

Abstract
Objective
Phenotypic plasticity of vascular smooth muscle cells (VSMCs) contributes to cardiovascular disease. Chondrocyte-like transformation of VSMCs associates with vascular calcification and underlies the formation of aortic cartilaginous metaplasia induced in mice by genetic loss of matrix Gla protein (MGP). Previous microarray analysis identified a dramatic down-regulation of Wnt16 in calcified MGP-null aortae, suggesting an antagonistic role for Wnt16 in the chondrogenic transformation of VSMCs.
Approach and Results
Wnt16 is significantly down-regulated in MGP-null aortae, before the histological appearance of cartilaginous metaplasia, and in primary MGP-null VSMCs. In contrast, intrinsic TGFβ is activated in MGP-null VSMCs and is necessary for spontaneous chondrogenesis of these cells in high-density micromass cultures. TGFβ3-induced chondrogenic transformation in wild-type VSMCs associates with Smad2/3-dependent Wnt16 down-regulation, but Wnt16 does not suppress TGFβ3-induced Smad activation. In addition, TGFβ3 inhibits Notch signaling in wild-type VSMCs and this pathway is down-regulated in MGP-null aortae. Exogenous Wnt16 stimulates Notch activity and attenuates TGFβ3-induced down-regulation of Notch in wild-type VSMCs, prevents chondrogenesis in MGP-null and TGFβ3-treated wild-type VSMCs, and stabilizes expression of contractile markers of differentiated VMSCs.
Conclusions
We describe a novel TGFβ-Wnt16-Notch signaling conduit in the chondrocyte-like transformation of VSMCs and identify endogenous TGFβ activity in MGP-null VSMCs as a critical mediator of chondrogenesis. Our proposed model suggests that the activated TGFβ pathway inhibits expression of Wnt16 which is a positive regulator of Notch signaling and a stabilizer of VSMC phenotype. These data advance the comprehensive mechanistic understanding of VSMC transformation and may identify a novel potential therapeutic target in vascular calcification.
Keywords: Wnt16, TGFβ, Notch, vascular smooth muscle, chondrogenesis, vascular calcification
Phenotypic plasticity of vascular smooth muscle (VSM) underlies various cardiovascular diseases. Loss of contractile phenotype and induction of genes characteristic for bone and cartilage cells in vascular smooth muscle cells (VSMCs) results in mineral deposition within the vascular wall known as vascular calcification 1. This condition afflicts aging and complicates atherosclerosis, hypercholesterolemia, end stage renal disease, and diabetes. Chondrogenic transformation of VSMCs leading to a rapid formation of calcifying cartilaginous metaplasia inside the vessel wall is also induced by genetic ablation of matrix Gla protein (MGP) 2 which is highly expressed in vascular tissue. Restoring expression of MGP in VSM, but not in the circulation, was sufficient to prevent the MGP-null vascular phenotype 3 indicating direct effects of MGP on phenotypic stability in VSMCs. In humans, a defective MGP gene has been linked to Keutel syndrome 4 – a rare inherited disease with abnormal calcification of cartilage and stenosis of pulmonary arteries, while polymorphism in MGP associates with elevated risk of vascular calcification 5. Molecular regulation of the osteochondrogenic transformation in VSMCs is strikingly similar to that in true bone formation and involves TGFβ/BMP 6 and β-catenin signaling 6, 7. In addition, Notch signaling is implicated in both vascular calcification 8 and bone formation 9. In bone, cross-talk between Notch, Wnt/β-catenin and TGFβ/BMP pathways has been well documented 10; however, there is still a limited understanding on the roles of individual ligands and on interactions between these pathways in VSM.
Ligands of the TGFβ superfamily, including several TGFβs and BMPs, have been described as potent inducers of osteochondrogenic differentiation in mesenchymal progenitors and in vascular cells 11, 12. Nevertheless, the actions of these ligands on VSMCs may not be quite as straightforward. For example BMP7 attenuates rather than enhances vascular calcium accrual in kidney disease 13 and BMPs 2/4 alone are insufficient to induce calcification in cultured VSMCs 8,14. Similarly, while activation of the canonical Wnt/β-catenin signaling pathway in many vascular calcification conditions, including heart valve disease 15, anticoagulant therapy with warfarin 16, loss of MGP 17 and diabetes 12 suggests a pro-calcific action of this pathway, genetic studies unexpectedly showed that Wnt7b acts to counteract vascular calcification in atherosclerosis 18. Likewise, despite the demonstrated importance of Notch signaling in vessel wall maturation and in vascular disease 19, and the antagonistic activity of Notch on osteochondrogenesis in progenitor and mesenchymal cells 20, in VSMCs Notch signaling has been shown to both promote 8,21 and prevent the induction of osteogenic pathways and mineral accumulation 22. Therefore, elucidating specific roles and integrating cues in the complex interactions of TGFβ, Notch, and β-catenin signaling pathways in VSMCs may be necessary to develop specific therapeutic strategies for vascular calcification.
In previous mechanistic analysis of vascular disease in MGP-null aortic tissue, we observed a dramatic reduction in the expression of Wnt16. While the role of Wnt16 in the vasculature has not been addressed previously, its expression in skeletal development is noticeably absent in the cartilaginous growth plate 23 while abundant in adjacent non-cartilaginous tissues such as the joint interzone 24 and perichondrium/periosteum 25. These observations imply that Wnt16 may block chondrogenesis and therefore the reduced expression of Wnt16 in the MGP-null aorta may contribute to chondrogenic transformation of VSMCs in the vessel wall. Although Wnt16 can activate the β-catenin pathway 26, in VSMCs, chondrogenic transformation occurs in the background of activated canonical β-catenin signaling 17 but reduced Wnt16 expression. Therefore, we hypothesized that this ligand acts instead in a “non-canonical” fashion by controlling activity of the Notch pathway as described in zebrafish development 27. We tested this hypothesis, analyzing the triggers and outcomes of reduced Wnt16 expression during chondrogenic transformation of VSMCs. Our data identify a novel TGFβ-Wnt16-Notch signaling network, where Wnt16 and Notch appear to prevent phenotypic instability in VSMCs and are down-regulated by TGFβ to promote/permit chondrogenesis VSM.
