

Abstract
From a small group of exotic compounds isolated only two decades ago, Cyclic Imine (CI) toxins have become a major class of marine toxins with global distribution. Their distinct chemical structure, biological mechanism of action, and intricate chemistry ensures that CI toxins will continue to be the subject of fascinating fundamental studies in the broad fields of chemistry, chemical biology, and toxicology. The worldwide occurrence of potent CI toxins in marine environments, their accumulation in shellfish, and chemical stability are important considerations in assessing risk factors for human health. This review article aims to provide an account of chemistry, biology, and toxicology of CI toxins from their discovery to the present day.
Introduction: CI Toxins as a new class of potent marine toxins
Marine toxins are a fascinating group of complex organic compounds that have had a lasting impact on human society. They pose a risk to human health through contamination of fish and shellfish – important components of a healthy human diet - and at the same time, they are sources of life-changing medicines. As an example, ciguatera is a debilitating condition caused by dinoflagellate toxins, ciguatoxins, that contaminate popular fish like black grouper, barracuda, and several species of snapper. Ciguatera is characterized by several symptoms, including vomiting, nausea, tingling, and cold allodynia, in which the sensations of heat and cold are reversed, and cold can stimulate a painful burning response. On occasion, these symptoms can reappear years after the original exposure and cause lifelong discomfort. There are about 50,000 cases of ciguatera reported annually worldwide.1 In another example, venom from the predatory snails of the genus Conus, conotoxins, have been developed as drug leads for neuropathic pain and other neurological conditions.2 Recent progress in the development of neuroactive marine natural products as biological probes has been reviewed.3 The appearance of microalgal toxins is increasing, possibly due to the rise in the ocean temperature that supports algal blooms. Detection of marine toxins in seafood is often hampered by inaccessibility of pure standards.
Non-protein-derived marine toxins with proven effect on humans have been historically divided into six classic groups based on symptoms, origin, and chemical structure: 1) paralytic shellfish poisoning (PSP, by saxitoxins), 2) Amnesic shellfish poisoning (ASP, by domoic acids), 3) Diarrheic shellfish poisoning (DSP, mainly by okadaic acids and dinophysistoxins), 4) Azaspiracid poisoning (AZP, by azaspiracids), 5) Neurotoxic shellfish poisoning (NSP, by brevetoxins) and 6) Ciguatera fish poisoning (CFP, by ciguatoxins).4 Since their discovery in mid-1990s, Cyclic Imine (CI) toxins (Figure 1), the subject of this article, form a new group of marine poisons that do not have proven record of acute toxicity towards humans. Nevertheless, their ever-increasing occurrence in warming oceanic environments and potent toxicity warrant vigilance in monitoring levels of CI toxins in marine products.
Figure 1.

Cyclic Imine toxins and closely related natural products identified as of May 2014.
Gymnodimine and pinnatoxin A are the first CI toxins that were structurally characterized, both, coincidentally, in 1995. Gymnodimine (2.0 mg) was isolated from 3 kg of oysters Tiostrea chilensis following the first recorded NSP incident on the North Island of New Zealand.5 The same study conclusively traced the origin of gymnodimine to a gymnodinoid dinoflagellate. Pinnatoxin A (PnTX A, 3.5 mg) was isolated from 45 kg of viscera of contaminated mussels Pinna muricata using bioassay-guided fractionation (i.p., mouse lethality).6 The algal source of PnTX A remained unknown for over two decades since its characterization.
Chemical Structure
The chemical structures of all CI toxins known to date are illustrated in Figures 1 and 2. These structures are unified by the presence of a characteristic spiroimine, or cyclic imine subunit that gives the name to this class of marine toxins. The spiroimine subunit is embedded within a large carbocyclic polyether framework that ranges in size from 14-membered for portimine and 16-membered for gymnodimines to 27-membered for pinnatoxins.
Figure 2.

Cyclic Imine toxins and closely related natural products identified as of May 2014.
The pinnatoxins and pteriatoxins form one of the largest subgroups of CI toxins. They also pose the highest risk for human health due to their potent oral toxicity, contrasting other CI toxins that loose their potency if administered orally (vide infra). A total of seven pinnatoxins and three pteriatoxins have been characterized to date. Shortly after the initial characterization of PnTX A (3 mg) along with 1.2 mg of PnTX B and C from 45 kg of the viscera of Pinna muricata, pinnatoxin D (2.0 mg) containing a γ-keto acid side chain at C33 was isolated from the same source.7 PnTX D features a somewhat modified macrocyclic skeleton with the addition of a methyl and a hydroxy group at C21 and C22, respectively, and the deletion of the 28-OH group. Three more pinnatoxins, PnTX E (1.9 mg), F (2.5 mg), and G (1.1 mg), extracted from Pacific oysters Crassostrea gigas and Pinna bicolor were reported in 2010.8 Pinnatoxins E and F share the macrocyclic unit of PnTX D but are distinct by the nature of the C33 substituent. Pinnatoxin G is most closely related to PnTX A, B, and C, again, with differences at the C33 position of the cyclohexene ring. In addition, accumulation of several 28-O-fatty acyl derivatives of pinnatoxins, mostly of PnTX G, was detected in Canadian mussels (Mytilus edulis) using LC-MS analysis.9 Pteriatoxins are the more scarcely studied members of the pinnatoxin subgroup. Pteriatoxins A-C (20 μg (A) and 8 μg (B,C)) were isolated as extremely toxic components of the viscera of Pteria penguin in 2001.10 These compounds bear a cysteine residue in the substituent at C33, and vary in the regio- and stereochemical features of the amino acid substituent.
Gymnodimines are among the smallest members of the CI family of toxins by molecular weight. The unsubstituted six-membered cyclic imine is one structural distinction of gymnodimines from other CI toxins. Four members have been described to date. These compounds are characterized by a labile 6,6-spirocyclic imine fragment embedded in a 16-membered carbocycle incorporating a tetrahydrofuran ring and one (GYM (A))5 or two (GYMs B11 and C)12 trisubstituted E-double bonds. The structure of 12-methyl gymnodimine C is not fully assigned, with the configuration of two stereocenters unknown.13
The spiroimine subunit of the spirolides hybridizes the seven-membered cyclic imine of pinnatoxins and the substituted cyclohexene of gymnodimines. More than 11 individual members of the spirolide subgroup have been characterized. One of the structural variations is found in the substitution pattern of the cyclic imine, which can carry either one or two methyl groups. Spirolides A14 and B15 with a C32-monomethyl substituted imine have been found to be unstable and readily hydrolysed to naturally occurring spirolides E and F,16 respectively, where the cyclic imine is replaced with the acyclic amino ketone subunit. Spirolides C,14 D,15 G,17, 18 H, and I19 all have 31,32-dimethyl substituted imine ring and appear to be substantially more stable to hydrolysis. The hydrolytic stability parallels the potency of these marine toxins. Another distinction in the structure of the spirolides is the ketal fragment. In contrast to pinnatoxins, the ketal unit of spirolides A-F possesses a 6,5,5-dispirotricyclic arrangement, while the bridged EF-ketal of pinnatoxins is absent, being replaced with a smaller allylic alcohol linker. Spirolides G and 20-methyl spirolide G have a homologous 6,6,5-dispiro bis-ketal, and the more recently discovered spirolides H and I contain a truncated dioxaspiro[4.5]decane monoketal unit.
In addition to the four major subgroups of CI toxins, other CI natural products include prorocentrolide,20 spiro-prorocentrimine,21 portimine,22 and symbioimine.23 The first two are among the largest members of the family by molecular weight and are not well studied. Portimine has been isolated recently from the newly discovered dinoflagellate Vulcanodinium rugosum. Symbioimine, a distantly related marine natural product with a tricyclic decahydro-2H-benzo[de]quinolinium structure, was isolated from the marine dinoflagellate Symbiodinium sp.24
Isolation and Algal origin
Pinnatoxins and pteriatoxins
The identification of the microalgal producer of pinnatoxins has been challenging and ultimately was correlated to the discovery of a new dinoflagellate from a French Mediterranean lagoon, Ingril, in 2011. A phylogenetic study based on LSU rDNA sequence data analysis confirmed that the taxon of the dinoflagellate is new and that it belongs to the order of Peridiniales. However, it was not affiliated with any particular family or a known genus, and it has been named Vulcanodinium rugosum (Figure 3).25 Using a high-quality synthetic reference standard in combination with LC-MS/MS analysis, samples derived from a culture of V. rugosum from Ingril Lagoon has confirmed it to be a producer of pinnatoxin G.26 Through morphological and phylogenetic analysis of algal samples from Japan, New Zealand, and Australia, V. rugosum has been determined to be the sole species producing PnTXs G, E, and F in these areas.27 In 2012, the occurrence of fatty acid esters of pinnatoxins A and G in mussels from Canada (Mytilus edulis), likely at the 28-OH position, has been described for the first time based on mass-spectrometric evidence. By analogy with other classes of toxins,28 it has been suggested that the modification likely occurred in the mussels rather than in the dinoflagellate source.9
Figure 3.

Contrast phase (A) and scanning electron micrographs (B and C) of the dinoflagellate Vulcanodinium rugosum. Note in (A), the cell nucleus (n). (B) Ventral view of the dinoflagellate. Note the small indentation in the c6 plate (white arrow). (C) Smaller form of the dinoflagellate revealing the arrangement of its thecal surface. Adapted with permission from Nézan and Chomérat (2011; Figures 4, 7 and 25). D) Producer of the spirolides, Alexandrium ostenfeldii.39 Scale bar = 20 μm.
Pteriatoxins have been isolated from 82 kg of the viscera of bivalve Pteria penguin.10 As of now, there is still no conclusive evidence for the natural source of pteriatoxins, and it is unclear whether they are produced directly by algae or by metabolic modification of pinnatoxins in higher organisms.8
Gymnodimines
The original isolation of gymnodimine from New Zealand oysters Tiostrea chilensis was immediately linked to a concurrent bloom of a gymnodinoid dinoflaggelate identified at the time as Gymnodinium cf. mikimotoi, which suggested a clear connection between these events. Analysis of a 60 L culture of G. cf. mikimotoi by HPLC, 1H NMR, and LC/MS methods suggested it to be the source of gymnodimine.5 Subsequently, the planktonic dinoflagellate Karenia selliformis, a new species from the genus Karenia (Dinophyceae),29 was unambiguously confirmed to produce gymnodimine and analogs gymnodimines B and C.11,12 In all likelihood, this is not a complete list of algal species that produce gymnodimine.30 Interestingly, the dinoflagellate Alexandrium peruvianum was reported to produce both the novel gymnodimine cogener 12-methylgymnodimine and 13-desmethyl spirolide C.13 This is the first account of the co-occurrence of gymnodimines and spirolides in a single microorganism, suggesting that common CI biosynthetic pathways exist between K. selliformis and A. peruvianum dinofliagellates, despite their belonging to distinct phylogenetic groups within the Dinophyceae (orders Gymnodiniales and Peridiniales, respectively).
Spirolides
All known spirolides have so far been linked to two producing organisms, dinoflagellate Alexandrium ostenfeldii14,17,19,31,32,33,34,40 and Alexandrium peruvianum.13,35,36,37 A. ostenfeldii is a gonyaulacoid38 dinoflagellate that has long been considered confined to Northern cold-temperature oceanic waters (Figure 3D). However, it is now known to be a globally distributed species. A. ostenfeldii is typically present as a background species at relative low concentrations of 100 to 1000 cells per L in combination with more populous bloom-forming phytoplankton.39 Geographically, the spirolides were first detected in the Atlantic coast of Nova Scotia (Canada) and subsequently in shellfish and phytoplankton extracts from European, North American, and South American coasts.
A recent LC/tandem MS analysis of cultured mussels sampled in 2002 and 2003 near Skjer, Norway, led to the detection of fatty acid metabolites of spirolide G, most likely formed in the mussels contaminated with phytoplankton A. ostenfeldii.40
Other CI natural products
As suggested by their names, prorocentrolide and spiro-prorocentrimine were found in related dinoflagellates Prorocentrum sp. – a neritic species that can be found in both temperate as well as tropical oceans, attached to the surface of brown and red algae and benthic debris. In 1988, prorocentrolide was detected in Prorocentrum lima,20 which is also the source of the DSP toxin okadaic acid and its esters.41 The CI toxin (70 mg) was isolated as an amorphous from a 1000 L culture of the dinoflagellate, which was originally collected at Sesoko Island, Okinawa, in 1985. Cultures of benthic Prorocentrum lima PL1 and an unknown species PM08 (100 L) from the Taiwanese coral reef waters were studied for the presence of toxins. The strain PL1 produced prorocentrolide analogs, 4-hydroxyprorocentrolide and 18-O-acetyl-4-hydroxyprorocentrolide. From PM08, a new compound, spiro-prorocentrimine (3 mg) was isolated along with prorocentrolide. As expected, okadaic acid and DTX-1 were also co-isolated in the same extract.21
Seafood Contamination and Acute Toxicity
The phytoplankton constitutes the basis of the marine food chain, and is an essential source of nutriments for filter-feeding bivalve mollusk (oysters, clams, mussels, and scallops), crustaceans, and finfish in marine environments, including aquacultures. Under favorable environmental condition during certain periods of the year, or in certain geographical areas, dinoflagellates K. selliformis, A. ostenfeldii / A. peruvianum and V. rugosum, which form part of the phytoplankton species of unicellular algae, may proliferate and produce CI phycotoxins. At present it is not clear why and how these dinoflagellates produce the CI toxins or what their biological function is. It is also unclear as to what triggers their massive proliferation in the so-called harmful algae blooms. Harmful algal blooms are worldwide natural phenomena with increasing occurrences. At present, the ecological dynamics of most toxic dinoflagellates producing phycotoxins are not well understood. In addition, existing models for predicting their proliferation are unreliable, and the general consensus is that there is a broad global increase in levels of toxic dinoflagellates spanning all geographic zones.42,43,44,45 Although the causes of this global increase remain unexplained, it is hypothesized to be linked to anthropogenic influence on climate change and eutrophication.46,47,48 Filter-feeding bivalves naturally ingest most dinoflagellate species and are thus exposed to a variety of toxic components. The accumulation and persistence of toxicity in bivalves is species-dependent, and the toxicity also varies according to the concentration of the dinoflagellate in the bloom and with the rates of acquisition and elimination of toxin.49,50 During active dinoflagellate blooms, CI phycotoxins may accumulate and concentrate in the digestive glands and edible tissues of shellfish, and act as a vector for the transfer of these chemical compounds into crabs, fish, birds, marine mammals, and ultimately to humans, endangering wildlife and human health.
