

Abstract
Synovial fibroblast proliferation is a hallmark of the invasive pannus in the rheumatoid joint. Activated protein C (APC) is a natural anticoagulant that exerts antiinflammatory and cyto-protective effects in various diseases via endothelial protein C receptor (EPCR) and proteinase-activated receptor (PAR)-mediated pathways. In this study, we investigated the effect and the underlying cellular signaling mechanisms of APC on proliferation of human rheumatoid synovial fibroblasts (RSFs). We found that APC stimulated proliferation of mouse dermal fibroblasts (MDFs) and normal human dermal fibroblasts (HDFs) by up to 60%, but robustly downregulated proliferation of RSFs. APC induced the phosphorylation of extracellular signal–regulated protein kinase (ERK) and enhanced expression of p21 and p27 in a dose-dependent manner in RSFs. The latter effect was inhibited by pre-treatment with the ERK inhibitors PD98059 and U0126 but not by p38 inhibitor SB203580. In addition, APC significantly downregulated tumor necrosis factor (TNF)α-stimulated cell proliferation and activation of p38, c-Jun NH2-terminal kinase (JNK) and Akt in RSFs. These results provide the first evidence that APC selectively inhibits proliferation and the inflammatory signaling pathways of RSFs. Thus, APC may reduce synovial hyperplasia and pannus invasion in rheumatoid arthritis.
INTRODUCTION
Rheumatoid synovial fibroblasts (RSFs) are one of the major cell types in the synovial pannus and contribute actively to inflammation in rheumatoid arthritis (RA) (1–3). These cells respond to cytokines, such as interleukin (IL)-1β and tumor necrosis factor (TNF)-α through the activation of multiple intracellular signaling pathways, including the mitogen-activated protein kinases (MAPKs) extra-cellular signal–regulated protein kinase (ERK), c-Jun NH2-terminal kinase (JNK) and p38, and the nuclear factor (NF)-κB system (2,4). Activation of these pathways may result in exaggerated proliferation and expression of multiple cytokines including IL-1β (5–7), IL-6 (8) and TNFα (6,7,9), as well as the secretion of matrix metalloproteinases (MMPs), which contribute to tissue destruction in RA (1–3). In addition, Akt activation prevents apoptosis and promotes cell survival of RSFs (10), and the level of phosphorylated (P)-Akt activity is increased by stimulation of primary RSFs with TNFα (10).
Cell cycle control mechanisms include sequential activation and inactivation of the cyclin-dependent kinases. The Cip/Kip family contains proteins such as p21/WAF1 and p27/KIP that bind to cyclin/cyclin-dependent kinase complexes and prevent kinase activation. These cyclin-dependent kinase inhibitors play a key role in RA pathogenesis by affecting proliferation and inflammatory cytokine production in human RSFs and mouse synovial fibroblasts (11,12).
Recombinant human activated protein C (APC) is approved by the U.S. Food and Drug Administration as a treatment for patients with severe sepsis; however, it was recently withdrawn from the market after the PROWESS-SHOCK trial in patients with septic shock showed no clinical benefit in subjects who had received APC compared with placebo (13). Nonetheless, numerous studies have shown that APC exerts antiinflammatory and cyto-protective properties in various organs and that these effects are often mediated by endothelial protein C receptor (EPCR) and proteinase-activated receptors (PARs) (14–17).
In all cells tested to date, including keratinocytes, endothelial cells and tenocytes, APC promotes proliferation via regulating MAPK pathways (17–19). Considering that synovial fibroblast proliferation is a hallmark of the invasive pannus in the RA joint and APC is significantly elevated in human RA synovial fluid compared with normal or osteoarthritic (OA) synovial fluids (20,21), it is possible that APC may contribute to the damage in RA. No data are available on the effects of APC on RSF proliferation. In this study, we investigated the effects of APC on RSF proliferation and, surprisingly, our results show that APC inhibits proliferation of RSFs and that it acts via induction of p21/p27 and activation of ERK1/2.
