

Abstract
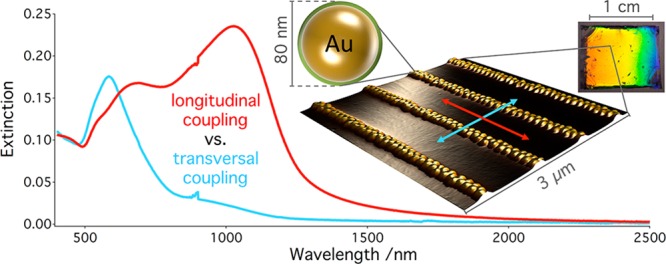
We present ensembles of surface-ordered nanoparticle arrangements, which are formed by template-assisted self-assembly of monodisperse, protein-coated gold nanoparticles in wrinkle templates. Centimeter-squared areas of highly regular, linear assemblies with tunable line width are fabricated and their extinction cross sections can be characterized by conventional UV/vis/NIR spectroscopy. Modeling based on electrodynamic simulations shows a clear signature of strong plasmonic coupling with an interparticle spacing of 1–2 nm. We find evidence for well-defined plasmonic modes of quasi-infinite chains, such as resonance splitting and multiple radiant modes. Beyond elementary simulations on the individual chain level, we introduce an advanced model, which considers the chain length distribution as well as disorder. The step toward macroscopic sample areas not only opens perspectives for a range of applications in sensing, plasmonic light harvesting, surface enhanced spectroscopy, and information technology but also eases the investigation of hybridization and metamaterial effects fundamentally.
Keywords: gold nanoparticle chains, protein coating, strong plasmon coupling, superradiant and subradiant modes, electromagnetic simulations
Though the investigation of plasmonic resonances is a classic topic of optics, the modern renaissance of plasmonic systems as tools for nanophotonics is intimately related to the study of strongly coupled plasmonic nanostructures. Strong coupling of localized plasmon resonances opens opportunities for tailoring near-fields and inducing drastic field enhancement, allows distance dependent tuning of absorption and scattering characteristics, and even enables the transition from localized resonances to guided plasmon polaritons.1 Consequently, a large range of applications, including surface-enhanced Raman spectroscopy (SERS),2−8 the fabrication of optical metamaterials,9−11 ultracompact optoelectronic circuitry,12−15 or plasmon-based light harvesting systems,16,17 has been targeted.
In all these applications and fundamental studies, the limited scalability with which strongly coupled plasmonic nanostructures can be fabricated is recognized as a bottleneck; many prospective applications such as plasmonic light harvesting require scaling to macroscopic areas at low costs. Similarly, experimental investigations of strongly coupling nanostructures are hampered by the fact that they have to be carried out on microscopic areas, which dramatically increases the instrumental efforts.
Compared to lithographically prepared structures, arrangements of nanoparticles produced by wet-chemical approaches are advantageous as these building blocks offer high crystallinity and can be synthesized in bulk processes that are scalable.18,19 Independent of the preparation method, strong coupling between plasmonic components is possible only for nanometer-sized gaps.20,21 Successful assembly of nanoparticles into such close-packed arrangements has been facilitated mainly by two techniques: chemical linking using either bifunctional molecules or specifically tailored DNA strands and convective assembly on lithographically structured templates.22−28 An advantage of these approaches is that the limited resolution of lithographic techniques, which ultimately determines the minimal gap size, can be overcome. Indeed, studies on close-packed assemblies combining single particle spectroscopy with high-resolution electron microscopy have contributed remarkably to a fundamental understanding of the optical properties of plasmonic nanoparticles and their arrangements.29−31 Besides small clusters,32,33 special attention has been dedicated to the investigation of linear nanoparticle chains,34,35 which show great potential for applications that require waveguiding below the diffraction limit.36 An important feature of these complex particle assemblies is their anisotropic optical response, in particular, a polarization dependency of the extinction cross section.
The fabrication of surface-supported particle assemblies with well-defined orientation is so far severely limited with regards to scalability. Chemical linking generally suffers from low yields and requires subsequent fractionation, whereas convective particle assembly demands template fabrication with nanoscale precision. Commonly employed techniques for template structuring, such as e-beam lithography or focused ion beam milling, run into the same scalability issues as direct lithographic fabrication of plasmonic nanostructures. Recently, we reported on the wrinkle-assisted assembly of nanoparticles as a lithography-free alternative for template-assisted self-assembly.37,38 This method circumvents typical scalability restrictions of conventional nanofabrication techniques and is capable of producing regular, close-packed assemblies as demonstrated for a variety of particle systems.39,40 However, the essential goal of creating assemblies of plasmonic nanoparticles that are both strongly coupling and at the same time homogeneous in their optical properties on macroscopic areas has not been achieved so far. In the present study, we demonstrate this crucial step toward the precise assembly of macroscopic scale arrays for the case of spherical gold nanoparticles that are coated with a nanoscopic protein shell. Utilizing a purely bottom-up, template-assisted approach, highly ordered, linear arrangements of closely spaced plasmonic nanoparticles, thus, can be fabricated with unprecedented uniformity over centimeter-squared areas. Strong plasmonic coupling between the gold nanoparticles gives rise to both dipolar and extremely separation-sensitive quadrupolar coupling modes. The well-defined plasmonic mode structure of the assembled nanoparticle chains is maintained on centimeter-squared areas, as we show by comparison of spectroscopic measurements and electrodynamic simulations. In particular, we find that effective interparticle distances (IPDs) of 1–2 nm can be homogeneously achieved over these areas and that consequently, collective resonances can be shifted up to a wavelength of 1500 nm, using particulate building blocks with an isolated plasmon resonance around 500 nm.