Using liquid chromatography coupled to mass spectrometry (LC-MS) or to tandem mass spectrometry (LC-MS/MS) a four-year retro-analysis survey revealed that at Ingril Lagoon (France), where the V. rugosum dinoflagellate was characterized, pinnatoxin G is a recurrent toxin in both mussels (Mytilus galloprovincialis) and clams (Venerupis decussata). High maximum concentrations of pinnatoxin G were observed in mussels every year, e.g., 261 μg kg−1 (in 2009), 1244 μg kg−1 (2010), 568 μg kg−1 (2011) and 631 μg kg−1 (2012) with an overall maximum of 1244 μg kg−1.26 So far, maximum concentrations have been reported to be below 110 μg kg−1 for whole shellfish flesh in Canada,9 below 120 μg kg−1 in Norway,121 and below 200 μg kg−1 in New Zealand.51 Thus, so far every year, the maximum concentration of pinnatoxin G observed in mussels at Ingril Lagoon exceeded all other previously reported levels. Interestingly, significant differences were observed between the accumulation of pinnatoxin G in mussels and in clams. On average, mussels contained seven- to eight-fold higher pinnatoxin G levels on the same sampling location (Ingril Lagoon, France), confirming that mussels are good sentinel species for pinnatoxins.26 Spirolide uptake and depuration have been studied experimentally by exposing oysters (Crassostrea gigas) to cultures of the toxic dinoflagellate A. ostenfeldii producing 13,19-didesmethyl spirolide C, 13-desmethyl spirolide C, 13-desmethyl spirolide D, and traces of spirolide D. After a four-day exposure, 83% of the total initial spirolide content was found in the digestive glands, and only 17% in remaining tissues, as determined by LC-MS/MS. Inflammatory responses were observed in the intestinal tract of oysters, but no mortality was reported. Detoxification of spirolides from digestive glands occurred within seven days and was improved by feeding oyster with non-toxic micro algae.52 Gymnodimine, has also been reported to be retained not only in the digestive gland of shellfish, but also in the remaining tissues, and is not readily depurated from shellfish. The long-time of residence in shellfish has been related to the lipo-soluble character of the phycotoxin.30,53,123 Contamination/detoxification studies in grooved carpet clams fed with dinoflagellate cultures of K. selliformis, as the source of gymnodimine, showed that clam’s filtration rate was impaired with no evidence of mortality. After 7 days of experimental contamination, 97% of gymnodimine was concentrated in the clam’s digestive glands, exhibiting a faster detoxification rate (5–7 days) in this tissue than from other tissues.54 The kinetics of detoxification of gymnodimine A in the digestive glands of toxic clams (Ruditapes decussatus) was shown to be biphasic, with an exponential decrease of 78% of the total amount during the first 12 days, followed by a slow decay between days 12 and 29 of about 7%. After 29 days, the amount of gymnodimine A remaining in clams corresponded to 15% of the initial.55
At present, there is no feasible means of preventing the uptake of CI toxins by shellfish, or of eliminating them immediately after harvesting. The situation is complicated further by the fact that shellfish may be contaminated at the same time by several phycotoxins from the same or distinct toxin family groups.121 To assess CI toxicity, most studies to date have been performed in rodents using mouse and rat bioassays, with toxins extracted either from cultured dinoflagellates, or from contaminated bivalves. Therefore, current knowledge on acute toxicities has been very much dependent on the performance of methods used for CI toxin extraction, even though in most cases chemically characterized and purified toxins have been used. Crude extracts of V. rugosum were recently reported to display significant cytotoxicity against mouse neuroblastoma Neuro2A cells and KB cells (a sub-line of the ubiquitous keratin-forming tumor cell line HeLa) after only 24 to 72 h of incubation, respectively. In the Caco-2 cell line (derived from human epithelial colorectal adenocarcinoma cells), 48 h after exposure to the crude extract of V. rugosum cell cycle arrest was observed accompanied by a dramatic increase in double strand DNA breaks indicating marked genotoxicity. In contrast, no reduction in cell viability was observed with purified PnTX G on the different cell lines examined.56 Fractionation and dereplication of extracts revealed the presence of a number of compounds, including nakijiquinone A, N-carboxy-methyl-smenospongine or stachybotrin A, when evaluated using full scan and tandem high resolution mass spectrometry in combination with the MarinLit™ database. In the extracts of V. rugosum, the identity of the compounds responsible for cytotoxicity was not determined.
The 13-desmethyl spirolide C (10–500 nM) was reported to exert no cytotoxicity for periods of up to 24 h in neuroblastoma BE(2)-M17 cells.108 Similarly, 13-desmethyl spirolide C did not produce obvious cytotoxicity when incubated for 24 h with various cultured mammalian cell lines including: liver HepG2, neuroblastoma NIE-115, adipocyte 3T3-L1, ovarian cancer SKOV-3, skeletal muscle C2C12, and macrophage RAW264.7 cells, as revealed by various indicators of cell viability such as lactate dehydrogenase, mitochondrial respiration, and lysosomal function.60 Gymnodimine (1–10μM) and analogs, when tested on Neuro2a cells, had variable effect on reducing cell viability, as determined by the MTT assay, and had no effect on the expression of several signal transduction proteins (c-Jun, ATF-2, ATF-3). However, pre-exposure of Neuro2a cells to gymnodimine for 24 h greatly sensitized these cells to the apoptotic action of okadaic acid.57
Thus, typical cytotoxicity tests are not appropriate for predicting the in vivo toxicity of CI toxins. The gold standards to assess the toxicity of phycotoxins have traditionally been in vivo methods, i.e., the mouse as well as the rat bioassay. In addition to ethical concerns, there is also a need for more reliable test methods, as has been discussed recently.58,59
CI toxins are considered to be “fast-acting toxins” because they exhibit a rapid onset of neurological symptoms followed by death, usually within 3–50 minutes after intraperitoneal (i.p.) injection in mouse. All CI toxins studied so far are rapidly absorbed, since mouse symptoms of intoxication are observed shortly after i.p. administration and are quite similar among various CI toxins investigated. These symptoms include: hyperactivity, piloerection, hyperextension of the back, stiffening and arching of the tail toward the head, tremors progressing to spams, paralysis and extension of the hind limbs, respiratory distress with pronounced abdominal breathing, tremors of the whole body and respiratory arrest.5,20,21,53, 55,60,61,93,94 In mice, the common sequence of symptoms was very much dependent on the CI toxin dose. Increasing the dose was linked to shorter times of observation for some of the symptoms, while others were not observed at all due to the rapid symptom transition. Mice given doses of CI toxins that were not lethal, but sufficient to produce toxicity (about 20–30% of the lethal dose 50), usually recovered rapidly with no perceptible long-term effects,93 suggesting that these compounds are rapidly detoxified and/or excreted.
The relative acute toxicities comparing i.p. injection, gavage, or voluntary consumption with mice were measured using crude extracts of mass dinoflagellate cultures containing pinnatoxins G, E, F and A, or pinnatoxin G only. These measurements revealed toxicity ratios of 1.0:1.8:4.5 for the former group of toxins, and 1.0:2.9:7.8 for pinnatoxin G.62 These results are similar to the ratios obtained for New Zealand dinoflagellate isolates.63 Further studies with purified samples of pinnatoxins E, F and G administered by i.p. injection revealed that PnTX F was about 4 times more toxic than PnTX E, and 3.1 times more potent that PnTX-G.64 These authors also reported that there is only a comparatively small differential between the i.p. and the per os routes of exposure (a factor of about 3 for PnTX-G). Ethanol was used as a delivery solvent, and toxin concentrations were different for i.p. and per os administration.64 If this high oral toxicity for PnTXs is confirmed with pure samples and lower quantities of the delivery solvent used to dissolve the toxins, a re-appraisal of the current process would be necessary to establish more precise “toxic equivalency factor” values.65 Currently, i.p. toxicities are used to estimate the relative oral toxicities of marine micro-algal toxins in seafood.
Reduction in oral toxicity vs. i.p. administration is more significant for the spirolides. The LD50 by oral administration via gavage is between 15 and 23 times lower than by i.p. injection in fed mice.60 By gavage, the state of alimentation has a marked effect. Fasted mice are more susceptible by factors between 1.3 and 3.3 than fed animals. It is likely that an increase in the rate of passage of cyclic imine toxins from the stomach to the small intestine is responsible for the higher toxicity observed.
The oral toxicity of gymnodimines has been reported to be significantly lower than the i.p. toxicity. The acute toxicity of gymnodimine A by i.p. administration was measured at about 10 times lower concentrations than that for gymnodimine B.94 The oral toxicity of gymnodimine A by gavage or absorption on dry mouse food was reported to be about 10–100-fold lower than by i.p. administration.93
The rank of potency for pinnatoxins in acute toxicity studies was reported to be the following: PnTX F > PnTX G > PnTX E for both i.p. and gavage administration.8,64 The rank of potency for spirolides in acute toxicity studies after i.p. administration was either spirolide C = 20-methyl spirolide G = 13-desmethyl spirolide C > 27-hydroxy-13-desmethyl spirolide C > 13,19-didesmethyl spirolide C > 27-oxo-13,19-didesmethyl spirolide C > spirolide A > spirolide B > spirolide E = spirolide F,16,60,66 or spirolide C > 27-hydroxy-13-desmethyl spirolide C = 13-desmethyl spirolide C > 13,19-didesmethyl spirolide C > 27-oxo-13,19-didesmethyl spirolide C = spirolide A > 20-methyl spirolide G > spirolide B > spirolide E = spirolide F.16,66,67 By oral administration, the rank of potency for the spirolides is spirolide C = 20-methyl spirolide G = 13-desmethyl spirolide C > spirolide A > spirolide B.60 Among various CI toxins, the rank of oral toxicity appears to be pinnatoxins > spirolides » gymnodimines.
Most studies on the acute toxicity of CI toxins have been carried out by a few groups using mouse bioassays to determine the lethal dose (LD50) values. However, there is a growing concern with respect to the use of mouse bioassays due to their poor specificity and ethical issues. Therefore, it is not considered an appropriate method for specific detection of CI toxins. Although it is crucial to have a uniform and validated approach for acute toxicity assessments, in most of the studies performed, there is a persistent lack of information on: (i) the purity of the CI toxins used, which points out the need to have an adequate supply of chemically-confirmed pure standards of the compounds to be tested, (ii) the characteristics of the mice used (strain, sex, inbred or outbreed), (iii) the animal house conditions (light/dark cycle), and (iv) the sanitary conditions (for bacteria and viruses) and animal health controls of the animals used. Furthermore, in some studies the validity of results obtained can be questioned due to the high concentrations of the solvent used to dissolve the CI toxins (5–10% ethanol for i.p. administration and 10–12.5% ethanol in per os administration studies). In some cases, the addition of 1% of Tween 60 to dilute the ethanolic solutions of CI toxins further complicates data interpretation. In some studies, CI toxins were administered intraperitoneally to mice of 18–22 g in a high bolus volume of 1 mL of the toxin dissolved in ethanol. Ethanol is well known to affect the rate of absorption of drugs and toxins. Therefore, the concentration of ethanol used has to be taken in consideration when designing further studies on acute toxicity of CI toxins. Because the amounts and compositions of the vehicle used for i.p. and per os administration were not identical, a more rigorous study is needed for a precise comparison between the various routes of administration for CI toxins.
Synthetic Chemistry and Chemical Stability of CI toxins
Studies directed at the total synthesis of pinnatoxins68 and spirolides69,70 have been reviewed previously. The aim of this section will be to provide a broader overview of the total synthesis efforts and an analysis of the most recent progress in the field. Insight into the chemical stability of CI toxins that emerged from these synthetic studies, sometimes quite striking, is also included.
Pinnatoxins
Pinnatoxin A received the most attention as a target for total chemical synthesis, with the first synthesis in the area completed by Kishi and co-workers in 1998.71 The effort defined the absolute configuration of the natural product and confirmed no toxic activity for its enantiomer. The synthesis produced 1.0 mg of (−)-pinnatoxin A by what to date remains the shortest synthetic route measured by its longest linear sequence of 38 steps. A total of about 73 chemical transformations from commercial starting materials were required. Scheme 1 provides a graphical overview of the total synthesis.
Scheme 1.