MATERIALS AND METHODS
Isolation and Culture of Human RSFs
Human RA synovial tissues were obtained during joint replacement surgery from patients with RA according to the American College of Rheumatology criteria (22) from 11 patients comprising 7 female patients (mean ± standard deviation [SD] age 68.2 ± 5.1 years) and 4 male patients (age 70 ± 4 years). Ethical approval was granted by the Royal North Shore Animal Care and Ethics Committee and Northern Sydney Health Human Research Ethics Committee, and the written consent of every patient was obtained. We compared RSFs with low metabolic dermal fibroblasts, as previously described (23,24).
RSFs, human dermal fibroblasts (HDFs) and mouse dermal fibroblasts (MDFs) were obtained from RA synovium, dermis of neonatal foreskin and C57BL/6J mouse skin, respectively, by enzymatic digestions as described previously (25). Briefly, tissues were minced into small pieces and digested with 2 mg/mL collagenase (Sigma-Aldrich, St. Louis, MO, USA) in Dulbecco’s modified Eagle medium (DMEM) containing 100 U/mL penicillin, 100 μg/mL streptomycin, 10 mmol/L HEPES (all from Gibco BRL/Life Technologies, Carlsbad, CA, USA) and 3.7 g/L NaHCO3 at 37°C for 3 h, followed by digestion with 0.25% trypsin and 0.02% ethylenediaminetetraacetic acid (EDTA) at 37°C for 30 min. The cells were cultured in DMEM containing 10% fetal bovine serum (FBS) (ICN, Aurora, OH, USA) in 75-cm2 flasks at 37°C in a humidified 5% CO2 atmosphere.
We used RSFs at passages 1–4 (one passage per experiment) (26). Cultured RSFs abundantly expressed the fibroblast-specific marker cadherin-11 (27) and expressed barely detectable levels of the myeloid cell marker CD11b (Supplementary Figure S1) at passage 1 and passage 4. Cells were plated onto 24-well plates (Corning, Corning, NY, USA) in DMEM containing 10% FBS. To avoid serum effects (24), confluent cells were preincubated in DMEM without FBS (basal medium) for 24 h and then used for experiments with or without recombinant TNFα (Pepro Tech, London, UK), APC (Xigris; Eli Lilly, Indianapolis, IN, USA) or SB203580, PD98059 and U0126 (Calbiochem, San Diego, CA, USA) as indicated in the figure legends, by using fresh basal media.
Quantitative Reverse Transcriptase–Polymerase Chain Reaction (qRT-PCR)
Total RNA was isolated from RSFs of passage 1 and passage 4 by using RNAzol (Molecular Research Centre, Cincinnati, OH, USA) according to the manufacturer’s instructions (http://www.mrcgene.com/rnazol.htm). RNA concentration was determined by NanoDrop spectrophotometry (Thermo Scientific; Scoresby, Australia) and reverse-transcribed into complementary DNA (cDNA) using the cDNA synthesis kit (Bioline; Taunton, MA, USA). Subsequently, qRT-PCR was performed with the Rotor-Gene 6000 Real-Time PCR machine (Corbett Life Science, Mortlake, Australia) by using ImmoMix (Bioline; Taunton) and SYBR Green dye (Qiagen, Hilden, Germany). The reaction mixture consisted of 1 μg cDNA template, 0.75 μL each of forward and reverse primer, 12.5 μL ImmoMix and 2.5 μL SYBR Green. Cycling conditions comprised an initial activation step at 95°C for 10 min followed by 45 amplification cycles of 95°C for 15 s (denaturation), 58°C for 20 s (primer annealing) and 20°C for 45 s (extension). Specificity of the amplification reactions was verified by melting curve analysis.