Colloidal Building Blocks
Spherical gold nanoparticles with an average diameter of 78 ± 4 nm were synthesized via a two-step seeded growth process.41 Template-assisted organization imposes several requirements on the colloidal interactions of the employed particles; long-range electrostatic attraction between the template and the particles has to be avoided and at the same time, the particles should maintain colloidal stability at elevated concentrations and possess hydrophilic surfaces.42 As prepared, the particles are coated by a layer of cetyltrimethylammonium bromide (CTAB), of which a large excess present in solution is necessary in order to ensure colloidal stability. During template-assisted assembly, this surfactant strongly adsorbs onto the surface of the used silica substrate, changing the wettability in an unfavorable fashion. Thus, the removal of CTAB is desirable for successful particle assembly.
If strongly coupled particle assemblies are targeted, a second aspect of the ligand coating becomes vital: the ligand layer ultimately limits the minimal interparticle distance. Indeed, earlier studies using CTAB-coated particles showed that only interparticle distances of 8 nm could be attained,39 which is comparable to twice the thickness of a CTAB layer.43 A suitable surface modification is achieved by ligand exchange with bovine serum albumin (BSA), which yields stable particles with a sufficiently thin shell and at the same time allows the complete removal of CTAB from the solution.44,45 For pH values above the isoelectric point of BSA, these particles have a negative zeta potential around −45 mV and can be used for the assembly process. Details on the employed gold nanoparticles can be found in the Supporting Information, including optical spectra in suspension before and after surface modification as well as pH dependent zeta potential measurements and a TEM characterization of the protein-coated particles (see Supporting Information, Figures S1 and S2). Whereas the dried protein shell itself is only few nanometers thick, interparticle distances even smaller than the resolution limit of our TEM can occur as the shell is soft and deformable. Thus, in close-packed assemblies very strong plasmonic coupling between the particles becomes feasible.
Nanoparticle Assembly
For templating we employed wrinkled elastomer stamps, which can be fabricated with the desired geometry by simple plasma oxidation of macroscopically strained PDMS stripes (see Figure 1A). Particle assembly into close-packed, linear chains is facilitated by spin coating. The subsequent transfer onto flat substrates can be accomplished effectively by wet contact printing (Figure 1A). Whereas a variety of parameters (pH, particle concentration, rotational acceleration and speed) influences the assembly structure formed during spin coating, we found adjustment of the wrinkle geometry to be the most reliable method for tuning the width of the particle chains. By varying the wrinkle periodicity in the range between 360 and 1060 nm, while maintaining adequate amplitudes for confinement, we were able to assemble the gold nanoparticles into close-packed single particle, dimer, and even tetramer lines. AFM images of the particle-filled PDMS stamps are included in Figure S3 in the Supporting Information.
Figure 1.

Assembly of nanoparticle chains: structured templates were fabricated by plasma oxidation of stretched PDMS stripes and subsequent relaxation (A). After spin coating the nanoparticle assemblies were transferred onto flat substrates by wet contact printing. Depending on the geometry of the employed wrinkles, single particle chains (B), dimer chains (C), or tetramer chains (D) were obtained with homogeneous surface coverage over centimeter-squared-scale areas. In reflection, the nanoparticle gratings diffract light (photographs).
Figure 1B–D display the morphology of the colloidal arrangements after transfer onto flat quartz substrates covering all length scales from nanoscopic (SEM), via microscopic (AFM) up to macroscopic (photography) dimensions. A quantitative transfer was achieved for all samples, yielding highly regular 1- and 2-dimensional nanoparticle chains in parallel orientation. The inset photographs in Figure 1B–D recorded under sideways illumination show strong iridescence due to diffraction by the periodic arrangement of the parallel chains. Clearly, the presented method enables the fabrication of nanoparticle chains with structural quality comparable to colloidal arrangements produced by intricate assembly techniques such as DNA strand linking or advanced lithographic templating.19,22,23,27,28 In contrast to those works, we achieved regular structure formation over centimeter-squared-scale areas, which for the first time allowed a correlation of extinction spectra, obtained by standard UV/vis/NIR spectroscopy, with simulated spectra of ideal particle chains.
Optical Characterization
For optical characterization of the particle assemblies, we utilized a commercial dual beam spectrometer configured in transmission geometry. For the entire spectral range probed, a halogen lamp was employed as the light source. Between the monochromator and the sample a rotatable polarizer was mounted. Light passing through the aligned samples was recorded by a detector array consisting of a photomultiplier tube covering the spectrum from 400 to 900 nm and a PbS detector for the near-infrared (NIR) regime between 900 and 2500 nm. Note that in the employed setup, the sample is irradiated under normal incidence, where grating effects that are responsible for the iridescent colors shown in Figure 1 are of minor importance. Indeed, under this illumination angle (Figure 2) the samples exhibit a grayish appearance, and despite the surface coverage not exceeding 25% of a dense particle monolayer on any substrate, the extinction caused by the assemblies is significant.
Figure 2.