Total synthesis of (−)-pinnatoxin A by Kishi and co-workers. (A) Schematic outline, a dot graph of the synthesis and central intermediates at points of convergence. (B) Selected key transformations in the synthesis.
Some of the more significant highlights of the synthesis include the putatively biomimetic intramolecular Diels-Alder (IMDA) reaction and the cyclic imine formation (Scheme 1B). The impressive macrocyclization constructs the 27-membered carbocycle of PnTX A simultaneously establishing the adjacent quaternary and tertiary stereogenic centers at the core of the spiroimine subunit. Although a total of eight products are possible, the desired diastereomer was isolated in 33% yield, along with two other isomers in a combined yield of 43%. The diene had to be immediately diluted upon preparation to avoid Diels-Alder dimerization. After removing the protecting groups on the amine and alcohol from the Diels-Alder macrocyclization product, the imine formation provided an early insight into the remarkable chemical stability of pinnatoxin A. An interconversion between the cyclic imine and the amino ketone forms of the natural product proved to be unusually difficult. In this case, imine formation required heating of the amino ketone at 200 °C for 1 h at 1–2 torr, which provided a 70% yield of PnTX A tert-butyl ester. Removal of the tert-butyl group upon treatment with trifluoroacetic acid at room temperature for 15 min afforded the enantiomer of the natural product. The last two steps of the synthesis underscore the stability of pinnatoxin A under a rather significant thermal or acidic stress.
Kishi and co-workers have subsequently described tour-de-force studies on the total synthesis and the complete structure elucidation for PnTXs B and C6 as well as pteriatoxins (PtTX) A-C.10b These important results appeared in 2006, eight years after the initial disclosure of the total synthesis of (−)-PnTX A.
In 2004, Hirama, Inoue, and co-workers reported a formal synthesis of pinnatoxin A, intercepting the amino ketone intermediate previously converted to the natural product in 2 steps by the Kishi group. After an extensive effort adopting a Diels-Alder strategy for the assembly of the cyclohexene ring G, recourse to an intramolecular alkylation of a nitrile anion with epoxide provided an ultimate solution to the synthetic problem. The set up for this step was costly in terms of the synthesis length, but the product contained all necessary functionalization and stereochemistry for the fabrication of the critical spiroimine fragment. Another important observation from the synthesis is the long-range hydrogen bonding between OH-10 and OH-24 groups that potentially controls stereoselectivity in the thermodynamic dispiroketalization reaction during the BCD-ketal assembly (Scheme 2B). X-ray crystallographic analysis of a closely related compound provided evidence for the proposed long-range hydrogen bonding.
Scheme 2.

Formal synthesis of (+)-pinnatoxin A Hirama, Inoue, and co-workers. (A) Schematic outline, a dot graph of the synthesis and central intermediates at points of convergence. (B) Selected key transformations in the synthesis.
The overall yield of the tert-butyl ester of the amino keto acid, the most advanced intermediate, is 0.024% over 56 steps in the longest linear sequence from glucose. It is not clear from the report how much material the synthesis provided. The yield from geranyl acetate is 0.020%, notably, comparable to that from glucose in spite of a substantially shorter linear sequence of 37 steps.
Some of the most notable lessons from the synthesis are summarized in Scheme 2B. Besides the ring G construction by the intramolecular nitrile alkylation, the stereochemistry in the thermodynamically controlled bis-ketalization for the tricyclic BCD subunit was enhanced by putative long-range hydrogen bonding maximized in non-polar solvents. A key convergence point was realized by an alkylation of an advance BCD-ketal-derived dithiane with a high-molecular-weight iodide. Subsequent 27-membered ring closure catalysed by the Grubbs II catalyst completed the carbocyclic framework of PnTX A. An additional 14 steps were needed to install the EF bridged ketal, C1 nitrogen, and C10 exo-methylene group. An illuminating observation at the final stages of the synthesis was the inability to form the imine ring from the C1 iminophosphorane, formed in situ by the reduction of the C1 azide with trimethylphosphine. The failure of this potentially powerful transformation, especially in light of its success in a truncated acyclic system,72,73 underscores the unusual chemistry of the 7-membered imine in PnTX A.
Two versions of the total synthesis of PnTX A were disclosed by the Zakarian group,74 of which only the latest will be summarized.75 One goal of the synthesis was to advance the enolate-based methodology for asymmetric synthesis, especially in sophisticated applications of the Ireland-Claisen rearrangement.76 Another was to provide a reliable supply for the scantly available toxins in the pinnatoxin subgroup, thereby permitting studies aimed at a deeper understanding of the mechanism of biological activity and toxicology of pinnatoxins. The first synthesis completed in 2008 furnished 3.5 mg of PnTX A, and enabled initial biological studies aimed at defining the biological target of the toxin. These studies stimulated the development of a larger-scale, reproducible total synthesis, which afforded over 40 mg of synthetic PnTX A along with 10 mg of PnTX G, in addition to a remaining stock of 0.30 g of an advanced intermediate, the azido triol depicted in Scheme 3B. This synthetic effort provided a long-term supply of the important natural product for our own and a variety of other research groups, as well as its commercialization.77 The second synthesis was completed in 1.1% overall yield in a longest linear sequence of 44 steps from citronellic acid, or 1.0% yield over 43 steps from acrolein and tert-butyl acetate.
Scheme 3.

Total synthesis of (+)-pinnatoxin A by Zakarian and co-workers. (A) Schematic outline, a dot graph of the synthesis and central intermediates at points of convergence. (B) Selected key transformations in the synthesis.
A highlight of the synthesis is the strategic application of a complex Ireland-Claisen rearrangement as a solution for stereocontrolled synthesis of the spiroimine subunit of pinnatoxins. At the onset, the stereoselective generation of the acyclic tetrasubstituted enolate was an unmet synthetic problem. The stereoselective enolization was critical for the powerful chirality transfer from the substrate to the product, establishing the stereochemistry at the quaternary C5 and the adjacent tertiary C31 positions in a single C—C bond-forming process. An effective solution for the problem was discovered by the use of a chiral Koga-type lithium amide, whereby chirality match between the lithium amide reagent and the substrate defines the geometric selectivity of enolization. A subsequent aldol cyclocondensation installed the C32-C33 double bond, completing the synthesis of ring G fragment.
By and large, the synthesis of the BCD-bisketal subunit followed the blueprint outlined in the Kishi’s and Hirama/Inoue’s syntheses. The stereoselectivity of ketalization was achieved under thermodynamic control in non-polar solvents using an acyclic substrate engineered to maximize both the anomeric and hydrogen-bonding stabilization in the desired diastereomer of the product. Fragment coupling and the formation of the 27-membered macrocycle formation was accomplished by 1) organolithium coupling of a complex C7-organolithium reagent to the C6-aldehyde, and 2) ring-closing metathesis (RCM) forging the C26-C27 double bond. Preserving the silyl ether at the C15 tertiary alcohol (ring B), a seemingly minor adjustment, increased the yield of the ring-closing metathesis from 57% to 75% at the expense of undesired metathesis pathways. Only five steps were needed to advance the RCM product to the key azido triol, of which 0.37 g were produced in 34% overall yield. The azido triol served as the stock supply of material to complete the synthesis of over 40 mg of PnTX A, 10 mg of PnTX G, ~3 mg of high-specific-activity radiolabelled [3H]-PnTX G,78 and other derivatives.
The group of Nakamura and Hashimoto described the fourth total synthesis of (+)-pinnatoxin A in 2008.79 The distinguishing transformations in the synthesis are the exo-selective intermolecular Diels-Alder (DA) reaction to set up the spiroimine subunit, Ru-catalyzed macrocycloisomerization reaction, and the late-stage imine formation from an amino keto precursor bearing a free C34 carboxylic acid (Scheme 4). Another notable highlight of the synthesis is the Michael addition-terminated spirocyclization during the assembly of the BCD-bisketal subunit of PnTX A.80 The synthesis furnished 0.9 mg of (+)-pinnatoxin A in 55 steps in the longest linear sequence with an overall yield of 0.27% from (S)-malic acid.
Scheme 4.

Total synthesis of (+)-pinnatoxin A by Nakamura, Hashimoto, and co-workers. (A) Schematic outline, a dot graph of the synthesis and central intermediates at points of convergence. (B) Selected key transformations in the synthesis.
In parallel to the intramolecular Diels-Alder variants in the previous synthetic efforts, the intermolecular Diels-Alder cycloaddition between the preassembled BCDEF-polyketal diene and the cyclic 7-membered exo-methylene lactone (ring A) was characterized by similar stereochemical challenges. The desired isomer was isolated in 34% yield along with 23% of the alternative exo-addition isomer and a combined 23% yield of two endo-cycloaddition isomers. Remarkably, the regioselectivity in the intermolecular DA reaction was complete. From the DA cycloadduct, 8 steps were required to advance to the enyne intermediate for the critical cycloisomerization step. The substrate proved to be surprisingly reactive (Scheme 4B). In the presence of [CpRu(MeCN)3]PF6 (10 mol %), the cycloisomerization was essentially complete within 15 min at 50 °C, providing the 27-membered macrocycle in 79% yield. The facility of this impressive macrocyclization may be indicative of the conformational predisposition of the complex substrate for large ring formation placing the reacting groups in proximity. With the macrocycle in place, the total synthesis of (+)-pinnatoxin A was completed in 11 steps. In route, formation of the imine ring A in the penultimate step by a direct cyclocondensation of the silyl-protected PnTX A amino ketone (Scheme 5) (chlorobenzene, 120 °C, 18 h, 74% yield) lends additional insight into the unusual energetics of the amino ketone–cyclic imine interchange in pinnatoxins.
Scheme 5.

PnTX A imine condensation/hydrolysis studies.
Interconversion between the open and closed ring A forms of PnTX A under aqueous conditions was examined in a separate study by the Zakarian group.81 Cyclization of (+)-PnTX A amino ketone, 5.1 mg of which was prepared by total synthesis, was studied at pH 7.3 and 4.1 in D2O (0.01 M) at various temperatures. At pH 7.3, the substrate showed remarkable stability and resistance to imine formation, with no change observed during monitoring by 1H NMR after heating at 100 °C for 72 h. At a more acidic pH of 4.1, no change was observed after 48 h at 20 °C and additional 19 h at 50 °C. However, partial decomposition with no formation of PnTX A was recorded after 24 h at 100 °C, with complete decomposition after 72 h at this temperature.
In the reverse direction, hydrolysis of (+)-PnTX A was studied at pH 1.5, 4.0, and 7.3 in D2O (0.01 M). Monitoring by 1H NMR at pH 4.0 for 48 h at 40 °C, acidification to pH 1.5, and an additional 24 h at 40 °C revealed no change. When (+)-PnTX A was heated at 100 °C in a KH2PO4 buffer (D2O, 0.01M, pH 7.3), slow but clean hydrolysis to the amino ketone was observed, reaching a conversion of 20% after 24 h.
The remarkable chemical and thermal stability of pinnatoxins suggests an explanation for only a moderate reduction in their oral vs. intraperitoneal toxicity in contrast to other CI toxins, at the same time indicating an increased risk that these toxins may pose for seafood contamination.
Spirolides
Although no total synthesis of any member of the spirolide subfamily has been reported to date, a number of synthetic studies have been described (Scheme 6). The Ishihara and Brimble groups made substantial progress to the dispirotricyclic fragment of spirolides using distinct chemistries. The Ishihara group developed an acid-catalyzed dispiroketalization of a 15,18,19-triketo substrate followed by methyllithium addition to establish the C19 tertiary alcohol.82 Subject to thermodynamic control, the initial cyclization afforded predominantly the undesired C15 epimer. Silylation of the tertiary alcohol reversed the isomer distribution, providing a 2:1 mixture favouring the desired diastereomer, which, if allowed to reach equilibrium, leads to the exclusive formation of the undesired C15-epimer. The Brimble group creatively exploited iterative photoinduced cyclizations to access the bisketal subunit. Starting from an elaborate dihydropyran substrate, oxidative radical cyclization upon irradiation in the presence of PhI(OAc)2 and I2 afforded dioxaspiro[5.6]decane ring system.83 After desilylation, the reaction was repeated to install the second ketal. Complete lack of stereocontrol in these cyclizations was ameliorated upon epoxidation of the double bond and in-situ acid-catalyzed equilibration, which afforded the desired bisketal as a single isomer. The C19 tertiary alcohol was produced in three additional steps. In both approaches, addition of the methyllithium or methylmagnesium bromide to the C19 ketone was highly stereoselective in favor of the desired isomer.
Scheme 6.

Synthetic studies directed towards the spirolides described in the literature.
Very recently, another effective approach to the spiroketal subunit of 13-desmethyl spirolide C was reported by the Landais and Desvergnes group.84 This approach utilizes a sila-Stetter reaction in a convergent assembly of the spiroketalization precursor. As in previous approaches, acid-catalyzed thermodynamic spiroketalization generally favors the undesired diastereomer.
The synthesis of the spiroimine subunit was studied by the Brimble85 and Zakarian groups. In the latter study, the fully elaborated precursor to the spiroimine was constructed by a highly diastereoselective Ireland-Claisen rearrangement (Scheme 6).86 The reaction established the quaternary center at the core of the spiroimine ring system along with other stereochemical features. The product was advanced to a 23-membered macrocyclic intermediate incorporating most of the structural information of the spirolides; however, the ring-closing metathesis in the last step of the sequence proved to be challenging.87 Only 25% conversion was achieved during formation of the C13-C14 double bond with an optimized substrate. The results suggest that perhaps another location for C—C formation during the assembly of the large carbocycle needs to be identified.
