

Abstract
Objective
Lp(a) is an independent, causal, genetic risk factor for cardiovascular disease and aortic stenosis. Current pharmacologic lipid-lowering therapies do not optimally lower Lp(a), particularly in patients with familial hypercholesterolemia (FH).
Approach and Results
In four Phase III trials, 382 patients on maximally tolerated lipid-lowering therapy were randomized 2:1 to weekly subcutaneous mipomersen 200 mg (n=256) or placebo (n=126) for 26 weeks. Populations included homozygous FH (HoFH), heterozygous FH (HeFH) with concomitant coronary artery disease (CAD), severe hypercholesterolemia (HC), and HC at high risk for CAD. Lp(a) was measured eight times between baseline and week 28 inclusive. Of the 382 patients, 57% and 44% had baseline Lp(a) levels >30 mg/dL and >50 mg/dL, respectively. In the pooled analysis, the mean percent decrease (median, interquartile range, IQR) in Lp(a) at 28 weeks was significantly greater in the mipomersen group compared with placebo (-26.4 (-42.8, 5.4) vs. -0.0 (10.7, 15.3), p<0.001). In the mipomersen group in patients with Lp(a) levels >30 mg/dL or >50 mg/dL, attainment of Lp(a) values ≤30 mg/dL or ≤50 mg/dL was most frequent in HoFH and severe HC patients. In the combined groups, modest correlations were present between percent change in apoB and Lp(a) (r=0.43, p<0.001) and LDL-C and Lp(a) (r=0.36, p<0.001) plasma levels.
Conclusions
Mipomersen consistently and effectively reduced Lp(a) levels in patients with a variety of lipid abnormalities and cardiovascular risk. Modest correlations were present between apoB and Lp(a) lowering but the mechanistic relevance mediating Lp(a) reduction is currently unknown.
Keywords: antisense oligonucleotide, lipoprotein(a), familial hypercholesterolemia, coronary artery disease, hypolipidemic agents, dyslipidemias
Introduction
Lipoprotein(a) [Lp(a)] is composed of apolipoprotein(a) covalently bound to apolipoprotein B-100 of LDL.1,2 Lp(a) levels vary substantially among individuals (<0.1 mg/dL to >250 mg/dL) with minimal dietary or environmental effect and display a wide racial distribution, with highest levels in people of African and South Asian origin, followed by Caucasians, Hispanics and East Asians, respectively.2-4 Approximately 30% of whites and 60-70% of blacks have what are considered elevated Lp(a) levels of >30 mg/dL. Recent epidemiologic, genome-wide association and Mendelian randomization studies strongly support the concept that Lp(a) is a causal, independent, genetic risk factor for myocardial infarction, stroke, peripheral arterial disease and calcific aortic stenosis.5-11
Lp(a) levels are elevated in patients with familial hypercholesterolemia (FH) compared with healthy individuals or unaffected family members and more so than would be predicted from LPA genotypes such as number of KIV-2 repeats.12 Elevated Lp(a) levels are a particularly strong and independent risk factor for CVD in this subgroup with concomitant elevation of low density lipoprotein cholesterol (LDL-C).12-18 Individuals with homozygous FH (HoFH) with 2 nonfunctional low-density lipoprotein-receptor (LDLR) alleles have approximately 2-fold higher levels of Lp(a) compared with heterozygous (HeFH) patients and higher risk of CVD.12,19 These data demonstrate a gene-dosage effect on Lp(a) despite the fact that human and murine turnover kinetic studies have failed to show that the LDLR is significantly involved in Lp(a) clearance, suggesting additional mechanisms for elevated levels.20,21
Established drug therapies to lower Lp(a) levels are essentially limited to niacin, although trials to assess clinical outcomes with niacin in patients with elevated Lp(a) levels have not been performed. In addition, lipoprotein apheresis is specifically approved in Germany for patients with progressive CVD, controlled LDL-C levels and Lp(a) levels >60 mg/dL, and is used on an ad hoc basis in the United States and other countries.22 With the established, independent clinical risk of elevated Lp(a) levels,23 new treatments are needed to reduce CVD in these patients. Mipomersen is a second-generation antisense oligonucleotide directed to liver messenger RNA (mRNA) of apolipoprotein B-100 (apoB) that reduces LDL-C.24-27 Since apoB is the essential structural component of all apoB containing lipoproteins, including LDL, inhibition of apoB synthesis may reduce levels of all apoB containing atherogenic lipoproteins, including Lp(a). In this study, we examined the effect of mipomersen on Lp(a) in 4 randomized, double-blind, placebo controlled phase 3 trials.
Materials and Methods
Materials and methods are available in the online-only Data Supplement.
Results
Trial Design
Datasets were obtained from phase III trials of mipomersen in 4 different populations: 1- HoFH (NCT00607373)—genetic confirmation of HoFH or a clinical diagnosis based on an untreated LDL-C >500 mg/dL together with either xanthoma before 10 years of age or evidence of HeFH in both parents24; 2- severe hypercholesterolemia (NCT00794664)—diagnosis of severe hypercholesterolemia having an LDL-C ≥200 mg/dL25; 3- HeFH with CAD (NCT00706849)—diagnosis of HeFH plus LDL C ≥100 mg/dL and triglycerides (TG) <200 mg/dL and a diagnosis of CAD26; 4- hypercholesterolemia at high risk for CAD per National Cholesterol Education Program Adult Treatment Panel III guidelines (NCT00770146)—LDL-C ≥100 mg/dL with TG <200 mg/dL.27 All 4 trials were conducted with the same study design (Figure 1) at 26 clinical centers in 6 countries. Each trial randomized patients 2:1 to weekly, subcutaneous injections of mipomersen 200 mg or placebo for 26 weeks on a background of maximally tolerated lipid-lowering therapy.
Figure 1. Study Design for Mipomersen Phase 3 Clinical Trials.
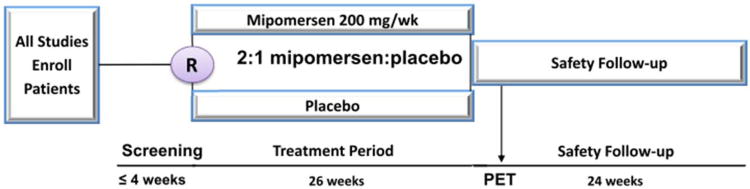
R=randomization; PET=Primary Efficacy Timepoint; week 28 or 2 weeks after the last dose.
Patient characteristics
The primary endpoints from each trial were previously reported.24-27 Patient characteristics are shown in Table 1 and included 382 patients. Most patients were middle-aged (mean age, 53 years) and overweight (mean BMI, 29 kg/m2), with 48.4% having metabolic syndrome and about one-sixth being current smokers. HoFH patients were younger and tended to be leaner compared to the other groups.
Table 1. Baseline patient characteristics.
| Variable | Pooled Phase 3 | HoFH | Severe HC | HeFH with CAD | HC at high risk for CAD |
|---|---|---|---|---|---|
| n=382 | n=51 | n=57 | n=123 | n=151 | |
| Male Gender, n (%) | 205 (53.7) | 22 (43.1) | 25 (43.9) | 78 (63.4) | 80 (53.0) |
| Age in years, mean (SD) | 53 (14.1) | 31 (12.4) | 50 (14.1) | 56 (9.6) | 59 (9.7) |
| BMI, mean (SD) | 29 (4.8) | 26 (5.3) | 29 (5.0) | 29 (4.2) | 30 (4.4) |
| * Hypertensive, yes, n (%) | 10 (2.6) | 0 (0.0) | 3 (5.3) | 4 (3.3) | 3 (2.0) |
| Metabolic Syndrome, yes, n (%) | 185 (48.4) | 8 (15.7) | 22 (38.6) | 45 (36.6) | 110 (72.8) |
| Current Smoker, n (%) | 65 (17.0) | 10 (19.6) | 9 (15.8) | 17 (13.8) | 29 (19.2) |
BMI=Body Mass Index
Hypertensive was defined as having the 3 parameters of 1) systolic blood pressure ≥140 mmHg, diastolic blood pressure ≥ 90 mmHg and previous use of blood pressure medication.
Prior/concomitant lipid lowering medications are shown in Table 2. About one-third of all patients were concomitantly taking a statin as monotherapy, another third were taking a statin plus niacin or ezetimibe or a combination of both, and another third were taking a combination of a statin and another lipid lowering medication.