MATERIALS AND METHODS
Materials and Methods are available in the online-only Data Supplement.
RESULTS
Wnt16 expression is down-regulated in the cartilaginous metaplasia of Mgp−/− arteries
A 47.7±4.9-fold down-regulation of Wnt16 mRNA was detected by real-time PCR in Mgp−/− (KO) aortae with extensive calcified cartilaginous metaplasia dissected from 4.5 week old mice, as compared to age-matched wild-type (WT) controls (Fig. 1A, N=6, p<0.001). Accordingly, an 88.6±3.5% reduction in Wnt16 protein levels was found by Western blot analysis (Fig. 1B, N=4, p<0.001). In primary MGP-null VSMCs (passage 2) that still express contractile markers 28, the level of Wnt16 mRNA is also significantly down-regulated (3.55±0.13-fold compared to wild-type VSMCs, p<0.001, Fig. 1C). in vivo, significantly reduced Wnt16 protein could be detected by immunostaining in both the calcified cartilaginous aortic lesions lacking expression of the contractile marker smooth muscle actin (smAct) in 4.5 week old KO mice (Supplemental Fig. I, A) and in the aortic wall of 7 day old KO animals in which vascular calcification is yet undetectable 29 and aortic VSMCs express smAct (Supplemental Fig. I, B). Together these results show that reduced Wnt16 expression precedes chondrogenic transformation in MGP-null VSMCs in vivo and in vitro, and therefore may contribute to this pathological process.
FIGURE 1. Wnt16 expression is down-regulated in MGP-deficient VSMCs.

A–B, Aortic Wnt16 expression in 4.5 week old Mgp−/− (KO) and wild-type (WT) mice analyzed by real-time PCR (A, N=6) and Western blot (B, N=4) C, Wnt16 mRNA in primary WT or KO VSMCs (N=3). ***, p<0.001.
Elevated Wnt16 attenuates spontaneous chondrogenic transformation in MGP-null VSMCs
For mechanistic in vitro studies on chondrogenic transformation, primary mouse VSMCs were cultured for 8 days as high-density cell micromasses. The micromass culture system is commonly used for mechanistic studies on mesenchymal chondrogenesis in vitro. Chondrogenesis is evident by the expression of chondrogenic gene markers including collagen type II, Sox9, and aggrecan and is histologically assessed by deposition of glycosaminoglycan (GAG)-rich extracellular matrix. Precartilage limb bud mesenchymal cells cultured in micromasses, spontaneously differentiate into chondrocytes, while various multipotential cell lines, including wild-type VSMCs 17, 29, require a stimulus for preferential chondrogenesis such as TGFβ proteins 30.
In contrast to wild-type cells, MGP-null VSMC micromasses deposit cartilaginous matrix even without TGFβ stimulation in plain DMEM (Fig. 2A, DMEM), and dexamethasone+ascorbic acid supplement further enchance this matrix deposition (DMEM-S, Fig. 2A). When MGP-null VSMC micromasses are cultured in medium preconditioned by Cos-7 cells transfected with Wnt16 plasmid, both the deposition of GAG-rich matrix and expression of Sox9, aggrecan (Agg) and collagen type II (Col II) observed in wild-type cells are supressed (Fig. 2B and C). The presence of active Wnt16 in the medium was confirmed by Western blot and by induction of the TCF/LEF-dependent β-catenin luciferase reporter in A10 VSMCs (Supplemental Fig. II). These data indicate that reduced Wnt16 expression in MGP-null VSMC micromasses is permissive for chondrogenic transformation, consistent with the proposed role for Wnt16 as an inhibitor of chondrogenisis.
FIGURE 2. Chondrogenic transformation in MGP-null (KO) VSMCs.

A, Deposition of glycosaminoglycan (GAG)-rich matrix detected by Alcian blue stain in micromasses of primary WT and KO VSMCs cultured in DMEM or DMEM-S (DMEM supplemented with ascorbic acid, dexamethasone, and ITS). Graph shows quantitation of extracted Alcian blue dye normalized to total cell number using Crystal violet (N=4). B–C, Wnt16 effect on GAG deposition (B) and expression of chondrogenic markers (C) in KO micromasses cultured in DMEM-S (N=3). Gene expression by real-time PCR normalized to beta-actin is expressed as fold change compared to non-transforming WT VSMCs. D–E, GAG deposition (D) and activation of Smad-dependent (TGFβ/BMP-sensitive) luciferase reporter cells (E) by secreted factors from WT and KO VSMCs in the presence or absence of inhibitors to TGFβ (bold face) or BMP (N=4). *, p<0.05; **, p<0.01.
Chondrogenic transformation in MGP-null VSMC micromasses is associated with elevated intrinsic TGFβ activity
Because chondrogenic transformation of wild-type VSMCs occurs in the presence of active TGFβ 17, spontaneous chondrogenesis in micromass cultures of MGP-null VSMCs suggested activation of intrinsic TGFβ activity. Indeed, inhibitors of TGFβRI/ALK5 receptors (LY2157299 and SB431542) completely abolished GAG deposition by these cells, while inhibitors of BMP signaling (Noggin and a specific inhibitor of ALK2/3 receptors LDN193189) had no effect (Fig. 2D). Further, TGFβ inhibitors attenuated the elevated expression of a Smad-dependent luciferase transgene induced by the medium conditioned by MGP-null VSMCs (Fig. 2E). In contrast, inhibitors of BMP signaling had no effect on Smad-dependent luciferase activity (Fig. 2E), although these inhibitors completely prevented induction of luciferase by purified BMP2 (Supplemental Fig. III). These results indicate secretion of active TGFβ isoform(s) by MGP-null VSMCs, but not of secreted BMPs. In agreement, downregulation of MGP, either in vivo or in vitro using shRNA approach, is sufficient to stimulate TGFβ signaling in VSMCs (see Online Data Supplement). Together, these data support the hypothesis that elevated endogenous TGFβ growth factors likely drive chondrogenic transformation of MGP-null VSMCs.