Further studies toward the synthesis of spirolides are justified as they can enable access to valuable biological probes, especially those incorporating radiolabels that can be used, for example, in investigations of their biodistribution in rodent mammalian models.
Gymnodimine
Several groups have reported synthetic studies toward the total synthesis of gymnodimine,88 of which only the Romo group disclosed success in completing the total synthesis.89 The Romo synthesis adopted a convergent strategy that separated the oxolane and spiroimine subunits within the macrocyclic structure of gymnodimine (Scheme 7). Upon completion, 1.9 mg of the natural product were produced in 30 steps (longest linear sequence) and 0.41% yield from 1,3-propanediol, or in 23 steps and 0.23% yield from acrylonitrile and diethyl malonate.
Scheme 7.

Total synthesis of (−)-gymnodimine by Romo and co-workers. (A) Schematic outline, a dot graph of the synthesis and central intermediates at points of convergence. (B) Selected key transformations in the synthesis.
A powerful catalytic asymmetric Diels-Alder reaction, used to construct the spiroimine precursor, is one of the highlights of the synthesis. The chiral copper-bis(oxazoline) catalyst provided the cycloadduct in 85% yield, 95:5 diastereoselectivity, and over 95% ee for the desired isomer. Thus, the Diels-Alder reaction is remarkably successful in establishing the challenging quaternary stereocenter at the core of the spirocylic ring system, as well as the adjacent tertiary center during the assembly of the cyclohexene ring of gymnodimine. The Murai group demonstrated that a similar approach is viable for the enantioselective synthesis of the azadispiro[5.6]dodec-9-ene ring systems found in spirolides and pinnatoxins, at least in simple model systems lacking the vicinal dimethyl substitution in the tetrahydroazepine ring.90
The initial stereochemical information in the oxolane ring was introduced using the Evans aldol reaction, and subsequent elaboration was aimed at joining the fragments by a convergent coupling using the Nozaki-Hiyama-Kishi reaction (7C, Scheme 7A). The 14-membered carbocyclic ring-closure was achieved by an extraordinary process involving lithium-iodine exchange and intramolcular acylation of the intermediate organolithium reagent with the N-tosyl lactam, producing the macrocylic ketone with concomitant opening of the lactam. The success of this remarkable transformation depended on optimized temperature control, where performing the reaction at 23 °C was found to be critical for success (Scheme 7B). At −78 °C, the major product was formed by proton quench of the complex organolithium intermediate. After N-trifuorocetylation of the product, the 4-tolylsulfonamide group was cleaved reductively with SmI2, and subsequent acid-catalyzed hydrolysis exposed the cyclohexanone for the ultimate appendage of the 2-methyl-2-butenolide unit. This operation was carried out by TiCl4-mediated aldol reaction of the ketone with 2-(trisiopropylsiloxy)-3-methylfuran. The mixture of stereoisomers produced (7D, Scheme 7A), was elaborated to gymnodimine by elimination of the C5 tertiary alcohol in the presence of thionyl chloride and triethyl amine and final deprotection under acidic conditions (TFA, CH2Cl2). The low efficiency of the final steps is a result of low region and stereoselectivity. However, the production of both C4 isomers of gymnodimine conclusively established the stereochemistry at C4 position in the butenolide unit.
Biological Activity and Modes of Action
Effect of CI Toxins at the Cellular Level
The first investigations on the mode of action of CI toxins suggested their potential activity on voltage-gated ion channels. Pinnatoxins and spirolide-B and D, for example, were initially proposed to act as calcium channel agonists,6,15,91,92 while gymnodimine was suggested to interact with sodium channels.5 Nevertheless, no convincing support for these mechanisms was demonstrated. Due to the severe and rapid neurological symptoms observed in mice after i.p injection of gymnodimine A,93 the mode of action of this CI toxin was studied more carefully at the cellular level. It has been found that, on isolated frog or mouse neuromuscular preparations, gymnodimine A induces a reversible time- and concentration-dependent blockage of twitch tension responses evoked by nerve stimulation without affecting directly-elicited muscle twitches.94 Similar effects were later observed with 13-desmethyl spirolide C95 and pinnatoxin F, with the latter toxin causing a complete abolition of the rat hemidiaphragm action potential responses evoked by nerve stimulation, while no effect was observed after an electrical stimulation.96 In addition, gymnodimine A blocks the miniature endplate potential in frog and mouse isolated neuromuscular preparations.94 Finally, the potential role of cholinergic receptors in the toxicity of 13-desmethyl spirolide C was also proposed, based on transcriptional analysis highlighting the upregulation of muscarinic and nicotinic receptor genes in rats intoxicated with this CI toxin.97 All of these results strongly suggested that gymnodimines, spirolides, and pinnatoxins may elicit their action by blocking endplate nAChRs.
Molecular Targets of CI Toxins
Interaction of CI Toxins with nAChRs
Nicotinic acetylcholine receptors (nAChRs) are prototypes of the ligand-gated ion channel (LGIC) family that include the excitatory 5-HT3 subtype of serotonine receptor, and the inhibitory receptors for glycine and γ-aminobutyric acid (GABAA and GABAC).98 The nAChRs consist of homo- or heteropentamers composed of the various subunits that have been identified so far (α1-α10, β1-β4, γ, δ, ε) that are arranged symmetrically around an axis perpendicular to the membrane delineating the ionic pore.99 The composition and stoichiometry of the subunits constituting the pentamer may have a profound impact on the receptor pharmacology, cation selectivity, desensitization kinetics, and spatial distribution. For example, the mature muscle subtype present at the neuromuscular junction comprised the 2α1, β1, γ, and δ subunit arrangement in Torpedo fish, while in most mammalian mature receptors the ε subunit replaces the γ of the embryonic form.
To demonstrate that the toxicity of CI toxins was due to their effect on the cholinergic system, and mainly due to their interaction with nAChRs located at the neuromuscular junction, electrophysiological experiments, binding studies, and structural analysis have been performed.
Patch-clamp recordings in Xenopus skeletal myocytes first revealed that gymnodimine A reversibly blocks the ACh-dependent nicotinic current in the preparation containing embryonic form of nAChRs with no voltage-dependency.94 Activity on muscle-type nAChRs was later demonstrated using Xenopus oocytes microtransplantated with Torpedo electrocyte membranes, which are known to contain a high density of receptors. Using this preparation, the high efficacy of gymnodimine A, 13-desmethyl spirolide C, and pinnatoxin A in blocking the ACh-elicited nicotinic currents in a concentration-dependent manner was demonstrated, with the 13-desmethyl spirolide C being the most potent (IC50 = 0.51 nM) as compared to gymnodimine A (IC50 = 2.8 nM) or pinnatoxin A (IC50 = 5.5 nM).75,100 Moreover, the reversibility of this antagonist effect was rapid with gymnodimine A and pinnatoxin A but extremely slow with 13-desmethyl spirolide C. In addition to their effect on musle-type receptors, functional electrophysiological studies also showed that CI toxins behave as antagonists on neuronal nAChRs subtypes. Thus, the dual-microelectrode voltage-clamp technique in oocytes expressing the human homomeric α7 or heteromeric α4β2 nAChR subtypes revealed that gymnodimine A, 13-desmethyl spirolide C, and pinnatoxin A decrease the peak amplitude of the ACh-evoked nicotinic currents in these Xenopus oocytes very efficiently.75,94,100 The inhibition constants for the potent action of pinnatoxin A shows remarkable selectivity for α7 (IC50 = 0.1 nM) receptor in comparison to the heteromeric α4β2 (IC50 = 30.4 nM) subtype (Figure 4).75
Figure 4.

Synthetic PnTX A blocks nAChRs in Xenopus Oocytes. A, B. ACh-evoked nicotinic currents recorded in Xenopus oocytes expressing the human α7 (A) and human α4β2 (B) nAChRs, before (black tracing), and after (red tracing) 125 pM (A) and 50 nM (B) PnTX A treatment, respectively. ACh was microperfused, as indicated by the lines above nicotinic current tracing, at concentrations corresponding to the half maximal activation of nAChRs previously determined experimentally (EC50; 350 μM for human α7 and 150 μM for human α4β2). The holding membrane potential during recordings was −60 mV. C. Concentration-dependent inhibition of ACh-evoked currents by synthetic PnTX A in Xenopus oocytes expressing the human α7 (red circles and curve) and human α4β2 (blue circles and curve) neuronal nAChRs, or having incorporated into their membranes the Torpedo α12β1γδ muscle type nAChR (orange circles and curve). Amplitudes of the ACh-current peak recorded at a holding membrane potential of −60 mV, in the presence of PnTX A were normalized to control currents and fitted to the Hill equation. Data with error bars represent the mean ± S.E.M. (n = 5).
The function of CI toxins was also recently studied using a calcium flux functional assay with various cell lines expressing different neuronal nAChR subtypes. 101 The antagonist activity of gymnodimine A and 13-desmethyl spirolide C was evaluated by measuring their inhibition potency on the nicotine-mediated calcium flux, confirming their high activity on α7 and α4β2 subtypes.
Competition binding experiments were used to determine the affinity constants of CI ligands under equilibrium conditions, and to characterize the mode of action of antagonist toxins (competitive, non-competitive, allosteric) according to the characteristics of the competition binding curves (Figure 5). As with the electrophysiological studies, gymnodimine A, pinnatoxin A and 13-desmethyl spirolide C were the three CI toxins that were studied using binding experiments with various nAChRs subtypes. Activity on muscle-type nAChRs was measured using Torpedo membrane preparation or HEK-293 cells transfected with human receptor subunits (α1, β1, γ, δ) and 125I-α-bungarotoxin as radiotracer. 75,100 The three CI toxins displace the tracer from the muscle-membrane preparations in a dose-dependent manner with the following the ranking of affinity constants: 13-desmethyl spirolide C (Ki = 0.08 nM) > gymnodimine A (Ki = 0.23 nM) > pinnatoxin A (Ki = 2.8 nM). The affinity constants for the CI toxins were also determined with five distinct neuronal nAChRs: the human or chick homopentameric α7 subtype with 125I-α-bungarotoxin as radiotracer, and the human heteropentameric α3β2, α4β2, α3β4, and α6β3β4α5 subtypes with 3H-epibatidine or 3H-nicotine as radiotracers.75,100,101 The CI toxins exhibit a broad specificity in interaction with these different neuronal nAChR subtypes with affinity constants ranging from 0.02 nM for 13-desmethyl spirolide C on α3β2 receptor14 to 70 nM for gymnodimine A on α4β2 subtype.101 Furthermore, as shown in Figure 3 for the α3β2 receptor, various CI toxins interact with different potencies on each receptor subtype, highlighting the important effect of chemical variations around the conserved spirocyclic imine on subtype specificity. With α3β2 receptor, the order of potency is: 13-desmethyl spirolide C > gymnodimine A > pinnatoxin A ≫ pinnatoxin AK. Pinnatoxin AK (PnTX AK) is a synthetic analog of PnTX A that contains an acyclic form of the imine ring (Scheme 5).75
Figure 5.

Competition binding curves of pinnatoxin A (PnTX A), pinnatoxin AK (PnTX AK), gymnomidine A (GYM A) and 13 desmethylspirolide C (13-DMS) on α3β2 nAChRs subtype expressed on HEK-293 cells that interact with 3H-epibatidine. The results are expressed as the ratio of specific radiotracer binding measured with (B) or without (Bo) competitive ligand expressed as a percentage. Curve fitting was based on non-linear regression analysis using the Hill equation. Data are mean values +/− SEM of at least three experiments.
Comparing the potency of natural and chemically modified CI toxins was instrumental in identifying the pharmacophore responsible for such efficient binding of these ligands with nAChRs. For example, as previously shown for the pinnatoxin A, disruption of the imine ring in PnTX AK resulted in a drastic loss of affinity of this toxin for all the nAChRs subtypes studied, revealing the crucial role of this imine subunit for the CI toxin-nAChR interaction.75 Furthermore, analogs of (−)-gymnodimine A with isolated spiroimine moieties have been recently synthesized and studied, revealing a significant decrease of potency for both theracemic and optically active derivatives in the inhibition of the ACh-evoked nicotinic currents in Xenopus oocytes incorporating the muscle-type or neuronal α4β2 nAChRs. These results highlight the important impact of structural features of the CI toxins in relation to their function on nicotinic signal transduction.75,81,102
One of the most informative, albeit challenging approaches to understanding the mode of action of CI toxins, especially regarding precise structural factors implicated in their potent binding to nAChRs, is through crystallographic studies of toxin-nAChR complexes. A highly valuable insight was provided recently by resolving the single crystal X-ray structures of gymnodimine A and 13-desmethyl spirolide C bound to Aplysia-Acetylcholine Binding Protein (AChBP), a structural and functional surrogate of the extracellular domain of nAChRs.100 These findings were further augmented by computational investigations of ligand-ion channel complexes with pinnatoxin A, AChBP, and several subtypes of nAChRs.75 In these studies, the high affinity and slow dissociation of these antagonists was clearly explained by their common location at the subunits interface within the pentameric receptor. The toxins are neatly imbedded within the nest of the aromatic side chains contributed by loops C and F (Figure 6), maximizing their surface complementarity. In addition, the toxin cores were centered within the binding pocket by hydrogen bonds between the protonated imine nitrogen of CI toxins and the carbonyl oxygen of the Trp147 in loop C of the receptor. Finally, the gymnodimine tetrahydrofuran or the spirolide bisspiroketal further anchor these CI toxins in the apical and membrane directions along the subunit interface, encompassing largely the binding surface of classical antagonists such as other alkaloid or peptide toxins. Furthermore, computational modeling of the interaction of pinnatoxin A with Torpedo, α7 or α4β2 receptors on one hand or gymnodimine A and 13-desmethyl spirolide C with α7 subtype on the other hand, suggested that these toxins interact similarly with these receptors as previously shown for the gymnodimine A and 13-desmethyl spirolide C with Aplysia-AChBP.75,101 The number of hydrogen bonds between CI toxins and variable residues in different receptor binding sites accounts for their different selectivity profiles. For example, the data help interpret the varying profile for pinnatoxin A, which has the Ki of 0.35 nM for α7 receptor and a fiftyfold weaker binding (Ki=15.6 nM) to the α4β2 receptor.75
Figure 6.
