

Abstract
Adoptive T cell transfer (ACT) has achieved clinical success in treating established cancer, particularly in combination with lymphodepleting regimens. Our group previously demonstrated that ACT following whole-body irradiation (WBI) promotes high-level T cell accumulation, regression of established brain tumors, and long-term protection from tumor recurrence in a mouse model of SV40 T antigen-induced choroid plexus tumors. Here we asked whether an approach that can promote strong donor T-cell responses in the absence of WBI might also produce this dramatic and durable tumor elimination following ACT. Agonist anti-CD40 antibody can enhance antigen-specific CD8+ T-cell responses and has shown clinical efficacy as a monotherapy in the setting of cancer. We show that anti-CD40 conditioning promotes rapid accumulation of tumor-specific donor CD8+ T cells in the brain and regression of autochthonous T antigen-induced choroid plexus tumors, similar to WBI. Despite a significant increase in the lifespan, tumors eventually recurred in anti-CD40-conditioned mice coincident with loss of T-cell persistence from both the brain and lymphoid organs. Depletion of CD8+ T cells from the peripheral lymphoid organs of WBI-conditioned recipients failed to promote tumor recurrence, but donor cells persisted in the brains long-term in CD8-depleted mice. These results demonstrate that anti-CD40 conditioning effectively enhances ACT-mediated acute elimination of autochthonous tumors, but suggest that mechanisms associated with WBI conditioning, such as the induction of long-lived T cells, may be critical for protection from tumor recurrence.
Electronic supplementary material
The online version of this article (doi:10.1007/s00262-014-1635-7) contains supplementary material, which is available to authorized users.
Keywords: CD8+ T cell, CD40 agonist, Choroid plexus tumor, SV40 T antigen transgenic mice, Whole-body irradiation
Introduction
Adoptive T-cell transfer (ACT) with CD8+ T cells has shown promise as a therapy for solid tumors, including metastases. The use of host conditioning regimens such as non-myeloablative chemotherapy and whole-body irradiation (WBI) prior to ACT has increased overall response rates to an impressive 50 % or higher and improved response durability [1, 2]. WBI has broad systemic effects, a subset of which are thought to be critical for therapeutic success of ACT [3–6]. Although predictors of therapeutic outcome remain elusive, certain “naïve-like” characteristics of donor T cells, such as increased telomere length and CD27 expression, correlate with success [7]. Additionally, long-term persistence of T cells following ACT has been associated with complete, durable remissions in clinical trials [2]. A current challenge is to broaden the applicability of ACT-based therapies, which require large numbers of ex vivo expanded T cells and are targeted to select patients.
WBI conditioning was shown previously to enhance ACT in mice that develop autochthonous tumors due to transgenic expression of the simian virus 40 (SV40) large T antigen (T Ag) oncoprotein within unique tissues [8–11]. In particular, WBI facilitates rapid and high-level accumulation of adoptively transferred T cells in the brains of SV11 mice bearing choroid plexus tumors [9, 10, 12]. Line SV11 mice express T Ag from the SV40 promoter, which selectively targets high-level oncoprotein expression in the choroid plexus of the brain and low levels in the kidney, although tumor formation is restricted to the choroid plexus [13]. T Ag expression in the choroid plexus begins within 14 days of birth and results in the appearance of microscopic papillomas by 35 days [14]. Tumors progress rapidly beginning at approximately 80 days of age, causing death at a mean age of 105 days [14, 15]. Due to low-level transgene expression in the thymus (unpublished observations), SV11 mice are immunologically tolerant to T Ag and unable to mount a CD8+ T-cell response toward the dominant T Ag determinants, including the immunodominant site IV determinant (residues 404–411) [8]. However, transfer of T Ag-specific donor CD8+ T cells into 80-day-old WBI-conditioned mice results in rapid, high-level T-cell accumulation within the brain, tumor elimination, T-cell persistence at the tumor site, and prevention of tumor recurrence [10]. These results raise the question of whether alternative approaches that trigger high-level T-cell accumulation at the tumor site can promote regression of autochthonous tumors, independent of the additional mechanisms associated with irradiation.
Agonist anti-CD40 antibodies promote strong anti-tumor CD8+ T-cell responses in vivo [16–19]. A member of the tumor necrosis factor receptor superfamily, CD40 is expressed on the surface of professional antigen-presenting cells (pAPC), as well as endothelial cells and some tumors [20]. Ligation with CD40 ligand (CD154), expressed by CD4+ T cells, results in the upregulation of major histocompatibility complex class II and costimulatory molecules on pAPCs and licenses these cells to trigger productive CD8+ T-cell activation and differentiation [21–23]. CD40 agonists mimic this signal and promote anti-tumor responses through mechanisms including induction of anti-tumor T-cell responses [16, 24], recruitment of tumoricidal myeloid cells [25], activation of tumor vasculature [26], and direct cytotoxicity of CD40-expressing tumors [27]. In clinical trials, anti-CD40 administration has resulted in objective responses [28], and this cancer immunotherapeutic agent is prioritized for investigation by the National Cancer Institute-supported Cancer Immunotherapy Trials Network [29]. The combination of anti-CD40 conditioning with other immune-based therapies has the potential to produce more significant anti-tumor effects [30]. In particular, combination with ACT has yet to be translated to human cancer patients. Anti-CD40 conditioning promotes the in vivo expansion of adoptively transferred T cells capable of controlling solid tumor progression in experimental models [18, 31–33]; however, the effects on immune surveillance and tumor recurrence have not been thoroughly investigated. Thus, anti-CD40 conditioning could potentially broaden the use of ACT therapy to cancer patients for whom lymphodepleting chemotherapy or WBI is contraindicated.
In the current study, we directly compared the therapeutic and immunological impact of WBI and anti-CD40 conditioning on ACT-mediated immunotherapy of autochthonous brain tumors. We show that anti-CD40 reproduced the initial T-cell accumulation and dramatic tumor elimination observed in WBI-conditioned mice while also significantly extending survival. However, WBI was superior in establishing both donor T-cell persistence and protection from tumor recurrence at late time points.
Materials and methods
Mice
SV11 mice [34] (C57BL/6-Tg(TAg)11Bri) were maintained as previously described [8] and used at 75–85 days of age in all experiments. B6.Cg-Tg(TcraY4,TcrbY4)2025Tdsc, or TCR-IV, mice express a T cell receptor (TCR) αβ pair specific for the H2-Kb-restricted site IV epitope [10]. For some experiments, TCR-IV males were bred with B6.PL-Thy1 a/CyJ (CD90.1) homozygous females to yield CD90.1+ donor cells. All mice were maintained in specific pathogen-free conditions at the Milton S. Hershey Medical Center animal facility. All animal protocols were approved by the Institutional Animal Care and Use Committee at the Penn State Hershey College of Medicine.