Table 2. Lipid lowering medications on randomization.
| Population | Therapies | Placebo n (%) | Mipomersen n (%) |
|---|---|---|---|
| Pooled Analysis | n | 126 | 256 |
|
| |||
| Statins alone | 42 (33.3) | 80 (31.3) | |
| Statin + Niacin | 3 (2.4) | 9 (3.5) | |
| Statin + Ezetimibe | 34 (27.0) | 81 (31.6) | |
| Statin + Niacin + Ezetimibe | 5 (4.0) | 11 (4.3) | |
| Statin + Other LLT(s) | 42 (33.3) | 73 (28.5) | |
| Other LLT(s) without Statin | 0 | 1 (0.4) | |
| None | 0 | 1 (0.4) | |
|
| |||
| HoFH | n | 17 | 34 |
|
| |||
| Statins alone | 6 (35.3) | 6 (17.6) | |
| Statin + Niacin | 0 | 1 (2.9) | |
| Statin + Ezetimibe | 6 (35.3) | 23 (67.6) | |
| Statin + Niacin + Ezetimibe | 1 (5.9) | 1 (2.9) | |
| Statin + Other LLT(s) | 4 (23.5) | 2 (5.9) | |
| Other LLT(s) without Statin | 0 | 0 | |
| None | 0 | 1 (2.9) | |
|
| |||
| Severe Hypercholesterolemia | n | 18 | 39 |
|
| |||
| Statins alone | 3 (16.7) | 9 (23.1) | |
| Statin + Niacin | 0 | 1 (2.6) | |
| Statin + Ezetimibe | 8 (44.4) | 14 (35.9) | |
| Statin + Niacin + Ezetimibe | 1 (5.6) | 0 | |
| Statin + Other LLT(s) | 6 (33.3) | 15 (38.5) | |
| Other LLT(s) without Statin | 0 | 0 | |
|
| |||
| HeFH with CAD | n | 41 | 82 |
|
| |||
| Statins alone | 3 (7.3) | 9 (11.0) | |
| Statin + Niacin | 1 (2.4) | 3 (3.7) | |
| Statin + Ezetimibe | 16 (39.0) | 37 (45.1) | |
| Statin + Niacin + Ezetimibe | 3 (7.3) | 8 (9.8) | |
| Statin + Other LLT(s) | 18 (43.9) | 24 (29.3) | |
| Other LLT(s) without Statin | 0 | 1 (1.2) | |
|
| |||
| Hypercholesterolemia at high risk for CAD | n | 50 | 101 |
|
| |||
| Statins alone | 30 (60.0) | 56 (55.4) | |
| Statin + Niacin | 2 (4.0) | 4 (4.0) | |
| Statin + Ezetimibe | 4 (8.0) | 7 (6.9) | |
| Statin + Niacin + Ezetimibe | 0 | 2 (2.0) | |
| Statin + Other LLT(s) | 14 (28.0) | 32 (31.7) | |
| Other LLT(s) without Statin | 0 | 0 | |
Includes lipid-lowering medications started before, and continued into, the treatment period. The treatment period spans the time during which the study treatment is administered until the later of the primary efficacy time point and 14 days beyond the last study medication date.
The cardiovascular history summary for patients enrolled in the Phase 3 trials are shown in Supplemental Table. In the HoFH population, about half of the patients had a history of cardiovascular disease/procedures. The placebo and mipomersen groups were similar with respect to cardiovascular history.
Baseline Lp(a) levels and changes following mipomersen therapy
The distributions of baseline Lp(a) levels by individual study show that Lp(a) values are progressively shifted rightward to higher levels with the severity of hypercholesterolemia (Figure 2), relative to what is normally seen in unselected populations, such as in the Copenhagen studies.28
Figure 2. Frequency distribution of baseline Lp(a) levels in the 4 randomized trials.
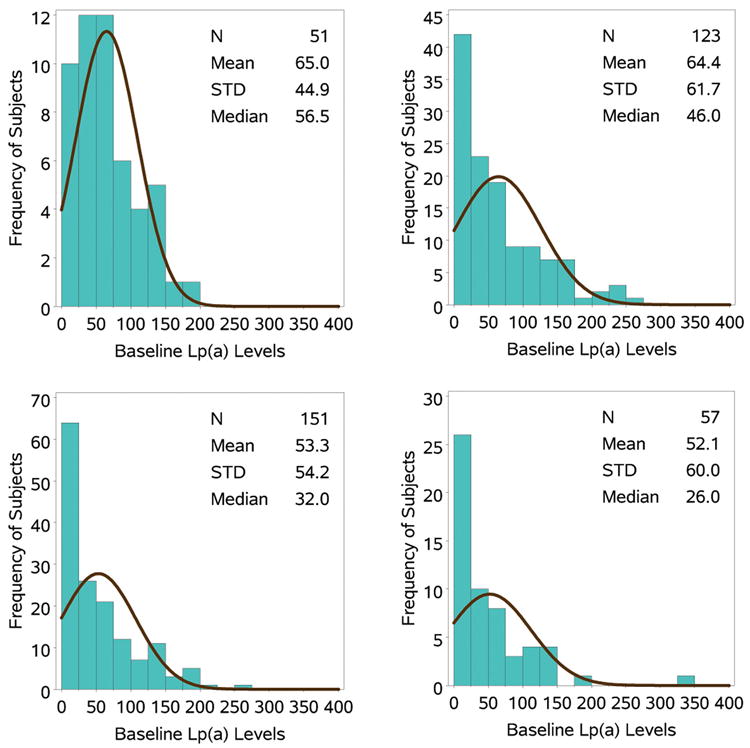
Histogram of the distribution of Lp(a) levels in the 4 studies. The black line represents the normal curve for the histogram.
Table 3 shows the baseline and percent change in Lp(a) over the 28 weeks of follow-up. In the HoFH and HeFH groups, median baseline Lp(a) levels were elevated (relative to values ≤30 mg/dL): HoFH 58.0 mg/dl in placebo and 56.3 mg/dL in mipomersen; HeFH, 52.5 mg/dL in placebo and 45.0 mg/dL in mipomersen. Following randomization in all 4 trials, the pooled analysis revealed that the mean percent change (median, interquartile range, IQR) in Lp(a) was significantly greater in the mipomersen group compared with placebo [-26.4 (Median: -26.4, IQR: -42.8, 5.4) vs. -0.9 (Median: 0.0, IQR: 10.7, 15.3), p<0.001]. The greatest reductions in median percent change were seen in the HoFH (31.8) and severe HC (39.1) populations.
Table 3. Effect of mipomersen on Lp(a) Levels.
| Lp(a) | ||||||
|---|---|---|---|---|---|---|
| Patient Population | Median (IQR) Baseline (mg/dL) | Median (IQR) PET (mg/dL) | Median % Change at PET | |||
| Placebo | Mipomersen | Placebo | Mipomersen | Placebo | Mipomersen | |
| HoFH N=51 | 58.0 (20.5,102.0) | 56.3 (36.0, 78.0) | 53.0 (14.0, 111.0) | 33.0 (23.0, 56.0) | -7.0 (-30.7, 7.5) | -31.8 (-45.5, -15.6) |
| Severe HC N=57 | 21.8 (13.0, 59.0) | 34.0 (12.5, 95.0) | 21.0 (12.0, 64.0) | 18.0 (5.0, 82.0) | -4.7 (-8.3, 7.9) | -39.1 (-57.7, -15.6) |
| HeFH with CAD N=123 | 52.5 (17.0, 108.0) | 45.0 (13.0, 93.0) | 51.0 (18.0, 108.0) | 34.5 (9.0, 56.0) | 0.0 (-8.0, 13.0) | -21.1 (-37.9, 0.0) |
| HC at high risk for CAD N=151 | 30.5 (10.5, 76.0) | 36.0 (10.0, 85.0) | 33.0 (10.0, 78.0) | 21.0 (6.0, 54.0) | 0.0 (-16.0, 17.6) | -25.6 (-40.0, -7.8) |
| Pooled Analysis* N=382 | 32.0 (13.0, 89.0) | 41.8 (13.3, 85.0) | 34.0 (14.0, 87.0) | 29.0 (9.0, 56.0) | 0.0 (-10.7, 15.3) | -26.4 (-42.8, -5.4) |
The temporal changes in Lp(a) are shown in Figure 3. A separation of the curves in the mipomersen versus placebo is evident in the pooled data analysis by week 5 and the curves continue to diverge until week 26. Similar changes were noted in all 4 individual trials. Figure 4 displays waterfall plots of individual patients for percent change in Lp(a) from baseline in the pooled analysis of all 4 trials. In the mipomersen group, a wide variability was noted in Lp(a) responses, with the greatest maximal reduction of -84.2% and the maximal increase was 97%. It is also noted that a few patients (10.5% total) in each study had increases in Lp(a), as a percent change from baseline, despite mipomersen treatment and a number of patients (46.0% total) had Lp(a) decreases in the placebo arm. Similar changes were noted in all 4 individual trials. It is to be emphasized that because of the broad distribution of baseline Lp(a) levels, small absolute changes in Lp(a), particularly in patients with low values, can be accentuated when analyzed as a percent change. The median and maximum % changes (increase and decrease) in Lp(a) for patients with baseline Lp(a) levels above 30 mg/dL was -28.2% (97% and -84.2%) respectively.