Primers used in the assay were designed and checked for specificity by using the National Center for Biotechnology Information BLAST search tool (http://www.ncbi.nlm.nih.gov/tools/primer-blast). The primer sequences for cadherin-11 (NM_001797; 163 bp) were 5′-ACCCTCACCATCAAAGTCTG-3′ (forward) and 5′-TCAGGGTCAC AAACAATACT-3′ (reverse) and CD11b (NM_001145808; 187 bp) were 5′-CAGAC AGGAAGTAGCAGCTCCT-3′ (forward) and 5′-CTGGTCATGTTGATGAAGGT GCT-3′ (reverse). The amplification efficiency of each primer set was between 95% and 105%. Data were analyzed using the standard curve for absolute quantification method, and results were expressed as gene copies/μg cDNA, as described in a previous study (28). Fourfold serial dilution cDNA standard curves (1, 250, 62.5, 15.626 and 0 μg) were prepared by using preadipocyte (cadherin-11) and monocyte cDNA (CD11b) at the same time as experimental samples and assessed in the same real-time PCR run.
Cell Proliferation Assay
Cell proliferation was performed as described previously (17) with modification. Briefly, (1 × 104 cells/well) confluent cells from 75-cm2 flasks were seeded into a 96-well microplate (Corning) to a final volume of 200 μL and incubated for 4 h to allow cells to attach and then preincubated in basal media for a further 12 h. Cells were then treated with test agents in serum-free conditions by using fresh basal media. To avoid fibroblast contact inhibition (26), after incubation for 24 h (29,30), culture medium was removed and cells were stained with 1 μg/mL crystal violet (Sigma-Aldrich) dissolved in phosphate-buffered saline (PBS). The unbound dye was removed by washing with tap water, and cells were left to completely dry overnight. Bound crystal violet was solubilized with 0.1% sodium dodecyl sulfate in PBS. The optical density of each well was determined at a wavelength of 550 nm. Results were expressed as relative to control.
Cell Morphology and Cell Death
Passage 4 RSFs were seeded into a 24-well plate (Corning) at 1 × 105 cells/well to a final volume of 250 μL and incubated for 24 h to allow cells to attach and then preincubated in basal media for a further 12 h. At 90% confluency, cells were treated with or without 10 μg/mL APC in fresh basal media for 24 h. Micrographs were taken by an inverted phase contrast microscope (Nikon Eclipse TE2000-U) fitted with a DS-Fi1 digital camera and DS-U2 camera control unit (Nikon, Tokyo, Japan). RSFs in culture with or without APC treatment appear elongated by phase light microscopy, sometimes oval or polygonal, with a few branched cytoplasmic processes (Supplementary Figure S2A). Cell death was measured by adding 10 μL 0.5% trypan blue (Sigma-Aldrich) in PBS to 100 μL cell supernatant, and cell viability was assessed by the trypan blue exclusion method on a hemocytometer and corrected for the total volume of media in the well.
Western Blotting
Western blotting was performed as described previously (17,31). The primary antibodies used were as follows: rabbit polyclonal antibodies directed against phosphorylated (P) forms of p38 and p38 (Santa Cruz Biotechnology, Santa Cruz, CA, USA); p21, p27, P-Akt (Ser473) and Akt; and P-p54, P-ERK1/2, P-ERK2 and ERK1/2 (Cell Signaling, Beverly, MA, USA). Immunoreactivity was detected by using the ECL detection system (Amersham Biosciences, Buckinghamshire, UK). Anti-human β-actin (Sigma-Aldrich) antibody was included to normalize for unequal loading. Protein band intensity was evaluated by densitometry by using image analysis.
Statistical Analysis
The data are expressed as the mean ± SD. Statistical analyses were performed by using the Student t test or analysis of variance (ANOVA) followed by the Bonferroni post hoc test (where appropriate). Statistical significance was accepted at the p < 0.05 level.
All supplementary materials are available online at www.molmed.org.
RESULTS
APC Inhibits Cultured Human RSF Proliferation
To determine the relative effect of APC on human RSF proliferation, we used normal MDFs and normal HDFs as controls. Treatment with APC (0.1–10 μg/mL) for 24 h stimulated the proliferation of normal MDFs and normal HDFs by up to 60% (Figure 1A). However, when RSFs were treated with APC for 24 h, their proliferation was inhibited by up to 30% (Figure 1A). To determine the effect of APC on TNFα-stimulated RSFs, two different treatment regimens were used. APC (10 μg/mL) was delivered either 30 min before TNFα (Figure 1B, far right bar) or at the same time as TNFα. Both methods of delivery completely reversed TNFα-stimulated proliferation (Figure 1B). APC at a 10 μg/mL concentration has no effect on cell death (Supplementary Figure S2B).