Host conditioning and adoptive T-cell transfer
Irradiation-conditioned mice were administered 4 Gy WBI on day −1 using a 60Co Gammacell irradiator (Nordion International) or an X-RAD 320ix biological X-ray irradiator (Precision X-Ray Inc.). Anti-CD40- and control-conditioned mice received 100 μg purified anti-CD40 (clone FGK45, BioXcell) or control rat IgG (Sigma) on days −1 and +1 by intraperitoneal (i.p.) injection. Whole-cell populations were recovered from spleens and axillary, brachial, superficial cervical, mesenteric, inguinal, and lumbar lymph nodes [35] of TCR-IV donor mice. CD8+ cells were enriched by autoMACS magnetic sorting using the manufacturer’s recommendations (Miltenyi Biotec), resulting in approximately 90 % pure CD8+TCR-IV+ T cells. 1 × 106 naïve TCR-IV T cells were administered intravenously in 200 μL PBS on day 0 of the experiments.
Lymphocyte isolation and flow cytometric analysis
On the day of analysis, spleens, superficial cervical lymph nodes (cLN), and brains were removed from euthanized mice following exsanguination. Spleens and cLNs were mechanically disrupted to obtain single-cell suspensions and red blood cells (RBCs) eliminated as previously described [8]. Brain lymphocytes were obtained as previously described [9]. Briefly, brains were minced with a razorblade and mixed by pipetting in RPMI-1640 with GlutaMAX™ (Gibco) supplemented with 2 % fetal bovine serum, 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM l-glutamine, 50 μM 2-mercaptoethanol, 10 mM HEPES, and 25 μg/mL pyruvic acid. Dispersed cells were placed on ice and large debris allowed to settle twice for 5 min, after which the supernatant containing cells was collected. The single-cell suspension was RBC-depleted and washed before centrifugation (500×g for 20 min) on a density gradient (Percoll, 70:32 %). Cells at the interface were removed with a Pasteur pipet, washed, and live cells counted by trypan blue exclusion. Fluorochrome-labeled antibodies were obtained from eBioscience (CD8α clone 53-6.7, CD90.1 clone HIS51, CD44 clone IM7, killer cell lectin-like receptor G1 (KLRG1) clone 2F1), BD Biosciences (CD45.2 clone 104, TCRβ clone H57-597), BioLegend (CD8β clone 53-5.8, CD62L clone MEL-14), and Tonbo Biosciences (CD8α clone 53-6.7). Site IV/Kb tetramers were prepared and used to stain site IV-specific T cells as previously described [10, 36]. Data were acquired using an LSR II SORP, LSRFortessa, or FACSCanto II (BD Biosciences) flow cytometer in the Penn State Hershey Flow Cytometry Core Facility. Data analyses were performed with FlowJo software (TreeStar Inc.).
Intracellular cytokine staining and degranulation assay
Isolated lymphocytes were incubated with T Ag site IV 404–411 peptide variant C411L (VVYDFLKL) or control herpes simplex glycoprotein B 498–505 peptide (SSIEFARL) for 5–6 h at 37 °C in the presence of Brefeldin A (cytokine staining; Sigma) or Brefeldin A and fluorescein isothiocyanate (FITC)-conjugated CD107a antibody (degranulation assay, clone 1D4B; BD Biosciences). Following incubation, cells were stained for surface markers as described above, then fixed and permeabilized using Cytofix/Cytoperm (BD Pharmingen). For cytokine staining, cells were subsequently incubated with FITC-conjugated anti-interferon (IFN)γ (clone XMG1.2; eBioscience) for 15 min at room temperature, washed 3 times, and analyzed by flow cytometry.
Survival analysis
Median lifespan was determined by monitoring mice for the development hydrocephalus and neurological symptoms indicative of advanced tumor development such as lethargy and ataxia [8]. Symptomatic mice were euthanized and Kaplan–Meier survival curves were created using GraphPad Prism software (GraphPad Prism Software, Inc.).
Histology and immunohistochemistry
After euthanasia, mice were perfused with PBS followed by 10 % neutral buffered formalin (NBF). Brains from perfused mice were removed and stored overnight in NBF and then transferred to 70 % ethanol. Fixed brains were paraffin embedded and representative coronal sections were collected throughout the brain. Brain sections were hematoxylin and eosin (H&E) stained and the maximum tumor diameter was determined for each mouse by light microscopy. T Ag immunohistochemistry (clones pAB901 and pAB419) was performed as described [37]. Images were captured using an Olympus BX51 microscope with a 2×, 20×, or 40× objective fitted with an Olympus DP71 digital camera and cellSens Standard 1.6 imaging software (Olympus). Histological sections were evaluated blindly by a board certified veterinary pathologist.
Anti-CD8 antibody production and in vivo T cell depletion
Anti-CD8 monoclonal antibody clone 2.43 [38] was produced in BD Cell MAb animal component-free medium (BD Biosciences) using CELLine reactor flasks (Corning) per the manufacturer’s recommendations. Concentrated antibodies were dialyzed into PBS, purity-verified by gel electrophoresis and aliquots stored at −20 °C. Beginning on day +20 of experiments, mice received weekly i.p. injection of 100 μg of control antibody (rat IgG; Sigma) or anti-CD8 antibody in 200 μL PBS.
Statistics
All statistical tests were performed using GraphPad Prism software. Unpaired Student’s t test was used to determine significance unless otherwise noted. A p value <0.05 was considered statistically significant and is indicated using *, **, ***, **** (p < 0.05, p < 0.01, p < 0.001, p < 0.0001, respectively).
Results
Host conditioning with anti-CD40 induces high-level T cell accumulation in the lymphoid organs and brains of SV11 mice at early time points
We initiated experiments at 80 days of age, when choroid plexus tumors fully or partially fill the ventricles but typically are not invasive [9]. Previously, WBI conditioning was found to accelerate accumulation of donor T cells within the brains of SV11 mice, which first appeared on day +5 following ACT [10]. We first compared the magnitude and kinetics of donor T-cell accumulation in SV11 mice conditioned with anti-CD40 agonist antibody or sublethal WBI followed by ACT with naïve CD8+ TCR-IV T cells that are reactive with the immunodominant H-2Kb-restricted site IV determinant of T Ag. We utilized naïve donor T cells in order to evaluate the impact of each conditioning regimen on initial T-cell activation, differentiation, and accumulation. While current clinical protocols utilize ex vivo expanded T cells for ACT, studies in mice have demonstrated improved efficacy of less-differentiated donor cells for ACT-based therapies [39], resulting in efforts to generate donor T cells with a younger phenotype [40, 41].