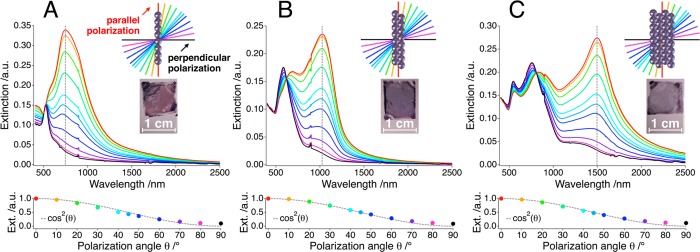
Optical characterization of nanoparticle chain assemblies: the polarization-dependent extinction spectra of single particle chains (A), dimer chains (B), and tetramer chains (C) display strong optical anisotropy with regard to the relative orientation between polarizer and assembled lines. Inset photographs illustrate the significant extinction caused by the nanoparticle films. The extinction at the absolute maxima follows a cos2 dependency as a function of the polarization angle θ (bottom).
Far-field UV/vis/NIR spectra recorded in air are displayed in Figure 2. For each morphology, 11 polarization angles between 0° and 90° relative orientation to the chain axis were probed. In contrast to isolated particles, which display a polarization-independent, single plasmon resonance peak at 520 nm (see Supporting Information, Figure S4) the microscopic organization of nanoparticles in parallel chains gives rise to a pronounced macroscopic optical anisotropy. Generally, for polarization perpendicular to the chains, extinction is limited mostly to the visible spectrum, whereas rotation of the polarization angle toward parallel orientation leads to increasing extinction in the near-infrared range. As shown in the lower section of Figure 2, the extinction efficiency of the pronounced NIR bands exhibit a clear cos2 dependence on the polarizer angle, which is typical for dipolar plasmonic modes.
Individual Chain Approximation
For an in-depth analysis of the measured spectra it is crucial to consider the probed surface area. In all measurements, the size of the illuminated spot was kept constant at approximately 1 × 10 mm2 corresponding to 28 000 single particle lines, 13 000 dimer lines, and 9 000 tetramer lines probed. Intriguingly, even with these huge ensembles we observe a distinct set of peaks for each chain morphology. We start the discussion of these features by considering individual chains and subsequently expand the model to assess the influence of defects (chain length distribution and interparticle distance fluctuations) as well as grating effects.
As the optical response of aligned nanoparticle chains is fully described by their extinction cross sections for parallel and perpendicularly polarized excitation, that is, their longitudinal and transversal modes, we will focus on the analysis of these two polarization states. In Figure 3, experimentally determined extinction spectra of single particle, dimer, and tetramer chain ensembles are compared to simulation data modeled for ideal chains via the generalized multisphere Mie theory (GMMT).46,47 The optical response of the investigated chain ensembles is largely determined by plasmonic coupling between adjacent nanoparticles, which allows a description via theoretical modeling of individual chains (see Figure 3).
Figure 3.

Experimental and modeled extinction cross sections of single particle (A), dimer (B), and tetramer chains (C): the spectra in the lower frames were modeled via GMMT for corresponding chain types consisting of 10 repeat units each. Blue curves in the experimental spectra illustrate the nongrating case for transversal polarization. FDTD modeled surface charge images of a single particle chain (D): the upper plot (1) features the dipolar transversal mode D (electric polarization vector E perpendicular to particle line, wave vector k perpendicular to particle line). The lower plot highlights the longitudinal subradiant and superradiant modes (2, 3). Black lines indicate the surface charge density integrated along the particle chain and perpendicular to it, respectively. Modeled surface charge images of a dimer chain (E): the left graph shows the transversal modes, namely, a dipolar D (5) and a quadrupolar QT coupling mode (4). The right graph displays the longitudinal quadrupolar coupling mode QL (6) and the radiant longitudinal modes (7, 8). The surface charge intensities were adjusted for better clarity.
Due to the limited resolution of SEM on nonconducting surfaces, a direct determination of the interparticle distance is not possible. We therefore took advantage of the well-known plasmon ruler concept, which allows estimating interparticle distances from the characteristic position of the energetically lowest resonance (superradiant mode).48 The spectral position of the superradiant mode is practically independent of the chain length above a threshold value of 10 repeat units, known as the “infinite chain limit”.34,49,50 Thus, we performed simulations for 10 repeat units and varied the interparticle distance. The best agreement was found for a gap size of 1.5 nm (see Figure 3). Further details on the selection of the parameters can be found in the Supporting Information in Figure S5 and S6. Despite the simplicity of this first approximation, we found a good agreement with the experimental data of the single particle and dimer chains (Figure 3). This elementary model also provides information beyond an estimation of the interparticle distance. The simulations predict the appearance of quadrupolar coupling modes for dimer and tetramer chains at approximately 530 nm, which are indeed visible in the experimental spectra. These modes will be discussed in detail later. In the case of the tetramer chains, the measured optical response is generally consistent with the simulations. However, the Fano-like resonances present in the modeled spectra at around 800 nm could not be observed experimentally. Fano resonances are very sensitive toward geometrical variations51 and most likely not resolved in the measured spectra due to averaging effects. Also, the experimental tetramer chain spectrum displays a stronger red shift of the superradiant mode than predicted by the simulations. For further discussion, we will therefore focus on the single particle and dimer chains, whose spectra exhibit remarkable agreement with the optical response of ideal colloidal assemblies modeled on the individual chain level.
