, expression of which was under the control of the regulatable nmt1 promoter (see Materials and Methods). This required the seven amino acid TEV cleavage site (sequence ENLYFQS) to be introduced into the Sp Pol3 protein in such a way that Sp Pol3 function was not compromised by the insertion. Initially, the TEV cleavage site was placed between the two ZnF modules (Sp Pol3-T1 protein), rather than N-terminal to both (Sp Pol3-T2), but the Sp Pol3-T1 protein was non-functional. Two further attempts (Sp Pol3-T3 and Sp Pol3-T4) at placing the cleavage site between the two fingers also met with failure (Fig. 2); both proteins were non-functional, even in the presence of elevated levels of Cdc1 (S. A. MacNeill, unpublished results). These results illustrate the sensitivity of this region of the protein to sequence insertions and highlight a significant limitation of the use of the TEV protease in this manner, that insertion of the target site renders the protein non-functional even in the absence of protease expression. In a parallel study (P. Fulton and S. A. MacNeill, unpublished), TEV cleavage sites introduced into the Cdc1 protein in a region that appeared more likely to be able to tolerate sequence insertions (based on multiple sequence alignments across species) also resulted in a significant diminution of protein function, with the mutant cells being viable at suboptimal growth temperatures (18–20°C) only.
Nevertheless, induction of TEV expression in pol3-T2 cells resulted in efficient cell cycle arrest and inhibition of DNA replication, graphically illustrating the importance of the ZnF modules for Sp Pol3 function (Fig. 1). No effects of TEV induction were seen in wild-type S.pombe cells, suggesting that there are no essential proteins in the fission yeast nucleus that contain cleavable TEV target sites. Consistent with this, BLAST searching suggests that the S.pombe genome does not have the capacity to encode proteins with the optimal TEV cleavage site ENLYFQS (S. A. MacNeill, unpublished observations).
By two-hybrid analysis, the second ZnF module of Sc Pol3, ZnF2, was shown to be both necessary and sufficient for Pol31 binding (Table 1A). Mutational analysis highlighted the importance of the conserved cysteine residues, which presumably coordinate the metal ion, in Pol31 binding (Fig. 3), as well as a number of other residues adjacent to the cysteines. In the case of Sp Pol3, similar results were obtained when each of the four cysteines was individually mutated (Table 1B). A detailed understanding of the structure of the ZnF region, ideally complexed with the B-subunit, is required before more specific conclusions can be drawn about the effects of mutating amino acids other than the cysteines on Pol3 function. Very recently, the solution structure of a zinc-bound 38mer peptide corresponding to the ZnF2 region of the human Pol α catalytic subunit has been solved by NMR (22). This reveals a novel helix–turn–helix structure, with two cysteine residues being located in the first helix and two at the end of a loop region that comprises three distinct turns. Coordination of a zinc atom by the cysteines facilitates formation of the loop. Whether this structure provides an adequate model for Pol3 ZnF2 remains to be seen; despite their common evolutionary history, there is little primary structure conservation between the Pol1 and Pol3 ZnF regions (S. A. MacNeill, unpublished observations).
While one function of the ZnF2 is clear, the role of the first zinc finger module ZnF1 is not. Sequence alignments of Pol3 proteins from different organisms indicate that the primary sequence of the first ZnF is not as well conserved as that of ZnF2 (Fig. 2B). There are frequent short insertions in the sequence, particularly in the region between the cysteine pairs, which vary in length from nine to 14 residues. Also, in two of the sequences shown in Figure 2B (Pol3 proteins from the fungi Neurospora crassa and Magnaporthe grisea), the final cysteine is replaced by an aspartic acid residue. However, as is the case with ZnF2, individual replacement of the conserved cysteines in ZnF1 with alanine abolishes Sp Pol3 function in vivo (M.-E. Beydon and S. A. MacNeill, unpublished), indicating that this ZnF module has an essential function. It is unclear to what extent this function is distinct from that of ZnF2, as certain point mutations within ZnF1 were able to disrupt binding to Cdc1 in two-hybrid assays (M.-E. Beydon and S. A. MacNeill, unpublished). Further analysis of the role of ZnF1 is in progress.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank our friends and colleagues for their help during the course of this work, in particular Dr K. Sugimoto (University of Nagoya) for plasmid pBTM116-Hys2, Professor P. Legrain (Institute Pasteur) for permission to use the Fromont-Racine two-hybrid library, Professor J. Beggs (University of Edinburgh) for supplying the library and appropriate yeast strains and for advice on their use, and Dr B. Smith (University of Glasgow) for structural discussions. J.S.G. was supported by a BBSRC Research Committee studentship. S.A.M. and L.F.C. were supported by a Wellcome Trust Senior Research Fellowship in Basic Biomedical Sciences. X.Y. and S.E.K. were supported by BBSRC grant 43/G15095.