As early as day +4 post-ACT, increased frequencies of TCR-IV T cells were detected in the spleens of mice that received anti-CD40 or WBI relative to unconditioned mice (Fig. 1a) and these proportions further increased by day +5. Similar results were observed in the tumor-draining cLNs (unpublished observations). Despite the lower frequency of TCR-IV T cells detected in the spleens of anti-CD40-conditioned mice, the absolute number of TCR-IV T cells was approximately tenfold higher than in WBI-conditioned mice by day +5 (Fig. 1b). Total splenocyte counts revealed a general increase in cellularity in anti-CD40-conditioned mice, in contrast to lymphodepletion in WBI-conditioned mice (Supplementary Fig. 1a). These results demonstrate that anti-CD40 dramatically increases donor T-cell accumulation in the lymphoid organs of SV11 mice.
Fig. 1.

Host conditioning with anti-CD40 induces high-level T cell accumulation in the lymphoid organs and brains of SV11 mice at early time points. Groups of mice received either anti-CD40, WBI, or no conditioning regimen with TCR-IV T-cell ACT. Representative plots show MHC tetramer staining (mean ± SEM) of TCR-IV T cells on days +4 and +5 in a spleen and c brain. Quantification of TCR-IV T-cell accumulation (mean ± SEM) in b spleen and d brain of mice that received ACT with the indicated treatments. n = 3 mice/group (except n = 2 for day +4 control group). Data shown are from one experiment and representative of two independent experiments. Asterisks above connecting lines indicate significant differences between time points. Asterisks next to vertical brackets indicate significant differences between treatment groups. *p < 0.05; **p < 0.01; ns not significant
Consistent with previous results [10], high-level T-cell accumulation in the brain was first detected on day +5 post-ACT in mice that received WBI conditioning (Fig. 1c), and this effect was duplicated in mice that received anti-CD40. Unconditioned mice accumulated few TCR-IV T cells in the brain at this early time point. The total number of TCR-IV T cells in the brain of anti-CD40-conditioned versus WBI-conditioned mice at day +5 was similar (Fig. 1d), indicating that accumulation in the brain is not proportional to accumulation in the periphery. Likewise, total cell accumulation in the brain was similar for WBI- and anti-CD40-conditioned mice (Supplementary Fig. 1b). Thus, both conditioning regimens promote early, high-level TCR-IV T-cell accumulation in the brain of tumor-bearing mice despite their differential effects on T-cell numbers in the lymphoid organs.
Anti-CD40-enhanced ACT promotes initial regression of established tumors
We asked whether T-cell accumulation in the brains of anti-CD40-conditioned mice was associated with tumor regression. Groups of mice received anti-CD40, control immunoglobulin (IgG), or WBI conditioning prior to the ACT and were euthanized on day +10 to assess tumor burden. Nine of ten mice in the anti-CD40-conditioned group showed evidence of tumor regression, either lacking detectable tumors or having only residual small lesions that displayed stromal condensation (Fig. 2a, b). All mice that received ACT with WBI conditioning had either small residual lesions or none at all, consistent with previous observations [10]. In contrast, tumors were detected in 100 % of control IgG-conditioned mice, which in some cases invaded into the brain parenchyma (Fig. 2a, b).
Fig. 2.
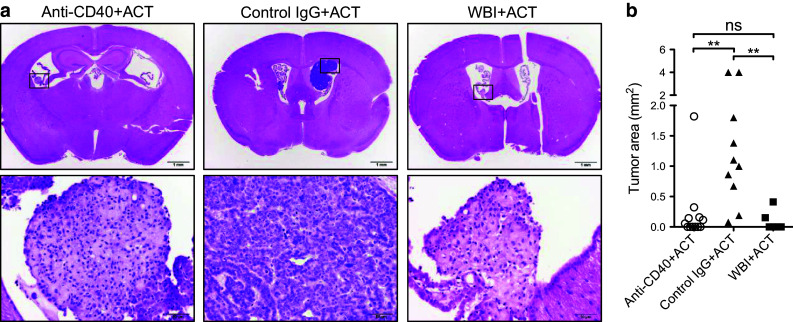
Anti-CD40-enhanced ACT promotes initial regression of established tumors. a H&E brain sections on day +10 post-ACT following conditioning with anti-CD40 (left), control IgG (middle), or WBI (right). Representative low-power images (top row, scale bar = 1 mm) and high-power images (bottom row, scale bar = 50 μm) that show established tumor refractory to therapy (middle column) or tumor stromal condensation indicative of tumor regression (left and right columns). b For each mouse, the largest cross-sectional tumor area (mm2) observed in H&E sections was plotted. Data are pooled from multiple experiments with a total of 6–10 mice/group. Statistical significance was determined using the Kruskal–Wallis test with Dunn’s multiple comparison test. **p < 0.01; ns not significant
Donor T cells contract dramatically in anti-CD40-conditioned SV11 mice
We evaluated T-cell accumulation on days +6 and +10 post-ACT to determine the kinetics of the T-cell response following recruitment into the brain on day +5. On day +6, TCR-IV T cells comprised a high percentage of CD8+ T cells in the spleens and brains of anti-CD40- and WBI-conditioned, but not control IgG-conditioned mice (Fig. 3a, b). These T cells were uniformly CD44hiCD62Llo, indicating that donor cells in all groups had undergone initial T-cell activation and differentiation (Supplementary Fig. 2a). Donor cell frequencies were reduced in the spleen by day +10, but the reduction was much more dramatic in anti-CD40-conditioned mice where T-cell frequencies dropped to levels observed in control IgG-conditioned mice (Fig. 3a). This reduced frequency was paralleled by a 48-fold decrease in total splenic TCR-IV T cells between days +6 and +10 (Fig. 3c, e), while the total number of splenic TCR-IV T cells remained constant in WBI-conditioned mice. TCR-IV cell contraction in the brain was less pronounced (Fig. 3d, f), with the frequency significantly decreasing only for anti-CD40-conditioned mice (Fig. 3b), but with similar numbers of donor T cells persisting by day +10 in both WBI- and anti-CD40-conditioned mice. These results demonstrate that anti-CD40 promotes high-level TCR-IV T-cell accumulation in the lymphoid organs of SV11 mice followed by dramatic contraction by day +10 when tumors have regressed. Contraction was less pronounced in the brain where a significant pool of donor T cells remained at day +10.
Fig. 3.