The physical meaning of different plasmonic modes can be seen from the surface charge distributions at the specific resonance frequencies (Figure 3D/E). In the case of the single particle chain exposed to a polarization perpendicular to the chain axis (transversal excitation), we observe one plasmonic mode (peak 1) only. As expected, this transversal mode is located close to the single particle resonance in air (520 nm). The surface charge distribution pattern identifies this resonance D as a nonradiant dipolar mode, which is characterized by its stronger absorption than scattering cross section. For a better understanding of the complex charge distributions we integrated the surface charge density along the polarization axis (black lines in Figure 3D and E). An exposure to a polarization parallel to the chain axis (longitudinal excitation) reveals several radiant modes (peaks 2 and 3), for which scattering is predominant over absorption. These so-called chain plasmon resonances, termed as super- and subradiant modes, feature charge density distributions along the main chain axis that can be described by standing wave patterns.20 Whereas the most prominent feature, the energetically lowest mode, is defined by the presence of two nodal points at the chain ends, the less pronounced subradiant modes possess additional nodes in the charge density distribution. For longitudinal excitation at the corresponding wavelengths, the chain exhibits a net dipole moment allowing the experimental detection of these resonances as bright modes.
Densely packed dimer chains display similar mode characteristics as single particle chains. The surface charge distributions modeled for a hexagonal arrangement are presented in Figure 3E. Hereby, an interparticle spacing of 1.5 nm was assumed. For transversal excitation, the additional particle line gives rise to a new spectral feature. Due to the close packing of the nanoparticles, the transversal resonance peak splits into two distinct modes: a dipolar coupling mode D near 600 nm (peak 5) and an energetically higher, quadrupolar coupling mode QT (peak 4). Compared to the dipolar mode the quadrupolar mode displays one more polarity change as depicted by the integrated surface charge density (black line). Further investigations of isolated dimers are included in the Supporting Information and prove that the spectral position of the quadrupolar mode is extremely distance-sensitive (visible only for interparticle distances below 3 nm) but nearly independent of the relative orientation (Supporting Information Figures S7, S8, and S9). In fact, the surface charge distribution of the longitudinal, quadrupolar coupling mode QL displays a close resemblance to the transversal mode QT and can be excited with the same energy (peak 6). Our experimental spectra clearly show the presence of the quadrupolar mode as a left shoulder near 530 nm for both transversal and longitudinal polarization. To the best of our knowledge, this is the first report of such a highly sensitive plasmonic coupling effect being observed experimentally by a macroscopic ensemble measurement. For longitudinal excitation, the dimer chain further exhibits bright radiant modes that display wave-like surface charge distributions similar to that of the single particle line. In direct comparison, the longitudinal modes are more red shifted for the dimer line, which can be attributed to stronger retardation.35 Though the charge distribution assigned to the superradiant mode (peak 8) of the dimer chains shows strong similarity to the single particle chain, the subradiant mode (peak 7) exhibits two knots of the surface charge distribution concentrated near the center of the assembly.
So far, we have dedicated our attention to individual particle chains with a fixed number of repeat units, fixed interparticle distance, and without interaction to neighboring chains. This simple approach is a good first approximation but cannot fully describe the macroscopic optical response of the fabricated chain ensembles. SEM investigations show that structural defects can be avoided only to a certain degree. During assembly processes based on controlled drying, attractive capillary interaction between adjacent particles, jumping menisci, and shrinking of the originally hydrated shell can induce distance variations along the chains. In the case of plasmonic particles these variations can have a pronounced influence on the optical properties of the assembled structures. We find two characteristic types of defects: first, occasionally particles in the chain are missing; second, within the chains the gap size is not constant. In the following, we thus extend our initial approach from individual chain simulations to modeling of larger ensembles consisting of chains with different lengths and variable interparticle distance.
Variation of the Chain Length
We therefore conducted simulations of chains containing up to 120 particles arranged in single particle and dimer lines. The results are displayed in Figure 4. For both chain morphologies, the position of the superradiant mode converges to a fixed value at around 10 repeat units. However, raising the chain length further does have a pronounced influence on the number and the spectral positions of the subradiant modes. With increasing chain length, the subradiant modes converge toward the energy level of the superradiant mode, which is accompanied by considerable spectral broadening. In consequence, for both morphologies the longitudinal spectra of very long chains (repeat units ≫10) are dominated by a single, broad peak. In this regime, the subradiant modes are observed as a wavy left shoulder of the respective superradiant mode. Thus, the common approximation of long particle chains by a simplified model with 10 repeat units can be considered valid only for certain resonances (here: D, QT, QL, and the superradiant mode), whereas the appearance of the subradiant modes changes with the number of repeat units even for much longer chains.
Figure 4.

GMMT simulations of the longitudinal modes calculated with a constant interparticle distance of 1.5 nm and varying chain lengths: extinction spectra of single particle (A) and dimer (B) lines are shown for chain lengths up to 60 repeat units. The normalized extinction is mapped for all chain lengths in the image plots below the graphs. For both chain types, the energy of the superradiant mode converges toward a fixed value around 10 repeat units. Additional subradiant modes that occur with increasing chain length converge to the spectral position of the superradiant mode.