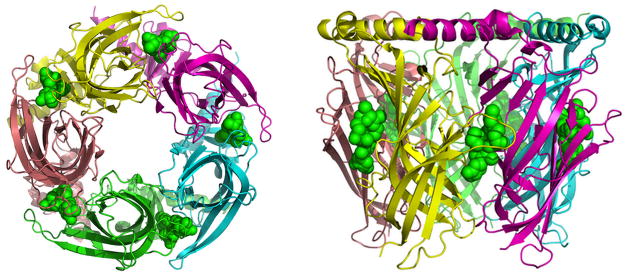
Aplysia-AChBP pentamer in complex with gymnodimine A. Bottom (left) and side (right) views of the X-ray crystallographic structure of gymnodimine A (in green, sphere representation) imbedded at the interfaces of the five subunits of the Aplysia-AChBP pentamer (pdb code 2X00).
Interaction of CI Toxins with Muscarinic Acetylcholine Receptors (mAChRs)
Even if all the results detailed above clearly demonstrate the ability of CI toxins to interact very efficiently with muscle- and neuronal-type nAChRs, the increase of mRNA levels of mAChRs after i.p injection of the spirolides97 and some of the neurological symptoms associated with their administration may suggest a potential role of muscarinic receptors in the physiological effect of these toxins.
The mAChRs belong to the class I (rhodopsin family) of the G-Protein Coupled Receptors (GPCRs) superfamily. More than 1000 different GPCRs have been identified in vertebrates, and they all possess a common structural organization constituted by seven transmembrane helices (TM1–TM7) connected by three intracellular (i1–i3) and three extracellular (e1–e3) loops. These receptors can be activated by a huge diversity of ligands and, despite the great chemical diversity of these endogenous ligands, the receptor activation process occurs via coupling to heterotrimeric guanine nucleotide-binding proteins and subsequent enzyme activation/inhibition or ion channel activity regulation.103 Five different mAChRs have been identified in mammals (M1–M5) and they varied by the main G-protein to which each subtype preferentially couples (Gq for M1, M3, M5 and Gi for M2 and M4), by the physiological responses they mediate, and by their distribution throughout the central and peripheral nervous systems. Peripheral receptors (mainly M3 or M2) are known to mediate the actions of ACh on parasympathetically innervated organs including heart, airways, gastrointestinal and urinary tracts, and exocrine or endocrine glands, whereas central mAChRs (M1, M4, M5) are implicated in various sensory, cognitive, vegetative, and motor function. 104 Recently, three X-ray structures of mAChRs have been solved in complex with antagonist (M2 subtype in complex with the 3-quinuclidinyl-benzilate (QNB),105 M3 subtype in presence of tiotropium),106 agonist, or positive allosteric modulator (M2 in complex with iperoxo and LY2119620 ligands).107
These structures offer important insight into the function, activation mechanism, and allosteric modulation of mAChRs, and provide a structural basis for the rational design of subtype-selective ligands with various functional properties for muscarinic receptors.
Only few studies described the pharmacological characterization of CI toxins on mAChRs. Using a human neuroblastoma cell model known to express various mAChRs subtypes, and the measure of the calcium response following ACh stimulation or the binding of 3H-QNB, the effect of 13-desmethyl spirolide C was investigated.108 At micromolar concentrations, this toxin reduced the maximum response elicited by ACh and inhibited the specific 3H-QNB binding, suggesting a weak interaction with some mAChRs subtypes. In addition, to define the potential interaction of gymnodimine A, 13-desmethyl spirolide C, and pinnatoxin A on mAChRs, the effect of these toxins on the 3H-QNB or 3H-N-methylscopolamine binding to TE671/RD cells, rat cortical membranes, or CHO cells stably expressing the five distinct human mAChRs subtypes was examined. The results showed no or low radiotracer displacement even with high CI toxin concentration (from 1 to 100 μM), suggesting that toxicity of these compounds cannot be directly related to their interaction with mAChRs.75,101
Activity of CI Toxins with Other Receptors
In the course of the identification of the mode of action of CI toxins, the pharmacological properties of these molecules was evaluated on other receptors than nAChRs or mAChRs. For example, in vitro assays showed that spirolides do not affect the NMDA, AMPA, and kainate receptors or do not inhibit serine/threonine phosphoprotein phosphatases.15 Pinnatoxin A was subject to an extensive screen to identify possible alternative targets, evaluate selectivity of its action on nAChRs, and probe the originally proposed modulation of calcium channels. Using 5.6 mg of PnTX A provided by total synthesis, the data for a primary binding assay with over 40 receptors, transporters, and ion channels were obtained (Table 2).75 No significant binding to any of the screened receptors was detected. Notably, at >10 mM concentration, no competitive binding to L-type calcium cannels from rat heart vs. nitrendipine was observed. Notable initial binding to dopamine D4 receptor was followed up by a secondary functional assay, which showed no effect at concentrations up to 10,000 nM (Figure 7).
Table 2.
Primary binding assay data for Pinnatoxin A at 10 μM concentration
| Receptor subtype | Source | 3H ligand | Mean % inhibition |
|---|---|---|---|
| 5-HT1A | Human (H), cloned | 8-OH-DPAT | −2.5 |
| 5-HT1B | H, cloned | GR-125743 / 5CT | −5.4 |
| 5-HT1D | H, cloned | GR-125743 / 5CT | −5.9 |
| 5-HT1E | H, cloned | 5HT | −0.5 |
| 5-HT2A | H, cloned | Ketanserin | 0.9 |
| 5-HT2B | H, cloned | LSD | −14.5 |
| 5-HT2C | Rat, cloned | Mesulergine | 38.7 |
| 5-HT3 | H, cloned | LY 278,584 | 2.4 |
| 5-HT5A | H, cloned | LSD | −6.9 |
| 5-HT6 | H, cloned | LSD | −18.3 |
| 5-HT7 | H, cloned | LSD | 0.0 |
| Alpha1A | H, cloned | Prazosin / 125I-heat | 27.8 |
| Alpha1B | H, cloned | Prazosin / 125I-heat | 8.7 |
| Alpha1D | H, cloned | 3H-Prazocin | 4.3 |
| Alpha2A | H, cloned | Clonidine (2nM) / 125Iodoclonidine | 1.7 |
| Alpha2B | H, cloned | Clonidine (2nM) / 125Iodoclonidine | 4.0 |
| Alpha2C | H, cloned | Clonidine (2nM) / 125Iodoclonidine | 12.6 |
| Beta1 | H, cloned | Dihydroalprenolol / 125Iodopindolol | −1.8 |
| Beta2 | H, cloned | Dihydroalprenolol / 125Iodopindolol | −4.0 |
| Beta3 | H, cloned | Dihydroalprenolol / 125Iodopindolol | −7.6 |
| BZP Rat Brain Site | Rat brain | Flunitrazepam | −0.9 |
| Calcium Channel | Rat heart | [3H]-Nitrendipine (0.1 nM) | Ki > 10,000 nM |
| D1 | H, cloned | SCH23390 | |
| D2 | H, cloned | N-Methylspiperone, NMSP | −5.9 |
| D3 | H, cloned | N-Methylspiperone, NMSP | 6.4 |
| D4 | H, cloned | N-Methylspiperone, NMSP | 63.4 |
| D5 | H, cloned | SCH23390 | 12.9 |
| DAT | H, cloned | WIN35428 | 5.9 |
| DOR | H, cloned | DADL | 10.0 |
| GABAA | Rat, forebrain | Muscimol | 39.5 |
| H1 | H, cloned | Pyrilamine | 51.2 |
| H2 | H, cloned | Tiotodine | 12.2 |
| KOR | Rat, cloned | U69593 | 6.7 |
| M1 | H, cloned | [3H]-QNB (0.5 nM) | −11.7 |
| M2 | H, cloned | [3H]-QNB (0.5 nM) | 8.5 |
| M3 | H, cloned | [3H]-QNB (0.5 nM) | 27.9 |
| M4 | H, cloned | [3H]-QNB (0.5 nM) | 5.0 |
| M5 | H, cloned | [3H]-QNB (0.5 nM) | −0.6 |
| MOR | H, cloned | DAMGO | −10.3 |
| NET | H, cloned | Nisoxetine | 11.7 |
| SERT | H, cloned | Citalopram | 12.2 |
| Sigma1 | GP rat brain | [3H]-Pentazocine (3 nM) | 26.6 |
| Sigma2 | PC12 | [3H]-DTG (3 nM) | −14.7 |
Figure 7.
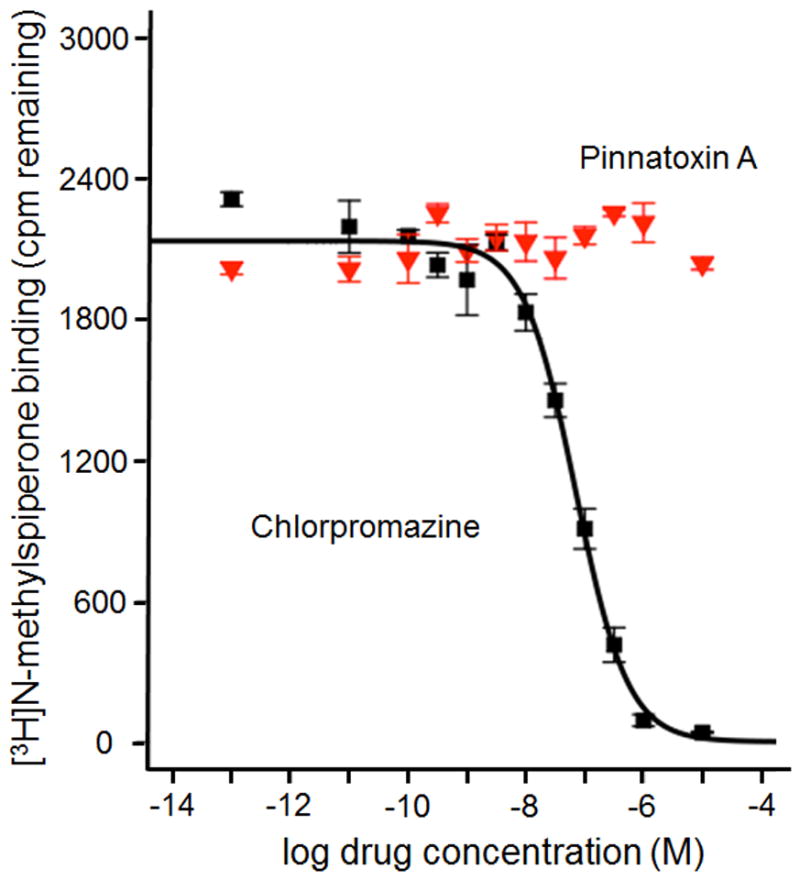
Competitive binding displacement of [3H]N-methylspiperone to D4 dopamine receptor by chlorpromazine (black squares, Ki = 31 nM) and lack of displacement by 10 μM PnTX A (red triangles).
Collectively, these experimental results reveal remarkable selectivity for the interaction of PnTX A with nicotinic acetylcholine receptors, with no significant binding to a wide range of other signaling proteins detected.
Activity of CI Toxins and Neurodegenerative Disease
Based on previous results demonstrating the benefit of nicotinic antagonist such as the α7 blocker methyllycaconitine on the level of β-amyloid peptide (Aβ),109 recent studies evaluated the potential activity of CI toxins in neurodegenerative disease models. First, the in vitro effect of long term exposure of cortical neurons to gymnodimine or 13-desmethyl spirolide C was shown to decrease the intracellular Aβ accumulation and the levels of the hyperphosphorylated isoforms of the tau protein. Moreover, these toxins also decreased the glutamate-induced neurotoxicity in vitro.110,111 In addition, in vivo studies with 13-desmethyl spirolide C on the 3xTg mouse, revealed that the intraperitoneal injection of the spirolide decreased the intracellular Aβ level in the hippocampus of treated versus non-treated mouse, highlighting the positive effects of the molecule in this well-known model of Alzheimer’s disease.112 These results suggest that CI toxins, by virtue of their interaction with nAChRs, may constitute a valuable tool in developing drugs for the treatment of neurodegenerative diseases.
Current Methods for Detection of CI Toxins
The CI toxins were first detected from shellfish extracts using the mouse bioassay developed for lipophilic toxins.113 However, because of ethical concerns and selectivity issues, liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) will replace mouse bioassays as the reference method for monitoring marine lipophilic toxins in Europe.114 LC-MS/MS provides sensitivity, selectivity, as well as structural and quantitative information for unambiguous and simultaneous monitoring of an array of phycotoxins including CI toxins. Importantly, LC-MS/MS-based detection methods depend on certified CI toxin standards for determining a) optimal chromatographic elution profiles, b) optimal mass fragmentation conditions for best MRM transitions, c) matrix effects, if any, and d) limits of detection and quantification.