Donor T cells contract dramatically in anti-CD40-conditioned SV11 mice. Groups of SV11 mice were treated as in Fig. 2 and euthanized on days +6 and +10 post-ACT. Accumulation of tetramer-IV+ T cells is shown as percentage of CD8+ fraction in a spleen and b brain and as total number of TCR-IV T cells in c spleen and d brain. Using sample means in parts c and d, the magnitude of TCR-IV T-cell contraction from day +6 to +10 was graphed for e spleen and f brain. Data are pooled from two experiments with a total of 3–6 mice/group. *p < 0.05; ***p < 0.001; ****p < 0.0001
We also found that the proportion of accumulating TCR-IV T cells that expressed the inhibitory receptor KLRG1 was slightly elevated in spleens and brains of anti-CD40-conditioned mice on day +6 (Supplementary Fig. 2b). By day +10, the KLRG1+ splenic subpopulation appeared elevated in both WBI- and anti-CD40-conditioned mice, but statistical significance was only achieved in anti-CD40-conditioned mice (Supplementary Fig. 2c). Meanwhile, KLRG1 expression in the brain remained low on day +10 in all groups and was significantly reduced in anti-CD40-conditioned mice (Supplementary Fig. 2c). These results suggest that accumulation of terminally differentiated or senescent T cells is significantly enhanced by anti-CD40 conditioning.
Anti-CD40-enhanced ACT promotes increased survival but not long-term surveillance against tumor recurrence
Survival of mice that received anti-CD40 conditioning plus TCR-IV ACT was significantly prolonged compared to mice that received TCR-IV ACT with control IgG (Fig. 4a, b). However, all mice that received anti-CD40 conditioning plus TCR-IV ACT eventually succumbed to tumor recurrence (median lifespan = 176 days of age). This was in contrast to mice that received WBI conditioning plus TCR-IV ACT, all of which survived until termination of the experiment at 215 days of age and appeared healthy and asymptomatic. A noninvasive tumor was observed in one mouse with papillary to focally solid architecture that did not resemble primary untreated tumors ([14] and supplementary Fig. 3a: Tumor recurrence). TCR-IV T cells were detected in the spleen and cLN of all surviving mice in this group (Supplementary Fig. 3b). Approximately, half of the site IV-specific CD8+ T cells in the spleen and cLN retained the ability to produce IFNγ and degranulate in response to specific antigen stimulation (Supplementary Fig. 3b). Thus, ACT promotes initial tumor regression and significantly extends survival in anti-CD40-conditioned mice, but tumors eventually recur in 100 % of mice. Conversely, enduring tumor control was observed in WBI-conditioned mice, which was associated with the persistence of functional TCR-IV T cells for over 130 days.
Fig. 4.

Anti-CD40-enhanced ACT promotes increased survival but short-term surveillance against tumor recurrence. a Groups of mice received the indicated conditioning with or without ACT and were monitored for tumor recurrence. The percentage of surviving mice versus age is plotted. b Statistical differences in survival were calculated using the log-rank test. Data are pooled from multiple experiments with 5–8 mice/group
Donor T cells fail to persist in anti-CD40-conditioned SV11 mice following acute tumor regression
We asked whether tumor recurrence in anti-CD40-conditioned mice correlated with loss of persisting TCR-IV T cells. Groups of mice were treated as described in Fig. 2 using CD90.1+ donor TCR-IV T cells. On day +30, TCR-IV T cells were apparent in both spleen and brain of WBI-conditioned mice (Fig. 5a, b). In contrast, only low frequencies of TCR-IV T cells were detected in anti-CD40- and control IgG-conditioned mice (Fig. 5a, b). Quantitative analysis in the spleen, cLN, and brain reinforced that TCR-IV T cells failed to persist in anti-CD40-conditioned mice (Fig. 5c). Within the spleen of WBI-conditioned mice, a subset of the persisting TCR-IV T cells were KLRG1+ (Fig. 5d), indicative of terminally differentiated effector cells and suggestive of ongoing immune surveillance. However, TCR-IV T cells persisting in brains from all groups lacked KLRG1 expression (Fig. 5e). Persisting TCR-IV T cells in spleens and brains of all mice expressed a similar CD44hiCD62Llo phenotype (Fig. 5d, e). These results illustrate that TCR-IV T cells fail to persist at significant levels in the brain and lymphoid organs of anti-CD40-conditioned mice despite their continued presence and signature of ongoing immunity in WBI-conditioned mice.
Fig. 5.
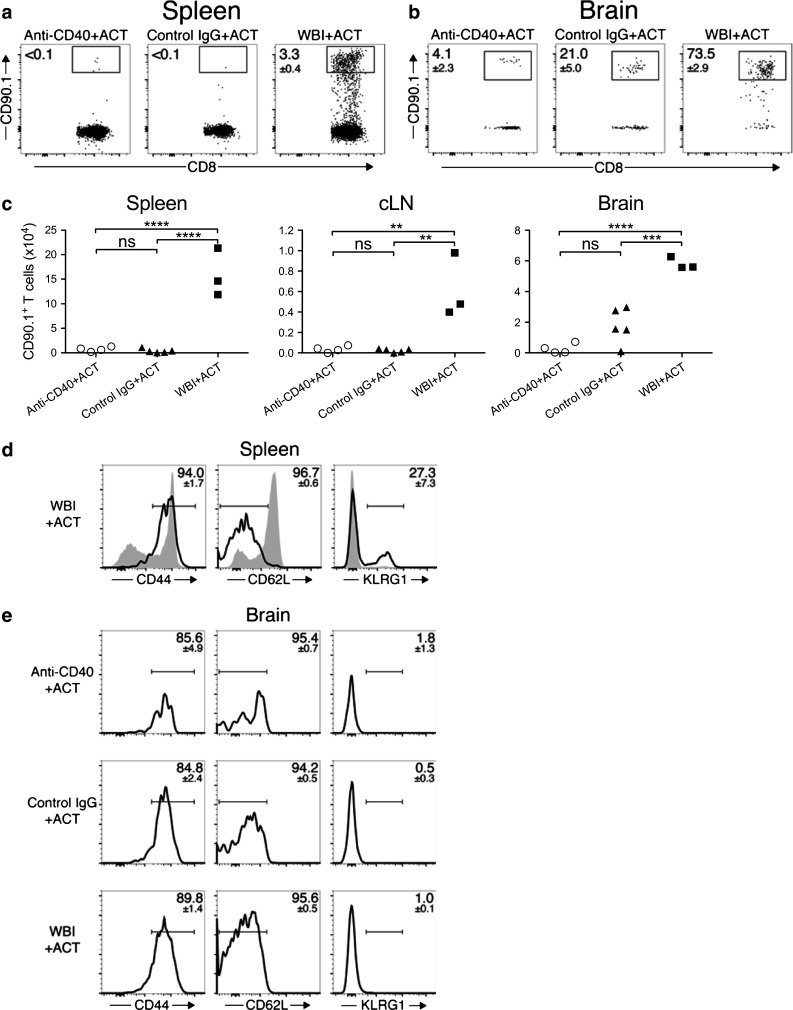
Donor T cells fail to persist in anti-CD40-conditioned SV11 mice following acute tumor regression. Groups of SV11 mice received either anti-CD40, control IgG, or WBI conditioning prior to ACT with CD90.1+ TCR-IV T cells. On day +30, cells from a spleens, cLN (not shown), and b brains were stained for CD90.1 and CD8. Values on dot plots indicate percent CD90.1+ of total CD8+ cells (mean ± SEM). c Total CD90.1+ cells in spleens, cLN, and brains on day +30 are plotted. Representative histograms of CD44, CD62L, and KLRG1 expression gated on live CD45.2+CD8+CD90.1+ TCR-IV T cells (open histogram) or CD45.2+CD8+CD90.1− T cells (filled histogram, spleen only) are shown on day +30 in d spleen and e brain. Values indicate the percent of TCR-IV T cells within the indicated gate (mean ± SEM). Samples with <25 tetramer-IV+ T cells were not included in phenotype analyses. Data are representative of 3–5 mice/group. Statistical significance was determined using one-way ANOVA with Bonferroni posttest. **p < 0.01; ***p < 0.001; ****p < 0.0001; ns not significant
Sustained peripheral depletion of CD8+ T cells following WBI-enhanced ACT does not promote tumor recurrence or eliminate donor T cells from the brain