Figure 3. Median change in Lp(a) mediated by mipomersen in the pooled data of the 4 trials and in individual trials.
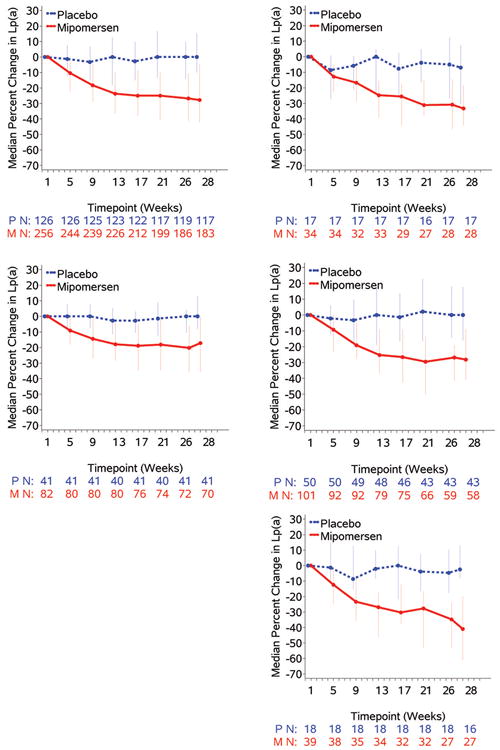
Effect of mipomersen (median (IQR) percent change) over time in the pooled 4 Phase III studies and in individual studies.
Figure 4. Waterfall plots of the percent change in Lp(a) from baseleine to the primary efficacy endpoint in the pooled data of the 4 trials and in individual trials.
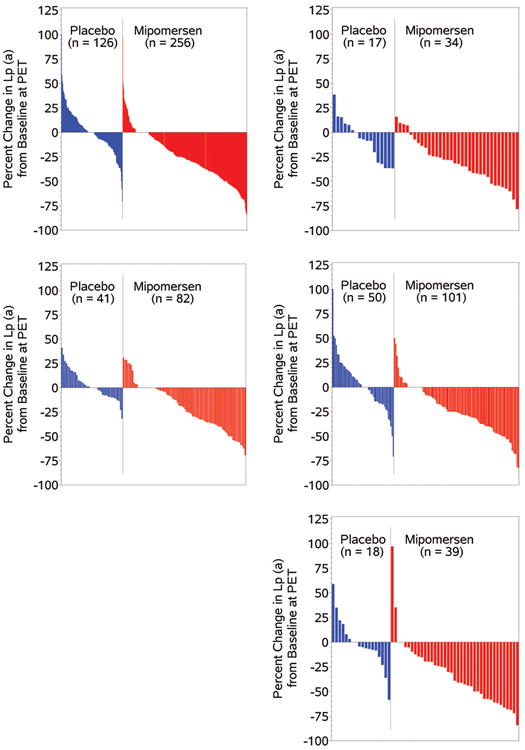
Waterfall plots depicting the distribution of the percent change in Lp(a) levels in reposnse to mipomersen and plabebo in the pooled 4 Phase III studies and in individual studies.
Percent of patients reaching Lp(a) levels ≤30 mg/dL and ≤50 mg/dL
It is generally accepted that elevated Lp(a) levels are defined as >30 mg/dL or >75 nmol/L, which approximates the 75th percentile of the Framingham population.29 Additionally, the European Atherosclerosis Society (EAS) suggested optimal Lp(a) levels should be <50 mg/dL, which represents the 80th percentile of northern European populations.28
Based on these parameters, we determined the percentage of patients reaching these goals as shown in Table 4. At baseline, 64 (50.8%) placebo patients and 152 (59.4%) mipomersen patients had Lp(a) levels >30 mg/dL, and 56 (44.4%) placebo patients and 111 (43.4%) mipomersen patients had Lp(a) levels >50 mg/dL. In the mipomersen group after 26 weeks of treatment, 10.9% of patients shifted from having Lp(a) levels >30 mg/dL to levels ≤30 mg/dL. The greatest reductions to ≤30 mg/dL were present in the HoFH (26.5%) and severe HC (15.4%) populations; smaller shifts were observed in the HeFH with CAD (8.5%) and HC at high risk for CAD populations (5.9%). Similarly, 15.6% of patients having baseline Lp(a) >50 mg/dL achieved Lp(a) ≤50 mg/dL on mipomersen, with the greatest shifts in the HoFH population (23.5%). Smaller shifts were observed in the severe HC (12.8%), HeFH with CAD (15.9%) and in the HC at high risk for CAD populations (13.9%). No patients in the placebo group with a baseline Lp(a) >30 mg/dL achieved≤30 mg/dL; only 2.4% of patients in the placebo group having an Lp(a) >50 mg/dL achieved Lp(a) levels ≤50 mg/dL.
Table 4. Shifts in baseline Lp(a) levels to below established thresholds.
| Placebo (n=126) n (%) | Mipomersen (n=256) n (%) | ||
|---|---|---|---|
| Shift of Lp(a) Above or Below 30 mg/dL | |||
|
| |||
| Baseline Lp(a)≤30 mg/dL to PET Lp(a)≤30 mg/dL | 60 (47.6) | 102 (39.8) | |
| Baseline Lp(a)≤30 mg/dL to PET Lp(a)>30 mg/dL | 2 (1.6) | 2 (0.8) | |
| Baseline Lp(a)>30 mg/dL to PET Lp(a)≤30 mg/dL | 0 | 28 (10.9) | |
| Baseline Lp(a)>30 mg/dL to PET Lp(a)>30 mg/dL | 64 (50.8) | 124 (48.4) | |
|
| |||
| Shift of Lp(a) Above or Below 50 mg/dL | |||
|
| |||
| Baseline Lp(a)≤50 mg/dL to PET Lp(a)≤50 mg/dL | 70 (55.6) | 144 (56.3) | |
| Baseline Lp(a)≤50 mg/dL to PET Lp(a)>50 mg/dL | 0 | 1 (0.4) | |
| Baseline Lp(a)>50 mg/dL to PET Lp(a)≤50 mg/dL | 3 (2.4) | 40 (15.6) | |
| Baseline Lp(a)>50 mg/dL to PET Lp(a)>50 mg/dL | 53 (42.1) | 71 (27.7) | |
Correlations between changes in Lp(a) with lipid and lipoprotein parameters
To address the question whether significant correlations existed between the baseline and % change in Lp(a) and lipid levels, Spearman correlations were generated in the mipomersen treated patients (Table 5). At baseline, Lp(a) levels were not correlated with total cholesterol, apoB, LDL-C, HDL-C and triglycerides. However, when evaluating the data as the percent change, modest but significant correlations were noted between the change in Lp(a) and the change in total cholesterol, apoB, and LDL-C. However, these correlations explain approximately 5-15% of the relationship. Furthermore, correction of LDL-C for Lp(a) cholesterol content did not appreciably change these relationships. Figure 5 displays the correlations between the changes in Lp(a), apoB, and LDL-C in the mipomersen and placebo groups. As expected, there was a strong linear correlation between percent reduction in LDL-C and apoB levels pre- and post-treatment (r=0.95, p<0.001) over 26 weeks. However, only modest correlations were noted between percent change in apoB and Lp(a) (r=0.43, p<0.001) and LDL-C and Lp(a) (r=0.36 p<0.001), respectively.
Table 5. Spearman correlations between baseline and percent change in Lp(a) and lipid and lipoprotein variables in the mipomersen treated patients.