Notwithstanding the lack of certified standards, the European Commission promoted the implementation and validation of inter-laboratory and in-house LC-MS/MS methods for simultaneous monitoring of internationally regulated lipophylic toxins, including gymnodimine A and 13-desmethyl spirolide C, in order to replace mouse bioassays by 2015.115,116,117,118,119,120,121,122 Toxin detection was performed using tandem quadrupole mass spectrometers in the multiple-reaction-monitoring mode, one of the most suitable methods for optimal quantitation of bioactive molecules, using one, two, and sometimes three specific ion transitions. The development of LC/MS-MS multitoxin detection protocols led to the discovery of CI toxins worldwide: gymnodimine A was detected in shellfish samples collected in New Zealand,123 Chinese,124 and Tunisian shellfish farms.125 Spirolide C and 13-desmethyl spirolide C were detected among other lipophylic toxins in phytoplankton samples collected in the East coast of Scotland.126 Pinnatoxin G, spirolide C, 13-desmethyl spirolide C, 13,19-didesmethyl spirolide C, and 20-methyl spirolide G were detected in Norwegian and French coasts.26,121,127 Mussel samples collected from natural banks and breeding areas located along Emilia Romagna coasts in Italy were contaminated with 13-desmethyl spirolide C, 13,19-didesmethyl spirolide C and 27-hydroxy-13,19-didesmethyl spirolide C. 128 The development of sub-2 μm particle size chromatographic columns, and the emergence of ultra-performance liquid chromatography-MS/MS (UPLC-MS/MS) improved the throughput of LC methods, ideal for routine monitoring of lipophilic phycotoxins and CI toxins.115,129,130
Is there room for bio-analytical methods? In spite of the technological advances, mass spectrometry remains mostly predictive and highly dependent on certified toxin standards. High resolution MS constitutes an alternative technique to determine the molecular mass, and provide structural data of phycotoxins and unknown bioactive molecules from toxic shellfish extracts with high accuracy and sensitivity.131,132 However, when certified standards are not available, uncertainty – a parameter that provides an interval within which the true value is believed to lie at a set level of confidence – becomes difficult to estimate and the data are qualitative at best.132 Total synthesis of CI toxins is of paramount importance for unambiguous detection and risk assessment. Numerous CI toxins possess the same molecular formula and similar molecular mass, but different structure and thus different elution profiles, mass fragmentation and, therefore, different antagonistic potency towards nAChRs.130 We believe that there is room for bio-analytical methods, since they are target-directed, they can detect all congeners of a given phycotoxin family, they can be used for discovering of novel phycotoxins and, in addition, they can be formatted for high throughput.
Since CI toxins act as potent antagonists of nAChRs, several non-radioactive ligand-binding assays based on their mechanism of action were developed. In fact, the affinity of CI toxins for muscle-type nAChRs was determined by a radioactive ligand-binding assay using Torpedo marmorata electrocyte membranes enriched in nAChR and 125I-α-bungarotoxin as tracer.75,94,100 However, the application of such methodology for the detection of CI toxins is limited to specialized laboratories, because of radioactive debris and radio-decomposition of the tracer.133
Several non-radioactive ligand-binding assays were developed for CI toxins detection: a) Fluorescence polarization assay is a direct method that detects changes in the emitted fluorescence yield by a fluorophore alone or in complex with a given receptor upon excitation by plane polarized light. With the use of Torpedo electrocyte membranes as a source of nAChR and Alexa Fluor 488-α-bungarotoxin as the fluorophore, the competitive inhibition between the fluorescent tracer and CI toxins to nAChR could be measured in solution. Gymnodimine A, 13-desmethyl spirolide C, and 13,19-didesmethyl spirolide C inhibited tracer binding in the nanomolar region using certified toxin standards and shellfish extracts.134,135,136,137 Okadaic acid, yessotoxin, and brevetoxin-2 did not interfere with this assay.134 The matrix effect of mussels, clams, cockles and scallops on the competitive fluorescence polarization assay was also assessed.136,137 b) Solid-phase receptor-based assay was developed using streptavidin-coated microplates (96- and 384-wells). 138, 139 Firstly, biotin-α-bungarotoxin is immobilized on the streptavidin-coated wells. The CI toxins competitively inhibits the binding of Torpedo-nAChR to the immobilized biotinylated tracer in a concentration dependent manner. Torpedo-nAChRs are subsequently detected with a primary anti-nAChR antibody. Finally, a secondary antibody conjugated to horseradish peroxidase will allow detection of Torpedo-nAChR by chemiluminescence, fluorescence, or calorimetry.138,139 c) Microsphere receptor-based assay using flow cytometry was recently developed.140 Torpedo-nAChR or AChBP from Lymnaea stagnalis –a structural surrogate of α7 nAChR – were immobilized on the surface of carboxylated microspheres. Following incubation of the coated microspheres with CI toxins and with biotinylated α-bungarotoxin, the mixture was filtered, and streptavidin-R-Phycoerythrin conjugate was added to detect the competitive binding inhibition by flow cytometry luminex. The advantage of this methodology is the use of microsphere beads possessing different intrinsic fluorescence. Therefore, more than one nAChR subtype could be simultaneously analyzed.140 d) Microplate receptor binding assay based on the use of Torpedo electrocyte membranes rich in nAChRs, biotin-α-bungarotoxin as non-radioactive tracer, and streptavidin-HRP for calorimetric detection was developed.130 The key step of this assay was the immobilization of Torpedo-nAChR on the surface of a 96-wells microplate providing a high throughput format for routine detection of neurotoxins targeting nAChRs in environmental samples, with minimal sample handling, high sensitivity, reduced matrix effect, and low cross-reactivity. The inhibition binding parameters of six CI toxins and their detection directly in shellfish extracts was confirmed by UPLC-MS/MS in a 14-min run. Such a multitoxin detection was possible by the availability of purified and synthetic CI toxin standards.68,74,75
The fact that the functional methods described here are based on the mechanism of action of CI toxins towards Torpedo-nAChR, they are highly sensitive, and will detect any competitive nicotinic ligands. The coupling of the microplate-receptor binding assay to mass spectrometry improved the selectivity and the read-out of this assay. The CI toxins tightly bound to the coated Torpedo-nAChRs were eluted from wells and analyzed by mass spectrometry, shortening the time between toxin screening and toxin identification.130 Taken together, any of these functional methods can be used to replace mouse bioassay for rapid detection of unknown and unanticipated neurotoxins targeting nAChRs related to harmful algal blooms, and can further facilitate LC-MS/MS analysis.
Concluding Remarks
From a handful of exotic compounds only about two decades ago, CI toxins are now a sizeable family of marine toxins with global distribution in marine environments. The list continues to expand due to discovery of new members in new locations. Much has been learned about their biological properties and mechanism of action. In what is still a relatively rare occurrence, considering their structural complexity, the vanishingly scarce natural supply of some of the major members, pinnatoxins A and G, was considerably enhanced by total chemical synthesis. Along with available natural spirolides, the access to synthetic pinnatoxins was instrumental in identification of their biological target, nAChRs, and defining structural parameters for the interaction of the toxins with the receptor in great detail. In addition, synthesis provides the source of exquisitely pure reference material for commercialization as homogeneous standards for analytical purposes, and of radiolabelled natural toxins for a variety of biological investigations, for instance, in vivo biodistribution studies.
Some of the intriguing questions remain to be answered. It is still unclear what risks for human health are presented by CI toxins, given their widespread occurrence as seafood contaminants. Besides their acute toxicity, are there long-term effects on human health upon long-term exposure due to potent binding of CI toxins to nAChRs in the central and peripheral nervous systems? What are the precise structural factors that are responsible for subtype selectivity in binding to nAChRs found for PnTX A and some other toxins? Answering these questions will define future directions in the field of this fascinating new group of marine toxins.
Supplementary Material
Table 1.
Lethal doses (LD50) for CI toxins by different routes of administration.
| PnTX E | PnTX F | PnTX G | SPX A | SPX C | 13-dmSPX Ca | 20-mSPX Gb | GYM | |
|---|---|---|---|---|---|---|---|---|
| intraperitoneal injection, LD50 (μg/kg) | 57 | 12.7 | 48 | 37 | 8.0 | 6.9 | 8.0 | 96 |
| gavage, fasted, LD50 (μg/kg) | 29.9 | 240 | 53 | 130 | 88 | |||
| gavage, fed, LD50 (μg/kg) | 2800 | 25.0 | 150 | 550 | 180 | 160 | 160 | 755 |
| voluntary consumption fasted, LD50 (μg/kg) | 50–77 | 1200 | 500 | 500–630 | 500 | |||
| voluntary consumption fed,c LD50 (μg/kg) | 50.0 | 400 | 1300 | 780 | 1000 | 630 | >7500 |
13-desmethyl spirolide C,
20-methyl spirolide G,
vehicle for voluntary consumption is either dry mousefood, moist mousefoof, or, in most cases, cream cheese
Acknowledgments
This material is based upon work supported by the U.S. National Institutes of Health (National Institute of General Medical Sciences) under Award 2R01GM077379-06, University of California Santa Barbara, the CNRS, and in part by grant Aquaneurotox ANR-12-ASTR-0037-01 from the Agence Nationale de la Recherche (France). Additional support was provided by kind gifts from Eli Lilly and Amgen.
Biography
Armen Zakarian received his PhD in Florida under the supervision of Professor Robert A. Holton. After postdoctoral studies with Professor Larry E. Overman at the University of California Irvine, he started his independent academic position in 2004. Since 2008, his research group has been located at the University of California Santa Barbara. Research in Zakarian group combines the areas of reaction development, complex molecule synthesis, natural products chemistry, bioorganic chemistry, and medicinal chemistry.
References
- 1.Recent case from New York City. http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6204a1.htm.
