

Abstract
To determine the role of surfactant protein C (SP-C) in host defense, SP-C-deficient (Sftpc−/−) mice were infected with the pulmonary pathogen Pseudomonas aeruginosa by intratracheal injection. Survival of young, postnatal day 14 Sftpc−/−ice was decreased in comparison to Sftpc+/+ mice. The sensitivity to Pseudomonas bacteria was specific to the 129S6 strain of Sftpc−/− mice, a strain that spontaneously develops interstitial lung disease-like lung pathology with age. Pulmonary bacterial load and leukocyte infiltration were increased in the lungs of Sftpc−/− mice 24 h after infection. Early influx of polymorphonuclear leukocytes in the lungs of uninfected newborn Sftpc−/− mice relative to Sftpc+/+ mice indicate that the lack of SP-C promotes proinflammatory responses in the lung. Mucin expression, as indicated by Alcian blue staining, was increased in the airways of Sftpc−/− mice following infection. Phagocytic activity of alveolar macrophages from Sftpc−/− mice was reduced. The uptake of fluorescent beads in vitro and the number of bacteria phagocytosed by alveolar macrophages in vivo was decreased in the Sftpc−/− mice. Alveolar macrophages from Sftpc−/− mice expressed markers of alternative activation that are associated with diminished pathogen response and advancing pulmonary fibrosis. These findings implicate SP-C as a modifier of alveolar homeostasis. SP-C plays an important role in innate host defense of the lung, enhancing macrophage-mediated Pseudomonas phagocytosis, clearance and limiting pulmonary inflammatory responses.
Pulmonary surfactant is a lipoprotein-rich complex that is required for normal pulmonary function and host defense. Pulmonary surfactant lowers surface tension to maintain alveolar stability during breathing and impedes infection by various pathogens. Surfactant homeostasis is maintained by precise regulation of synthesis, secretion, and recycling by alveolar type II epithelial cells and its degradation by alveolar macrophages (1, 2). Conditions that compromise surfactant function are associated with respiratory distress. These include surfactant insufficiency due to premature birth or inactivation of surfactant due to lung injury or infection. Mutation in genes essential for surfactant homeostasis including Sftpb, Sftpc Gmcsf, and Abca3 causes acute or chronic lung disease (2–5).
Pulmonary surfactant protein C (SP-C)4 is synthesized in type II cells as a 21-kDa precursor (proSP-C) that is proteolytically processed to the active secreted peptide. The mature form of SP-C is a 35 aa peptide that is distinguished by an extended hydrophobic valine rich domain from residues 9–23. In a lipid environment, SP-C forms an α helical structure that spans lipid bilayers. The hydrophobic nature of SP-C is further enhanced by palmitoylation of two adjacent N-terminal cysteine residues. SP-C increases the rate of adsorption of lipids into a surface film and alters the organization of lipid acyl side chains (2, 6). Synthetic lipid preparations containing only SP-C restore the dynamic compliance and oxygenation of lungs in surfactant depleted animal models (7). Surfactant extracts enriched in SP-C and phospholipids are used to treat neonatal respiratory distress syndrome in newborn infants (8–10).
Surfactant proteins play important roles in innate host defense of the lung. An elaborate host defense system suppresses infection from inhaled microorganisms while minimizing inflammation. The innate pulmonary defense system consists of physical, cellular, and biochemical barriers. The surfactant layer acts as both a barrier and a source of proteins that modulate inflammatory signaling in the alveolus and enhance the elimination of pathogens. For example, surfactant protein A (SP-A) and surfactant protein D (SP-D) bind to various infectious microorganisms as well as to LPS and other bacterial and fungal components to enhance uptake and killing by phagocytic cells (11). SP-C binds to the lipid A component of bacterial LPS and to CD14, a component of the cellular LPS signaling complex (12, 13). SP-C inhibits LPS interactions with macrophages in vitro, potentially reducing overt phagocytic cell activity in the alveolus (14). These in vitro findings imply that SP-C neutralizes LPS to influence subsequent inflammatory stimuli in the alveolar compartment.
Mutations in the gene encoding SP-C (Sftpc) have been linked to hereditary forms of interstitial lung disease (ILD). Mutations that alter the structure of the proSP-C precursor predominate, whereas a smaller number of patients lack a primary mutation yet still have a selective deficiency of mature SP-C (15, 16). Clinical findings in SP-C-dependent ILD are highly variable even among affected individuals from the same family (17). Morphological changes due to SP-C deficiency include cellular infiltrates, alveolar remodeling, airspace loss, and fibrosis. Episodes of respiratory insufficiency in SP-C-deficient individuals are often preceded by pulmonary infection from a variety of viral or bacterial sources (16, 17). These findings suggest that the inability to process the aberrant proSP-C or loss of SP-C in the alveolus predisposes the lungs to infection. The SP-C gene-targeted mice (Sftpc−/−) on a 129S6 background develop a progressive lung injury with features similar to those of familial ILD in humans (18, 19). To determine whether SP-C plays a role in innate host defense of the lung, Sftpc−/− mice were challenged with the pulmonary pathogen Pseudomonas aeruginosa. P. aeruginosa is a common pathogen associated with pulmonary infection in cystic fibrosis and chronic diseases requiring ventilator support (20). In the present study we demonstrate that Sftpc−/− mice are susceptible to pulmonary infection by P. aeruginosa.
Materials and Methods
Animal husbandry
Sftpc−/− mice were generated by targeted gene inactivation as previously described (18, 19). Sftpc−/− mice were previously bred onto 129S6/SvEvTac (129S6) and FVB/N backgrounds to generate congenic 129S6 and FVB/N Sftpc−/− lines. Mice were maintained in a barrier facility and housed in sterilized cages with access to sterilized food, water, and filtered air. Lung homogenates were negative for culture of bacteria and fungus. Sentinel mice in the colony were not infected by bacterial or viral pathogens. All animals were handled under aseptic conditions. Animal studies were performed under protocols approved by the Institutional Animal Care and Use Committee of the Children’s Hospital Research Foundation (Cincinnati, OH).