Wnt16 down-regulation is central to TGFβ-induced chondrogenic transformation in VSMCs
In vitro, TGFβ1, TGFβ2 and TGFβ3 have similar pro-chondrogenic activities on wild-type VSMCs (Supplemental Fig. VI) and in vivo TGFβ2 and TGFβ3 are expressed in the media of arteries 31 acting on VSMCs. To study the role of Wnt16 in the pro-chondrogenic effect of TGFβ, we analyzed TGFβ3-treated micromasses of rat A10 and primary mouse wild-type VSMCs that were not deficient in MGP 17, 29. The TGFβ–induced chondrogenic transformation was accompanied by a significant 10.8±2.7-fold down-regulation of Wnt16 mRNA (Fig. 3A, p<0.001). In addition, a 48 hour exposure of monolayer wild-type VSMCs to TGFβ3 also resulted in the down-regulation of Wnt16 (Supplemental Fig. VII, A), further supporting the notion that loss of Wnt16 precedes chondrogenic transformation in VSMCs. Inhibition of Wnt16 expression is Smad-dependent and can be prevented by selective inhibitors of Smad2/3 signaling SB431542 32 and LY2157299 33 (Fig. 3B), while BMP inhibitors have no effect.
FIGURE 3. Wnt16 attenuates TGFβ-induced chondrogenesis in wild-type VSMCs.

A–B, Expression of Wnt16 mRNA in TGFβ3-induced A10 VSMC micromasses compared to non-stimulated micromasses after 8 days (A, N=4) or 48 hours in the presence or absence of specific inhibitors to TGFβ (bold face) or BMP signaling (B, N=3). C–D, Exogenous Wnt16 effect on deposition of GAG matrix (C, graph shows quantitation of N=4) and induction of chondrogenic markers (D, N=3) in TGFβ3-induced micromasses of WT VSMCs. E, Wnt16 effect on Smad-dependent luciferase reporter in A10 cells treated for 48 hours with TGFβ3 (N=3). *, p<0.05; **, p<0.01; ***, p<0.001.
To restore Wnt16 levels in TGFβ3-treated cells, which synthesize GAG, micromasses were cultured in medium conditioned by Cos-7 cells with forced expression of Wnt16. The Wnt16-conditioned medium caused a 75% reduction in the deposition of GAG-enriched matrix (Fig. 3C and Supplemental Fig. VII, B) and attenuated the enhanced expression of chondrogenic markers (Fig. 3D) compared to medium conditioned by control mock-transfected Cos-7 cells. Importantly, exogenous Wnt16 did not attenuate TGFβ3-induced activation of the Smad-dependent luciferase reporter in VSMCs (Fig. 3E) demonstrating that Wnt16 is a downstream target of TGFβ signaling rather than an upstream antagonist of this pathway. indeed, Wnt16 alone did not induce GAG synthesis in wils-type VSMCs in the absence of TGFβ3. These data suggest that loss of Wnt16 may destabilize vascular phenotype allowing for chondrogenic transformation. This is further supported by both loss-of-function and gain-of-function studies in VSMCs (see Online Data Supplement).
TGFβ-Wnt16-Notch cross-talk in VSMCs
Recently, Wnt16 was shown to act as a non-canonical activator of Notch signaling in hematopoetic differentiation 27. In VSMCs, Wnt16 alone stimulated Notch activity in rat A10 cells stably expressing the Notch-dependent RBP-Jκ-responsive luciferase construct (termed A10-Notch VSMCs) (Fig 4A). In contrast, Notch inhibitor DAPT had no effect on Wnt16 expression (Fig. 4B) at concentrations that efficiently down-regulated expression of Notch target genes (Supplemental Fig. IX, A). These results agree with Notch being a downstream target of Wnt16.
FIGURE 4. Wnt16 regulates Notch signaling in wild-type VSMCs.
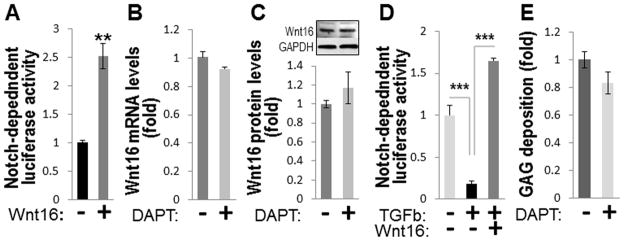
A, Fold change in activity of Notch-dependent luciferase reporter in A10 VSMCs overexpressing Wnt16 compared to to mock-transfected cells cultured for 72 hours in plain DMEM (N=4, normalized to total lactate dehydrogenase). B, Expression of Wnt16 mRNA (left) and protein (right) in A10 VSMCs treated with Notch inhibitor DAPT (N=3). C, Activity of Notch-dependent luciferase reporter in 14 day TGFβ3-induced chondrogenic micromasses of A10 VSMCs in the presence or absence of exogenous Wnt16 (N=4). D, GAG deposition by A10 VSMC micromasses in the presence or absence of Notch inhibitor DAPT. **, p<0.01; ***, p<0.001.