- 2.Alonso D, Khalil Z, Satkunanthan N, Livett BG. Mini Rev in Med Chem. 2012;3:785–787. doi: 10.2174/1389557033487746. [DOI] [PubMed] [Google Scholar]
- 3.Sakai R, Swanson GT. Nat Prod Rep. doi: 10.1039/c3np70083f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aune T. Risk Assessment of Marine Toxins. In: Botana L, editor. Seafood and Freshwater Toxins. 2. Taylor & Francis Group, LLC; 2008. pp. 3–20. [Google Scholar]
- 5.Seki T, Satake M, Mackenzie L, Kaspar HF, Yasumoto T. Tetrahedron Lett. 1995;36:7093–7096. [Google Scholar]
- 6.Uemura D, Chou T, Haino T, Nagatsu A, Fukuzawa S, Zheng S, Chen H. J Am Chem Soc. 1995;117:1155–1156. [Google Scholar]
- 7.Chou T, Haino T, Kuramoto M, Uemura D. Tetrahedron Lett. 1996;37:4027–4030. [Google Scholar]
- 8.Selwood AI, Miles CO, Wilkins AL, van Ginkel R, Munday R, Rise F, McNabb P. J Agric Food Chem. 2010;58:6532–6542. doi: 10.1021/jf100267a. [DOI] [PubMed] [Google Scholar]
- 9.McCarron P, Rourke WA, Hardstaff W, Pooley B, Quilliam MA. J Agric Food Chem. 2012;60:1437–1446. doi: 10.1021/jf204824s. [DOI] [PubMed] [Google Scholar]
- 10.(a) Takada N, Umemura N, Suenaga K, Uemura D. Tetrahedron Lett. 2001;42:3495–3497. [Google Scholar]; (b) Hao J, Matsuura F, Kishi Y, Kita M, Uemura D, Asai N, Iwashita T. J Am Chem Soc. 2006;128:7742–7743. doi: 10.1021/ja061893j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miles CO, Wilkins AL, Stirling DJ, MacKenzie AL. J Agric Food Chem. 2000;48:1373–1376. doi: 10.1021/jf991031k. [DOI] [PubMed] [Google Scholar]
- 12.Miles CO, Wilkins AL, Stirling DJ, MacKenzie AL. J Agric Food Chem. 2003;51:4838–4840. doi: 10.1021/jf030101r. [DOI] [PubMed] [Google Scholar]
- 13.Van Wagoner RM, Misner I, Tomas CR, Wright JLC. Tetrahedron Lett. 2011;52:4243–4246. [Google Scholar]
- 14.Hu T, Burton IW, Cembella AD, Curtis JM, Quilliam MA, Walter JA, Wright JLC. J Nat Prod. 2001;64:308–312. doi: 10.1021/np000416q. [DOI] [PubMed] [Google Scholar]
- 15.Hu T, Curtis JM, Oshima Y, Quilliam MA, Walter JA, Watson-Wright WM, Wright JLC. J Chem Soc, Chem Commun. 1995:2139–2141. [Google Scholar]
- 16.Hu T, Curtis JM, Walker JA, Wright JLC. Tetrahedron Lett. 1996;37:7671–7674. [Google Scholar]
- 17.MacKinnon SL, Walter JA, Quilliam MA, Cembella AD, LeBlanc P, Burton IW, Hardstaff WR, Lewis NI. J Nat Prod. 1996;69:983–987. doi: 10.1021/np050220w. [DOI] [PubMed] [Google Scholar]
- 18.Aasen J, MacKinnon SL, LeBlanc P, Walter JA, Hovgaard P, Aune T, Quilliam MA. Chem Res Toxicol. 2005;18:509–515. doi: 10.1021/tx049706n. [DOI] [PubMed] [Google Scholar]
- 19.Roach JS, LeBlanc P, Lewis NI, Munday R, Quilliam MA, MacKinnon SL. J Nat Prod. 2009;72:1237–1240. doi: 10.1021/np800795q. [DOI] [PubMed] [Google Scholar]
- 20.Torigoe K, Murata M, Yasumoto T, Iwashita T. J Am Chem Soc. 1988;110:7876–7877. [Google Scholar]
- 21.Lu CK, Lee CH, Huang R, Chou HN. Tetrahedron Lett. 2001;42:1713–1716. [Google Scholar]
- 22.Selwood AI, Wilkins AL, Munday R, Shi F, Rhodes LL, Holland PT. Terahedron Lett. 2013;54:4705–4707. [Google Scholar]
- 23.Kita M, Kondo M, Koyama T, Yamada K, Matsumoto T, Lee K, Woo J, Uemura D. J Am Chem Soc. 2004;126:4794–4795. doi: 10.1021/ja049277f. [DOI] [PubMed] [Google Scholar]
- 24.Kita M, Kondo M, Koyama T, Yamada K, Matsumoto T, Lee K, Woo J, Uemura D. J Am Chem Soc. 2004;126:4794–4795. doi: 10.1021/ja049277f. [DOI] [PubMed] [Google Scholar]
- 25.Nézan E, Chomerat N. Vulcanodinium rugosum gen. Nov., sp. Nov. (dinophyceae): A new marine dinoflagellate from French mediterranean coast. Cryptogam Algol. 2011;32:3–18. [Google Scholar]
- 26.Hess P, Abadie E, Hervé F, Berteaux T, Séchet V, Aráoz R, Molgó J, Zakarian A, Sibat M, Rundberget T, Miles CO, Amzil Z. Toxicon. 2013;75:16–26. doi: 10.1016/j.toxicon.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rhodes L, Smith K, Selwood A, McNabb P, Munday R, Suda S, Molenaar S, Hallegraff G. Phycologia. 2011;50:624–628. [Google Scholar]
- 28.Wilkins AL, Rehmann N, Torgersen T, Rundberget T, Keogh M, Petersen D, Hess P, Rise F, Miles CO. J Agric Food Chem. 2006;54:5672–5678. doi: 10.1021/jf060396j. [DOI] [PubMed] [Google Scholar]
- 29.Haywood AJ, Steidinger KA, Truby EW, Bergquist PR, Bergquist PL, Adamson J, MacKenzie L. J Phycol. 2004;40:165–179. [Google Scholar]
- 30.Biré R, Krys S, Frémy JM, Dragacci S, Stirling DJ, Kharrat R. J Nat Toxins. 2002;11:269–275. [PubMed] [Google Scholar]
- 31.Cembella AD, Quilliam MA. Nat Toxins. 1999;7:197–206. doi: 10.1002/1522-7189(200009/10)7:5<197::aid-nt62>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 32.Cembella AD, Quilliam MA, Lewis NI. Phycologia. 2000;39:67–74. [Google Scholar]
- 33.Ciminiello P, Dell’Aversano C, Fattorusso E, Magno S, Tartaglione L, Cangini M, Pompei M, Boni L. Toxicon. 2006;47:597–604. doi: 10.1016/j.toxicon.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 34.Ciminiello P, Dell’Aversano C, Fattorusso E, Forino M, Grauso L, Tartaglione L, Guerrini F, Pistocchi R. J Nat Prod. 2007;70:1878–1883. doi: 10.1021/np0703242. [DOI] [PubMed] [Google Scholar]
- 35.Touzet N, Franco JM, Raine R. Harmful Algae. 2008;7:782–797. [Google Scholar]
- 36.Tatters AO, Van WRM, Wright JLC, Tomas CR. Harmful Algae. 2012;19:160–168. [Google Scholar]
- 37.Tomas CR, van Wagoner R, Tatters AO, Hall S, White K, Wright JLC. Harmful Algae. 2012;17:54–63. [Google Scholar]
- 38.For tabulation of dinoflagellates see, for example: http://tolweb.org/notes/?note_id=4796
- 39.Kremp A, Lindholm T, Dreßler N, Erler K, Gerdts G, Eirtovaara S, Leskinen E. Harmful Algae. 2009;8:318–328. [Google Scholar]
- 40.Aasen JAB, Hardstaff W, Aune T, Quilliam MA. Rapid Commun Mass Spectrom. 2006;20:1531–1537. doi: 10.1002/rcm.2501. [DOI] [PubMed] [Google Scholar]
- 41.Yasumoto T, Seino N, Murakami Y, Murata M. Biol Bull. 1987;172:128–131. [Google Scholar]
- 42.Hallegraeff GM. Phycologia. 1993;32:79–99. [Google Scholar]
- 43.Smayda TJ. J Oceanogr. 2002;58:281–294. [Google Scholar]
- 44.Van Dolah FM. Environ Health Perspect. 2000;108:133–141. doi: 10.1289/ehp.00108s1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hallegraeff GM. J Phycol. 2010;46:220–235. [Google Scholar]
- 46.Jørgensen BB, Richardson K. Coastal and Estuarine Studies. Vol. 52. American Geophysical Union; Washington, D.C: 1996. Eutrophication in coastal marine ecosystems; pp. 1–272. [Google Scholar]
- 47.Smith VH. Environ Sci Pollut Res. 2003;10:126–139. doi: 10.1065/espr2002.12.142. [DOI] [PubMed] [Google Scholar]
- 48.Howarth RW, Marino R. Limnol Oceanogr. 2006;51(1 part 2):364–376. [Google Scholar]
- 49.Shumway SE. J World Aquacult Soc. 1990;21:65–104. [Google Scholar]
- 50.Lesser MP, Shumway SE. J Shellfish Res. 1993;12:377–381. [Google Scholar]
- 51.McNabb PS, McCoubrey DJ, Rhodes L, Smith K, Selwood AI, Munday R, Holland PT. Harmful Algae. 2012;13:34–39. [Google Scholar]
- 52.Medhioub W, Lassus P, Truquet P, Bardouil M, Amzil Z, Sechet V, Sibat M, Soudant P. Aquaculture. 2012;358–359:108–115. [Google Scholar]
- 53.Stirling DJ. N Z J Mar Freshwater Res. 2001;35:851–857. [Google Scholar]
- 54.Medhioub W, Guéguen M, Lassus P, Bardouil M, Truquet P, Sibat M, Medhioub N, Soudant P, Kraiem M, Amzil Z. Harmful Algae. 2010;9:200–207. [Google Scholar]
- 55.Marrouchi R, Dziri F, Belayouni N, Hamza A, Benoit E, Molgó J, Kharrat R. Mar Biotechnol. 2010;12:579–585. doi: 10.1007/s10126-009-9245-7. [DOI] [PubMed] [Google Scholar]
- 56.Geiger M, Desanglois G, Hogeveen K, Fessard V, Leprêtre T, Mondeguer F, Guitton Y, Hervé F, Séchet V, Grovel O, Pouchus YF, Hess P. Marine Drugs. 2013;11:3350–3371. doi: 10.3390/md11093350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dragunow M, Trzoss M, Brimble MA, Cameron R, Beuzenberg V, Holland P, Mountfort D. Environmental Toxicology and Pharmacology. 2005;20:305–312. doi: 10.1016/j.etap.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 58.Daneshian M, Botana LM, Dechraoui Bottein MY, Buckland G, Campàs M, Dennison N, Dickey RW, Diogène J, Fessard V, Hartung T, Humpage A, Leist M, Molgó J, Quilliam MA, Rovida C, Suarez-Isla BA, Tubaro A, Wagner K, Zoller O, Dietrich D. ALTEX. 2013;30:487–545. doi: 10.14573/altex.2013.4.487. [DOI] [PubMed] [Google Scholar]
- 59.Botana LM. Chemical Research in Toxicology. 2012;25:1800–1804. doi: 10.1021/tx3001863. [DOI] [PubMed] [Google Scholar]
- 60.Munday R, Quilliam MA, LeBlanc P, Lewis N, Gallant P, Sperker SA, Ewart HS, MacKinnon SL. Toxins (Basel) 2012;4:1–14. doi: 10.3390/toxins4010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Richard D, Arsenault E, Cembella A, Quilliam M. In: Harmful Algal Blooms. Hallegraeff GM, Blackburn SI, Bolch C, Lewis RJ, editors. Intergovernmental Oceanographic Commission of UNESCO; Paris, France: 2001. pp. 383–386. [Google Scholar]
- 62.Rhodes L, Smith K, Selwood A, McNabb P, Molenaar S, Munday R, Wilkinson C, Hallegraeff G. New Zealand Journal of Marine and Freshwater Research. 2011;145:703–709. [Google Scholar]
- 63.Rhodes L, Smith K, Selwood A, McNabb P, van Ginkel R, Holland P, Munday R. Harmful Algae. 2010;9:384–389. [Google Scholar]
- 64.Munday R, Selwood AI, Rhodes L. Toxicon. 2012;60:995–999. doi: 10.1016/j.toxicon.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 65.EFSA - The European Food Safety Authority. Scientific opinion of the panel on contaminants in the food chain on a request from the European Commission on marine biotoxins in shellfish. Summary on Regulated Marine Biotoxins. EFSA J. 2009;1306:1–23. [Google Scholar]
- 66.Ciminiello P, Dell’Aversano C, Dello Iacovo E, Fattorusso E, Forino M, Grauso L, Tartaglione L, Guerrini F, Pezzolesi L, Pistocchi R. Toxicon. 2010;56:1327–1333. doi: 10.1016/j.toxicon.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 67.Otero P, Alfonso A, Rodriguez P, Rubiolo JA, Cifuentes JM, Bermúdez R, Vieytes MR, Botana LM. Food Chem Toxicol. 2012;50:232–237. doi: 10.1016/j.fct.2011.10.062. [DOI] [PubMed] [Google Scholar]
- 68.Beaumont S, Ilardi EA, Tappin NDC, Zakarian A. Eur J Org Chem. 2010:5743–5765. doi: 10.1002/ejoc.201000842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.O’Connor PD, Brimble MA. Nat Prod Rep. 2007;24:869–885. doi: 10.1039/b700307m. [DOI] [PubMed] [Google Scholar]
- 70.Guéret SM, Brimble MA. Nat Prod Rep. 2010;27:1350–1366. doi: 10.1039/c005400n. [DOI] [PubMed] [Google Scholar]
- 71.McCauley JA, Nagasawa K, Lander PL, Mischke SG, Semones MA, Kishi Y. J Am Chem Soc. 1998;120:7647–7648. [Google Scholar]
- 72.Pelc MJ, Zakarian A. Org Lett. 2005;7:1629–1631. doi: 10.1021/ol050321l. [DOI] [PubMed] [Google Scholar]
- 73.Wang J, Sakamoto S, Kumada K, Nitta A, Noda T, Oguri H, Hirama M. Synlett. 2003:891–893. [Google Scholar]
- 74.Stivala CE, Zakarian A. J Am Chem Soc. 2008;130:3774–3776. doi: 10.1021/ja800435j. [DOI] [PubMed] [Google Scholar]
- 75.Araoz R, Servent D, Molgó J, Iorga BI, Fruchart-Gaillard C, Benoit E, Gu Z, Stivala CE, Zakarian A. J Am Chem Soc. 2011;133:10499–10511. doi: 10.1021/ja201254c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.For recent examples, see: Magauer T, Martin HJ, Mulzer J. Angew Chem Int Ed. 2009;48:6032–6036. doi: 10.1002/anie.200900522.ref 14 therein. Magauer T, Martin HJ, Mulzer J. Chem Eur J. 2010;16:507–519. doi: 10.1002/chem.200902226.