Figure 1.
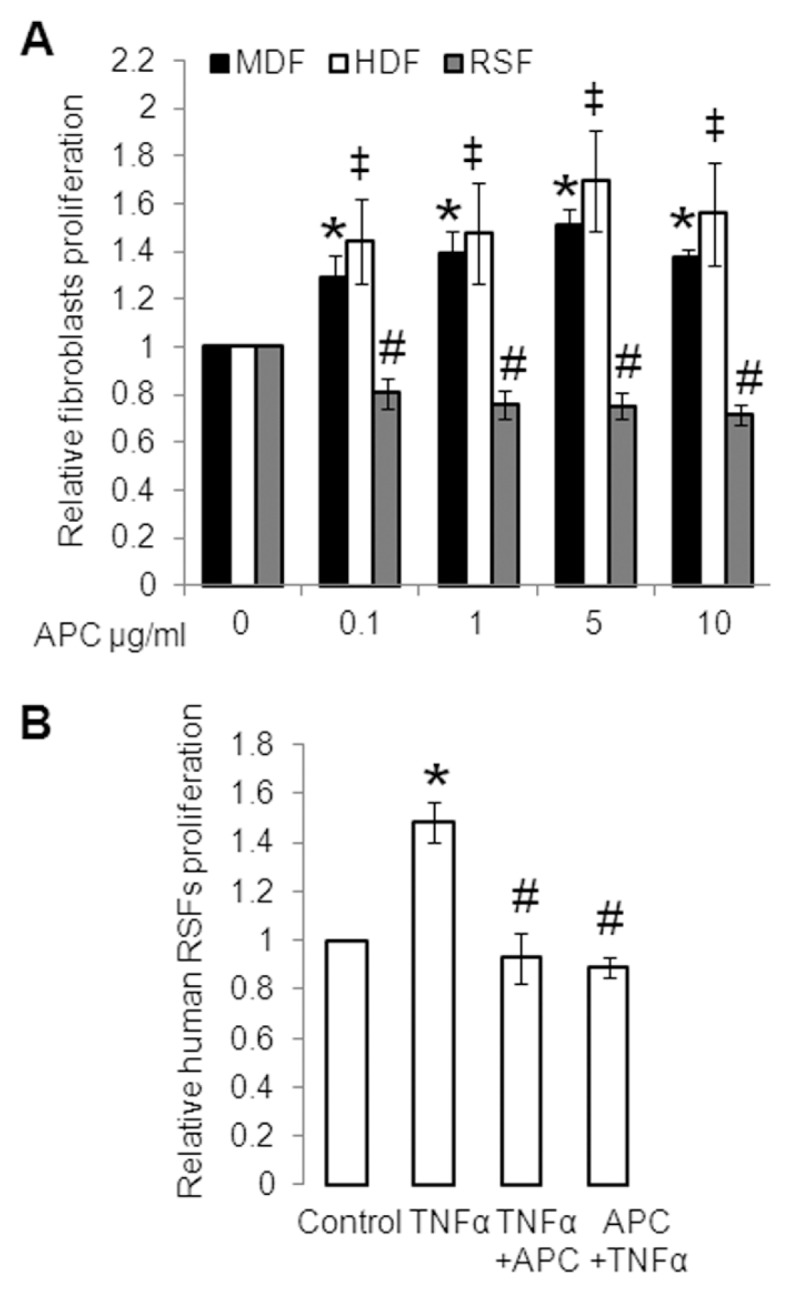
Inhibition of primary cultured human RSF proliferation by APC. (A) Effect of APC on proliferation of normal MDFs, normal HDFs and human cultured RSFs, measured using a crystal violet assay. MDFs, HDFs and RSFs were treated with 0 (control), 0.1, 1, 5 and 10 μg/mL APC. Controls were defined as 1. Values shown are mean ± SD from four (MDFs and HDFs) and three (RSFs) separate experiments. *‡#p < 0.05 versus control with the use of ANOVA followed by Bonferroni post hoc test. (B) RSFs were treated with or without TNFα (2 ng/mL) or APC (10 μg/mL), added after (third bar) or before (last bar) TNFα. Proliferation was measured by crystal violet assay. Values are shown as mean ± SD; n = 3; *p < 0.05 versus control; #p < 0.05 versus TNFα-treated RSFs, with the use of a paired t test.