The presence of KLRG1+ TCR-IV T cells at day +30 in the spleens of WBI-conditioned mice suggested that peripheral effector cells may contribute to long-term tumor control. Thus, we depleted CD8+ cells from the lymphoid organs of WBI-conditioned mice that received TCR-IV ACT by weekly injection of anti-CD8 antibody beginning on day +20. Flow cytometric analysis of blood samples showed near complete CD8+ T-cell depletion (Supplementary Fig. 4a–b). Mice were monitored for 12 weeks post-ACT during which time all appeared healthy without symptoms of tumor recurrence. On day +80, mice were euthanized to assess TCR-IV T-cell levels and tumor burden. Anti-CD8 injections reduced TCR-IV T-cell levels in spleen and cLN, although the latter was not statistically significant due to variation in the control IgG-treated group (Fig. 6a). Inspection of brain sections revealed only small lesions in 6/6 mice in the CD8-depleted group (Fig. 6b and supplementary Fig. 4c). Similar results were obtained in the control-treated group, in which 5/6 mice had only residual lesions and one mouse developed a small mass. T Ag-expressing choroid plexus cells were still detectable in representative sections from both groups (Fig. 6c), indicating that extended survival of WBI-conditioned mice was not due to loss of oncogene expression. The experiment was repeated using CD90.1+ TCR-IV T cells to evaluate T-cell persistence in the brain. Despite reduced donor cell levels in the periphery, total numbers of brain-resident TCR-IV T cells were similar between treatment groups (Fig. 6d). Thus, long-term depletion of tumor-specific T cells from the circulation of SV11 mice following tumor regression does not promote tumor recurrence or eliminate persisting donor T cells from the brain.
Fig. 6.

Sustained peripheral depletion of CD8+ T cells following WBI-enhanced ACT does not promote tumor recurrence or eliminate donor T cells from the brain. WBI-conditioned mice received ACT with TCR-IV T cells. Beginning on day +20, groups of mice received weekly injections of control antibody or depleting anti-CD8. a Quantification of tetramer-IV+ T cells on day +80 in spleen and cLN. b The cross-sectional area of the largest lesion in each brain was calculated and plotted. n = 6 mice/group. c Immunohistochemical staining for SV40 T antigen on brain sections from mice in part b. d In a second experiment using CD90.1+ TCR-IV T cells, mice were harvested at day +80 post-ACT and CD90.1+ T cells were quantified in all three tissues. n = 4 mice/group. **p < 0.01; ***p < 0.001; ns not significant
Discussion
Our findings indicate that anti-CD40 conditioning facilitates acute regression of established tumors in combination with ACT but imply that additional immune manipulation is required to establish continuous immune surveillance when tumor recurrence is likely. The combination of anti-CD40 with ACT was previously shown to enhance the magnitude of tumor-specific T-cell responses, but had only a minimal impact on tumor progression [18, 31, 33, 42] unless additional immune modulators, such as interleukin (IL)-2 or immunization, were provided [31–33]. Our results demonstrate that agonist anti-CD40 alone not only facilitates rapid, high-level donor CD8+ T-cell accumulation systemically and at the tumor site but also promotes regression of established autochthonous tumors. This robust response may be explained in part by the choice of target antigen, as targeting the weaker and less stable T Ag site V determinant with this approach previously failed to induce significant regression of choroid plexus tumors unless mice received multiple rounds of immunization [31]. Likewise, Cho et al. [32] found that using a combination of anti-CD40, tumor peptide, poly:ICLC, and recombinant IL-2 immune complexes with ACT was most effective against antigenic determinants expressed at relatively high-levels on the tumor cells. The tumor type and microenvironment may also play a dominant role in the success of the observed response. Anti-CD40-conditioning plus ACT targeting the site I determinant of T Ag in mice that develop insulinomas induced only a modest delay in tumor progression despite initial T-cell accumulation at the tumor site [18]. Whether T cells targeting site IV would promote effective tumor regression in anti-CD40-conditioned mice bearing insulinomas remains to be determined.
Acute TCR-IV T-cell accumulation reached tenfold higher levels in the spleens of anti-CD40- compared to WBI-conditioned SV11 mice (Fig. 1). The basis for this difference could be explained by more efficient antigen presentation or increased support for T-cell proliferation within anti-CD40-conditioned mice, among other explanations. In support of the latter, we found that total cell counts were dramatically increased in the lymphoid organs of anti-CD40-conditioned mice relative to control mice (Supplementary Fig. 1), as previously observed [16]. Despite such dramatic differences in the periphery, the number of donor T cells that initially accumulated in the brain was similar among anti-CD40- and WBI-conditioned mice, suggesting that only a threshold of activated T cells need be reached in the periphery to achieve a therapeutic level of T cells in the brain.
We demonstrate that anti-CD40-enhanced ACT significantly increased the lifespan of SV11 mice but did not recapitulate the extended immune surveillance against tumor recurrence achieved in WBI-conditioned mice. Thus, tumor regression is not predictive of protection from tumor recurrence (Figs. 2, 4). Rather, T-cell persistence observed in WBI-conditioned mice was associated with long-term progression-free survival. Demonstration that tumors do not recur in WBI-conditioned mice following depletion of peripheral CD8+ T cells (Fig. 6) suggests that brain-resident TCR-IV T cells, which were resistant to antibody-based depletion, may provide long-term immune surveillance in the setting of ongoing T Ag expression, although this remains to be proven. These results raise the issue of why long-lived CD8+ T cells were not maintained in anti-CD40-conditioned mice despite recruitment of an equivalent number of TCR-IV T cells into the brain at early time points. Alternatively, protection from tumor recurrence may be T-cell independent once tumors have been eliminated.