Because Notch signaling is antagonistic for chondrogenesis 20, 34, we tested the hypothesis that Wnt16 may prevent phenotypic transformation in VSMCs by sustaining Notch signaling. In chondrogenic micromasses of A10-Notch VSMCs, TGFβ3 alone caused a 75% down-regulation of Notch-dependent luciferase (Fig. 4C), while exogenous Wnt16 (secreted by transfected Cos-7 cells) attenuated this inhibition and even slightly induced Notch-dependent reporter (Fig. 4C) in agreement with its non-canocial role as a Notch activator. Similarly to rapid inhibition of Wnt16 in VSMCs, TGFβ3 significantly down-regulated several key components of Notch signaling after a short 48 hour exposure (Supplemental Fig. IX, B), indicating that the TGFβ3-induced inhibition of both Wnt16 expression and Notch signaling precedes chondrogenic differentiation. In contrast, specific Notch inhibitor DAPT was insufficient to stimulate GAG synthesis by A10 micromasses in the absence of TGFβ3 (Fig. 4D), indicating antagonistic actions of TGFβ and Notch pathways in the chondrogenic transformation of VSMCs.
Notch signaling is repressed in MGP-null VSMCs
Lastly, we analyzed the activity of Notch signaling in the cartilaginous MGP-null arterial tissue from 4.5 week old mice. Expression of several ligands, receptors and molecular targets of this pathway were significantly reduced in the MGP-null aortae compared to wild-type tissue (Fig. 5A, B). Further, a 90% decrease in expression of the Notch-dependent (RBP-Jκ-responsive) luciferase transgene was detected in primary mouse MGP-deficient VSMCs compared to wild-type cells (Fig. 5C, p<0.05).
FIGURE 5. Notch signaling is repressed in MGP-null vascular smooth muscle.
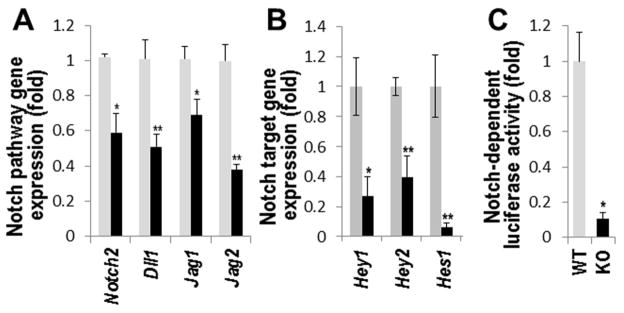
A–B, Real-time PCR analysis of Notch ligands and receptors (A) and down-stream Notch targets (B) in aortae from 4.5 week old WT (grey bars) and KO (black bars) mice (N=3). C, Relative luciferase activity in primary mouse WT or KO VSMCs expressing Notch-dependent luciferase transgene (N=3). *, p<0.05; **, p<0.01.
DISCUSSION
Chondrogenic transformation and calcification of vascular cells complicates various conditions including renal disease 35, however the underlying molecular mechanisms underlying are still incompletely understood. This study on chondrogenic transformation in the background of MGP deficiency identified Wnt16 as a novel regulator of the chondrogenic phenotypic switch in VSMCs. We propose that normal Wnt16 expression supports the contractile phenotype in VSM via Notch signaling (Fig. 6A). We show that when TGFβ is activated via either exogenous addition or the loss of MGP, this causes a rapid decrease in Wnt16 expression, leading to suppression of Notch signaling and permitting the subsequent chondrogenic transformation of VSMCs which in turn is stimulated by additional downstream effects of TGFβ (Fig. 6B).
FIGURE 6. Model for integrated TGFβ-Wnt16-Notch signaling network in VSMC transformation.
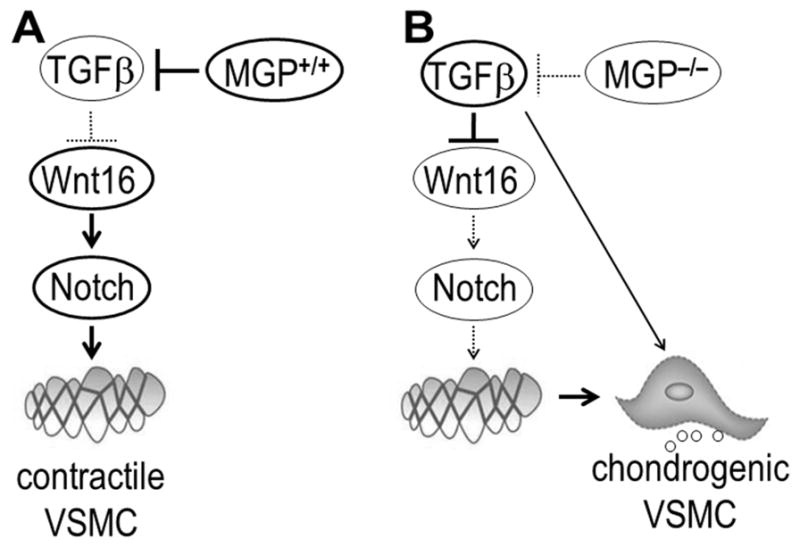
A, In wild-type (Mgp+/+) aortic tissue, MGP represses TGFβ production in VSMCs, Wnt16 is abundant, and Notch signaling is active supporting a stable, contractile phenotype of VSMCs. B, In the aortae of MGP-null (Mgp−/−) mice, TGFβ production is elevated leading to down-regulation of Wnt16 and repression of Notch signaling, and allowing for chondrogenic transformation of VSMCs by additional TGFβ-mediated events.