| Baseline | ApoB | Total Cholesterol | LDL-C | Corrected LDL-C | HDL-C | Triglycerides | |
|---|---|---|---|---|---|---|---|
| Lp(a) | Spearman correlation coefficient | 0.29 | 0.11 | 0.10 | -0.15 | -0.02 | -0.06 |
| p-value | <.0001 | 0.093 | 0.093 | 0.014 | 0.74 | 0.34 | |
| ApoB | Spearman correlation coefficient | 0.93 | 0.94 | 0.82 | -0.21 | 0.15 | |
| p-value | <.0001 | <.0001 | <.0001 | 0.0006 | 0.014 | ||
| Total Cholesterol | Spearman correlation coefficient | 0.96 | 0.91 | -0.04 | 0.10 | ||
| p-value | <.0001 | <.0001 | 0.50 | 0.094 | |||
| LDL-C | Spearman correlation coefficient | 0.94 | -0.17 | -0.02 | |||
| p-value | <.0001 | 0.01 | 0.77 | ||||
| Corrected LDL-C | Spearman correlation coefficient | -0.20 | 0.01 | ||||
| p-value | 0.001 | 0.89 | |||||
| HDL-C | Spearman correlation coefficient | -0.15 | |||||
| p-value | 0.017 | ||||||
| Lp(a) | Spearman correlation coefficient | 0.43 | 0.37 | 0.36 | 0.30 | 0.08 | 0.16 |
| p-value | <.0001 | <.0001 | <.0001 | <.0001 | 0.20 | 0.01 | |
| ApoB | Spearman correlation coefficient | 0.94 | 0.95 | 0.92 | 0.03 | 0.57 | |
| p-value | <.0001 | <.0001 | <.0001 | 0.58 | <.0001 | ||
| Total Cholesterol | Spearman correlation coefficient | 0.96 | 0.95 | 0.16 | 0.54 | ||
| p-value | <.0001 | <.0001 | 0.011 | <.0001 | |||
| LDL-C | Spearman correlation coefficient | 0.98 | 0.09 | 0.46 | |||
| p-value | <.0001 | 0.17 | <.0001 | ||||
| Corrected LDL-C | Spearman correlation coefficient | 0.09 | 0.44 | ||||
| p-value | 0.13 | <.0001 | |||||
| HDL-C | Spearman correlation coefficient | -0.14 | |||||
| p-value | 0.027 |
LDL-C was corrected for Lp(a) cholesterol content by subtracting 0.3*Lp(a) mass from measured LDL-C [LDL-C corr = LDL-C - 0.3*Lp(a)].
Figure 5. Correlations between percent change in Lp(a), apoB and LDL-C.
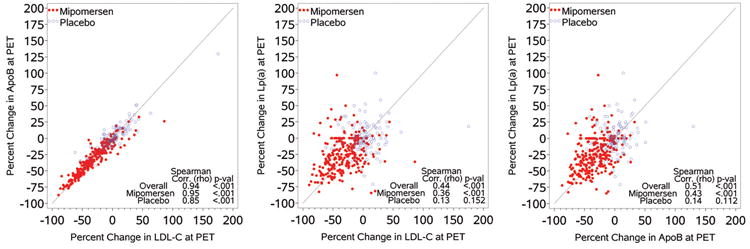
Spearman Correlations (r-values) between LDL-C and apoB (A), LDL-C and Lp(a) (B) and apoB and Lp(a) (C) in the mipomersen and placebo groups in the pooled 4 Phase III studies.
Predictors of the change in Lp(a) levels
The relationship between baseline patient characteristics and effect of mipomersen in Lp(a) lowering was assessed by evaluating patients that achieved Lp(a) <= median % change or > median % change [-26.4%] (Table 6). This analysis reveals that the only factors predicting >median % change were white race, lower baseline total cholesterol and LDL-C levels and particularly lower achieved % change in total cholesterol and LDL-C levels. Other baseline variables and medications did not predict change in Lp(a) levels.
Table 6. Predictors of the change in Lp(a) levels in the pooled 4 Phase III studies.
| Patients achieving % change in Lp(a) <= median % change [-26.4] (N=128) | Patients achieving % change in Lp(a) > median % change [-26.4] (N=128) | p-Value | |
|---|---|---|---|
| Age, mean (SD) | 53.6 (16.09) | 53.2 (12.21) | 0.83 |
|
| |||
| Gender, n (%) | |||
| Male | 62 (48.4) | 72 (56.3) | 0.26 |
| Female | 66 (51.6) | 56 (43.8) | |
|
| |||
| Ethnicity, n (%) | |||
| Hispanic or Latino | 5 (3.9) | 16 (12.5) | 0.02 |
| Not Hispanic or Latino | 123 (96.1) | 112 (87.5) | |
|
| |||
| Race, n (%) | |||
| White | 98 (76.6) | 119 (93.0) | 0.0002 |
| Black | 18 (14.1) | 7 (5.5) | |
| Asian | 7 (5.5) | 2 (1.6) | |
| Other | 5 (3.9) | 0 | |
|
| |||
| BMI, mean (SD) | 29.1 (5.0) | 29.1 (5.0) | 0.92 |
|
| |||
| Waist/Hip Ratio, mean (SD) | 0.92 (0.08) | 0.93 (0.07) | 0.42 |
|
| |||
| Metabolic syndrome, n (%) | |||
| No | 58 (45.3) | 71 (55.5) | 0.13 |
| Yes | 70 (54.7) | 57 (44.5) | |
|
| |||
| Tobacco, n(%) | |||
| Current | 18 (14.1) | 24 (18.8) | 0.57 |
| Non-current | 39 (30.5) | 39 (30.5) | |
| Never | 71 (55.5) | 65 (50.8) | |
|
| |||
| Alcohol, n(%) | |||
| Current | 74 (57.8) | 78 (60.9) | 0.68 |
| Non-current | 20 (15.6) | 22 (17.2) | |
| Never | 34 (26.6) | 28 (21.9) | |
|
| |||
| Hypertension, n(%) | |||
| No | 123 (96.1) | 125 (97.7) | 0.72 |
| Yes | 5 (3.9) | 3 (2.3) | |
|
| |||
| Baseline Lipid Lowering Medications, n (%) | |||
| Statins | 41 (32.0) | 39 (30.5) | 0.89 |
| Statin + Niacin | 5 (3.9) | 4 (3.1) | 1.00 |
| Statin + Ezetimibe | 36 (28.1) | 45 (35.2) | 0.28 |
| Statin + Niacin + Ezetimibe | 7 (5.5) | 4 (3.1) | 0.54 |
| Statin + Other LLT(s) | 38 (29.7) | 35 (27.3) | 0.78 |
| Other LLT(s) without Statin | 0 | 1 (0.8) | |
|
| |||
| Total Cholesterol at Baseline (mg/dL), mean (SD) | 289.6 (136.6) | 256.7 (111.4) | 0.036 |
|
| |||
| Total Cholesterol at PET (mg/dL), mean (SD) | 203.7 (111.4) | 205.5 (93.4) | 0.89 |
|
| |||
| Total Cholesterol % change, mean (SD) | -29.4 (19.4) | -18.2 (17.4) | <0.0001 |
|
| |||
| LDL Cholesterol at Baseline (mg/dL), mean (SD) | 213.9 (138.0) | 181.5 (111.8) | 0.040 |
|
| |||
| LDL Cholesterol at PET (mg/dL), mean (SD) | 133.0 (111.3) | 132.9 (93.5) | 0.99 |
|
| |||
| LDL Cholesterol % change, mean (SD) | -39.4 (27.5) | -25.2 (22.6) | <0.0001 |
|
| |||
| HDL Cholesterol at Baseline (mg/dL), mean (SD) | 48.4 (14.1) | 49.2 (13.4) | 0.62 |
|
| |||
| HDL Cholesterol at PET (mg/dL), mean (SD) | 49.8 (14.9) | 51.0 (13.7) | 0.51 |
|
| |||
| HDL Cholesterol % change, mean (SD) | 5.4 (23.3) | 5.1 (17.4) | 0.94 |
|
| |||
| Triglyceride at Baseline (mg/dL), mean (SD) | 136.9 (69.8) | 128.2 (62.9) | 0.30 |
|
| |||
| Triglyceride at PET (mg/dL), mean (SD) | 104.4 (65.0) | 107.9 (61.5) | 0.66 |
|
| |||
| Triglyceride % change, mean (SD) | -20.1 (34.9) | -10.9 (37.3) | 0.068 |
|
| |||
| Lp(a) at Baseline (mg/dL), mean (SD) | 60.4 (49.0) | 59.2 (68.5) | 0.87 |
Discussion
This pooled analysis of four phase III randomized trials shows that mipomersen consistently lowers plasma Lp(a) levels by a median of 26.4% compared with placebo. This was shown across various groups of patients with different etiologies of hypercholesterolemia, including patients with HoFH and HeFH who had Lp(a) levels that were 2-3-fold higher than non-FH patients. In the mipomersen treated group, a wide variability was noted in Lp(a) lowering responses. Modest correlations were present between apoB and Lp(a) lowering but the mechanistic relevance to Lp(a) lowering of this observation is currently unknown.
Based on a large number of epidemiologic, genome-wide association, and Mendelian randomization studies, Lp(a) is generally considered an independent, causal, genetic risk factor for CVD and calcific aortic stenosis.1,2,5,6,10,17,30-36 In fact, in the genome-wide association studies, its relationship with CAD is as strong, if not stronger, than other lipid and inflammatory genes.33 Lp(a) confers cardiovascular risk through its LDL moiety and through its pro-atherogenic, pro-inflammatory and potentially pro-thrombotic properties, which may be due in part to the oxidized phospholipids (OxPL) bound to Lp(a). In addition, Lp(a) can induce macrophage apoptosis, a key factor in plaque vulnerability.37-42
A randomized trial of Lp(a) lowering to assess clinical outcomes has not been performed to date, mainly due to the lack of specific or effective therapies. The European Atherosclerosis Society (EAS) and the National Lipid Association recommend screening for elevated Lp(a) in patients with CVD, recurrent events, family history of premature CVD, and high to intermediate risk for CVD.28,43-45 These recommendations were reinforced by the results of a recent prospective, observational study performed specifically in patients with elevated Lp(a) (>60 mg/dL) on lipid-lowering medications and prior history of CVD, showing that apheresis prevented the recurrence of major atherosclerotic events.22 Hence, it is generally recommended that patients with FH be screened for elevated Lp(a) levels.46 Although there are no randomized trials to support it, the EAS recommends that desirable levels of Lp(a) should be under the 80th percentile, or ≤50 mg/dL.