Bacterial infection
A clinical isolate of mucoid P. aeruginosa from a single freezer stock was used to minimize variation in virulence throughout the study. Bacteria were grown overnight in tryptic soy broth at 37°C with continuous shaking. Broth cultures were centrifuged, and the pelleted bacteria washed and re-suspended in 1 ml of sterile PBS. The concentration of the inoculum was determined by quantitative culture on tryptic soy blood agar. Administration of P. aeruginosa was performed by intratracheal inoculation of bacteria diluted in PBS. Mice were anesthetized with isofluorane and the trachea exposed by a small anterior midline incision. A 30-gauge needle attached to a tuberculin syringe was inserted into the trachea, and 50 or 100 μl of inoculum dispensed into the lung of postnatal day 14 (PND14) pups or adult mice, respectively. Sterile PBS was injected as a control. The incision was closed with one drop of Nexaband. For in vivo macrophage phagocytic assays, mice were instilled with 8 × 105 P. aeruginosa that constitutively express bacterial GFP, a gift of Dr. T. Machem (University of California, Berkeley, CA).
Survival and bacterial clearance
To determine an effective bacterial dose 14-day-old Sftpc+/+ and Sftpc−/− mice were given 105–107 bacteria intratracheally. Mortality occurred at the 1 × 107 dose. Therefore, PND14 mice were intratracheally injected with 1–4 × 107 bacteria and placed in warmed incubators. Cages were checked at 1-h intervals for the first 4 – 6 h and then monitored continuously for changes in survival. For clearance studies, PND14 mice were administered 2–4 × 106 bacteria. Quantitative cultures of lung homogenates were obtained 4 and 24 h postinoculation at which time mice were given a lethal i.p. injection of ketamine/xylazine/acepromazine. The abdomen was opened and the animal exsanguinated by transection of the inferior vena cava to minimize pulmonary hemorrhage. Lungs were removed, weighed, and homogenized in 1 ml of sterile PBS. Aliquots of the homogenates were serially diluted and plated on tryptic soy-blood agar plates to quantitate the surviving bacteria. Bacterial colony counts were normalized to milligrams of lung wet weight per milliliter of recovered homogenate.
Lung histology
Lungs were inflated via a tracheal cannula at 20 cm of pressure with 10% buffered formalin and removed en bloc from the thorax. Each lobe was bisected, dehydrated, and embedded in paraffin. The 5-micron tissue sections were cut and stained with H&E or Alcian blue (Poly Scientific).
Bronchoalveolar lavage
For adult mice, lungs were lavaged three times with 1 ml of sterile PBS. For PND14 mice, the lungs were lavaged three times with 350-μl aliquots. bronchoalveolar lavage fluid (BALF) was centrifuged at 1250 rpm for 5 min, and the cell pellet resuspended in 1 ml of PBS. Total cell counts were obtained using a hemocytometer and differential cell counts were made on cytospin preparations using Diff-Quik stain (Scientific Products).
Macrophage phagocytic assays
Phagocytic activity of alveolar macrophages isolated from BALF was assessed by fluorescent bead uptake in vitro. Lungs from PND14 mice were lavaged with cold Hanks’ buffer and placed on ice. Lavage from six mice of each genotype was pooled, and cells were pelleted and resuspended in 1 ml of DMEM of 5% FBS with antibiotics. Cell counts were determined and 5 × 105 cells plated in 6-well dishes. Cells were allowed to adhere for 15 min, the medium and nonadherent cells removed, and the medium replaced. Examination of Diff-Quik-stained adherent cells indicated that the cultures were macrophages and free of polymorphonuclear leukocytes or lymphocytes. Fluorescent beads were added at a ratio of 100 beads per cell. The wells were gently washed after 1.5 h to remove nonadherent cells. The remaining cells were dislodged by scraping and collected by low speed centrifugation (2000 × g for 5 min). Cells were resuspended in 1 ml of PBS with 0.004% trypan blue to quench background fluorescence. FACS analysis was used to identify fluorescent-positive cells. The phagocytic index was calculated as the mean fluorescence multiplied by the number of gated events. Incremental peaks in fluorescence intensity corresponded to the number of beads internalized per cell.
For determination of phagocytic activity in vivo, mice were instilled with 8 × 105 fluorescent bacteria, P. aeruginosa that constitutively express bacterial GFP, and bronchoalveolar lavage performed 4 h postinfection. BALF was analyzed from individual mice or pooled from four mice for each genotype. BALF cells were pelleted, washed, and resuspended in 500 μl of PBS for FACS analysis. Event recording gates were set to exclude nonmacrophage cells based upon size and complexity. BALF from mice instilled with nonfluorescent P. aeruginosa was used in FACS determinations as a control for nonspecific autofluorescence.
Identification of protein differences in Sftpc+/+ and Sftpc−/− alveolar macrophages
Macrophages were collected from BALF of adult mice by selective adherence to tissue culture dishes, washed, and lysed in situ with 100 μl of SDS lysis buffer (50 mM Tris-HCl (pH 7.4)) supplemented with protease inhibitors (Sigma-Aldrich) and protein determine by bicinchoninic acid assay for cell lysate and cell-free BALF. BALF protein (5 μg) was dried, resuspended in Laemmli buffer, boiled 5 min, and loaded on 8–16% polyacrylamide gels (Invitrogen). Gels were silver stained, and select bands that differed in abundance were extracted and submitted for trypsin digest. Fragments were analyzed using a Bruker MALDI-TOF mass spectrometer (University of Cincinnati Proteomics and Mass Spectrometry Core Facility). Unique tryptic fragments were searched against the Profound database.
Results
Establishment of Pseudomonas infection model in Sftpc−/− (129S6) mice
On the 129S6 background, Sftpc−/− mice have normal lung morphology at birth but develop lung pathology consistent with ILD associated with hereditary SP-C deficiency. PND14 mice were chosen to assess pathogen susceptibility before development of overt pathological changes in the lungs of Sftpc−/− mice. PND14 also represented the earliest age at which intratracheal inoculation of bacteria and recovery of lavage fluid was reproducible.