In VSMCs with reduced MGP (from either shRNA or genetic ablation), we detect activated TGFβ signaling, including induction of multiple TGFβ targets, secretion of factors able to activate Smad2/3-dependent luciferase reporter cells, and detection of phosphorylated Smad2 in MGP-null aortic tissue. To the best of our knowledge, this is the first evidence for activation of the TGFβ/ALK5 pathway in VSMCs by MGP loss, providing new insights into MGP-null vascular disease and into other maladies with altered levels of functional MGP. Of note, our data suggest a limited role for BMPs in the chondrogenic transformation of VSMCs. Indeed, neither forced expression of BMP2 under the α-smooth muscle actin promoter 36 nor BMP4 infusion by osmotic pump 37 led to the formation of cartilaginous metaplasia characteristic for MGP-null vascular disease. In vitro, TGFβ1-3 isoforms possess similar pro-chondrogenic activities in high-density VSMC cultures. In TGFβ3-induced chondrogenic transformation in VSMCs, we identify the down-regulation of Wnt16 as an early event mediated via an ALK5/Smad2/3-dependent mechanism. Physiological relevance of these studies is supported by the pattern of TGFβ3 and TGFβ3 expression in the media and adventitia of the greater arteries, while TGFβ1 is abundant in vascular endothelium 31. Specific genetic disruption of each TGFβ protein leads to discrete phenotypes with a major cardiovaslucar phenotype observed in TGFβ2-knockout mice 38. In contrast to the pro-chondrogenic effects of all TGFβ isoforms in VSMC miscromasses, in progenitor cells TGFβ1 activates expression of smooth muscle markers 39 indicating that TGFβ effects may be cell type- and/or isoform-specific.
Exogenous Wnt16 prevents the TGFβ3-induced chondrogenesis in wild-type micromasses and spontaneous chondrogenesis in MGP-null cells. Although Wnt16 is capable of β-catenin activation 26, down-regulation of Wnt16 in MGP-null aortae occurs in the background of activated canonical β-catenin signaling 17. Therefore, we propose that in VSMCs Wnt16 acts instead via a non-canonical conduit involving Notch activation, as has been also described in keratinocytes 42, cancer cells 43 and developing somites 27, while the β-catenin pathway is largely regulated by other ligands including canonical Wnts 1, 3 and 7 17 and enzyme transglutaminase 2 7, 16. Importantly, Wnt16 is not an antagonist of TGFβ. Instead, our data show that Wnt16 regulates Notch signaling which in turn promotes contractile VSMC phenotype 19. Inhibition of Wnt16 by TGFβ represses Notch signaling, permitting the TGFβ-induced chondrogenic transformation program in VSMCs. A similar correlation between activated TGFβ and attenuated Notch signaling was described in de-differentiating VSMCs of the aneurysmal aortic wall 40 indicating a potentially broad involvement of TGFβ-Notch antagonism in vascular pathologies. We detected a similar reduction in Notch receptors, ligands and target In both MGP-null aortae and in TGFβ-treated VSMCs. In contrast, in endothelial cells MGP ablation leads to enhanced expression of Notch ligands Jagged 1 and 2 and activation of BMP signaling 45. These findings suggest that in vivo the TGFβ and BMP signaling pathways may synergistically contribute to blood vessel abnormalities by regulating phenotypic transformations in distinct vascular cell types.
While inhibition of Wnt16-mediated Notch activity is critical in TGFβ-induced chondrogenic transformation of VSMCs, inhibition of Notch alone, in the absence of TGFβ, was not sufficient to achieve chondrogenic transformation in VSMCs. Further, genetic ablation of Wnt16 does not associate with vascular abnormalities in mice with normal endogenous levels of vascular TGFβ 41; and since the potentially redundant ligand Wnt14 is not discernible in vascular tissue 24, this lack of a vascular phenotype may not be attributed to compensation. These observations indicate that inhibition of Wnt16 and Notch by TGFβ acts in concert with other TGFβ-dependent mechanisms to promote chondrogenesis. Nonetheless, re-expression of Wnt16 is sufficient to prevent TGFβ-induced VSMC chondrogenesis, underscoring the importance of Wnt16 suppression by TGFβ in permitting this phenotypic switch.
In our studies, alterations in Wnt16 levels in wild-type VSMCs do not induce expression of osteogenic genes, althgough missense mutations in Wnt16 in patients 26 as well as genetic ablation in mice 41 associate with low bone mass and easier fractures. It is plausible that the bone phenotype may result from systemic rather than local effects of reduced Wnt16 which is expressed in various tissues including hematopoietic and lymphoid progenitors 44. Our results show that Wnt16 sustains Notch activity and thus maintains VSMC phenotype. A similar mechanism may be involved in skeletal chondrogenesis, in which both Wnt16 expression and Notch activity are excluded from cartilaginous areas 20, 23. This study may be the first direct demonstration of Wnt16 in suppression of chondrogenic differentiation in phenotypically plastic cells.
There are naturally limitations to our study. In this work, we investigate the role of Wnt16 in TGFβ-mediated chondrogenic transformation of VSMCs, whereas in vivo other signaling pathways are also involved including activated β-catenin signaling and endothelium-derived elevated BMPs which contribute to MGP-null disease 17, 46. Further, in this study we did not address the role of signaling beyond Alk5-Smad2/3-Wnt16-Notch pathway in pro-chondrogenic TGFβ effects on VSMCs. A plausible contributor is the MAPK-mediated conduit known as an important mediator of TGFβ1 action in VSMC 47. Nonetheless, the novel TGFβ-Wnt16-Notch axis described in this study may be relevant to multiple cardiovascular diseases in which TGFβ is implicated including hypertension, restenosis, atherosclerosis, cardiac hypertrophy and heart failure 48.
Supplementary Material
SIGNIFICANCE.
Chondrocyte-like transformation of VSMCs underlies the formation of cartilaginous metaplasia that associates with vascular calcification caused by genetic defects in matrix Gla protein (MGP) in human patients and in animal models. Here we identify Wnt16 as a novel regulator of VSMC phenotype, and show that Wnt16 is critical for the integration of TGFβ and Notch signaling pathways. We also show activation of endogenous TGFβ rather than BMP signaling in MGP-null VSMCs. Our finding that TGFβ promotes chondrogenic transformation expands the current BMP-centric view of MGP-null arterial disease. These results advance our understanding of the complex mechanisms underlying vascular calcification and may provide new insights into the molecular mechanisms of the cartilage and bone formation and homeostasis.