Could the high-level of donor T-cell accumulation achieved in anti-CD40-conditioned mice promote loss of the responding cells? These cells may be subjected to sustained high-level activation if they remain in the environment of anti-CD40-conditioned mice, which may be indicated by the increased frequencies of KLRG1-expressing cells observed early in the response (Supplementary Fig. 2). Although unknown for the current study, exhaustion of donor T cells has been shown to coincide with tumor relapse [43] and may precede donor T-cell loss in anti-CD40-conditioned SV11 mice. Of note, deletion of endogenous T cells was observed in human patients that received multiple doses of anti-CD40 [44]. The authors suggested that frequent dosing of anti-CD40 may lead to T-cell hyperstimulation and apoptosis. This conclusion is consistent with a study showing that continuous provision of agonist anti-CD40 can suppress the induction of collagen-induced arthritis in mice [45]. However, suppression was achieved only if anti-CD40 was provided after collagen administration. Thus, altering the timing of anti-CD40 administration relative to ACT could potentially improve the durability of anti-tumor immune surveillance.
Kedl et al. [42] previously showed that anti-CD40 as a monotherapy promoted early deletion of endogenous tumor-specific T cells but that T cells were protected from deletion by immunization with antigen-expressing vaccinia virus. Additionally, provision of toll-like receptor ligands has been shown to enhance the development of T-cell memory in anti-CD40-conditioned mice in vaccine and tumor models [46, 47], perhaps through induction of type I IFN which improves responses to survival cytokines such as IL-7 and IL-15 [48]. Notably, microbial products such as lipopolysaccharide can translocate across the gut lumen following WBI conditioning [6], triggering innate immune cells that may contribute to T-cell persistence. Recently, Zhang et al. [49] demonstrated that IL-15Rα is upregulated on multiple cell types including CD8+ T cells, dendritic cells, and B cells following anti-CD40 administration. Provision of exogenous IL-15, important for memory T-cell survival [50] was required for successful control of transplantable prostate tumors. This finding raises the possibility that in the current study, limited IL-15 precludes TCR-IV T-cell persistence. Indeed, anti-CD40 promotes the systemic expansion of multiple immune cell types, which may increase competition for survival cytokines between donor T cells and host immune cells. Meanwhile, the lymphodepleting effects of WBI create an environment in which increased cytokine availability, including IL-15, supports the differentiation and survival of donor T cells [3]. The combination of ACT with a cocktail of immune-modulatory agents, including anti-CD40, has been shown to reduce initial tumor burden and promote durable T-cell responses in a melanoma mouse model [32]. Taken together, these studies suggest that use of combination therapy in which other immune modulators are included with anti-CD40 may lead to prolonged immune surveillance.
While WBI plus ACT was sufficient to mediate long-term progression-free survival in SV11 mice, immunotherapy against more resistant tumors may benefit from combining WBI with anti-CD40. As demonstrated, WBI provides limited control of tumor progression in the absence of ACT (Fig. 4). However, WBI may enhance tumor antigen availability through direct induction of immunogenic tumor cell death. While this mechanism has been primarily investigated using higher doses of irradiation [51–53], a recent study provides evidence that immunogenic cell death may be induced using lower doses of irradiation as used in the current study [54]. Therefore, WBI and anti-CD40 could play complementary roles in the setting of ACT, with WBI increasing tumor antigen availability for APCs that become licensed for enhanced T-cell priming by anti-CD40 [55]. WBI might also improve T-cell persistence when used in combination with anti-CD40 by increasing donor T-cell access to survival cytokines through lymphodepletion [3]. Conversely, combining anti-CD40 with WBI may be detrimental, as anti-CD40-mediated T-cell depletion may override the pro-survival effects of WBI. These questions remain to be evaluated.
In clinical trials, T cells expanded ex vivo to provide large doses for reinfusion into cancer patients are characterized by terminal differentiation and reduced potential to persist and self-renew [1]. Recently the potential for dedifferentiation of patient-derived T cells from a terminal-effector toward a naïve- or stem-like phenotype has been highlighted [56]. The addition of anti-CD40 to WBI conditioning could potentially reduce the demand for high numbers of expanded T cells, resulting in transfer of less-differentiated T cells that are desirable for use in ACT. Collectively, our results indicate that anti-CD40 conditioning can promote strong anti-tumor effects by ACT and raise the possibility of utilizing this approach alone or in combination with other immune interventions to achieve significant clinical benefits.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We thank Jeremy Haley for outstanding technical assistance and Penn State Hershey Flow Cytometry Core Facility personnel for support with data acquisition. This work was supported by research Grant RO1 CA025000 from the National Cancer Institute, National Institutes of Health (to Todd Schell). Portions of this work were previously published and presented in an abstract and poster at the Translational Research Cancer Centers Consortium in Seven Springs, PA in February of 2014.
Conflict of interest
All authors declare that they have no conflict of interest.
Abbreviations
- ACT
Adoptive T-cell transfer
- cLN
Superficial cervical lymph nodes
- FITC
Fluorescein isothiocyanate
- H&E
Hematoxylin and eosin
- IFN
Interferon
- IL
Interleukin
- i.p.