Survival and bacterial clearance in Sftpc−/− mice
To determine an appropriate bacterial dose, 129S6 Sftpc+/+ and Sftpc−/− mice were inoculated by intratracheal injection with concentrations of P. aeruginosa ranging from 106 to 108 CFU and monitored for 24 h. Bacteria were poorly tolerated by the Sftpc−/− mice. Six hours after bacterial instillation, the Sftpc−/− mice were inactive and physically unresponsive to mild stimulation in comparison to the wild-type mice that remained active. The survival of PND14 Sftpc−/− mice was decreased when challenged with 107 P. aeruginosa (Fig. 1). In contrast, there was no difference in survival of adult 129S6 Sftpc+/+ and Sftpc−/− mice infected with P. aeruginosa. To determine whether SP-C deficiency altered bacterial clearance, PND14 mice were inoculated with a smaller dose of bacteria (2–4 × 106). Twenty-four hours after instillation, the lungs were removed and homogenized, and bacteria quantified by plating serial dilutions of the lung homogenates on tryptic soy-blood agar plates. By inspection of the excised lungs, injury was more extensive in Sftpc−/− than in Sftpc+/+ mice noted by extensive discoloration. Bacterial colony counts were 24-fold higher in the lung homogenates from PND14 129S6 Sftpc−/− mice (Fig. 2). When age-matched congenic FVB/N Sftpc+/+ and Sftpc−/− mice were similarly infected, there was no difference in the colony counts from lung homogenates. Based upon the reduced survival of the 129S6 Sftpc−/− mice, 2–4 × 106 bacteria were used for subsequent clearance and phagocytic assays and all studies completed in the 129S6 strain of Sftpc−/− mice.
FIGURE 1.
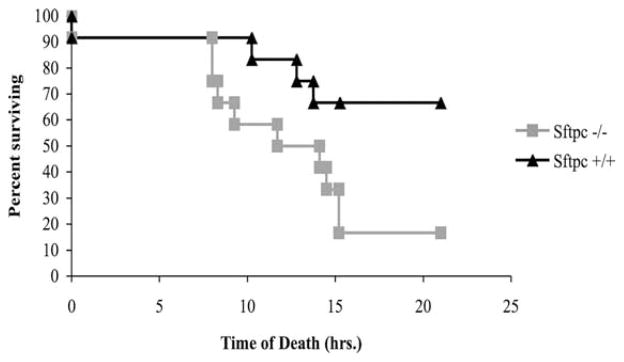
Decreased survival of Sftpc−/− mice following intratracheal challenge with P. aeruginosa. The survival of 2-wk-old mice was monitored after intratracheal instillation of 1 × 107 bacteria. Bacteria were poorly tolerated for the Sftpc−/− mice (n = 12 mice of each genotype). p = 0.017 determined by Kaplan-Meier survival.
FIGURE 2.
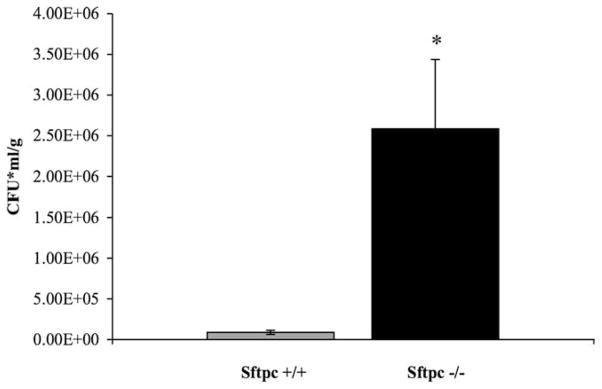
SP-C is required for normal clearance of P. aeruginosa. Concentrations of P. aeruginosa were determined 24 h after intratracheal administration of 1–4 × 107 bacteria to mice at 2 wk of age. Bacterial counts were significantly increased in the PND14 Sftpc−/− mice (n = 5 or 6 mice per genotype per experiment). *, p < 0.001, by t test.
Increased inflammation following P. aeruginosa challenge
Increased inflammation was observed in histological sections of lungs from PND14 Sftpc+/+ and Sftpc−/− mice 24 h postinfection. Tissue and airspace infiltrates contained neutrophils and enlarged macrophages. The cellular infiltration was more extensive in the lungs of Sftpc−/− mice and included areas of complete consolidation (Fig. 3). Airway inflammation occurred in response to P. aeruginosa infection and included distinct goblet cell hyperplasia. Mucin production was assessed by Alcian blue histochemistry (Fig. 4). Alcian blue staining cells were present in the airways of Sftpc−/− mice before exposure to bacteria. Following infection, the number of Alcian blue-positive cells and the intensity of staining increased dramatically in the airway epithelia of Sftpc−/− mice relative to the Sftpc+/+ airways (Fig. 4, B and D).
FIGURE 3.

Increased inflammation in the lungs of Sftpc−/− mice after infection with P. aeruginosa. Lungs of 2-wk-old Sftpc+/+ and Sftpc−/− mice were assessed before (A and B), and 24 h after (C and D) administration of P. aeruginosa. Areas of subtle interstitial thickening and inflammatory cells were infrequently seen in Sftpc−/− lungs before challenge (B). Extensive granulocytic infiltrates were observed in the lungs of infected Sftpc−/− mice (D) compared with infected control mice (C). Arrows indicate bronchiolar epithelium. Final magnification is shown at ×77.
FIGURE 4.

Goblet cell hyperplasia in lungs of Sftpc−/− mice in response to P. aeruginosa challenge. Lung sections were stained with Alcian blue before (A and B) and after (C and D) infection. Alcian blue staining was not detected in lungs of Sftpc+/+ mice (A). Alcian blue-positive staining was present in cells of the conducting airways of 2-wk-old Sftpc−/− mice (B). Alcian blue staining was detected in small clusters of goblet cells along the bronchiolar epithelium of Sftpc+/+ lungs following infection (C). In contrast, the number and intensity of Alcian blue staining cells were increased in the lungs of Sftpc−/− mice after infection (D). Final magnification is shown at ×77 (A and B) and at ×280 (C and D).