Acknowledgments
We are grateful to Prof. Dell’Accio (Queen Mary University of London) for providing the Wnt 16 expression vector. We would like to thank Sean Daugherty and Institute for Genomic Sciences at University of Maryland for help with normal massively parallel sequencing and data analysis.
Sources of Funding: This study was supported by the NIH grants R01HL099305 (M.N.) and T32AR007592 (K.B.).
Non-standard Abbreviations
- VSM
vascular smooth muscle
- VSMC
vascular smooth muscle cell
- MGP
matrix Gla protein
- GAG
glycosaminoglycan
Footnotes
Disclosure: Dr. Nurminskaya is currently an employee of the National Institutes of Health. This work was prepared while she was employed at the University of Maryland. The opinions expressed in this article are the author’s own and do not reflect the view of the National Institutes of Health, the Department of Health and Human Services, or the United States government.
References
- 1.Vattikuti R, Towler DA. Osteogenic regulation of vascular calcification: an early perspective. Am J Physiol Endocrinol Metab. 2004;286:E686–E696. doi: 10.1152/ajpendo.00552.2003. [DOI] [PubMed] [Google Scholar]
- 2.Luo G, Ducy P, McKee MD, Pinero GJ, Loyer E, Behringer RR, Karsenty G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78–81. doi: 10.1038/386078a0. [DOI] [PubMed] [Google Scholar]
- 3.Murshed M, Schinke T, McKee MD, Karsenty G. Extracellular matrix mineralization is regulated locally; different roles of two gla-containing proteins. J Cell Biol. 2004;165:625–630. doi: 10.1083/jcb.200402046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Munroe PB, Olgunturk RO, Fryns JP, Van ML, Ziereisen F, Yuksel B, Gardiner RM, Chung E. Mutations in the gene encoding the human matrix Gla protein cause Keutel syndrome. Nat Genet. 1999;21:142–144. doi: 10.1038/5102. [DOI] [PubMed] [Google Scholar]
- 5.Herrmann SM, Whatling C, Brand E, Nicaud V, Gariepy J, Simon A, Evans A, Ruidavets JB, Arveiler D, Luc G, Tiret L, Henney A, Cambien F. Polymorphisms of the human matrix gla protein (MGP) gene, vascular calcification, and myocardial infarction. Arterioscler Thromb Vasc Biol. 2000;20:2386–2393. doi: 10.1161/01.atv.20.11.2386. [DOI] [PubMed] [Google Scholar]
- 6.Bostrom KI, Rajamannan NM, Towler DA. The regulation of valvular and vascular sclerosis by osteogenic morphogens. Circ Res. 2011;109:564–577. doi: 10.1161/CIRCRESAHA.110.234278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beazley KE, Deasey S, Lima F, Nurminskaya MV. Transglutaminase 2-Mediated Activation of beta-Catenin Signaling Has a Critical Role in Warfarin-Induced Vascular Calcification. Arterioscler Thromb Vasc Biol. 2012;32:123–130. doi: 10.1161/ATVBAHA.111.237834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shimizu T, Tanaka T, Iso T, Matsui H, Ooyama Y, Kawai-Kowase K, Arai M, Kurabayashi M. Notch signaling pathway enhances bone morphogenetic protein 2 (BMP2) responsiveness of Msx2 gene to induce osteogenic differentiation and mineralization of vascular smooth muscle cells. J Biol Chem. 2011;286:19138–19148. doi: 10.1074/jbc.M110.175786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Long F. Targeting intercellular signals for bone regeneration from bone marrow mesenchymal progenitors. Cell Cycle. 2008;7:2106–2111. doi: 10.4161/cc.7.14.6257. [DOI] [PubMed] [Google Scholar]
- 10.Andersson ER, Sandberg R, Lendahl U. Notch signaling: simplicity in design, versatility in function. Development. 2011;138:3593–3612. doi: 10.1242/dev.063610. [DOI] [PubMed] [Google Scholar]
- 11.Grainger DJ, Metcalfe JC, Grace AA, Mosedale DE. Transforming growth factor-beta dynamically regulates vascular smooth muscle differentiation in vivo. J Cell Sci. 1998;111:2977–2988. doi: 10.1242/jcs.111.19.2977. [DOI] [PubMed] [Google Scholar]
- 12.Shao JS, Cai J, Towler DA. Molecular mechanisms of vascular calcification: lessons learned from the aorta. Arterioscler Thromb Vasc Biol. 2006;26:1423–1430. doi: 10.1161/01.ATV.0000220441.42041.20. [DOI] [PubMed] [Google Scholar]
- 13.Mathew S, Davies M, Lund R, Saab G, Hruska KA. Function and effect of bone morphogenetic protein-7 in kidney bone and the bone-vascular links in chronic kidney disease. Eur J Clin Invest. 2006;36 (Suppl 2):43–50. doi: 10.1111/j.1365-2362.2006.01663.x. [DOI] [PubMed] [Google Scholar]
- 14.Mikhaylova L, Malmquist J, Nurminskaya M. Regulation of in vitro vascular calcification by BMP4, VEGF and Wnt3a. Calcif Tissue Int. 2007;81:372–381. doi: 10.1007/s00223-007-9073-6. [DOI] [PubMed] [Google Scholar]