- 77.http://techtransfer.universityofcalifornia.edu/NCD/21833.html
- 78.Unpublished results
- 79.Nakamura E, Kikuchi F, Hashimoto S. Angew Chem Int Ed. 2008;47:7091–7094. doi: 10.1002/anie.200802729. [DOI] [PubMed] [Google Scholar]
- 80.Nakamura S, Inagaki J, Sugimoto T, Ura Y, Hashimoto S. Tetrahedron. 2002;58:10375–10386. [Google Scholar]
- 81.Jackson JJ, Stivala CE, Iorga BI, Molgó J, Zakarian A. J Org Chem. 2012;77:10435–10440. doi: 10.1021/jo301632d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ishihara J, Ishizaka T, Suzuki T, Hatakeyama S. Tetrahedron Lett. 2004;45:7855–7858. [Google Scholar]
- 83.(a) Caprio V, Brimble MA, Furkert DP. Tetrahedron. 2001;57:4023–4034. [Google Scholar]; (b) Furkert DP, Brimble MA. Org Lett. 2002;4:3655, 3658. doi: 10.1021/ol026605c. [DOI] [PubMed] [Google Scholar]; (c) Meilert K, Brimble MA. Org Biomol Chem. 2006;4:2184–2192. doi: 10.1039/b604334h. [DOI] [PubMed] [Google Scholar]; (d) Meilert K, Brimble MA. Org Lett. 2005;7:3497–3500. doi: 10.1021/ol051260u. [DOI] [PubMed] [Google Scholar]
- 84.Labarre-Lainé J, Periñan I, Desvergnes V, Landais Y. Chem Eur J. 2014;20 doi: 10.1002/chem.201402894. dx.doi.org/10.1002/chem.201402894. [DOI] [PubMed] [Google Scholar]
- 85.(a) Brimble MA, Trzoss M. Tetrahedron. 2004;60:5613–5622. [Google Scholar]; (b) Trzoss M, Brimble MA. Synlett. 2003:2042–2046. [Google Scholar]
- 86.Stivala CE, Zakarian A. Org Lett. 2009;11:839–842. doi: 10.1021/ol8027797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stivala CE, Gu Z, Smith LL. Org Lett. 2012;14:804–807. doi: 10.1021/ol203342e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.(a) Johannes JW, Wenglowsky S, Kishi Y. Org Lett. 2005;7:3997–4000. doi: 10.1021/ol051553n. [DOI] [PubMed] [Google Scholar]; (b) Kong K, Moussa Z, Romo D. Org Lett. 2005;7:5127–5130. doi: 10.1021/ol051840r. [DOI] [PubMed] [Google Scholar]; (c) White JD, Quaranta L, Wang G. J Org Chem. 2007;72:1717–1728. doi: 10.1021/jo062396o. [DOI] [PubMed] [Google Scholar]; (d) White JD, Wang G, Quaranta L. Org Lett. 2003;5:4983–4986. doi: 10.1021/ol035939e. [DOI] [PubMed] [Google Scholar]; (e) Qin Y, Stivala CE, Zakarian A. Angew Chem Int Ed. 2007;46:7466–7469. doi: 10.1002/anie.200702142. [DOI] [PubMed] [Google Scholar]
- 89.(a) Kong K, Romo D, Lee C. Angew Chem Int Ed. 2009;48:7402–7405. doi: 10.1002/anie.200903432. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kong K, Moussa Z, Lee C, Romo D. J Am Chem Soc. 2011;133:19844–19856. doi: 10.1021/ja207385y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ishihara J, Horie M, Shimada Y, Tojo S, Murai A. Synlett. 2002:403–406. [Google Scholar]
- 91.Kita M, Uemura D. Progress in molecular and subcellular biology. 2006;43:25–51. doi: 10.1007/978-3-540-30880-5_2. [DOI] [PubMed] [Google Scholar]
- 92.Zheng SF, Huang S, Chen X, Tan J, Zuo Z, Peng J, Xie R. Chin J Mar Drugs. 1990;9:33–35. [Google Scholar]
- 93.Munday R, Towers NR, Mackenzie L, Beuzenberg V, Holland PT, Miles CO. Toxicon. 2004;44:173–178. doi: 10.1016/j.toxicon.2004.05.017. [DOI] [PubMed] [Google Scholar]
- 94.Kharrat R, Servent D, Girard E, Ouanounou G, Amar M, Marrouchi R, Benoit E, Molgó J. J Neurochem. 2008;107:952–963. doi: 10.1111/j.1471-4159.2008.05677.x. [DOI] [PubMed] [Google Scholar]
- 95.Aráoz R, Servent D, Ouanounou G, Benoit E, Molgó J. Biol Res. 2009;42:R–118. [Google Scholar]
- 96.Hellyer SD, Selwood AI, Rhodes L, Kerr DS. Toxicon. 2011;58:693–699. doi: 10.1016/j.toxicon.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 97.Gill S, Murphy M, Clausen J, Richard D, Quilliam M, MacKinnon S, LaBlanc P, Mueller R, Pulido O. Neurotoxicology. 2003;24:593–604. doi: 10.1016/S0161-813X(03)00014-7. [DOI] [PubMed] [Google Scholar]
- 98.Corringer PJ, Le Novere N, Changeux JP. Annual review of pharmacology and toxicology. 2000;40:431–458. doi: 10.1146/annurev.pharmtox.40.1.431. [DOI] [PubMed] [Google Scholar]
- 99.Lukas RJ, Changeux JP, Le Novere N, Albuquerque EX, Balfour DJ, Berg DK, Bertrand D, Chiappinelli VA, Clarke PB, Collins AC, Dani JA, Grady SR, Kellar KJ, Lindstrom JM, Marks MJ, Quik M, Taylor PW, Wonnacott S. Pharmacological reviews. 1999;51:397–401. [PubMed] [Google Scholar]
- 100.Bourne Y, Radic Z, Aráoz R, Talley TT, Benoit E, Servent D, Taylor P, Molgó J, Marchot P. Proc Natl Acad Sci USA. 2010;107:6076–6081. doi: 10.1073/pnas.0912372107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hauser TA, Hepler CD, Kombo DC, Grinevich VP, Kiser MN, Hooker DN, Zhang JH, Mountfort D, Selwood A, Akireddy SR, Letchworth SR, Yohannes D. Neuropharmacology. 2012;62:2239–2250. doi: 10.1016/j.neuropharm.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 102.Duroure L, Jousseaume T, Araoz R, Barre E, Retailleau P, Chabaud L, Molgó J, Guillou C. Org Biomol Chem. 2011;9:8112–8118. doi: 10.1039/c1ob06257c. [DOI] [PubMed] [Google Scholar]
- 103.Bockaert J, Pin JP. The EMBO journal. 1999;18:1723–1729. doi: 10.1093/emboj/18.7.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Caulfield MP. Pharmacology & Therapeutics. 1993;58:319–379. doi: 10.1016/0163-7258(93)90027-b. [DOI] [PubMed] [Google Scholar]
- 105.Haga K, Kruse AC, Asada H, Yurugi-Kobayashi T, Shiroishi M, Zhang C, Weis WI, Okada T, Kobilka BK, Haga T, Kobayashi T. Nature. 2012;482:547–551. doi: 10.1038/nature10753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kruse AC, Hu J, Pan AC, Arlow DH, Rosenbaum DM, Rosemond E, Green HF, Liu T, Chae PS, Dror RO, Shaw DE, Weis WI, Wess J, Kobilka BK. Nature. 2012;482:552–556. doi: 10.1038/nature10867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, Hubner H, Pardon E, Valant C, Sexton PM, Christopoulos A, Felder CC, Gmeiner P, Steyaert J, Weis WI, Garcia KC, Wess J, Kobilka BK. Nature. 2013;504:101–106. doi: 10.1038/nature12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wandscheer CB, Vilariño N, Espina B, Louzao MC, Botana LM. Chem Res Toxicol. 2010;23:1753–1761. doi: 10.1021/tx100210a. [DOI] [PubMed] [Google Scholar]
- 109.Mousavi M, Hellstrom-Lindahl E. Neurochem Int. 2009;54:237–244. doi: 10.1016/j.neuint.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 110.Alonso E, Vale C, Vieytes MR, Laferla FM, Gimenez-Llort L, Botana LM. Cell Physiol Biochem. 2011;27:783–794. doi: 10.1159/000330086. [DOI] [PubMed] [Google Scholar]
- 111.Alonso E, Vale C, Vieytes MR, Laferla FM, Gimenez-Llort L, Botana LM. Neurochem Int. 2011;59:1056–1065. doi: 10.1016/j.neuint.2011.08.013. [DOI] [PubMed] [Google Scholar]
- 112.Alonso E, Otero P, Vale C, Alfonso A, Antelo A, Gimenez-Llort L, Chabaud L, Guillou C, Botana LM. Curr Alzheimer Res. 2013;10:279–289. doi: 10.2174/1567205011310030007. [DOI] [PubMed] [Google Scholar]
- 113.Yasumoto T, Oshima Y, Yamaguchi M. Bulletin of the Japanese Society of Scientific Fisheries. 1978;44:1249–1255. [Google Scholar]
- 114.COMMISSION REGULATION. Official Journal of the European Union. 2011;54:3–7. [Google Scholar]
- 115.Fux E, McMillan D, Bire R, Hess P. Journal of Chromatography A. 2007;1157:273–280. doi: 10.1016/j.chroma.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 116.García-Altares M, Diogène J, de la Iglesia P. J Chromatogr A. 2013;1275:48–60. doi: 10.1016/j.chroma.2012.12.021. [DOI] [PubMed] [Google Scholar]
- 117.Gerssen A, van Olst EHW, Mulder PPJ, de Boer J. Analytical and Bioanalytical Chemistry. 2010;397:3079–3088. doi: 10.1007/s00216-010-3886-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gerssen A, Mulder PPJ, McElhinney MA, de Boer J. Journal of Chromatography A. 2009;1216:1421–1430. doi: 10.1016/j.chroma.2008.12.099. [DOI] [PubMed] [Google Scholar]
- 119.Chapela MJ, Reboreda A, Vieites JM, Cabado AG. Journal of Agricultural and Food Chemistry. 2008;56:8979–8986. doi: 10.1021/jf801572j. [DOI] [PubMed] [Google Scholar]
- 120.Krock B, Tillmann U, John U, Cembella A. Analytical and Bioanalytical Chemistry. 2008;392:797–803. doi: 10.1007/s00216-008-2221-7. [DOI] [PubMed] [Google Scholar]
- 121.Rundberget T, Aasen JAB, Selwood AI, Miles CO. Toxicon. 2011;58:700–711. doi: 10.1016/j.toxicon.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 122.McNabb P, Selwood AI, Holland PT. Journal of Aoac International. 2005;88:761–772. [PubMed] [Google Scholar]
- 123.MacKenzie L, Holland P, McNabb P, Beuzenberg V, Selwood A, Suzuki T. Toxicon. 2002;40:1321–1330. doi: 10.1016/s0041-0101(02)00143-5. [DOI] [PubMed] [Google Scholar]
- 124.Liu RY, Liang YB, Wu XL, Xu DY, Liu YJ, Liu L. Toxicon. 2011;57:1000–1007. doi: 10.1016/j.toxicon.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 125.Ben Naila I, Hamza A, Gdoura R, Diogene J, de la Iglesia P. Harmful Algae. 2012;18:56–64. [Google Scholar]
- 126.Hummert C, Ruhl A, Reinhardt K, Gerdts G, Luckas B. Chromatographia. 2002;55:673–680. [Google Scholar]
- 127.Amzil Z, Sibat M, Royer F, Masson N, Abadie E. Marine Drugs. 2007;5:168–179. doi: 10.3390/md504168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ciminiello P, Dell’Aversano C, Fattorusso E, Forino M, Tartaglione L, Boschetti L, Rubini S, Cangini M, Pigozzi S, Poletti R. Toxicon. 2010;55:280–288. doi: 10.1016/j.toxicon.2009.07.037. [DOI] [PubMed] [Google Scholar]
- 129.Shen Q, Gong L, Baibado JT, Dong W, Wang Y, Dai Z, Cheung HY. Talanta. 2013;116:770–775. doi: 10.1016/j.talanta.2013.07.042. [DOI] [PubMed] [Google Scholar]
- 130.Aráoz R, Ramos S, Pelissier F, Guérineau V, Benoit E, Vilariño N, Botana LM, Zakarian A, Molgó J. Analytical Chemistry. 2012;84:10445–10453. doi: 10.1021/ac3027564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Blay P, Hui JPM, Chang JM, Melanson JE. Analytical and Bioanalytical Chemistry. 2011;400:577–585. doi: 10.1007/s00216-011-4772-2. [DOI] [PubMed] [Google Scholar]
- 132.Domenech A, Cortes-Francisco N, Palacios O, Franco JM, Riobo P, Llerena JJ, Vichi S, Caixach J. Journal of Chromatography A. 2014;1328:16–25. doi: 10.1016/j.chroma.2013.12.071. [DOI] [PubMed] [Google Scholar]
- 133.Aráoz R, Herdman M, Rippka R, Ledreux A, Molgó J, Changeux JP, de Marsac NT, Nghiem HO. Toxicon. 2008;52:163–174. doi: 10.1016/j.toxicon.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 134.Vilariño N, Fonfría ES, Molgó J, Aráoz R, Botana LM. Analytical Chemistry. 2009;81:2708–2714. doi: 10.1021/ac900144r. [DOI] [PubMed] [Google Scholar]
- 135.Fonfria ES, Vilarino N, Molgo J, Araoz R, Otero P, Espina B, Louzao MC, Alvarez M, Botana LM. Analytical Biochemistry. 2010;403:102–107. doi: 10.1016/j.ab.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 136.Fonfria ES, Vilariño N, Espina B, Louzao MC, Alvarez M, Molgó J, Aráoz R, Botana LM. Analytica Chimica Acta. 2010;657:75–82. doi: 10.1016/j.aca.2009.10.027. [DOI] [PubMed] [Google Scholar]
- 137.Otero P, Alfonso A, Alfonso C, Aráoz R, Molgó J, Vieytes MR, Botana LM. Analytica Chimica Acta. 2011;701:200–208. doi: 10.1016/j.aca.2011.05.034. [DOI] [PubMed] [Google Scholar]
- 138.Rodriguez LP, Vilariño N, Molgó J, Aráoz R, Antelo A, Vieytes MR, Botana LM. Analytical Chemistry. 2011;83:5857–5863. doi: 10.1021/ac200423s. [DOI] [PubMed] [Google Scholar]
- 139.Rodriguez LP, Vilariño N, Molgó J, Aráoz R, Botana LM. Toxicon. 2013;75:35–43. doi: 10.1016/j.toxicon.2013.06.015. [DOI] [PubMed] [Google Scholar]
- 140.Rodriguez LP, Vilariño N, Molgó J, Aráoz R, Louzao MC, Taylor P, Talley T, Botana LM. Analytical Chemistry. 2013;85:2340–2347. doi: 10.1021/ac3033432. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.