To assess the amount of defects in our assemblies, we studied the single particle and dimer lines via SEM and evaluated their chain length distributions. Though plasmonic coupling has been demonstrated for interparticle distances as large as 5× the particle radius for gold particles, the observed spectral shift decays almost exponentially with increasing gap size.52,53 Significant chain coupling is only feasible for distances below one particle radius (see Supporting Information, Figure S10). Consequently, we have chosen a maximum interparticle distance of 40 nm as the cutoff criterion for the statistical analysis. The determined chain length distributions are displayed in Figure 5. Clearly, the vast majority of the nanoparticles is located inside chain fragments containing more than 10 repeat units (84% in case of single particle lines and 96% for dimer lines). It must be noted that the maximum chain length measurable in the highly magnified SEM micrographs is limited to about 6 μm, for which the content of long chains may even be underestimated. Especially for the dimer chains, the limiting effect of finite image sizes is reflected in the statistical analysis by an accumulation of counts around 60 dimers (Figure 5B). However, due to the discussed convergence of the radiant modes, the impact of even higher chain lengths on the optical properties is minimal.
Figure 5.
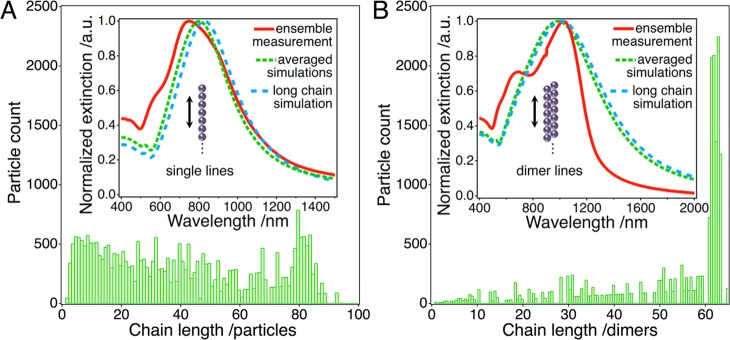
Statistical chain length distributions of single particle (A) and dimer lines (B) obtained from SEM images: the inset plots compare measured and modeled spectra. Besides GMMT simulations of individual long chains (120 particles, blue), averaged extinction spectra (green) calculated from the experimentally determined chain length distributions are shown.
Utilizing the previously established library of simulated spectra (Figure 4), we calculated ensemble extinction spectra of single particle and dimer lines by averaging based on the experimentally determined chain length distributions. The results are displayed in the insets of Figure 5 and compared to the measured optical responses as well as the simulated extinction spectra of extremely long, individual chains containing 120 particles. For the single particle lines, the averaged spectrum nearly matches the spectrum of an individual, long chain displaying little spectral broadening and only a minor blue shift. Further, this averaged ensemble spectrum provides an adequate description of the spectral position and form of the dominant longitudinal peak observed for the macroscopic ensemble. The averaging results cannot reproduce the left shoulder of the superradiant mode visible in the measured spectrum. Similar observations arise for the dimer lines. Again, the measured and modeled spectra are in excellent agreement regarding the position of the superradiant mode. However, the measurements clearly display a spectral modulation in the form of an additional peak near 700 nm not predicted by the averaged simulations as well as an unexpectedly sharp decay of the energetically lowest mode in the NIR region.
Possible reasons for these features are collective phenomena, dark mode excitation, or disorder. As the position of the spectral notch at 800 nm and the fast decay around 1200 nm both correspond to the periodicity of the line ensemble (d = 770 nm, see Figure 3B), grating effects are the most likely cause of these features. Grating effects due to the periodic arrangement of the nanoparticle lines manifest themselves in the transversal modes as deviations from the exponential decay. The expected spectral response in the absence of grating effects is illustrated for the transversal modes by the blue curves in Figure 3. Qualitatively similar effects are expected for the peak shapes in the longitudinal modes. This could explain the sharp decay of the superradiant mode. Further discussion is included in the Supporting Information (Figure S11). Another possible explanation could be the excitation of dark modes, which results in a similar signature with a spectral notch.54,55 These interesting additional features will be subject to future investigations.
Impact of Gap Size Variations within the Particle Chains
In the final section, we investigate the influence of irregular interparticle distances on the optical properties of individual chains of coupling nanoparticles and the respective chain ensembles. For this purpose, the spectral responses of linear assemblies containing 10 repeat units were modeled for an array of configurations with varying interparticle distances randomly distributed along the chains. The resulting longitudinal extinction spectra are illustrated for a selection of single particle and dimer chain ensembles with different interparticle distance variability in Figure 6A and B next to the corresponding gap size distributions. As references the spectra of chains modeled with a constant gap size of 1.5 nm (in green color) are included as well as the measured spectra (in orange color). A red shift compared to the reference could be observed only when allowing gap sizes smaller than 1.5 nm. Larger interparticle distances lead to an overall blue shift accompanied by spectral broadening. Thus, the spectra of individual chains react sensitively to variations of the gap sizes.
Figure 6.

Influence of disorder inside single particle (A) and dimer (B) chains with 10 repeat units: extinction spectra for randomly varied gap sizes (gray), averaged spectra (red/blue), and constant 1.5 nm gap size (green) are plotted next to the corresponding statistical distributions of the gap size. The position of the superradiant mode is correlated to the amount of disorder and allows estimating the gap size inside the fabricated single particle (C) and dimer (D) chains. All spectra were calculated using GMMT.
The relation of the superradiant mode λsuper to the mean interparticle distance (IPD) is of general interest. We describe the relationship in Figure 6C and D by the allometric power law
The coefficients a, b, and c are listed in the Supporting Information (Table S2). Since the peak position of the superradiant mode is largely independent of the chain length above the “infinite chain limit”, a comparison with the experimentally observed lowest energy peak enables an estimation of the effective gap size. For the single particle lines (peak located at 750 nm), our model yields a dominant gap size close to 2 nm. The superradiant mode of the dimer chains (peak located at 1025 nm) corresponds to an even smaller effective gap size of approximately 1 nm. This more sophisticated model is in excellent agreement with our first approximation assuming a fixed interparticle distance. Including the disorder in the particle chain calculations, we obtained a robust criterion to prove the realization of strongly coupled plasmonic modes over centimeter scale dimensions.