- 15.Caira FC, Stock SR, Gleason TG, McGee EC, Huang J, Bonow RO, Spelsberg TC, McCarthy PM, Rahimtoola SH, Rajamannan NM. Human degenerative valve disease is associated with up-regulation of low-density lipoprotein receptor-related protein 5 receptor-mediated bone formation. J Am Coll Cardiol. 2006;47:1707–1712. doi: 10.1016/j.jacc.2006.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beazley KE, Banyard D, Lima F, Deasey SC, Nurminsky DI, Konoplyannikov M, Nurminskaya MV. Transglutaminase Inhibitors Attenuate Vascular Calcification in a Preclinical Model. Arterioscler Thromb Vasc Biol. 2013;33:43–51. doi: 10.1161/ATVBAHA.112.300260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beazley KE, Lima F, Borras T, Nurminskaya M. Attenuation of Chondrogenic Transformation in Vascular Smooth Muscle by Dietary Quercetin in the MGP-Deficient Mouse Model. PLoS One. 2013;8:e76210. doi: 10.1371/journal.pone.0076210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng SL, Shao JS, Behrmann A, Krchma K, Towler DA. Dkk1 and MSX2-Wnt7b signaling reciprocally regulate the endothelial-mesenchymal transition in aortic endothelial cells. Arterioscler Thromb Vasc Biol. 2013;33:1679–1689. doi: 10.1161/ATVBAHA.113.300647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fouillade C, Monet-Lepretre M, Baron-Menguy C, Joutel A. Notch signalling in smooth muscle cells during development and disease. Cardiovasc Res. 2012;95:138–146. doi: 10.1093/cvr/cvs019. [DOI] [PubMed] [Google Scholar]
- 20.Watanabe N, Tezuka Y, Matsuno K, Miyatani S, Morimura N, Yasuda M, Fujimaki R, Kuroda K, Hiraki Y, Hozumi N, Tezuka K. Suppression of differentiation and proliferation of early chondrogenic cells by Notch. J Bone Miner Metab. 2003;21:344–352. doi: 10.1007/s00774-003-0428-4. [DOI] [PubMed] [Google Scholar]
- 21.Shimizu T, Tanaka T, Iso T, Doi H, Sato H, Kawai-Kowase K, Arai M, Kurabayashi M. Notch signaling induces osteogenic differentiation and mineralization of vascular smooth muscle cells: role of Msx2 gene induction via Notch-RBP-Jk signaling. Arterioscler Thromb Vasc Biol. 2009;29:1104–1111. doi: 10.1161/ATVBAHA.109.187856. [DOI] [PubMed] [Google Scholar]
- 22.Nigam V, Srivastava D. Notch1 represses osteogenic pathways in aortic valve cells. J Mol Cell Cardiol. 2009;47:828–834. doi: 10.1016/j.yjmcc.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Witte F, Dokas J, Neuendorf F, Mundlos S, Stricker S. Comprehensive expression analysis of all Wnt genes and their major secreted antagonists during mouse limb development and cartilage differentiation. Gene Expr Patterns. 2009;9:215–223. doi: 10.1016/j.gep.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 24.Guo X, Day TF, Jiang X, Garrett-Beal L, Topol L, Yang Y. Wnt/beta-catenin signaling is sufficient and necessary for synovial joint formation. Genes Dev. 2004;18:2404–2417. doi: 10.1101/gad.1230704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kronenberg HM. The role of the perichondrium in fetal bone development. Ann N Y Acad Sci. 2007;1116:59–64. doi: 10.1196/annals.1402.059. [DOI] [PubMed] [Google Scholar]
- 26.Hendrickx G, Boudin E, Fijalkowski I, Nielsen TL, Andersen M, Brixen K, Van HW. Variation in the Kozak sequence of WNT16 results in an increased translation and is associated with osteoporosis related parameters. Bone. 2014;59:57–65. doi: 10.1016/j.bone.2013.10.022. [DOI] [PubMed] [Google Scholar]
- 27.Clements WK, Kim AD, Ong KG, Moore JC, Lawson ND, Traver D. A somitic Wnt16/Notch pathway specifies haematopoietic stem cells. Nature. 2011;474:220–224. doi: 10.1038/nature10107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Speer MY, Li X, Hiremath PG, Giachelli CM. Runx2/Cbfa1, but not loss of myocardin, is required for smooth muscle cell lineage reprogramming toward osteochondrogenesis. J Cell Biochem. 2010;110:935–947. doi: 10.1002/jcb.22607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beazley KE, Reckard S, Nurminsky D, Lima F, Nurminskaya M. Two Sides of MGP Null Arterial Disease: chondrogenic lesions dependent on transglutaminase 2 and elastin fragmentation associated with induction of adipsin. J Biol Chem. 2013;288:31400–31408. doi: 10.1074/jbc.M113.495556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Denker AE, Nicoll SB, Tuan RS. Formation of cartilage-like spheroids by micromass cultures of murine C3H10T1/2 cells upon treatment with transforming growth factor-beta 1. Differentiation. 1995;59:25–34. doi: 10.1046/j.1432-0436.1995.5910025.x. [DOI] [PubMed] [Google Scholar]
- 31.Molin DGM, DeRuiter MC, Doetschamn T, Suciv HM, Poelmann RE, Gittehnerger-de Groot A. TGFb2 does not affect neural creast cell migration but is a key player in vascular remodeling during embryogenesis. In: Artman MD, Benson W, Srivastava D, Nakazawa M, editors. Cardiovascular Development and Congenital Malformations: Molecular & Genetic Mechanisms. 1. John Wiley and sons; 2008. pp. 148–50. [Google Scholar]
- 32.Inman GJ, Nicolas FJ, Callahan JF, Harling JD, Gaster LM, Reith AD, Laping NJ, Hill CS. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol. 2002;62:65–74. doi: 10.1124/mol.62.1.65. [DOI] [PubMed] [Google Scholar]