The wide variability in baseline plasma Lp(a) levels, as noted here in all 4 trials, is mainly due to the LPA gene 2. The variability in plasma Lp(a) levels is attributed to: 1) polymorphic differences in kringle IV-type 2 repeats of which there are more than 40 isoforms (∼50%); 2) single nucleotide polymorphisms in coding and non-coding sequences that can increase or decrease Lp(a) levels (∼40%); and 3) undefined mechanisms (∼10%). In addition, the LPA gene is under transcriptional regulation by a variety of factors, such as the estrogen and farnesoid X receptors, transforming growth factor, interleukin-6, and fibroblast growth factor that may increase or decrease plasma levels under physiologic or pathophysiologic conditions.47 Interestingly, in the placebo group, there was also significant variability on repeat testing. The day-to-day or seasonal variability of Lp(a) levels has not been well studied, but levels seem to fluctuate around a genetically-determined level. Lp(a) is an acute-phase reactant and levels may increase acutely 50-100%, such as during percutaneous intervention or following acute myocardial infarction.48-50 Low fat diets,51,52 garlic supplements53,54 and different types and doses of statins may exert modest increases in Lp(a),55-59 including in children with HeFH,60 although these findings are not entirely consistent.
Plasma Lp(a) concentrations are largely determined by lipoprotein synthesis rather than clearance.2 The LDLR does not appear to play a significant role in Lp(a) catabolism but this has only been studied in small numbers of patients and in mice that do not have the LPA gene.12,20,21 A large number of studies show an LDLR gene-dose effect on Lp(a) levels in patients with FH,12-18 as also noted here. A priori, this might suggest that defective clearance of LDL, which underlies the metabolic defect in HoFH or HeFH, would also be responsible for elevated Lp(a) levels seen in these subjects. However, this contradicts the in vivo turnover studies in humans and mice20,21 that demonstrate that the LDLR is not directly involved with Lp(a) clearance. Interestingly, a re-analysis of these data suggests that the fractional catabolic rate of Lp(a) may be slightly reduced in HoFH patients, however, this does not seem to explain the significantly elevated Lp(a) levels in FH patients.12 Alternative mechanisms must be operative to explain the high Lp(a) levels in subjects with defective or null LDLR mutations.19
Apo(a) is covalently linked with apoB of LDL to form a mature Lp(a). However, it is not established if Lp(a) is formed within the hepatocyte, as suggested recently by an in vivo kinetic study,61 or at the hepatocyte surface after the independent secretion of apo(a) and LDL, or even formed in the circulation by secreted free apo(a) binding to LDL.2 Mipomersen acts by specifically binding to apoB mRNA in the hepatocyte, thus reducing translation and synthesis of apoB. As a consequence, mipomersen reduces hepatic apoB synthesis and consequently LDL formation.24,26 We speculate that a reduced pool of LDL particles (in the hepatocyte, at the surface or even in plasma) would then lead to decreased formation of Lp(a), though the site, or sites (in the hepatocyte, at the surface or even in plasma), at which this occurs remains to be determined. Insight into this suggested scheme comes from studies in which mipomersen was administered to Lp(a) transgenic mice that express both human apoB-100 and human apo(a). Mipomersen profoundly lowered hepatic apoB-100 synthesis and plasma apoB-100 levels in these mice. Importantly, mipomersen reduced circulating Lp(a) levels by ∼75%.62 However, hepatic apo(a) mRNA and plasma apo(a) levels were not significantly reduced, suggesting that reduced availability of newly synthesized apoB was rate limiting in inhibiting generation of Lp(a) particles in this murine model. By analogy to humans, the mipomersen-induced inhibition of hepatic apoB synthesis may have led to the lack of availability of a specific pool of apoB in the form of newly synthesized LDL in the hepatocyte, or on the hepatocyte surface, or even in plasma, thus limiting generation of Lp(a) particles, which would be translated to reduced plasma Lp(a) levels. Alternative methods to lower Lp(a) in these high risk patients may include directly targeting Lp(a). In a recently completed Phase I study63, dose dependent reductions of Lp(a) up to 78%, and their associated pro-inflammatory oxidized phospholipids,64 were present with ISIS-APO(a)Rx targeting apolipoprotein (a).
The major predictors of the change in Lp(a) >median were white race and lower baseline and particularly, a lower percent change in LDL-C in response to mipomersen. For example, in the pooled data, patients with >median Lp(a) reductions, had a 25.2% decrease in LDL-C, whereas those with ≤median Lp(a) had a 39.4% reductions in LDL-C. Furthermore, no correlations were noted at baseline between Lp(a) and TC, LDL-C, HDL-C, but correlations existed in the percent change of these variables. This suggests that the effect of mipomersen on LDL-C and Lp(a) lowering may be interrelated through common pathways. Clearly, additional studies are necessary to further elucidate the mechanisms by which Lp(a) is synthesized and catabolized, which should help clarify how mipomersen reduces plasma Lp(a) levels in humans.
LDL-C and Lp(a) are independently and additively associated with the risk of CVD. This is especially valid for HoFH and HeFH where both lipoproteins are elevated in comparison with normolipidemic individuals. Mipomersen has been approved in the USA, as an adjunct to lipid-lowering medications and diet to reduce LDL-C, apoB, TC, and non-HDL-C in patients with HoFH. Although mipomersen has not been evaluated in reducing CVD, other studies of LDL lowering in patients with HoFH and HeFH, such as apheresis and statin therapy, have shown a reduction in CVD events.65 In addition, elevated Lp(a) levels are an independent predictor of CVD in men and women with FH, particularly those with a receptor-negative mutation in LDLR gene.19 The Lp(a) lowering effect of mipomersen, along with the LDL-C reduction, may favorably impact CVD risk in this very high risk population.66
The preponderance of data to date support the concept that Lp(a) represents a potential target for further reducing overall CVD risk,67 but whether the pharmacological lowering of Lp(a) reduces CVD risk remains to be proved. Current pharmacological lipid lowering therapies, except niacin, generally do not lower Lp(a).67 In fact, statins that increase LDLR expression either have a neutral or Lp(a) raising effect.37 Niacin reduces Lp(a) by 20-30% in a dose-dependent manner (67). Apheresis is the most efficacious treatment available to reduce Lp(a) (up to 75% acutely and ∼40% in time-averaged Lp(a) reduction).68 PCSK9 inhibitors and CETP inhibitors in clinical trials also lower Lp(a) by 25-40%, also by unknown mechanisms. A specific antisense oligonucleotide to apo(a) was recently shown to reduce plasma Lp(a) levels in a phase I study in normal volunteers up to 78%.63 Clinical trials of these novel agents that lower Lp(a) by new mechanisms may allow testing of the hypothesis that lowering Lp(a) levels may further reduce CVD risk. Determining whether lowering plasma Lp(a) is effective in reducing CVD events is one of the major challenges for improved treatment and prevention of CVD.
Supplementary Material
Significance.
Epidemiologic, genome-wide association, and Mendelian randomization studies have shown that Lp(a) is an independent, causal, genetic risk factor for CVD and calcific aortic stenosis. A randomized trial of Lp(a) lowering to assess clinical outcomes has not been performed to date, mainly due to the lack of specific or effective therapies. This study shows the following: 1-patients with familial hypercholesterolemia have significantly elevated Lp(a) levels; 2- mipomersen, an antisense oligonucleotide to apolipoprotein B-100, consistently lowers plasma Lp(a) levels by a median of 26.4% compared with placebo in several groups of patients with different ranges of hypercholesterolemia; and 3- the Lp(a) lowering effect was mildly related to LDL-C lowering, suggesting that the effect on LDL-C and Lp(a) lowering is partially interrelated through common pathways but that the mechanisms underlying the Lp(a) lowering of mipomersen are not fully defined. Whether the combined LDL-C and Lp(a) lowering effect of mipomersen leads to improved clinical outcomes awaits future studies.
Acknowledgments
The authors wish to acknowledge the contributions of the investigators and site coordinators for their diligence in data acquisition. Barbara Rinehart, MS of ReSearch Pharmaceutical Sciences assisted with manuscript preparation.