Conclusion
Our study highlights the ability of wrinkle-assisted particle assembly for precise nanofabrication over macroscopic areas, when combined with appropriate soft spacers. Already for the simple case of linear arrangements, strong coupling attributable to interparticle distances of 1–2 nm induces a remarkable shift of the main resonance from around 500 nm up to 1500 nm. This opens exciting perspectives for light harvesting because almost the entire range of the solar spectrum can be covered with one simple particulate building block.56 Likewise, we expect the assemblies to be of great interest for applications in surface-enhanced spectroscopy. Already with interparticle distances of 8 nm, high SERS enhancement factors of 108 were observed for similar arrangements,39 and SERS effects are expected to increase by orders of magnitude upon further minimization of the gap size. Finally, the strong coupling we have observed should impact on plasmonic waveguiding properties, especially because the assemblies can be fabricated with macroscopic lengths.
Beyond the specific example of parallel lines, this concept can be expanded to more complex geometries. We have demonstrated in earlier work for polymeric model particles that wettability structuring of substrates provides control over the transfer.42 Hydrophobic surface modifications effectively passivate the substrates against particle attachment. Thus, wettability structuring on the micron scale is a convenient way for breaking symmetries and enables the transition from infinite chains to arrangements of finite length (plasmonic polymers), which in turn has direct consequences on their scattering and absorption properties.34,35,57 The transfer printing process can be applied multiple times to the same substrate.42 Thus, not only are fishnet architectures attainable,38 but different particle types can also be combined with each other in a well-defined fashion.58,59 Concerning the particle types, this concept is indeed generic because the protein coating governs the colloidal interactions and wetting as well as the interparticle spacing, rather than the nature of the core. We would like to point out that we have limited our fabrication to centimeter-squared areas, but the procedure is readily scalable. The concept in this article involves exclusively macroscopic processing steps, namely, macroscopic strain and relaxation, plasma treatment, suspension drying, and printing transfer. We therefore believe that the approach presented here opens new avenues for both basic scientific investigations of strong coupling effects and a range of practical applications.
Acknowledgments
The research leading to these results has received funding from the European Research Council under the European Union’s Seventh Framework Programme (FP/2007-2013) / ERC Grant Agreement no. METAMECH-306686. We further acknowledge the German Research Foundation (DFG) for funding within the collaborative research center SFB 840. M.T. was supported by the Elite Network Bavaria within the framework of the Elite Study Program “Macromolecular Science” and funded via a grant for Ph.D. candidates according to Bavarian elite promotion law (BayEFG). Financial support from U.S. Department of Energy, Office of Basic Energy Sciences, Division of Materials Sciences and Engineering under Award # DE-FG02-09ER46604 (simulations) is gratefully acknowledged by T.K. and V.T. We thank the IT service center at the University of Bayreuth (btrzx3 cluster) and the Partnership for an Advanced Computing Environment (PACE) at Georgia Tech for computer resources. The authors further thank Dr. Nicolás Pazos-Pérez, Dr. Heiko Schoberth, Dr. Thorsten Schumacher, and Dr. Wolfgang Häfner for fruitful discussions as well as Dr. Markus Drechsler, Martina Heider, and Carmen Kunert for electron microscopy measurements.
Supporting Information Available