APC Differentially Regulates p21/p27 in Cultured HDFs versus RSFs
In HDFs, APC (24-h treatment) had little effect on p21 protein expression, but downregulated p27 expression dose-dependently compared with control (Figure 2A). In contrast, in RSFs, APC robustly increased protein expression of p21 and p27 in a dose-dependent manner (Figure 2B). These data agree with the proliferation results in Figure 1 and suggest that inhibition of RSF proliferation by APC is linked to increased p21/p27 expression.
Figure 2.

APC acts differentially on cultured HDFs and RSFs in p21 and p27 induction. HDFs (A) and RSFs (B) were treated with APC for 24 h, and p21 and p27 were measured by Western blotting. β-Actin was used as a loading control. The band intensity of the proteins was normalized with β-actin, and controls were defined as 1. Values are the mean ± SD of two separate experiments.
Increased Induction of p21/p27 and Inhibition of Proliferation in RSFs by APC are ERK Dependent
When RSFs were preincubated with an inhibitor to ERK (PD98059), the increase in p21/p27 was reversed, whereas the p38 inhibitor SB203580 had a minimal effect (Figure 3A). Treatment of RSFs with APC alone (10 μg/mL) for 30 min strongly induced activation of ERK1/2 and ERK2 in a dose-dependent manner (Figure 3B and Supplementary Figure S3A, respectively), while it inhibited activation of p38 (Supplementary Figure S3A). APC also down-regulated activation of p38 at 15 and 60 min (Supplementary Figure S3B). In addition, 1-h pretreatment with PD98059 and U0126 significantly blocked the inhibition of RSF proliferation by APC (Figure 3C and Supplementary Figure S4, respectively), whereas the p38 inhibitor SB203580 had no effect (Figure 3D). These results suggest that APC inhibits proliferation and induces cell cycle inhibitory proteins p21 and p27 via ERK in RSFs.
Figure 3.

APC inhibits proliferation via ERK in RSFs. (A) RSFs were pretreated for 1 h with SB203580 (10 μmol/L) or PD98059 (10 μmol/L) and then treated without (control) or with APC (10 μg/mL) for 24 h. p21 and p27 were assessed by Western blotting. β-Actin was used as a loading control. The band intensity of the proteins was normalized with β-actin, and controls were defined as 1. Values are shown as mean ± SD; n = 3; *p < 0.05 versus control; #p < 0.05 versus APC treated RSFs, with the use of paired t test. (B) RSFs were treated without or with APC for 30 min; ERK1/2 and phosphorylation (P) of ERK1/2 were assessed by Western blotting. (C, D) RSFs were pretreated for 1 h without or with PD98059 (1, 5 μmol/L) (C) and SB203580 (1, 5 μmol/L) (D) and then treated with or without APC (10 μg/mL) for 24 h. Proliferation was measured by crystal violet assay. Controls were defined as 1. Values shown are mean ± SD; n = 4; *p < 0.05 versus control; #p < 0.05 versus APC-treated RSFs, with the use of a t test.
APC Downregulates TNFα-Stimulated P-p38, P-JNK and P-Akt in Human RSFs
To determine the effects of APC on TNFα-stimulated MAPKs and Akt, two different treatment regimens were used: APC was either added 30 min before or at the same time as TNFα (data not shown). Using RSFs, TNFα significantly stimulated P-p38 (Figure 4A) and P-JNK (Figure 4B) but had no significant stimulatory effect on P-ERK1/2 (Figure 4D), compared with control. Pretreatment with APC significantly inhibited TNFα-stimulated P-p38 (Figure 4A) and P-JNK (Figure 4B) and conversely activated P-ERK1/2 (Figure 4D). In addition to the MAPKs, TNFα induced Akt activation, and this was completely reversed by APC (Figure 4C).