Total cell counts were increased in BALF from adult and PND14 mice 4 h postinfection (Fig. 5). Cell counts from BALF of infected adult Sftpc−/− mice were 2-fold higher than cell counts in Sftpc+/+ mice (Fig. 5A). Cell counts from BALF of infected PND14 Sftpc−/− mice were increased 4-fold over PND14 Sftpc+/+ mice (Fig. 5C). Neutrophils accounted for the majority of the cells in BALF after infection (Fig. 5, B and D). Large, foamy macrophages were identified in cytospin preparations from the BALF of Sftpc−/− mice. Neutrophil counts were also elevated in the BALF of uninfected adult and PND14 Sftpc−/− mice relative to BALF from Sftpc+/+ mice, consistent with a pre-established chronic low level inflammation due to SP-C deficiency (Fig. 5, B and D). In both Sftpc+/+ and Sftpc−/− mice, total protein levels in BALF increased 3 fold following infection consistent with increased injury. The BALF from Sftpc−/− or Sftpc+/+ mice (n = 6) was collected 4 h postinfection with Pseudomonas, pooled, and screened using specific ELISA kits (R&D Systems) for IL-1β, IL-4, IL-6, IL-13, and with an Ab array to 20 inflammatory cytokines (Ray Biotech) to profile changes in inflammatory mediators. Surprisingly there were no observable differences in cytokine levels between the two groups of mice (data not shown).
FIGURE 5.

Increased inflammatory cells in BALF from Sftpc−/− mice after infection. Total cell counts were elevated 4 h after challenge with bacteria. Total cell number was significantly higher (*, p = 0.035) in the BALF of adult Sftpc−/− mice (A) or (*, p = 0.03) the BALF of PND14 Sftpc−/− mice (C), compared with with Sftpc+/+ mice. Neutrophils comprise the majority of BALF cells after P. aeruginosa infection in adult Sftpc−/− mice (B) and more significantly (*, p = 0.01) in the young PND14 Sftpc−/− mice (D), compared with Sftpc+/+ mice. Although the percentage of neutrophils were similar, total neutrophils were increased in lungs of uninfected Sftpc−/− mice compared with Sftpc+/+ mice.
SP-C does not opsonize P. aeruginosa
SP-C preparations were mixed with bacteria before infection to determine whether direct interaction of SP-C with bacteria enhances phagocytosis or clearance. Bacterial colony counts from lung homogenates 24 h postinfection were not altered by pretreatment of the bacteria with a phospholipid preparation containing 2.5% purified human SP-C (Fig. 6). Thus, SP-C does not directly kill or enhance the uptake of bacteria to Sftpc−/− mice.
FIGURE 6.

SP-C does not restore phagocytic killing of P. aeruginosa. Bacteria were mixed with a synthetic phospholipid mixture or purified SP-C with phospholipids before infection of 2-wk-old Sftpc−/− mice. Bacterial counts were determined 24 h after infection. SP-C did not enhance bacterial clearance.
Phagocytic activity is reduced in alveolar macrophages from Sftpc−/− mice
Changes in alveolar macrophage morphology were observed in the lungs of Sftpc−/− mice. A subpopulation of the enlarged macrophages with numerous cytoplasmic inclusions is characteristic of Sftpc−/− mice. Multinucleated giant cells were observed in the alveolar spaces. Increased cell size and granularity of Sftpc−/− macrophages was verified by flow cytometry. To assess phagocytic activity, macrophages were isolated from PND14 mice and incubated with fluorescent-labeled beads in vitro. After 1.5 h, macrophages were recovered, and the uptake of beads determined by FACS analysis. The phagocytic index of Sftpc−/− macrophages was 64% of the index calculated for Sftpc+/+ macrophages (Fig. 7A). Phagocytic activity was evaluated in vivo after administration of fluorescent P. aeruginosa. Macrophage-associated fluorescence was determined in cells isolated from BALF 4 h after administration. Nonfluorescent P. aeruginosa were instilled in separate mice to control for nonspecific fluorescence. The phagocytic index of macrophages recovered from Sftpc−/− mice was reduced (Fig. 7B), supporting the concept that the lack of SP-C causes macrophage dysfunction leading to impaired bacterial uptake.
FIGURE 7.

Phagocytic activity of macrophages from Sftpc−/− mice. A, In vitro phagocytosis. Alveolar macrophages were collected from PND14 mice and incubated with fluorescent beads. The internalization was determined by FACS analysis. Peaks of descending magnitude indicate smaller populations of cells containing an increased number of beads. The phagocytic index of Sftpc+/+ and Sftpc−/− macrophages is shown (right) of the FACS profile (n = 3 mice). p = 0.04. B, In vivo phagocytosis. Phagocytosis by alveolar macrophages was determined by FACS analysis after instillation of P. aeruginosa that constitutively express bacterial GFP. The open peak corresponds to specific GFP fluorescence generated by ingestion of GFP expressing P. aeruginosa. The filled peak is nonspecific fluorescence produced by wild-type P. aeruginosa instilled into separate control mice. Phagocytosis of bacteria by Sftpc−/− macrophages was reduced (n = 3 mice). Mean fluorescence intensity is summarized (right) for macrophage-associated fluorescence in cells. p = 0.01. C and D, Sftpc−/− macrophages express markers of alternative activation. Arginase I gene (Arg I) expression in Sftpc+/+ and Sftpc−/− macrophages was assessed by RT-PCR of cDNA. Relative expression was normalized to β-actin expression (C). Gel electrophoresis identified a 40-kDa protein that was increased in macrophages and BALF from Sftpc−/− mice (D). The band was collected for sequence analysis by mass spectroscopy. The sequence of peptide fragments that match the protein Ym1 from both the macrophages and the BALF samples is shown below the gel image.
Sftpc−/− macrophages express markers associated with alternative activation
Alveolar macrophages from Sftpc−/− and Sftpc+/+ mice were analyzed for changes in gene expression or protein levels of molecules associated with an alternatively activated phenotype. By RT-PCR analysis, arginase I expression was increased 4-fold in Sftpc−/− macrophages (Fig. 7C). Increased arginase activity in macrophages contributes to matrix remodeling by promoting polyamine and subsequent collagen synthesis. The protein composition of Sftpc−/− and Sftpc+/+ macrophages was compared by gel electrophoresis of either whole cell lysates or BALF supernatant following cell isolation. Several abundant proteins were detected in Sftpc−/− macrophages after silver staining of the gel. Selected bands were excised from the gel, and the sequence determined for peptides generated by trypsin digestion. The peptide sequence generated from a prominent band of ~40 kDa had identity with the murine chitinase protein Ym1 (Fig. 7D). Sequence of 12 peptides from the macrophage sample matched with Ym1, covering 35% of the primary sequence. Sequence of eight peptides from the BALF sample matched Ym1 covering 28% of the primary sequence. The sequence of five tryptic fragments common to the whole macrophage lysate and supernatant bands are shown below the gel in Fig. 7D. Increased chitinase expression is associated with asthma and epithelial inflammation, and Ym1 is thought to contribute to innate defense.