Additional information on the employed preparation methods and experimental procedures, nanoparticle characterization, AFM images of colloidal arrangements prior to transfer, an optical spectrum of randomly adsorbed particles, simulated spectra of gold nanoparticles, modeling of nanoparticle dimers by GMMT and FDTD, GMMT simulations of nanoparticle chains with different gap sizes, modeling of 2D grating effects. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Halas N. J.; Lal S.; Chang W.-S.; Link S.; Nordlander P. Chem. Rev. 2011, 111, 3913–3961. [DOI] [PubMed] [Google Scholar]
- Sharma B.; Frontiera R. R.; Henry A.-I.; Ringe E.; Van Duyne R. P. Mater. Today 2012, 15, 16–25. [Google Scholar]
- Nie S.; Emory S. R. Science 1997, 275, 1102–1106. [DOI] [PubMed] [Google Scholar]
- Camden J. P.; Dieringer J. A.; Wang Y.; Masiello D. J.; Marks L. D.; Schatz G. C.; Van Duyne R. P. J. Am. Chem. Soc. 2008, 130, 12616–12617. [DOI] [PubMed] [Google Scholar]
- Yan B.; Thubagere A.; Premasiri W. R.; Ziegler L. D.; Dal Negro L.; Reinhard B. M. ACS Nano 2009, 3, 1190–1202. [DOI] [PubMed] [Google Scholar]
- Lee A.; Andrade G. F. S.; Ahmed A.; Souza M. L.; Coombs N.; Tumarkin E.; Liu K.; Gordon R.; Brolo A. G.; Kumacheva E. J. Am. Chem. Soc. 2011, 133, 7563–7570. [DOI] [PubMed] [Google Scholar]
- Alba M.; Pazos-Perez N.; Vaz B.; Formentin P.; Tebbe M.; Correa-Duarte M. A.; Granero P.; Ferré-Borrull J.; Alvarez R.; Pallares J.; Fery A.; de Lera Á. R.; Marsal L. F.; Alvarez-Puebla R. A. Angew. Chem., Int. Ed. 2013, 52, 6459–6463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodiyath R.; Malak S. T.; Combs Z. A.; Koenig T.; Mahmoud M. A.; El-Sayed M. A.; Tsukruk V. V. J. Mater. Chem. A 2013, 1, 2777–2788. [Google Scholar]
- Kawata S.; Ono A.; Verma P. Nat. Photonics 2008, 2, 438–442. [Google Scholar]
- Pryce I. M.; Kelaita Y. A.; Aydin K.; Atwater H. A. ACS Nano 2011, 5, 8167–8174. [DOI] [PubMed] [Google Scholar]
- Boltasseva A.; Atwater H. A. Science 2011, 331, 290–291. [DOI] [PubMed] [Google Scholar]
- Maier S. A.; Kik P. G.; Atwater H. A.; Meltzer S.; Harel E.; Koel B. E.; Requicha A. A. G. Nat. Mater. 2003, 2, 229–232. [DOI] [PubMed] [Google Scholar]
- Kriesch A.; Burgos S. P.; Ploss D.; Pfeifer H.; Atwater H. A.; Peschel U. Nano Lett. 2013, 13, 4539–4545. [DOI] [PubMed] [Google Scholar]
- Solis D.; Paul A.; Olson J.; Slaughter L. S.; Swanglap P.; Chang W.-S.; Link S. Nano Lett. 2013, 13, 4779–4784. [DOI] [PubMed] [Google Scholar]
- König T. A. F.; Ledin P. A.; Kerszulis J.; Mahmoud M. A.; El-Sayed M. A.; Reynolds J. R.; Tsukruk V. V. ACS Nano 2014, 8, 6182–6192. [DOI] [PubMed] [Google Scholar]
- Reineck P.; Lee G. P.; Brick D.; Karg M.; Mulvaney P.; Bach U. Adv. Mater. 2012, 24, 4750–4755. [DOI] [PubMed] [Google Scholar]
- Atwater H. A.; Polman A. Nat. Mater. 2010, 9, 205–213. [DOI] [PubMed] [Google Scholar]
- Huang J.-S.; Callegari V.; Geisler P.; Brüning C.; Kern J.; Prangsma J. C.; Wu X.; Feichtner T.; Ziegler J.; Weinmann P.; Kamp M.; Forchel A.; Biagioni P.; Sennhauser U.; Hecht B. Nat. Commun. 2010, 1, 150. [DOI] [PubMed] [Google Scholar]
- Cui Y.; Björk M. T.; Liddle J. A.; Sönnichsen C.; Boussert B.; Alivisatos A. P. Nano Lett. 2004, 4, 1093–1098. [Google Scholar]
- Willingham B.; Link S. Opt. Express 2011, 19, 6450–6461. [DOI] [PubMed] [Google Scholar]
- Gupta M. K.; König T.; Near R.; Nepal D.; Drummy L. F.; Biswas S.; Naik S.; Vaia R. A.; El-Sayed M. A.; Tsukruk V. V. Small 2013, 9, 2979–2990. [DOI] [PubMed] [Google Scholar]
- Barrow S. J.; Funston A. M.; Wei X.; Mulvaney P. Nano Today 2013, 8, 138–167. [Google Scholar]
- Tan S. J.; Campolongo M. J.; Luo D.; Cheng W. Nat. Nanotechnol. 2011, 6, 268–276. [DOI] [PubMed] [Google Scholar]
- Mirkin C. A.; Letsinger R. L.; Mucic R. C.; Storhoff J. J. Nature 1996, 382, 607–609. [DOI] [PubMed] [Google Scholar]
- Alivisatos A. P.; Johnsson K. P.; Peng X. G.; Wilson T. E.; Loweth C. J.; Bruchez M. P.; Schultz P. G. Nature 1996, 382, 609–611. [DOI] [PubMed] [Google Scholar]
- Yin Y.; Lu Y.; Gates B.; Xia Y. J. Am. Chem. Soc. 2001, 123, 8718–8729. [DOI] [PubMed] [Google Scholar]
- Kraus T.; Malaquin L.; Schmid H.; Riess W.; Spencer N. D.; Wolf H. Nat. Nanotechnol. 2007, 2, 570–576. [DOI] [PubMed] [Google Scholar]
- Fan J. A.; Bao K.; Sun L.; Bao J.; Manoharan V. N.; Nordlander P.; Capasso F. Nano Lett. 2012, 12, 5318–5324. [DOI] [PubMed] [Google Scholar]