REFERENCES
- 1.Hindges R. and Hübscher,U. (1997) DNA polymerase δ, an essential enzyme for DNA transactions. Biol. Chem., 378, 345–362. [DOI] [PubMed] [Google Scholar]
- 2.Waga S. and Stillman,B. (1998) The DNA replication fork in eukaryotic cells. Annu. Rev. Biochem., 67, 721–751. [DOI] [PubMed] [Google Scholar]
- 3.MacNeill S.A. and Burgers,P.M.J. (2000) Chromosomal DNA replication in yeast: enzymes and mechanisms. In Fantes,P. and Beggs,J. (eds), The Yeast Nucleus. Oxford University Press, Oxford, pp. 19–57. [Google Scholar]
- 4.Dua R., Levy,D.L. and Campbell,J.L. (1999) Analysis of the essential functions of the C-terminal protein/protein interaction domain of Saccharomyces cerevisiae pol ε and its unexpected ability to support growth in the absence of the DNA polymerase domain. J. Biol. Chem., 274, 22283–22288. [DOI] [PubMed] [Google Scholar]
- 5.Kesti T., Flick,K., Keranen,S., Syvaoja,J.E. and Wittenberg,C. (1999) DNA polymerase ε catalytic domains are dispensable for DNA replication, DNA repair and cell viability. Mol. Cell, 3, 679–685. [DOI] [PubMed] [Google Scholar]
- 6.Feng W. and D’Urso,G. (2001) Schizosaccharomyces pombe cells lacking the amino-terminal catalytic domains of DNA polymerase ε are viable but require the DNA damage checkpoint control. Mol. Cell. Biol., 21, 4495–4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waga S. and Stillman,B. (1994) Anatomy of a DNA replication fork revealed by reconstitution of SV40 DNA replication in vitro. Nature, 369, 207–212. [DOI] [PubMed] [Google Scholar]
- 8.Waga S., Bauer,G. and Stillman,B. (1994) Reconstitution of complete SV40 DNA replication with purified replication factors. J. Biol. Chem., 269, 10923–10934. [PubMed] [Google Scholar]
- 9.MacNeill S.A., Moreno,S., Reynolds,N., Nurse,P. and Fantes,P.A. (1996) The fission yeast Cdc1 protein, a homolog of the small subunit of DNA polymerase δ, binds to Pol3 and Cdc27. EMBO J., 15, 4613–4628. [PMC free article] [PubMed] [Google Scholar]
- 10.Zuo S., Gibbs,E., Kelman,Z., Wang,T.S., O’Donnell,M., MacNeill,S.A. and Hurwitz,J. (1997) DNA polymerase δ isolated from Schizosaccharomyces pombe contains five subunits. Proc. Natl Acad. Sci. USA, 94, 11244–11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reynolds N., Watt,A., Fantes,P.A. and MacNeill,S.A. (1998) Cdm1, the smallest subunit of DNA polymerase δ in the fission yeast Schizosaccharomyces pombe, is non-essential for growth and division. Curr. Genet., 34, 250–258. [DOI] [PubMed] [Google Scholar]
- 12.Zuo S., Bermudez,V., Zhang,G., Kelman,Z. and Hurwitz,J. (2000) Structure and activity associated with multiple forms of Schizosaccharomyces pombe DNA polymerase δ. J. Biol. Chem., 275, 5153–5162. [DOI] [PubMed] [Google Scholar]
- 13.Podust V.N., Chang,L.S., Ott,R., Dianov,G.L. and Fanning,E. (2002) Reconstitution of human DNA polymerase δ using recombinant baculoviruses: the p12 subunit potentiates DNA polymerizing activity of the four-subunit enzyme. J. Biol. Chem., 277, 3894–3901. [DOI] [PubMed] [Google Scholar]
- 14.MacNeill S.A., Baldacci,G., Burgers,P.M. and Hubscher,U. (2001) A unified nomenclature for the subunits of eukaryotic DNA polymerase δ. Trends Biochem. Sci., 26, 16–17. [DOI] [PubMed] [Google Scholar]
- 15.Simon M., Giot,L. and Faye,G. (1991) The 3′ to 5′ exonuclease activity located in the DNA polymerase δ subunit of Saccharomyces cerevisiae is required for accurate replication. EMBO J., 10, 2165–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldsby R.E., Lawrence,N.A., Hays,L.E., Olmsted,E.A., Chen,X., Singh,M. and Preston,B.D. (2001) Defective DNA polymerase δ proofreading causes cancer susceptibility in mice. Nature Med., 7, 638–639. [DOI] [PubMed] [Google Scholar]