Figure 4.

APC downregulates TNFα-stimulated phosphorylation (P) of MAPKs p38, JNK and Akt in RSFs. RSFs were treated without (control [Cont.]) or with TNFα (2 ng/mL) in the presence or absence of APC (10 μg/mL). P-p38, p38 (A), P-JNK (B), P-Akt, Akt (C) and P-ERK1/2, ERK-1/2 (D) were detected by Western blotting. p38 or β-actin were used as a loading control. The band intensity of the proteins was normalized with p38 or β-actin, and TNFα was defined as 1. Values are shown as mean ± SD; n = 3, except (D), where the middle bar represent n = 4 and last bar represent n = 2 ; *p < 0.05 versus control; #p < 0.05 versus TNFα-treated RSF, with the use of a paired t test. APC pre, APC pretreatment.
DISCUSSION
This is the first report to show that APC can inhibit cell proliferation. This effect directly opposes the stimulatory effect of APC on other cells including endothelial cells, keratinocytes (14,17,18), normal MDFs and HDFs. The reason why APC selectively inhibits RSF proliferation is not entirely clear, but may relate to the native rate of proliferation in each cell type. RSFs play an active role in joint destruction in RA by contributing to synovial inflammation (1,2) and pannus invasion (1,3). A critical feature of these cells is excessive proliferation, which expands the synovial pannus to cause progressive cartilage destruction (3,32). In addition to increasing differentiation and migration of mesenchymal stem cells, imbalance between proliferation and apoptosis of RSFs largely contributes to this cellular excess (3). Although studies have failed to find evidence of markedly increased proliferation of RSFs in the RA synovium (33), distinct clonal expansion of cultured RSF has been reported (34). More interestingly, RSFs retain their resistance to apoptosis in vitro(35) and are able to invade and “metastasize” in vivo(32,36). In contrast, normal MDFs and HDFs (24,37) are relatively slow-growing cells unless activated in situations such as wound healing. Our results show that APC appears to selectively control the cell proliferation regulators p21 and p27 by replenishing their very low levels in RSFs. Cyclin-dependent kinase inhibitors play a vital role in controlling excessive proliferation and inflammation in RA. Gene transfer of p21 ameliorates arthritis by exerting antiproliferative effects and antiinflammatory effects in human RSFs (11,38). Loss of p21 expression in RSFs and prevention of p27 upregulation in hypoxic RSFs contribute to excessive invasion and subsequent joint destruction (12,39). In addition, p21 knockout (KO) mice exhibit increased and prolonged inflammatory arthritis and elevated cytokine levels, and p21 mediates a reduction in arthritis severity and suppression of cytokines production via inhibition of p38-MAPK in murine RA model (40).