Discussion
The increased susceptibility of Sftpc−/− mice to infection by the pulmonary pathogen P. aeruginosa was demonstrated in vivo. Bacterial clearance and survival was decreased and pulmonary inflammation was increased in Sftpc−/− mice compared with Sftpc+/+ mice after intratracheal delivery of P. aeruginosa. SP-C deficiency was associated with changes in macrophage activation state, supporting the critical role of SP-C in the maintenance and innate defense in the lung. These findings indicate that SP-C plays an important role in innate defense of the lung during Pseudomonas-induced pneumonia. The present study was designed to determine whether the susceptibility to P. aeruginosa was related to direct effects of SP-C on opsonization, to factors related to the inflammatory state of the lung, or to changes in macrophage function in Sftpc−/− mice.
The progressive pulmonary disease seen in 129S6 Sftpc−/− mice shares features with the familial SP-C-associated ILD found in humans (2, 19). Although a severe pulmonary disorder develops in 129S6 Sftpc−/− mice, there is virtually no pathology in the lungs of Sftpc−/− mice of the FVB/N background unless lung injury is induced in these mice. This strain-specific effect in mice suggests that other genes modify the severity of the SP-C-deficient phenotype. A role for modifier genes is also suggested in human disease because phenotypic heterogeneity is seen in SP-C-associated ILD among affected family members for whom injury can range from mild pulmonary insufficiency to severe fibrosis (17). The reports of bacterial and viral infections associated with exacerbations in SP-C-deficient patients supports the hypothesis that SP-C deficiency increases the effect of pathogens on lung inflammation and repair. For the current study the young mice were chosen for infection before the onset of observed inflammation or histopathologic changes to minimize the inflammatory effects seen in Sftpc−/− mice in pathogen clearance. PND14 129S6 Sftpc−/− mice had reduced survival and clearance of P. aeruginosa when compared with infected Sftpc+/+ mice. There was no difference in survival or clearance between PND14 FVB/N Sftpc+/+ and Sftpc−/− mice (data not shown). The impaired response of Sftpc−/− to Pseudomonas challenge was specific to the 129S6 Sftpc−/− strain that developed spontaneous lung injury with age. In contrast, there was no difference in survival between infected adult 129S6 Sftpc+/+ and Sftpc−/− mice. The selective susceptibility of the PND14 129S6 mice may be related to total lung macrophage content in which the less mature lungs have a smaller total resident macrophage population to respond to the Pseudomonas challenge. Alternatively, there may age-dependent differences in macrophage mobilization that alter the response. The rapid clearance of Pseudomonas in adult mouse lung and rapid injury when large bacteria inocula are used is problematic in other models of Pseudomonas-related lung disease. An outcome of the current study may be to improve Pseudomonas lung interactions by using young mice. The strain-specific response to P. aeruginosa challenge indicates that genetic factors modify pathogen susceptibility as well as the development of SP-C-deficient ILD.
The genetic factors affecting the host response to microbial infection among different inbred strains of mice are largely undefined. Comparison of susceptibility between the 129 and FVB mouse strains is even more limited. The 129 mice have been reported to be one of the most highly susceptible inbred strains to viral pulmonary pathogens (20). In a survey of the immediate response to P. aeruginosa in infection in 11 strains of adult inbred mice, 129 and FVB mice strains had similar clearance and inflammation at 6 h postinfection (21). The lack of strain sensitivity in that study is similar to the response of adult 129S6 and FVB/N mice in the current study. Effects of strain background in younger mice were not reported.
The genetic influence of the 129S6 background on susceptibility in young Sftpc−/− mice was unanticipated but reflects the variable disease progression that occurs among Sftpc−/− family members and among family members that carry the same cystic fibrosis mutation (22). Susceptibility to infection may be influenced by early activation of inflammatory signaling in the lungs of 129S6 Sftpc−/− mice. Microarray comparison of gene expression in the lungs of day 1 newborn 129S6 Sftpc+/+ and Sftpc−/− mice identified increased expression of host defense-related genes (>1.6-fold increase, p > 0.05 on triplicate samples), including chitinase 3-like 1 that is a family member with Ym1 as identified in this study, Ecsit a TLR accessory protein, Rhamm that is induced during oxidant-induced lung inflammation (23), and two antiproteases (data not shown). These initial findings suggest that there is activation of inflammation-related genes in the unchallenged 129S6 Sftpc−/− lung at birth. Lung microarray comparison between 129S6 and FVB/N strains of Sftpc−/− mice may identify sets of genes that contribute to SP-C-deficient disease.
The pulmonary inflammatory response to Pseudomonas infection in PND14 129S6 Sftpc−/− mice was vigorous and included increased cell infiltration and goblet cell hyperplasia. Pulmonary mucins are heterogeneous glycoproteins; increased mucin production is associated with colonization by P. aeruginosa in cystic fibrosis and chronic obstructive pulmonary disease patients (24–26). P. aeruginosa has been shown to adhere to respiratory mucins (27) and directly stimulate increased mucin synthesis by airway epithelial cells (28). Mucin production by airway epithelial cells is activated by MMP9 activity (29). Macrophages from the lungs of adult 129S6 Sftpc−/− mice were previously shown to have increased MMP9 activity (19). Therefore, the increased MMP9 levels produced by Sftpc−/− alveolar macrophages could influence mucin production and bacterial retention.