- 17.Goldsby R.E., Hays,L.E., Chen,X., Olmsted,E.A., Slayton,W.B., Spangrude,G.J. and Preston,B.D. (2002) High incidence of epithelial cancers in mice deficient for DNA polymerase δ proofreading. Proc. Natl Acad. Sci. USA, 99, 15560–15565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simon M., Giot,L. and Faye,G. (1993) A random mutagenesis procedure—application to the pol3 gene of Saccharomyces cerevisiae. Gene, 127, 139–144. [DOI] [PubMed] [Google Scholar]
- 19.Giot L., Chanet,R., Simon,M., Facca,C. and Faye,G. (1997) Involvement of the yeast DNA polymerase δ in DNA repair in vivo. Genetics, 146, 1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giot L., Simon,M., Dubois,C. and Faye,G. (1995) Suppressors of thermosensitive mutations in the DNA polymerase δ gene of Saccharomyces cerevisiae. Mol. Gen. Genet., 246, 212–222. [DOI] [PubMed] [Google Scholar]
- 21.Mizuno T., Yamagishi,K., Miyazawa,H. and Hanaoka,F. (1999) Molecular architecture of the mouse DNA polymerase α–primase complex. Mol. Cell. Biol., 19, 7886–7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Evanics F., Maurmann,L., Yang,W.W. and Bose,R.N. (2003) Nuclear magnetic resonance structures of the zinc finger domain of human DNA polymerase α. Biochim. Biophys. Acta, 1651, 163–171. [DOI] [PubMed] [Google Scholar]
- 23.Hollenberg S.M., Sternglanz,R., Cheng,P.F. and Weintraub,H. (1995) Identification of a new family of tissue-specific basic helix–loop–helix proteins with a two-hybrid system. Mol. Cell. Biol., 15, 3813–3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moreno S., Klar,A. and Nurse,P. (1991) Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol., 194, 795–823. [DOI] [PubMed] [Google Scholar]
- 25.Sambrook J. and Russell,D.W. (2001) Molecular Cloning: A Laboratory Manual, 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 26.Prentice H.L. (1992) High-efficiency transformation of Schizosaccharomyces pombe by electroporation. Nucleic Acids Res., 20, 621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fromont-Racine M., Rain,J.C. and Legrain,P. (1997) Toward a functional analysis of the yeast genome through exhaustive two-hybrid screens. Nature Genet., 16, 277–282. [DOI] [PubMed] [Google Scholar]
- 28.Francesconi S., Park,H. and Wang,T.S.F. (1993) Fission yeast with DNA polymerase δ temperature-sensitive alleles exhibits cell-division cycle phenotype. Nucleic Acids Res., 21, 3821–3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morrison A., Christensen,R.B., Alley,J., Beck,A.K., Bernstine,E.G., Lemontt,J.F. and Lawrence,C.W. (1989) REV3, a Saccharomyces cerevisiae gene whose function is required for induced mutagenesis, is predicted to encode a nonessential DNA polymerase. J. Bacteriol., 171, 5659–5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Randall R.E., Hanke,T., Young,D. and Southern,J.A. (1993) Two-tag purification of recombinant proteins for the construction of solid matrix–antibody–antigen (SMAA) complexes as vaccines. Vaccine, 11, 1247–1252. [DOI] [PubMed] [Google Scholar]
- 31.Ho J.C., Warr,N.J., Shimizu,H. and Watts,F.Z. (2001) SUMO modification of Rad22, the Schizosaccharomyces pombe homologue of the recombination protein Rad52. Nucleic Acids Res., 29, 4179–4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tanaka K., Nishide,J., Okazaki,K., Kato,H., Niwa,O., Nakagawa,T., Matsuda,H., Kawamukai,M. and Murakami,Y. (1999) Characterization of a fission yeast SUMO-1 homologue, pmt3p, required for multiple nuclear events, including the control of telomere length and chromosome segregation. Mol. Cell. Biol., 19, 8660–8672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang T.S.F. (1996) Cellular DNA polymerases. In DePamphilis,M.L. (ed.), DNA Replication in Eukaryotic Cells. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 461–493.
- 34.Makiniemi M., Pospiech,H., Kilpelainen,S., Jokela,M., Vihinen,M. and Syvaoja,J.E. (1999) A novel family of DNA polymerase-associated B subunits. Trends Biochem. Sci., 24, 14–16. [DOI] [PubMed] [Google Scholar]