Sources of Funding: Drs. Tsimikas and Witztum are partially funded by grants from the National Institutes of Health R01-HL119828, P01-HL088093, P01-HL055798, R01-HL093767 and U54-HL119893.
Abbreviations
- ApoB
apolipoprotein B-100
- FH
familial hypercholesterolemia
- HC
Hypercholesterolemia
- HeFH
heterozygous familial hypercholesterolemia
- HoFH
homozygous familial hypercholesterolemia
- LDLR
low-density lipoprotein receptor
- LLT
lipid-lowering therapies
- Lp(a)
lipoprotein(a)
- mRNA
messenger ribonucleic acid
- PET
primary efficacy timepoint
Footnotes
Disclosure: Drs. Tsimikas and Witztum are co-inventors of and receive royalties from patents owned by the University of California San Diego on oxidation-specific antibodies and mimotopes. Dr. Tsimikas has a dual appointment at UCSD and as an employee at Isis Pharmaceuticals, Inc., and was aconsultant to Sanofi-Aventis, Genzyme Corporation and Pfizer. Dr. Witztum has received honoraria for consulting for Isis Pharmaceuticals, Inc., and Intercept Pharmaceuticals. Dr Santos has the following disclosures: receiving honoraria for consulting and speaking activities for Amgen, Aegerion, Astra Zeneca, Boehringer-Ingelheim, Bristol Myers Squibb, Biolab, Genzyme, Pfizer, Eli-Lilly, Jansen, Novartis, Merck, Regeneron, and Unilever. Dr. Raal has received modest speaker or honoraria from Amgen, Sanofi-Aventis, AstraZeneca and Pfizer. Alberico L. Catapano has received honoraria from for consulting and speaking activities for Amgen, Aegerion, Astra Zeneca, Bristol Myers Squibb, Genzyme, Pfizer, Novartis, Merck, Regeneron, Sanofi, and Sigma Tau. Dr. Steinhagen-Thiessen has received honoraria for lecturing and consulting in the last 3 years from MSD Sharp & Dohme, Fresenius Medical Care, Sanofi, La Roche, Amgen and Pfizer.
References
- 1.Tsimikas S, Hall JH. Lipoprotein(a) as a potential causal genetic risk factor of cardiovascular disease: A rationale for increased efforts to understand its pathophysiology and develop targeted therapies. J Am Coll Cardiol. 2012;60:716–721. doi: 10.1016/j.jacc.2012.04.038. [DOI] [PubMed] [Google Scholar]
- 2.Kronenberg F, Utermann G. Lipoprotein(a): Resurrected by genetics. J Int Med. 2013;273:6–30. doi: 10.1111/j.1365-2796.2012.02592.x. [DOI] [PubMed] [Google Scholar]
- 3.Marcovina SM, Albers JJ, Wijsman E, Zhang Z, Chapman NH, Kennedy H. Differences in lp[a] concentrations and apo[a] polymorphs between black and white americans. J Lipid Res. 1996;37:2569–2585. [PubMed] [Google Scholar]
- 4.Tsimikas S, Clopton P, Brilakis ES, Marcovina SM, Khera A, Miller ER, de Lemos JA, Witztum JL. Relationship of oxidized phospholipids on apolipoprotein B-100 particles to race/ethnicity, apolipoprotein(a) isoform size, and cardiovascular risk factors: Results from the dallas heart study. Circulation. 2009;119:1711–1719. doi: 10.1161/CIRCULATIONAHA.108.836940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Erqou S, Kaptoge S, Perry PL, Di Angelantonio E, Thompson A, White IR, Marcovina SM, Collins R, Thompson SG, Danesh J. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302:412–423. doi: 10.1001/jama.2009.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301:2331–2339. doi: 10.1001/jama.2009.801. [DOI] [PubMed] [Google Scholar]
- 7.Li Y, Luke MM, Shiffman D, Devlin JJ. Genetic variants in the apolipoprotein(a) gene and coronary heart disease. Circ Cardiovasc Genet. 2011;4:565–573. doi: 10.1161/CIRCGENETICS.111.959601. [DOI] [PubMed] [Google Scholar]
- 8.Gurdasani D, Sjouke B, Tsimikas S, Hovingh GK, Luben RN, Wainwright NW, Pomilla C, Wareham NJ, Khaw KT, Boekholdt SM, Sandhu MS. Lipoprotein(a) and risk of coronary, cerebrovascular, and peripheral artery disease: The epic-norfolk prospective population study. Arterioscler Thromb Vasc Biol. 2012;32:3058–3065. doi: 10.1161/ATVBAHA.112.255521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bertoia ML, Pai JK, Lee JH, Taleb A, Joosten MM, Mittleman MA, Yang X, Witztum JL, Rimm EB, Tsimikas S, Mukamal KJ. Oxidation-specific biomarkers and risk of peripheral artery disease. J Am Coll Cardiol. 2013;61:2169–2179. doi: 10.1016/j.jacc.2013.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thanassoulis G, Campbell CY, Owens DS, et al. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. 2013;368:503–512. doi: 10.1056/NEJMoa1109034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kamstrup PR, Tybjaerg-Hansen A, Nordestgaard BG. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J Am Coll Cardiol. 2014;63:470–477. doi: 10.1016/j.jacc.2013.09.038. [DOI] [PubMed] [Google Scholar]
- 12.Kraft HG, Lingenhel A, Raal FJ, Hohenegger M, Utermann G. Lipoprotein(a) in homozygous familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2000;20:522–528. doi: 10.1161/01.atv.20.2.522. [DOI] [PubMed] [Google Scholar]
- 13.Seed M, Hoppichler F, Reaveley D, McCarthy S, Thompson GR, Boerwinkle E, Utermann G. Relation of serum lipoprotein(a) concentration and apolipoprotein(a) phenotype to coronary heart disease in patients with familial hypercholesterolemia. N Engl J Med. 1990;322:1494–1499. doi: 10.1056/NEJM199005243222104. [DOI] [PubMed] [Google Scholar]
- 14.Hopkins PN, Stephenson S, Wu LL, Riley WA, Xin Y, Hunt SC. Evaluation of coronary risk factors in patients with heterozygous familial hypercholesterolemia. Am J Cardiol. 2001;87:547–553. doi: 10.1016/s0002-9149(00)01429-6. [DOI] [PubMed] [Google Scholar]
- 15.Holmes DT, Schick BA, Humphries KH, Frohlich J. Lipoprotein(a) is an independent risk factor for cardiovascular disease in heterozygous familial hypercholesterolemia. Clinical chemistry. 2005;51:2067–2073. doi: 10.1373/clinchem.2005.055228. [DOI] [PubMed] [Google Scholar]
- 16.Orso E, Ahrens N, Kilalic D, Schmitz G. Familial hypercholesterolemia and lipoprotein(a) hyperlipidemia as independent and combined cardiovascular risk factors. Atherosclerosis. Supplements. 2009;10:74–78. doi: 10.1016/S1567-5688(09)71816-1. [DOI] [PubMed] [Google Scholar]
- 17.Jansen AC, van Aalst-Cohen ES, Tanck MW, Trip MD, Lansberg PJ, Liem AH, van Lennep HW, Sijbrands EJ, Kastelein JJ. The contribution of classical risk factors to cardiovascular disease in familial hypercholesterolaemia: Data in 2400 patients. J Intern Med. 2004;256:482–490. doi: 10.1111/j.1365-2796.2004.01405.x. [DOI] [PubMed] [Google Scholar]
- 18.Nenseter MS, Lindvig HW, Ueland T, Langslet G, Ose L, Holven KB, Retterstol K. Lipoprotein(a) levels in coronary heart disease-susceptible and -resistant patients with familial hypercholesterolemia. Atherosclerosis. 2011;216:426–432. doi: 10.1016/j.atherosclerosis.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 19.Alonso R, Andres E, Mata N, Fuentes-Jimenez F, Badimon L, Lopez-Miranda J, Padro T, Muniz O, Diaz-Diaz JL, Mauri M, Ordovas JM, Mata P, investigators S. Lipoprotein(a) levels in familial hypercholesterolaemia: An important predictor for cardiovascular disease independent of the type of LDL-receptor mutation. J Am Coll Cardiol. 2014 doi: 10.1016/j.jacc.2014.01.063. [DOI] [PubMed] [Google Scholar]
- 20.Jamieson DG, Usher DC, Rader DJ, Lavi E. Apolipoprotein(a) deposition in atherosclerotic plaques of cerebral vessels. A potential role for endothelial cells in lesion formation. Am J Pathol. 1995;147:1567–1574. [PMC free article] [PubMed] [Google Scholar]