- 33.Dituri F, Mazzocca A, Peidro FJ, Papappicco P, Fabregat I, De SF, Paradiso A, Sabba C, Giannelli G. Differential Inhibition of the TGF-beta Signaling Pathway in HCC Cells Using the Small Molecule Inhibitor LY2157299 and the D10 Monoclonal Antibody against TGF-beta Receptor Type II. PLoS One. 2013;8:e67109. doi: 10.1371/journal.pone.0067109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fujimaki R, Toyama Y, Hozumi N, Tezuka K. Involvement of Notch signaling in initiation of prechondrogenic condensation and nodule formation in limb bud micromass cultures. J Bone Miner Metab. 2006;24:191–198. doi: 10.1007/s00774-005-0671-y. [DOI] [PubMed] [Google Scholar]
- 35.Neven E, Persy V, Dauwe S, De ST, De Broe ME, D’Haese PC. Chondrocyte rather than osteoblast conversion of vascular cells underlies medial calcification in uremic rats. Arterioscler Thromb Vasc Biol. 2010;30:1741–750. doi: 10.1161/ATVBAHA.110.204834. [DOI] [PubMed] [Google Scholar]
- 36.Nakagawa Y, Ikeda K, Akakabe Y, Koide M, Uraoka M, Yutaka KT, Kurimoto-Nakano R, Takahashi T, Matoba S, Yamada H, Okigaki M, Matsubara H. Paracrine osteogenic signals via bone morphogenetic protein-2 accelerate the atherosclerotic intimal calcification in vivo. Arterioscler Thromb Vasc Biol. 2010;30:1908–1915. doi: 10.1161/ATVBAHA.110.206185. [DOI] [PubMed] [Google Scholar]
- 37.Miriyala S, Gongora Nieto MC, Mingone C, Smith D, Dikalov S, Harrison DG, Jo H. Bone morphogenic protein-4 induces hypertension in mice: role of noggin, vascular NADPH oxidases, and impaired vasorelaxation. Circulation. 2006;113:2818–2825. doi: 10.1161/CIRCULATIONAHA.106.611822. [DOI] [PubMed] [Google Scholar]
- 38.Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, Cardell EL, Doetschman T. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development. 1997;124:2659–2670. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kurpinski K, Lam H, Chu J, Wang A, Kim A, Tsay E, Agrawal S, Schaffer DV, Li S. Transforming growth factor-beta and notch signaling mediate stem cell differentiation into smooth muscle cells. Stem Cells. 2010;28:734–742. doi: 10.1002/stem.319. [DOI] [PubMed] [Google Scholar]
- 40.Doyle AJ, Redmond EM, Gillespie DL, Knight PA, Cullen JP, Cahill PA, Morrow DJ. Differential expression of Hedgehog/Notch and transforming growth factor-beta in human abdominal aortic aneurysms. J Vasc Surg. 2014;(14):10. doi: 10.1016/j.jvs.2014.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Medina-Gomez C, Kemp JP, Estrada K, Eriksson J, Liu J, Reppe S, Evans DM, Heppe DH, Vandenput L, Herrera L, Ring SM, Kruithof CJ, Timpson NJ, Zillikens MC, Olstad OK, Zheng HF, Richards JB, St PB, Hofman A, Jaddoe VW, Smith GD, Lorentzon M, Gautvik KM, Uitterlinden AG, Brommage R, Ohlsson C, Tobias JH, Rivadeneira F. Meta-analysis of genome-wide scans for total body BMD in children and adults reveals allelic heterogeneity and age-specific effects at the WNT16 locus. PLoS Genet. 2012;8:e1002718. doi: 10.1371/journal.pgen.1002718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Teh MT, Blaydon D, Ghali LR, Briggs V, Edmunds S, Pantazi E, Barnes MR, Leigh IM, Kelsell DP, Philpott MP. Role for WNT16B in human epidermal keratinocyte proliferation and differentiation. J Cell Sci. 2007;120(Pt 2):330–339. doi: 10.1242/jcs.03329. [DOI] [PubMed] [Google Scholar]
- 43.Binet R, Ythier D, Robles AI, Collado M, Larrieu D, Fonti C, Brambilla E, Brambilla C, Serrano M, Harris CC, Pedeux R. WNT16B is a new marker of cellular senescence that regulates p53 activity and the phosphoinositide 3-kinase/AKT pathway. Cancer Res. 2009;69:9183–9191. doi: 10.1158/0008-5472.CAN-09-1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Corrigan PM, Dobbin E, Freeburn RW, Wheadon H. Patterns of Wnt/Fzd/LRP gene expression during embryonic hematopoiesis. Stem Cells Dev. 2009;18:759–772. doi: 10.1089/scd.2008.0270. [DOI] [PubMed] [Google Scholar]
- 45.Yao Y, Yao J, Radparvar M, Blazquez-Medela AM, Guihard PJ, Jumabay M, Bostrom KI. Reducing Jagged 1 and 2 levels prevents cerebral arteriovenous malformations in matrix Gla protein deficiency. Proc Natl Acad Sci U S A. 2013;110:19071–19076. doi: 10.1073/pnas.1310905110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yao Y, Jumabay M, Ly A, Radparvar M, Cubberly MR, Bostrom KI. A role for the endothelium in vascular calcification. Circ Res. 2013;113:495–504. doi: 10.1161/CIRCRESAHA.113.301792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Douillet CD, Velarde V, Christopher JT, Mayfield RK, Trojanowska ME, Jaffa AA. Mechanisms by which bradykinin promotes fibrosis in vascular smooth muscle cells: role of TGF-beta and MAPK. Am J Physiol Heart Circ Physiol. 2000;279:H2829–H2837. doi: 10.1152/ajpheart.2000.279.6.H2829. [DOI] [PubMed] [Google Scholar]
- 48.Ruiz-Ortega M, Rodriguez-Vita J, Sanchez-Lopez E, Carvajal G, Egido J. TGF-beta signaling in vascular fibrosis. Cardiovasc Res. 2007;74:196–206. doi: 10.1016/j.cardiores.2007.02.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.