Intraperitoneal
- KLRG1
Killer cell lectin-like receptor G1
- NBF
Neutral buffered formalin
- pAPC
Professional antigen-presenting cell
- RBC
Red blood cell
- SV40
Simian virus 40
- T Ag
SV40 large T antigen
- TCR
T-cell receptor
- WBI
Whole-body irradiation
References
- 1.Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenberg SA, Yang JC, Sherry RM, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17:4550–4557. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gattinoni L, Finkelstein SE, Klebanoff CA, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202:907–912. doi: 10.1084/jem.20050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klebanoff CA, Khong HT, Antony PA, Palmer DC, Restifo NP. Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol. 2005;26:111–117. doi: 10.1016/j.it.2004.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reits EA, Hodge JW, Herberts CA, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. 2006;203:1259–1271. doi: 10.1084/jem.20052494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paulos CM, Wrzesinski C, Kaiser A, et al. Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling. J Clin Investig. 2007;117:2197–2204. doi: 10.1172/JCI32205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang J, Khong HT, Dudley ME, El-Gamil M, Li YF, Rosenberg SA, Robbins PF. Survival, persistence, and progressive differentiation of adoptively transferred tumor-reactive T cells associated with tumor regression. J Immunother. 2005;28:258–267. doi: 10.1097/01.cji.0000158855.92792.7a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schell TD, Mylin LM, Georgoff I, Teresky AK, Levine AJ, Tevethia SS. Cytotoxic T-lymphocyte epitope immunodominance in the control of choroid plexus tumors in simian virus 40 large T antigen transgenic mice. J Virol. 1999;73:5981–5993. doi: 10.1128/jvi.73.7.5981-5993.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schell TD, Tevethia SS. Control of advanced choroid plexus tumors in SV40 T antigen transgenic mice following priming of donor CD8(+) T lymphocytes by the endogenous tumor antigen. J Immunol. 2001;167:6947–6956. doi: 10.4049/jimmunol.167.12.6947. [DOI] [PubMed] [Google Scholar]
- 10.Tatum AM, Mylin LM, Bender SJ, Fischer MA, Vigliotti BA, Tevethia MJ, Tevethia SS, Schell TD. CD8+T cells targeting a single immunodominant epitope are sufficient for elimination of established SV40 T antigen-induced brain tumors. J Immunol. 2008;181:4406–4417. doi: 10.4049/jimmunol.181.6.4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ward-Kavanagh LK, Zhu J, Cooper TK, Schell TD. Whole-body irradiation increases the magnitude and persistence of adoptively transferred T cells associated with tumor regression in a mouse model of prostate cancer. Cancer Immunol Res. 2014;2:777–788. doi: 10.1158/2326-6066.CIR-13-0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yorty JL, Tevethia SS, Schell TD. Rapid accumulation of adoptively transferred CD8+ T cells at the tumor site is associated with long-term control of SV40 T antigen-induced tumors. Cancer Immunol Immunother. 2008;57:883–895. doi: 10.1007/s00262-007-0424-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Dyke T, Finlay C, Levine AJ (1985) A comparison of several lines of transgenic mice containing the SV40 early genes. In: Cold Spring Harbor symposia on quantitative biology, vol 50, pp 671–678 [DOI] [PubMed]
- 14.Van Dyke TA, Finlay C, Miller D, Marks J, Lozano G, Levine AJ. Relationship between simian virus 40 large tumor antigen expression and tumor formation in transgenic mice. J Virol. 1987;61:2029–2032. doi: 10.1128/jvi.61.6.2029-2032.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roy EJ, Cho BK, Rund LA, Patrick TA, Kranz DM. Targeting T cells against brain tumors with a bispecific ligand-antibody conjugate. Int J Cancer. 1998;76:761–766. doi: 10.1002/(SICI)1097-0215(19980529)76:5<761::AID-IJC23>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 16.French RR, Chan HT, Tutt AL, Glennie MJ. CD40 antibody evokes a cytotoxic T-cell response that eradicates lymphoma and bypasses T-cell help. Nat Med. 1999;5:548–553. doi: 10.1038/5505. [DOI] [PubMed] [Google Scholar]
- 17.Diehl L, den Boer AT, Schoenberger SP, van der Voort EI, Schumacher TN, Melief CJ, Offringa R, Toes RE. CD40 activation in vivo overcomes peptide-induced peripheral cytotoxic T-lymphocyte tolerance and augments anti-tumor vaccine efficacy. Nat Med. 1999;5:774–779. doi: 10.1038/10495. [DOI] [PubMed] [Google Scholar]
- 18.Otahal P, Knowles BB, Tevethia SS, Schell TD. Anti-CD40 conditioning enhances the T(CD8) response to a highly tolerogenic epitope and subsequent immunotherapy of simian virus 40 T antigen-induced pancreatic tumors. J Immunol. 2007;179:6686–6695. doi: 10.4049/jimmunol.179.10.6686. [DOI] [PubMed] [Google Scholar]
- 19.Staveley-O’Carroll K, Schell TD, Jimenez M, Mylin LM, Tevethia MJ, Schoenberger SP, Tevethia SS. In vivo ligation of CD40 enhances priming against the endogenous tumor antigen and promotes CD8+ T cell effector function in SV40 T antigen transgenic mice. J Immunol. 2003;171:697–707. doi: 10.4049/jimmunol.171.2.697. [DOI] [PubMed] [Google Scholar]
- 20.Eliopoulos AG, Young LS. The role of the CD40 pathway in the pathogenesis and treatment of cancer. Curr Opin Pharmacol. 2004;4:360–367. doi: 10.1016/j.coph.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 21.Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. 1998;393:474–478. doi: 10.1038/30989. [DOI] [PubMed] [Google Scholar]
- 22.Bennett SR, Carbone FR, Karamalis F, Flavell RA, Miller JF, Heath WR. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. 1998;393:478–480. doi: 10.1038/30996. [DOI] [PubMed] [Google Scholar]
- 23.Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. T-cell help for cytotoxic T lymphocytes is mediated by CD40–CD40L interactions. Nature. 1998;393:480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- 24.van Mierlo GJ, den Boer AT, Medema JP, van der Voort EI, Fransen MF, Offringa R, Melief CJ, Toes RE. CD40 stimulation leads to effective therapy of CD40(−) tumors through induction of strong systemic cytotoxic T lymphocyte immunity. Proc Natl Acad Sci USA. 2002;99:5561–5566. doi: 10.1073/pnas.082107699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beatty GL, Chiorean EG, Fishman MP, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–1616. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hamzah J, Nelson D, Moldenhauer G, Arnold B, Hammerling GJ, Ganss R. Vascular targeting of anti-CD40 antibodies and IL-2 into autochthonous tumors enhances immunotherapy in mice. J Clin Investig. 2008;118:1691–1699. doi: 10.1172/JCI33201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hess S, Engelmann H. A novel function of CD40: induction of cell death in transformed cells. J Exp Med. 1996;183:159–167. doi: 10.1084/jem.183.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vonderheide RH, Flaherty KT, Khalil M, et al. Clinical activity and immune modulation in cancer patients treated with CP-870,893, a novel CD40 agonist monoclonal antibody. J Clin Oncol. 2007;25:876–883. doi: 10.1200/JCO.2006.08.3311. [DOI] [PubMed] [Google Scholar]