- 21.Cain WJ, Millar JS, Himebauch AS, Tietge UJF, Maugeais C, Usher D, Rader DJ. Lipoprotein [a] is cleared from the plasma primarily by the liver in a process mediated by apolipoprotein [a] J Lipid Res. 2005;46:2681–2691. doi: 10.1194/jlr.M500249-JLR200. [DOI] [PubMed] [Google Scholar]
- 22.Leebmann J, Roeseler E, Julius U, Heigl F, Spitthoever R, Heutling D, Breitenberger P, Maerz W, Lehmacher W, Heibges A, Klingel R. Lipoprotein apheresis in patients with maximally tolerated lipid-lowering therapy, lipoprotein(a)-hyperlipoproteinemia, and progressive cardiovascular disease: Prospective observational multicenter study. Circulation. 2013;128:2567–2576. doi: 10.1161/CIRCULATIONAHA.113.002432. [DOI] [PubMed] [Google Scholar]
- 23.Willeit P, Kiechl S, Kronenberg F, Witztum JL, Santer P, Mayr M, Xu Q, Mayr A, Willeit J, Tsimikas S. Discrimination and net reclassification of cardiovascular risk with lipoprotein(a): Prospective 15-year outcomes in the bruneck study. J Am Coll Cardiol. 2014;64:851–860. doi: 10.1016/j.jacc.2014.03.061. [DOI] [PubMed] [Google Scholar]
- 24.Raal FJ, Santos RD, Blom DJ, Marais AD, Charng MJ, Cromwell WC, Lachmann RH, Gaudet D, Tan JL, Chasan-Taber S, Tribble DL, Flaim JD, Crooke ST. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: A randomised, double-blind, placebo-controlled trial. Lancet. 2010;375:998–1006. doi: 10.1016/S0140-6736(10)60284-X. [DOI] [PubMed] [Google Scholar]
- 25.McGowan MP, Tardif JC, Ceska R, Burgess LJ, Soran H, Gouni-Berthold I, Wagener G, Chasan-Taber S. Randomized, placebo-controlled trial of mipomersen in patients with severe hypercholesterolemia receiving maximally tolerated lipid-lowering therapy. PloS One. 2012;7:e49006. doi: 10.1371/journal.pone.0049006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stein EA, Dufour R, Gagne C, Gaudet D, East C, Donovan JM, Chin W, Tribble DL, McGowan M. Apolipoprotein b synthesis inhibition with mipomersen in heterozygous familial hypercholesterolemia: Results of a randomized, double-blind, placebo-controlled trial to assess efficacy and safety as add-on therapy in patients with coronary artery disease. Circulation. 2012;126:2283–2292. doi: 10.1161/CIRCULATIONAHA.112.104125. [DOI] [PubMed] [Google Scholar]
- 27.Thomas GS, Cromwell WC, Ali S, Chin W, Flaim JD, Davidson M. Mipomersen, an apolipoprotein B synthesis inhibitor, reduces atherogenic lipoproteins in patients with severe hypercholesterolemia at high cardiovascular risk: A randomized, double-blind, placebo-controlled trial. J Am Coll Cardiol. 2013;62:2178–2184. doi: 10.1016/j.jacc.2013.07.081. [DOI] [PubMed] [Google Scholar]
- 28.Nordestgaard BG, Chapman MJ, Ray K, et al. Lipoprotein(a) as a cardiovascular risk factor: Current status. Eur Heart J. 2010;31:2844–2853. doi: 10.1093/eurheartj/ehq386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marcovina SM, Koschinsky ML, Albers JJ, Skarlatos S. Report of the national heart, lung, and blood institute workshop on lipoprotein(a) and cardiovascular disease: Recent advances and future directions. Clin Chem. 2003;49:1785–1796. doi: 10.1373/clinchem.2003.023689. [DOI] [PubMed] [Google Scholar]
- 30.Tregouet DA, Konig IR, Erdmann J, et al. Genome-wide haplotype association study identifies the slc22a3-lpal2-lpa gene cluster as a risk locus for coronary artery disease. Nat Genet. 2009;41:283–285. doi: 10.1038/ng.314. [DOI] [PubMed] [Google Scholar]
- 31.Clarke R, Peden JF, Hopewell JC, et al. Genetic variants associated with lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361:2518–2528. doi: 10.1056/NEJMoa0902604. [DOI] [PubMed] [Google Scholar]
- 32.Erqou S, Thompson A, Di Angelantonio E, Saleheen D, Kaptoge S, Marcovina S, Danesh J. Apolipoprotein(a) isoforms and the risk of vascular disease: Systematic review of 40 studies involving 58,000 participants. J Am Coll Cardiol. 2010;55:2160–2167. doi: 10.1016/j.jacc.2009.10.080. [DOI] [PubMed] [Google Scholar]
- 33.Schunkert H, Konig IR, Kathiresan S, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43:333–338. doi: 10.1038/ng.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Helgadottir A, Gretarsdottir S, Thorleifsson G, et al. Apolipoprotein(a) genetic sequence variants associated with systemic atherosclerosis and coronary atherosclerotic burden but not with venous thromboembolism. J Am Coll Cardiol. 2012;60:722–729. doi: 10.1016/j.jacc.2012.01.078. [DOI] [PubMed] [Google Scholar]
- 35.Kamstrup PR, Tybjaerg-Hansen A, Nordestgaard BG. Extreme lipoprotein(a) levels and improved cardiovascular risk prediction. J Am Coll Cardiol. 2013;61:1146–1156. doi: 10.1016/j.jacc.2012.12.023. [DOI] [PubMed] [Google Scholar]
- 36.Hung MY, Witztum JL, Tsimikas S. New therapeutic targets for calcific aortic valve stenosis: The lipoprotein(a)-lipoprotein-associated phospholipase a2-oxidized phospholipid axis. J Am Coll Cardiol. 2014;63:478–480. doi: 10.1016/j.jacc.2013.08.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taleb A, Witztum JL, Tsimikas S. Oxidized phospholipids on apolipoprotein B-100 (oxpl/apoB) containing lipoproteins: A biomarker predicting cardiovascular disease and cardiovascular events. Biomarkers Med. 2011;5:673–694. doi: 10.2217/bmm.11.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spence JD, Koschinsky ML. Mechanisms of lipoprotein(a) pathogenicity. Arterioscler Thromb Vasc Biol. 2012;32:1550–1551. doi: 10.1161/ATVBAHA.112.251306. [DOI] [PubMed] [Google Scholar]
- 39.Leibundgut G, Witztum JL, Tsimikas S. Oxidation-specific epitopes and immunological responses: Translational biotheranostic implications for atherosclerosis. Curr Opin Pharmacol. 2013;13:168–179. doi: 10.1016/j.coph.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ravandi A, Leibundgut G, Hung MY, Patel M, Hutchins PM, Murphy RC, Prasad A, Mahmud E, Miller YI, Dennis EA, Witztum JL, Tsimikas S. Release and capture of bioactive oxidized phospholipids and oxidized cholesteryl esters during percutaneous coronary and peripheral arterial interventions in humans. J Am Coll Cardiol. 2014;63:1961–1971. doi: 10.1016/j.jacc.2014.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Dijk RA, Kolodgie F, Ravandi A, Leibundgut G, Hu PP, Prasad A, Mahmud E, Dennis E, Curtiss LK, Witztum JL, Wasserman BA, Otsuka F, Virmani R, Tsimikas S. Differential expression of oxidation-specific epitopes and apolipoprotein(a) in progressing and ruptured human coronary and carotid atherosclerotic lesions. J Lipid Res. 2012;53:2773–2790. doi: 10.1194/jlr.P030890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Choi SH, Yin H, Ravandi A, Armando A, Dumlao D, Kim J, Almazan F, Taylor AM, McNamara CA, Tsimikas S, Dennis EA, Witztum JL, Miller YI. Polyoxygenated cholesterol ester hydroperoxide activates tlr4 and syk dependent signaling in macrophages. PLoS One. 2013;8:e83145. doi: 10.1371/journal.pone.0083145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Myers GL, Christenson RH, Cushman M, Ballantyne CM, Cooper GR, Pfeiffer CM, Grundy SM, Labarthe DR, Levy D, Rifai N, Wilson PW. National academy of clinical biochemistry laboratory medicine practice guidelines: Emerging biomarkers for primary prevention of cardiovascular disease. Clin Chem. 2009;55:378–384. doi: 10.1373/clinchem.2008.115899. [DOI] [PubMed] [Google Scholar]
- 44.Catapano AL, Chapman J, Wiklund O, Taskinen MR. The new joint eas/esc guidelines for the management of dyslipidaemias. Atherosclerosis. 2011;217:1. doi: 10.1016/j.atherosclerosis.2011.06.011. [DOI] [PubMed] [Google Scholar]