The surfactant proteins SP-A and SP-D play important roles in innate defense of a variety of pulmonary pathogens including P. aeruginosa. SP-A and SP-D null mice were found to have impaired clearance of mucoid P. aeruginosa (30, 31). SP-A was shown to opsonize and enhance phagocytic clearance of mucoid P. aeruginosa (32). SP-D also binds directly to P. aeruginosa and enhances phagocytosis (33). These findings demonstrated that SP-A and SP-D directly contribute to limiting a Pseudomonas infection. P. aeruginosa secretes proteases that degrade both SP-A and SP-D (34, 35). Thus proteolytic degradation of SP-A or SP-D might contribute to the increased mortality of Sftpc−/− mice in the current study. By Western blot analysis SP-A and SP-D levels were unchanged in the BALF of Sftpc+/+ and Sftpc−/− mice before or after infection (data not shown). When a purified human SP-C/phospholipid mixture was incubated with the P. aeruginosa before instillation, there was no increase in bacterial clearance by Sftpc−/− mice. Thus, SP-C does not appear to augment P. aeruginosa clearance by opsonization as do SP-A and SP-D. It is unlikely that the observed impaired clearance of P. aeruginosa is an opsonization defect. Our finding that SP-C does not enhance host defense by opsonization is consistent with reports suggesting that SP-C may confer alveolar protection by neutralizing inhaled inflammatory compounds or microorganisms. SP-C was shown to bind to bacterial LPS and reduce macrophage release of cytokines in vitro (14). It is thus conceivable that the lack of SP-C alters the alveolar microenvironment by increasing the exposure of the alveolar epithelium or sentinal alveolar phagocytes to inflammatory stimuli. The pulmonary inflammation and ILD-like injury that develops with age in 129S6 Sftpc−/− mice may arise from chronic alveolar inflammation when SP-C does not sequester inhaled proinflammatory agents.
A prominent feature of familial SP-C ILD is extensive alveolar infiltration and accumulation of macrophages. The SP-C patient index cases were initially classified as desquamative interstitial pneumonitis and chronic pneumonitis of infancy to reflect the macrophage injury (15). The pneumonitis-like histopathology of both the affected human and adult 129S6 Sftpc−/− lungs suggest an impaired macrophage response as a common link. The alveolar macrophages from 129S6 Sftpc−/− mice had altered morphology and reduced phagocytosis in vitro and in vivo and were previously shown to have increased metalloproteinase activity (19). The impaired phagocytosis and increased MMP9 activity suggested that the status of the Sftpc−/− macrophage had shifted from the classic protective function to a repair and remodeling function termed the alternatively activated macrophage (AAM) (36). The alternative activation state is associated with markers affecting cell-cell and cell-matrix interactions and other markers with only partially defined function. Murine AAM produce and secrete Ym1, a 42-kDa protein with chitinase sequence homology. Ym1 is thought to contribute to innate defense through lectin-like binding to glucosamine oligosaccharides and to heparan sulfate components of the extracellular matrix (37, 38). Ym1 protein was abundant in isolated alveolar macrophages and in the BALF collected from 129S6 Sftpc−/− mice. AAM have increased expression of the arginase I gene (ArgI) that alters arginine metabolism and macrophage function (38). ArgI mRNA expression was increased in the 129S6 Sftpc−/− macrophages. ArgI converts arginine to precursors of polyamines and collagen used in extracellular matrix production, tissue remodeling, and eventual fibrosis. ArgI activity depletes arginine from the NO synthetic pathway thus reducing macrophage bacteriocidal activity (38, 39). The increased Ym1 protein and ArgI expression of Sftpc−/− macrophages is consistent with an AAM phenotype and a reduced ability to suppress infection. The presence of herpesvirus has been linked to familial pulmonary fibrosis and idiopathic pulmonary fibrosis (40). Pulmonary macrophages acquire an AAM phenotype in herpesvirus-positive patients with pulmonary fibrosis and in mice with either herpesvirus-induced lung fibrosis or bleomycin-induced lung fibrosis (41, 42). Macrophages positive for ArgI and Ym1 localized to fibrotic lung tissue of herpes virus-infected mice and ArgI-positive macrophages associated with fibrotic regions of lungs from bleomycin-treated mice (42). Similarly ArgI-positive macrophages were associated with fibroblastic foci of idiopathic pulmonary fibrosis patients (41). Collectively these data implicate AAM as a shared feature between experimentally induced fibrotic lung disease, clinical idiopathic pulmonary fibrosis, and the ILD of Sftpc−/− mice, wherein fibrosis develops with age. Characteristics of macrophages from affected SP-C patients have not been reported.
SP-A, SP-D, and GM-CSF null mice have macrophages with altered morphology and impaired phagocytosis of P. aeruginosa yet apparently do not develop populations of AAM or fibrosis (30, 31, 43). Thus the loss of SP-C appears to elicit a distinct alveolar injury that modifies both macrophage function and airway cell response to challenge. The susceptibility of Sftpc−/− mice to Pseudomonas challenge supports the emerging concept that chronic Ag stimulation may underlie rare familial and idiopathic pulmonary fibrosis. The current findings indicate that SP-C deficiency perturbs the activation state of alveolar macrophages resulting in decreased phagocytosis and clearance of P. aeruginosa. SP-C deficiency per se renders mice susceptible to pulmonary Pseudomonas infection as well as replicating the progressive ILD and fibrosis seen in human SP-C deficiency.
Acknowledgments
We thank Dusti Folger, Teah Witt, and William Hull for technical assistance with experiments, Ann Maher for manuscript preparation, and Chenxia Duan for purification of human SP-C.
Footnotes
This work was supported by Grants HL50046 and HL61646 (to S.W.G., J.A.W., and D.R.P.), HL58795 (to T.R.K.), and by the Parker B. Francis Foundation (to A.P.S.).