ERK activation is primarily triggered by certain receptors to cytokines and growth factors, whereas lipopolysaccharide, proinflammatory cytokines and osmotic shock activate p38 and ultraviolet light, protein synthesis inhibitors and cytokines such as TNFα stimulate the JNK pathway (2). Interestingly, our results show that the increased protein levels of p21 and p27 triggered by APC were abrogated by inhibition of ERK but not by p38 inhibition, suggesting that ERK is required for induction of p21 and p27 by APC in nonstimulated RSFs. Although ERK and MEK1/2 inhibitors have been shown to be beneficial in collagen-induced arthritis (41,42), a recent study showed that P-ERK was not increased in collagen-induced arthritis (43). The capacity of pharmacological inhibitors to MEK1/MEK2 in cell-based experiments is discordant (44,45). In addition to MEK1/2, both U0126 and PD98059 have been shown to inhibit MKK5 in vitro and in vivo, and U0126 has been stated to inhibit as many as 25 kinases (44,45). To minimize the risk of nonspecific effects in this study, we used both PD98059 and U0126. ERK signaling is usually associated with stimulation of cell proliferation; however, in our study, the ERK inhibitors PD98059 and U0126 block the antiproliferative effect of APC. Similar results have been described in cancer research. The active-site inhibitor mTOR (KU63794) causes a marked increase in ERK activation yet inhibits cancer cell proliferation more effectively than rapamycin, which does not affect ERK activation (46). Further, mTORC1 inhibition can activate ERK in vitro, in mouse models and in cancer patients, yet it downregulates cell proliferation, cancer growth and Ki 67 expression in vivo(47). The amplitude of the ERK1/2 signal plays a key role in regulating the cell cycle (48). Moderate ERK1/2 activation is required for the expression of cyclin D1 and cell cycle reentry, whereas high-intensity ERK1/2 activation can induce cell cycle arrest or senescence as a result of the high expression of p21CIP1(49). Consistent with our results, TNF-dependent activation markers relevant to RA are relatively insensitive to ERK inhibition in human RSFs (50). In agreement, IL-1β, but not TNFα, significantly stimulates ERK activation in human RSFs and rabbit synovial fibroblasts (51,52). Nonetheless, the effect of TNFα on RA inflammation appears to be dose-dependent (53), and when used in vitro at considerably higher doses (15–50 ng/mL) than the current study, TNFα can stimulate ERK activation (54,55). Anti-TNFα treatment can prevent RA joint destruction without affecting the clinical signs and symptoms of inflammation (56–58). In our previous studies, the ERK inhibitor PD98059 did not suppress IL-1β–stimulated MMP-13 or MMP-1 and −3 production in human RA chondrocytes (59) or human RSFs (25), respectively, whereas it significantly suppressed MMP-13 in OA chondrocytes (59), suggesting that ERK may contribute more to OA than RA pathogenesis.
TNFα stimulates proliferation of synovial fibroblasts and activates signaling molecules such as Akt and MAPKs (2,10), which leads to increased production of inflammatory cytokines that contributes to the pathogenesis of RA (2,4,60). Consistent with previous work (51,52,61), we found that TNFα significantly stimulated the phosphorylation of p38 and JNK in human RSFs. APC reversed the effect of TNFα on p38, JNK and Akt (Figures 4A–C) in human RSFs. In agreement, our previous studies showed that APC completely suppresses lipopolysaccharide-stimulated NF-κB activation, a pivotal regulator of inflammation, hyperplasia and tissue destruction in RA (62), and attenuates TNFα protein by >40% in RA monocytes (63).
MAPK specific inhibitor studies in vitro, in vivo and in clinical trials suggest that the most relevant MAPKs in RA pathogenesis appear to be p38 and JNK (4,64,65). Recent studies show that the p38 network predicts the response of anti-TNFα therapy (66) and JNK activation predicts the development of erosive arthritis (67). In addition, Akt is critical for survival of RSFs (10), by regulating cellular activation, chemotaxis and inflammatory responses (68).
Mechanism(s) of action of APC on ERK induction and downregulation of TNFα-stimulated p38, JNK and Akt in RSFs remain to be elucidated. In our previous studies, we reported that APC receptors EPCR, PAR-1 and PAR-2 are expressed by RA synovium (63,69) and APC inhibits the migration and activation of RA monocytes via EPCR (63). It is possible that ERK induction and down-regulation of TNFα-stimulated p38, JNK and Akt in RSFs by APC will also be mediated by EPCR and PARs.
However, further studies are required using synovial fibroblasts from normal and OA patients as well as RA synovium from different joints to confirm the selective effects of APC. This study provides the first evidence that APC may inhibit inflammation in the rheumatoid joint by acting on the synovial fibroblasts. As summarized in Figure 5, APC acts on RSFs to inhibit proliferation via p21, p27 and ERK and reduce TNFα-induced inflammatory signals of JNK, Akt and p38. This evidence, together with our previous report (63), suggest that, through its cyto-protective actions on signaling molecules, APC prevents proliferation and invasion in RA and impedes the inflammatory cascade to limit arthritic disease (14).