- 29.Cheever MA. Twelve immunotherapy drugs that could cure cancers. Immunol Rev. 2008;222:357–368. doi: 10.1111/j.1600-065X.2008.00604.x. [DOI] [PubMed] [Google Scholar]
- 30.Khong A, Nelson DJ, Nowak AK, Lake RA, Robinson BW. The use of agonistic anti-CD40 therapy in treatments for cancer. Int Rev Immunol. 2012;31:246–266. doi: 10.3109/08830185.2012.698338. [DOI] [PubMed] [Google Scholar]
- 31.Ryan CM, Staveley-O’Carroll K, Schell TD. Combined anti-CD40 conditioning and well-timed immunization prolongs CD8+ T cell accumulation and control of established brain tumors. J Immunother. 2008;31:906–920. doi: 10.1097/CJI.0b013e318189f155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cho HI, Reyes-Vargas E, Delgado JC, Celis E. A potent vaccination strategy that circumvents lymphodepletion for effective antitumor adoptive T-cell therapy. Cancer Res. 2012;72:1986–1995. doi: 10.1158/0008-5472.CAN-11-3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu C, Lewis CM, Lou Y, et al. Agonistic antibody to CD40 boosts the antitumor activity of adoptively transferred T cells in vivo. J Immunother. 2012;35:276–282. doi: 10.1097/CJI.0b013e31824e7f43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brinster RL, Chen HY, Messing A, van Dyke T, Levine AJ, Palmiter RD. Transgenic mice harboring SV40 T-antigen genes develop characteristic brain tumors. Cell. 1984;37:367–379. doi: 10.1016/0092-8674(84)90367-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dunn TB. Normal and pathologic anatomy of the reticular tissue in laboratory mice, with a classification and discussion of neoplasms. J Natl Cancer Inst. 1954;14:1281–1433. [PubMed] [Google Scholar]
- 36.Mylin LM, Schell TD, Roberts D, Epler M, Boesteanu A, Collins EJ, Frelinger JA, Joyce S, Tevethia SS. Quantitation of CD8(+) T-lymphocyte responses to multiple epitopes from simian virus 40 (SV40) large T antigen in C57BL/6 mice immunized with SV40, SV40 T-antigen-transformed cells, or vaccinia virus recombinants expressing full-length T antigen or epitope minigenes. J Virol. 2000;74:6922–6934. doi: 10.1128/JVI.74.15.6922-6934.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goodwin EM, Zhong Q, Abendroth CS, Ward-Kavanagh LK, Schell TD, Cooper TK. Anaplastic renal carcinoma expressing SV40 T antigen in a female TRAMP mouse. Comp Med. 2013;63:338–341. [PMC free article] [PubMed] [Google Scholar]
- 38.Sarmiento M, Glasebrook AL, Fitch FW. IgG or IgM monoclonal antibodies reactive with different determinants on the molecular complex bearing Lyt 2 antigen block T cell-mediated cytolysis in the absence of complement. J Immunol. 1980;125:2665–2672. [PubMed] [Google Scholar]
- 39.Klebanoff CA, Gattinoni L, Palmer DC, et al. Determinants of successful CD8+ T-cell adoptive immunotherapy for large established tumors in mice. Clin Cancer Res. 2011;17:5343–5352. doi: 10.1158/1078-0432.CCR-11-0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Forget MA, Huon Y, Reuben A, Grange C, Liberman M, Martin J, Mes-Masson AM, Arbour N, Lapointe R. Stimulation of Wnt/ss-catenin pathway in human CD8+ T lymphocytes from blood and lung tumors leads to a shared young/memory phenotype. PLoS One. 2012;7:e41074. doi: 10.1371/journal.pone.0041074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gattinoni L, Zhong XS, Palmer DC, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. 2009;15:808–813. doi: 10.1038/nm.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kedl RM, Jordan M, Potter T, Kappler J, Marrack P, Dow S. CD40 stimulation accelerates deletion of tumor-specific CD8+ T cells in the absence of tumor-antigen vaccination. Proc Natl Acad Sci USA. 2001;98:10811–10816. doi: 10.1073/pnas.191371898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goding SR, Wilson KA, Xie Y, et al. Restoring immune function of tumor-specific CD4+ T cells during recurrence of melanoma. J Immunol. 2013;190:4899–4909. doi: 10.4049/jimmunol.1300271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ruter J, Antonia SJ, Burris HA, Huhn RD, Vonderheide RH. Immune modulation with weekly dosing of an agonist CD40 antibody in a phase I study of patients with advanced solid tumors. Cancer Biol Ther. 2010;10:983–993. doi: 10.4161/cbt.10.10.13251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mauri C, Mars LT, Londei M. Therapeutic activity of agonistic monoclonal antibodies against CD40 in a chronic autoimmune inflammatory process. Nat Med. 2000;6:673–679. doi: 10.1038/76251. [DOI] [PubMed] [Google Scholar]
- 46.Wells JW, Cowled CJ, Farzaneh F, Noble A. Combined triggering of dendritic cell receptors results in synergistic activation and potent cytotoxic immunity. J Immunol. 2008;181:3422–3431. doi: 10.4049/jimmunol.181.5.3422. [DOI] [PubMed] [Google Scholar]
- 47.Ahonen CL, Doxsee CL, McGurran SM, Riter TR, Wade WF, Barth RJ, Vasilakos JP, Noelle RJ, Kedl RM. Combined TLR and CD40 triggering induces potent CD8+ T cell expansion with variable dependence on type I IFN. J Exp Med. 2004;199:775–784. doi: 10.1084/jem.20031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hervas-Stubbs S, Mancheno U, Riezu-Boj JI, et al. CD8 T cell priming in the presence of IFN-alpha renders CTLs with improved responsiveness to homeostatic cytokines and recall antigens: important traits for adoptive T cell therapy. J Immunol. 2012;189:3299–3310. doi: 10.4049/jimmunol.1102495. [DOI] [PubMed] [Google Scholar]
- 49.Zhang M, Ju W, Yao Z, et al. Augmented IL-15Ralpha expression by CD40 activation is critical in synergistic CD8 T cell-mediated antitumor activity of anti-CD40 antibody with IL-15 in TRAMP-C2 tumors in mice. J Immunol. 2012;188:6156–6164. doi: 10.4049/jimmunol.1102604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang X, Sun S, Hwang I, Tough DF, Sprent J. Potent and selective stimulation of memory-phenotype CD8+ T cells in vivo by IL-15. Immunity. 1998;8:591–599. doi: 10.1016/S1074-7613(00)80564-6. [DOI] [PubMed] [Google Scholar]
- 51.Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, Lord EM. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol. 2005;174:7516–7523. doi: 10.4049/jimmunol.174.12.7516. [DOI] [PubMed] [Google Scholar]
- 52.Obeid M, Panaretakis T, Joza N, Tufi R, Tesniere A, van Endert P, Zitvogel L, Kroemer G. Calreticulin exposure is required for the immunogenicity of gamma-irradiation and UVC light-induced apoptosis. Cell Death Differ. 2007;14:1848–1850. doi: 10.1038/sj.cdd.4402201. [DOI] [PubMed] [Google Scholar]
- 53.Apetoh L, Ghiringhelli F, Tesniere A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–1059. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 54.Golden EB, Frances D, Pellicciotta I, Demaria S, Helen Barcellos-Hoff M, Formenti SC. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Oncoimmunology. 2014;3:e28518. doi: 10.4161/onci.28518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Labeur MS, Roters B, Pers B, Mehling A, Luger TA, Schwarz T, Grabbe S. Generation of tumor immunity by bone marrow-derived dendritic cells correlates with dendritic cell maturation stage. J Immunol. 1999;162:168–175. [PubMed] [Google Scholar]
- 56.Crompton JG, Sukumar M, Restifo NP. Uncoupling T-cell expansion from effector differentiation in cell-based immunotherapy. Immunol Rev. 2014;257:264–276. doi: 10.1111/imr.12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.