Abbreviations used in this paper: SP-C, surfactant protein C; SP-A, surfactant protein A; SP-D surfactant protein D; ILD, interstitial lung disease; BALF, bronchoalveolar lavage fluid; MMP, matrix metalloproteinase; PND14, postnatal day 14; AAM, alternatively activated macrophage.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Goerke J. Pulmonary surfactant: functions and molecular composition. Biochim Biophys Acta. 1998;1408:79–89. doi: 10.1016/s0925-4439(98)00060-x. [DOI] [PubMed] [Google Scholar]
- 2.Whitsett JA, Weaver TE. Hydrophobic surfactant proteins in lung function and disease. N Engl J Med. 2002;347:2141–2148. doi: 10.1056/NEJMra022387. [DOI] [PubMed] [Google Scholar]
- 3.Nogee LM. Genetics of pediatric interstitial lung disease. Curr Opin Pediatr. 2006;18:287–292. doi: 10.1097/01.mop.0000193310.22462.1f. [DOI] [PubMed] [Google Scholar]
- 4.Whitsett JA, Wert SE, Trapnell BC. Genetic disorders influencing lung formation and function at birth. Hum Mol Genet. 2004;13:R207–R215. doi: 10.1093/hmg/ddh252. [DOI] [PubMed] [Google Scholar]
- 5.Whitsett JA. Genetic disorders of surfactant homeostasis. Paediatr Respir Rev. 2006;7(Suppl 1):S240–S242. doi: 10.1016/j.prrv.2006.04.191. [DOI] [PubMed] [Google Scholar]
- 6.Beers MF, Mulugeta S. Surfactant protein C biosynthesis and its emerging role in conformational lung disease. Annu Rev Physiol. 2005;67:663–696. doi: 10.1146/annurev.physiol.67.040403.101937. [DOI] [PubMed] [Google Scholar]
- 7.Davis AJ, Jobe AH, Hafner D, Ikegami M. Lung function in premature lambs and rabbits treated with a recombinant SP-C surfactant. Am J Respir Crit Care Med. 1998;157:553–559. doi: 10.1164/ajrccm.157.2.97-08019. [DOI] [PubMed] [Google Scholar]
- 8.Jobe AH. Pulmonary surfactant therapy. N Engl J Med. 1993;328:861–868. doi: 10.1056/NEJM199303253281208. [DOI] [PubMed] [Google Scholar]
- 9.Jobe AH. Mechanisms to explain surfactant responses. Biol Neonate. 2006;89:298–302. doi: 10.1159/000092866. [DOI] [PubMed] [Google Scholar]
- 10.Jobe AH. Which surfactant for treatment of respiratory-distress syndrome. Lancet. 2000;355:1380–1381. doi: 10.1016/S0140-6736(00)02130-9. [DOI] [PubMed] [Google Scholar]
- 11.Wright JR. Immunoregulatory functions of surfactant proteins. Nat Rev Immunol. 2005;5:58–68. doi: 10.1038/nri1528. [DOI] [PubMed] [Google Scholar]
- 12.Augusto LA, Li J, Synguelakis M, Johansson J, Chaby R. Structural basis for interactions between lung surfactant protein C and bacterial lipopolysaccharide. J Biol Chem. 2002;277:23484–23492. doi: 10.1074/jbc.M111925200. [DOI] [PubMed] [Google Scholar]
- 13.Augusto LA, Synguelakis M, Johansson J, Pedron T, Girard R, Chaby R. Interaction of pulmonary surfactant protein C with CD14 and lipopolysaccharide. Infect Immun. 2003;71:61–67. doi: 10.1128/IAI.71.1.61-67.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Augusto LA, Synguelakis M, Espinassous Q, Lepoivre M, Johansson J, Chaby R. Cellular antiendotoxin activities of lung surfactant protein C in lipid vesicles. Am J Respir Crit Care Med. 2003;168:335–341. doi: 10.1164/rccm.200212-1440OC. [DOI] [PubMed] [Google Scholar]
- 15.Nogee LM, Dunbar AE, III, Wert SE, Askin F, Hamvas A, Whitsett JA. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med. 2001;344:573–579. doi: 10.1056/NEJM200102223440805. [DOI] [PubMed] [Google Scholar]
- 16.Amin RS, Wert SE, Baughman RP, Tomashefski JF, Jr, Nogee LM, Brody AS, Hull WM, Whitsett JA. Surfactant protein deficiency in familial interstitial lung disease. J Pediatr. 2001;139:85–92. doi: 10.1067/mpd.2001.114545. [DOI] [PubMed] [Google Scholar]
- 17.Thomas AQ, Lane K, Phillips J, III, Prince M, Markin C, Speer M, Schwartz DA, Gaddipati R, Marney A, Johnson J, et al. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med. 2002;165:1322–1328. doi: 10.1164/rccm.200112-123OC. [DOI] [PubMed] [Google Scholar]
- 18.Glasser SW, Burhans MS, Korfhagen TR, Na CL, Sly PD, Ross GF, Ikegami M, Whitsett JA. Altered stability of pulmonary surfactant in SP-C-deficient mice. Proc Natl Acad Sci USA. 2001;98:6366–6371. doi: 10.1073/pnas.101500298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glasser SW, Detmer EA, Ikegami M, Na CL, Stahlman MT, Whitsett JA. Pneumonitis and emphysema in SP-C gene targeted mice. J Biol Chem. 2003;278:14291–14298. doi: 10.1074/jbc.M210909200. [DOI] [PubMed] [Google Scholar]
- 20.Anh DB, Faisca P, Desmecht DJ. Differential resistance/susceptibility patterns to pneumovirus infection among inbred mouse strains. Am J Physiol. 2006;291:L426–L435. doi: 10.1152/ajplung.00483.2005. [DOI] [PubMed] [Google Scholar]
- 21.Wilson KR, Napper JM, Denvir J, Sollars VE, Yu HD. Defect in early lung defence against Pseudomonas aeruginosa in DBA/2 mice is associated with acute inflammatory lung injury and reduced bactericidal activity in naïve macrophages. Microbiology. 2007;153:968–979. doi: 10.1099/mic.0.2006/002261-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cutting GR. Modifier genetics: cystic fibrosis. Annu Rev Genomics Hum Genet. 2005;6:237–260. doi: 10.1146/annurev.genom.6.080604.162254. [DOI] [PubMed] [Google Scholar]