- Novo C.; Funston A. M.; Pastoriza-Santos I.; Liz-Marzán L. M.; Mulvaney P. Angew. Chem., Int. Ed. 2007, 46, 3517–3520. [DOI] [PubMed] [Google Scholar]
- Tcherniak A.; Ha J. W.; Dominguez-Medina S.; Slaughter L. S.; Link S. Nano Lett. 2010, 10, 1398–1404. [DOI] [PubMed] [Google Scholar]
- Funston A. M.; Novo C.; Davis T. J.; Mulvaney P. Nano Lett. 2009, 9, 1651–1658. [DOI] [PubMed] [Google Scholar]
- Barrow S. J.; Wei X.; Baldauf J. S.; Funston A. M.; Mulvaney P. Nat. Commun. 2012, 3, 1275. [DOI] [PubMed] [Google Scholar]
- Pazos-Perez N.; Wagner C. S.; Romo-Herrera J. M.; Liz-Marzán L. M.; García de Abajo F. J.; Wittemann A.; Fery A.; Alvarez-Puebla R. A. Angew. Chem., Int. Ed. 2012, 51, 12688–12693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrow S. J.; Funston A. M.; Gómez D. E.; Davis T. J.; Mulvaney P. Nano Lett. 2011, 11, 4180–4187. [DOI] [PubMed] [Google Scholar]
- Slaughter L. S.; Willingham B. A.; Chang W.-S.; Chester M. H.; Ogden N.; Link S. Nano Lett. 2012, 12, 3967–3972. [DOI] [PubMed] [Google Scholar]
- Solis D.; Willingham B.; Nauert S. L.; Slaughter L. S.; Olson J.; Swanglap P.; Paul A.; Chang W.-S.; Link S. Nano Lett. 2012, 12, 1349–1353. [DOI] [PubMed] [Google Scholar]
- Lu C.; Möhwald H.; Fery A. Soft Matter 2007, 3, 1530–1536. [DOI] [PubMed] [Google Scholar]
- Schweikart A.; Fortini A.; Wittemann A.; Schmidt M.; Fery A. Soft Matter 2010, 6, 5860–5863. [Google Scholar]
- Pazos-Perez N.; Ni W.; Schweikart A.; Alvarez-Puebla R. A.; Fery A.; Liz-Marzan L. M. Chem. Sci. 2010, 1, 174–178. [Google Scholar]
- Müller M.; Karg M.; Fortini A.; Hellweg T.; Fery A. Nanoscale 2012, 4, 2491–2499. [DOI] [PubMed] [Google Scholar]
- Pazos-Perez N.; Garcia de Abajo F. J.; Fery A.; Alvarez-Puebla R. A. Langmuir 2012, 28, 8909–8914. [DOI] [PubMed] [Google Scholar]
- Hanske C.; Müller M. B.; Bieber V.; Tebbe M.; Jessl S.; Wittemann A.; Fery A. Langmuir 2012, 28, 16745–16750. [DOI] [PubMed] [Google Scholar]
- Wang H.; Levin C. S.; Halas N. J. J. Am. Chem. Soc. 2005, 127, 14992–14993. [DOI] [PubMed] [Google Scholar]
- Strozyk M. S.; Chanana M.; Pastoriza-Santos I.; Pérez-Juste J.; Liz-Marzán L. M. Adv. Funct. Mater. 2012, 22, 1436–1444. [Google Scholar]
- Chanana M.; Gil P. R.; Correa-Duarte M. A.; Liz-Marzán L. M.; Parak W. J. Angew. Chem., Int. Ed. 2013, 52, 4179–4183. [DOI] [PubMed] [Google Scholar]
- Xu Y. Appl. Opt. 1995, 34, 4573–4588. [DOI] [PubMed] [Google Scholar]
- Gouesbet G.; Grehan G. J. Opt. A: Pure Appl. Opt. 1999, 1, 706–712. [Google Scholar]
- Sönnichsen C.; Reinhard B. M.; Liphardt J.; Alivisatos A. P. Nat. Biotechnol. 2005, 23, 741–745. [DOI] [PubMed] [Google Scholar]
- Harris N.; Arnold M. D.; Blaber M. G.; Ford M. J. J. Phys. Chem. C 2009, 113, 2784–2791. [Google Scholar]
- Slaughter L. S.; Wang L.-Y.; Willingham B. A.; Olson J. M.; Swanglap P.; Dominguez-Medina S.; Link S. Nanoscale 2014, 6, 11451–11461. [DOI] [PubMed] [Google Scholar]
- Hentschel M.; Dregely D.; Vogelgesang R.; Giessen H.; Liu N. ACS Nano 2011, 5, 2042–2050. [DOI] [PubMed] [Google Scholar]
- Su K. H.; Wei Q. H.; Zhang X.; Mock J. J.; Smith D. R.; Schultz S. Nano Lett. 2003, 3, 1087–1090. [Google Scholar]
- Müller M. B.; Kuttner C.; König T. A. F.; Tsukruk V. V.; Förster S.; Karg M.; Fery A. ACS Nano 2014, 8, 9410–9421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N.; Hentschel M.; Weiss T.; Alivisatos A. P.; Giessen H. Science 2011, 332, 1407–1410. [DOI] [PubMed] [Google Scholar]
- Gómez D. E.; Teo Z. Q.; Altissimo M.; Davis T. J.; Earl S.; Roberts A. Nano Lett. 2013, 13, 3722–3728. [DOI] [PubMed] [Google Scholar]
- Karg M.; König T. A. F.; Retsch M.; Stelling C.; Reichstein P. M.; Honold T.; Thelakkat M.; Fery A.. Mater. Today, DOI: 10.1016/j.mattod.2014.10.036. [DOI] [Google Scholar]
- König T.; Kodiyath R.; Combs Z. A.; Mahmoud M. A.; El-Sayed M. A.; Tsukruk V. V. Part. Part. Syst. Charact. 2014, 31, 274–283. [Google Scholar]
- Horn A.; Hiltl S.; Fery A.; Böker A. Small 2010, 6, 2122–2125. [DOI] [PubMed] [Google Scholar]
- Hiltl S.; Schürings M.-P.; Balaceanu A.; Mayorga V.; Liedel C.; Pich A.; Böker A. Soft Matter 2011, 7, 8231–8238. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.