- 45.Davidson MH, Ballantyne CM, Jacobson TA, et al. Clinical utility of inflammatory markers and advanced lipoprotein testing: Advice from an expert panel of lipid specialists. J Clin Lipidol. 2011;5:338–367. doi: 10.1016/j.jacl.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 46.Nordestgaard BG, Chapman MJ, Humphries SE, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: Guidance for clinicians to prevent coronary heart disease: Consensus statement of the european atherosclerosis society. Eur Heart J. 2013;34:3478–3490. doi: 10.1093/eurheartj/eht273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoover-Plow J, Huang M. Lipoprotein(a) metabolism: Potential sites for therapeutic targets. Metabolism: clinical and experimental. 2013;62:479–491. doi: 10.1016/j.metabol.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tsimikas S, Bergmark C, Beyer RW, Patel R, Pattison J, Miller E, Juliano J, Witztum JL. Temporal increases in plasma markers of oxidized low-density lipoprotein strongly reflect the presence of acute coronary syndromes. J Am Coll Cardiol. 2003;41:360–370. doi: 10.1016/s0735-1097(02)02769-9. [DOI] [PubMed] [Google Scholar]
- 49.Tsimikas S, Lau HK, Han KR, Shortal B, Miller ER, Segev A, Curtiss LK, Witztum JL, Strauss BH. Percutaneous coronary intervention results in acute increases in oxidized phospholipids and lipoprotein(a): Short-term and long-term immunologic responses to oxidized low-density lipoprotein. Circulation. 2004;109:3164–3170. doi: 10.1161/01.CIR.0000130844.01174.55. [DOI] [PubMed] [Google Scholar]
- 50.Mbewu AD, Durrington PN, Bulleid S, Mackness MI. The immediate effect of streptokinase on serum lipoprotein(a) concentration and the effect of myocardial infarction on serum lipoprotein(a), apolipoproteins a1 and b, lipids and c-reactive protein. Atherosclerosis. 1993;103:65–71. doi: 10.1016/0021-9150(93)90040-2. [DOI] [PubMed] [Google Scholar]
- 51.Faghihnia N, Tsimikas S, Miller ER, Witztum JL, Krauss RM. Changes in lipoprotein(a), oxidized phospholipids, and LDL subclasses with a low-fat high-carbohydrate diet. J Lipid Res. 2010;51:3324–3330. doi: 10.1194/jlr.M005769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Silaste ML, Rantala M, Alfthan G, Aro A, Witztum JL, Kesaniemi YA, Horkko S. Changes in dietary fat intake alter plasma levels of oxidized low-density lipoprotein and lipoprotein(a) Arterioscler Thromb Vasc Biol. 2004;24:498–503. doi: 10.1161/01.ATV.0000118012.64932.f4. [DOI] [PubMed] [Google Scholar]
- 53.Budoff MJ, Ahmadi N, Gul KM, Liu ST, Flores FR, Tiano J, Takasu J, Miller E, Tsimikas S. Aged garlic extract supplemented with b vitamins, folic acid and l-arginine retards the progression of subclinical atherosclerosis: A randomized clinical trial. Prev Med. 2009;49:101–107. doi: 10.1016/j.ypmed.2009.06.018. [DOI] [PubMed] [Google Scholar]
- 54.Ahmadi N, Tsimikas S, Hajsadeghi F, Saeed A, Nabavi V, Bevinal MA, Kadakia J, Flores F, Ebrahimi R, Budoff MJ. Relation of oxidative biomarkers, vascular dysfunction, and progression of coronary artery calcium. Am J Cardiol. 2010;105:459–466. doi: 10.1016/j.amjcard.2009.09.052. [DOI] [PubMed] [Google Scholar]
- 55.Tsimikas S, Witztum JL, Miller ER, Sasiela WJ, Szarek M, Olsson AG, Schwartz GG. High-dose atorvastatin reduces total plasma levels of oxidized phospholipids and immune complexes present on apolipoprotein B-100 in patients with acute coronary syndromes in the miracl trial. Circulation. 2004;110:1406–1412. doi: 10.1161/01.CIR.0000141728.23033.B5. [DOI] [PubMed] [Google Scholar]
- 56.Choi SH, Chae A, Miller E, Messig M, Ntanios F, DeMaria AN, Nissen SE, Witztum JL, Tsimikas S. Relationship between biomarkers of oxidized low-density lipoprotein, statin therapy, quantitative coronary angiography, and atheroma: Volume observations from the reversal (reversal of atherosclerosis with aggressive lipid lowering) study. J Am Coll Cardiol. 2008;52:24–32. doi: 10.1016/j.jacc.2008.02.066. [DOI] [PubMed] [Google Scholar]
- 57.Ky B, Burke A, Tsimikas S, Wolfe ML, Tadesse MG, Szapary PO, Witztum JL, FitzGerald GA, Rader DJ. The influence of pravastatin and atorvastatin on markers of oxidative stress in hypercholesterolemic humans. J Am Coll Cardiol. 2008;51:1653–1662. doi: 10.1016/j.jacc.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 58.Yoshida H, Shoda T, Yanai H, Ikewaki K, Kurata H, Ito K, Furutani N, Tada N, Witztum JL, Tsimikas S. Effects of pitavastatin and atorvastatin on lipoprotein oxidation biomarkers in patients with dyslipidemia. Atherosclerosis. 2013;226:161–164. doi: 10.1016/j.atherosclerosis.2012.10.069. [DOI] [PubMed] [Google Scholar]
- 59.Kostner GM, Gavish D, Leopold B, Bolzano K, Weintraub MS, Breslow JL. Hmg coa reductase inhibitors lower LDL cholesterol without reducing lp(a) levels. Circulation. 1989;80:1313–1319. doi: 10.1161/01.cir.80.5.1313. [DOI] [PubMed] [Google Scholar]
- 60.Rodenburg J, Vissers MN, Wiegman A, Miller ER, Ridker PM, Witztum JL, Kastelein JJ, Tsimikas S. Oxidized low-density lipoprotein in children with familial hypercholesterolemia and unaffected siblings: Effect of pravastatin. J Am Coll Cardiol. 2006;47:1803–1810. doi: 10.1016/j.jacc.2005.12.047. [DOI] [PubMed] [Google Scholar]
- 61.Frischmann ME, Ikewaki K, Trenkwalder E, Lamina C, Dieplinger B, Soufi M, Schweer H, Schaefer JR, Konig P, Kronenberg F, Dieplinger H. In vivo stable-isotope kinetic study suggests intracellular assembly of lipoprotein(a) Atherosclerosis. 2012;225:322–327. doi: 10.1016/j.atherosclerosis.2012.09.031. [DOI] [PubMed] [Google Scholar]
- 62.Merki E, Graham MJ, Mullick AE, Miller ER, Crooke RM, Pitas RE, Witztum JL, Tsimikas S. Antisense oligonucleotide directed to human apolipoprotein B-100 reduces lipoprotein(a) levels and oxidized phospholipids on human apolipoprotein B-100 particles in lipoprotein(a) transgenic mice. Circulation. 2008;118:743–753. doi: 10.1161/CIRCULATIONAHA.108.786822. [DOI] [PubMed] [Google Scholar]
- 63.Viney NJ, Graham M, Crooke R, Hughes S, Singleton W. Evaluation of isis apo(a)rx, an antisense inhibitor to apolipoprotein(a), in healthy volunteers. Circulation. 2013;128:A14196. [Google Scholar]
- 64.Tsimikas S, Duff GW, Berger PB, Rogus J, Huttner K, Clopton P, Brilakis E, Kornman KS, Witztum JL. Pro-inflammatory interleukin-1 genotypes potentiate the risk of coronary artery disease and cardiovascular events mediated by oxidized phospholipids and lipoprotein(a) J Am Coll Cardiol. 2014;63:1724–1734. doi: 10.1016/j.jacc.2013.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sniderman AD, Tsimikas S, Fazio S. The severe hypercholesterolemia phenotype: Clinical diagnosis, management, and emerging therapies. J Am Coll Cardiol. 2014 doi: 10.1016/j.jacc.2014.01.060. [DOI] [PubMed] [Google Scholar]
- 66.Santos RD, Duell PB, East C, Guyton JR, Moriarty PM, Chin W, Mittleman RS. Long-term efficacy and safety of mipomersen in patients with familial hypercholesterolaemia: 2-year interim results of an open-label extension. Eur Heart J. 2013 doi: 10.1093/eurheartj/eht549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kolski B, Tsimikas S. Emerging therapeutic agents to lower lipoprotein (a) levels. Curr Opin Lipidol. 2012;23:560–568. doi: 10.1097/MOL.0b013e3283598d81. [DOI] [PubMed] [Google Scholar]
- 68.McGowan MP. Emerging low-density lipoprotein (LDL) therapies: Management of severely elevated LDL cholesterol--the role of LDL-apheresis. J Clin Lipidol. 2013;7:S21–26. doi: 10.1016/j.jacl.2013.03.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.