- 23.Zaman A, Cui Z, Foley JP, Zhao H, Grimm PC, Delisser HM, Savani RC. Expression and role of the hyaluronan receptor RHAMM in inflammation after bleomycin injury. Am J Respir Cell Mol Biol. 2005;33:447–454. doi: 10.1165/rcmb.2004-0333OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sadikot RT, Blackwell TS, Christman JW, Prince AS. Pathogen-host interactions in Pseudomonas aeruginosa pneumonia. Am J Respir Crit Care Med. 2005;171:1209–1223. doi: 10.1164/rccm.200408-1044SO. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Randell SH, Boucher RC University of North Carolina Virtual Lung Group . Effective mucus clearance is essential for respiratory health. Am J Respir Cell Mol Biol. 2006;35:20–28. doi: 10.1165/rcmb.2006-0082SF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Livraghi A, Randell SH. Cystic fibrosis and other respiratory diseases of impaired mucus clearance. Toxicol Pathol. 2007;35:116–129. doi: 10.1080/01926230601060025. [DOI] [PubMed] [Google Scholar]
- 27.Lillehoj EP, Hyun SW, Kim BT, Zhang XG, Lee DI, Rowland S, Kim KC. Muc1 mucins on the cell surface are adhesion sites for Pseudomonas aeruginosa. Am J Physiol. 2001;280:L181–L187. doi: 10.1152/ajplung.2001.280.1.L181. [DOI] [PubMed] [Google Scholar]
- 28.Kohri K, Ueki IF, Shim JJ, Burgel PR, Oh YM, Tam DC, Dao-Pick T, Nadel JA. Pseudomonas aeruginosa induces MUC5AC production via epidermal growth factor receptor. Eur Respir J. 2002;20:1263–1270. doi: 10.1183/09031936.02.00001402. [DOI] [PubMed] [Google Scholar]
- 29.Deshmukh HS, Case LM, Wesselkamper SC, Borchers MT, Martin LD, Shertzer HG, Nadel JA, Leikauf GD. Metalloproteinases mediate mucin 5AC expression by epidermal growth factor receptor activation. Am J Respir Crit Care Med. 2005;171:305–314. doi: 10.1164/rccm.200408-1003OC. [DOI] [PubMed] [Google Scholar]
- 30.LeVine AM, Kurak KE, Bruno MD, Stark JM, Whitsett JA, Korfhagen TR. Surfactant protein-A-deficient mice are susceptible to Pseudomonas aeruginosa infection. Am J Respir Cell Mol Biol. 1998;19:700–708. doi: 10.1165/ajrcmb.19.4.3254. [DOI] [PubMed] [Google Scholar]
- 31.Giannoni E, Sawa T, Allen L, Wiener-Kronish J, Hawgood S. Surfactant Proteins A and D enhance pulmonary clearance of Pseudomonas aeruginosa. Am J Respir Cell Mol Biol. 2006;34:704–710. doi: 10.1165/rcmb.2005-0461OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mariencheck WI, Savov J, Dong Q, Tino MJ, Wright JR. Surfactant protein A enhances alveolar macrophage phagocytosis of a live, mucoid strain of P. aeruginosa. Am J Physiol. 1999;277:L777–L786. doi: 10.1152/ajplung.1999.277.4.L777. [DOI] [PubMed] [Google Scholar]
- 33.Restrepo CI, Dong Q, Savov J, Mariencheck WI, Wright JR. Surfactant protein D stimulates phagocytosis of Pseudomonas aeruginosa by alveolar macrophages. Am J Respir Cell Mol Biol. 1999;21:576–585. doi: 10.1165/ajrcmb.21.5.3334. [DOI] [PubMed] [Google Scholar]
- 34.Mariencheck WI, Alcorn JF, Palmer SM, Wright JR. Pseudomonas aeruginosa elastase degrades surfactant proteins A and D. Am J Respir Cell Mol Biol. 2003;28:528–537. doi: 10.1165/rcmb.2002-0141OC. [DOI] [PubMed] [Google Scholar]
- 35.Beatty AL, Malloy JL, Wright JR. Pseudomonas aeruginosa degrades pulmonary surfactant and increases conversion in vitro. Am J Respir Cell Mol Biol. 2005;32:128–134. doi: 10.1165/rcmb.2004-0276OC. [DOI] [PubMed] [Google Scholar]
- 36.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 37.Chang N-C, Hung S-I, Hwa K-Y, Kato I, Chen J-E, Liu C-H, Chang AC. A macrophage protein, Ym1, transiently expressed during inflammation is a novel mammalian lectin. J Biol Chem. 2001;276:17497–17506. doi: 10.1074/jbc.M010417200. [DOI] [PubMed] [Google Scholar]
- 38.Nair MG, Guild KJ, Artis D. Novel effector molecules in type 2 inflammation: lessons drawn from helminth infection and allergy. J Immunol. 2006;177:1393–1399. doi: 10.4049/jimmunol.177.3.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Noël W, Raes G, Hassanzadeh Ghassabeh G, De Baetselier P, Beschin A. Alternatively activated macrophages during parasite infections. Trends Parasitol. 2004;20:126–133. doi: 10.1016/j.pt.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 40.Tang Y-W, Johnson JE, Browning PJ, Cruz-Gervis RA, Davis A, Graham BS, Brigham KL, Oates JA, Jr, Loyd JE, Stecenko AA. Herpesvirus DNA is consistently detected in lungs of patients with idiopathic pulmonary fibrosis. J Clin Microbiol. 2003;41:2633–2640. doi: 10.1128/JCM.41.6.2633-2640.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mora AL, Torres-González E, Rojas M, Corredor C, Ritzenthaler J, Xu J, Roman J, Brigham K, Stecenko A. Activation of alveolar macrophages via the alternative pathway in herpesvirus-induced lung fibrosis. Am J Respir Cell Mol Biol. 2006;35:466–473. doi: 10.1165/rcmb.2006-0121OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Endo M, Oyadomari S, Terasaki Y, Takeya M, Suga M, Mori M, Gotoh T. Induction of arginase I and II in bleomycin-induced fibrosis of mouse lung. Am J Physiol. 2003;285:L313–L321. doi: 10.1152/ajplung.00434.2002. [DOI] [PubMed] [Google Scholar]
- 43.Ballinger MN, Paine R, III, Serezani CHC, Aronoff DM, Choi ES, Standiford TJ, Toews GB, Moore BB. Role of granulocyte macrophage colony-stimulating factor during Gram-negative lung infection with Pseudomonas aeruginosa. Am J Respir Cell Mol Biol. 2006;34:766–774. doi: 10.1165/rcmb.2005-0246OC. [DOI] [PMC free article] [PubMed] [Google Scholar]