Figure 5.
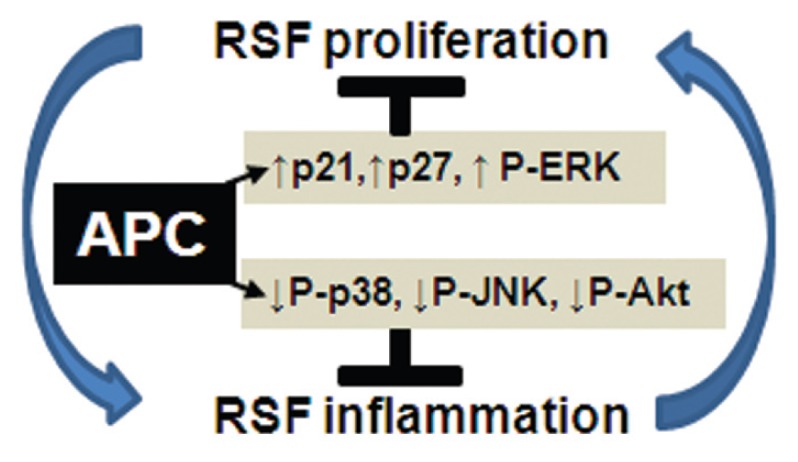
The proposed mechanism of action of APC on RSFs, as shown in this study. RSF proliferation contributes to inflammation, which further promotes a cyclic perpetuation in RA. APC acts on signaling molecules that inhibit both RSF proliferation and inflammation.
CONCLUSION
A key finding of this study is that APC can inhibit proliferation of RA synovial fibroblasts from patients in vitro, and this effect is mediated by an ERK-dependent increase of the cell cycle regulators p21 and p27. Furthermore, APC downregulates TNFα-induced proliferation and inflammatory signaling pathways p38, JNK-MAPK and Akt of RSFs.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by Arthritis Australia, University of Sydney Bridging Grant, The Rebecca Cooper Foundation and Cancer Surgery Research Foundation (CanSur). We thank Professor Ross Smith and Dr. Aiqun Xue, Cancer Surgery Group, Department of Surgery, and Dr. Meilang Xue, Sutton Laboratory, for helpful critique and Yee-Ka Agnes Chan for technical help.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
C Jackson holds patents for the use of APC in cutaneous wound healing.
REFERENCES
- 1.Pap T, Meinecke I, Muller-Ladner U, Gay S. Are fibroblasts involved in joint destruction? Ann. Rheum. Dis. 2005;64(Suppl 4):iv52–4. doi: 10.1136/ard.2005.042424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sweeney SE, Firestein GS. Mitogen activated protein kinase inhibitors: where are we now and where are we going? Ann. Rheum. Dis. 2006;65(Suppl 3):iii83–8. doi: 10.1136/ard.2006.058388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol. 2013;9:24–33. doi: 10.1038/nrrheum.2012.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thalhamer T, McGrath MA, Harnett MM. MAPKs and their relevance to arthritis and inflammation. Rheumatology (Oxford) 2008;47:409–14. doi: 10.1093/rheumatology/kem297. [DOI] [PubMed] [Google Scholar]
- 5.Bucala R, Ritchlin C, Winchester R, Cerami A. Constitutive production of inflammatory and mitogenic cytokines by rheumatoid synovial fibroblasts. J Exp Med. 1991;173:569–74. doi: 10.1084/jem.173.3.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jung YO, et al. Synergism of toll-like receptor 2 (TLR2), TLR4, and TLR6 ligation on the production of tumor necrosis factor (TNF)-alpha in a spontaneous arthritis animal model of interleukin (IL)-1 receptor antagonist-deficient mice. Immunol Lett. 2009;123:138–43. doi: 10.1016/j.imlet.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 7.Neumann E, Lefevre S, Zimmermann B, Gay S, Muller-Ladner U. Rheumatoid arthritis progression mediated by activated synovial fibroblasts. Trends Mol Med. 2010;16:458–68. doi: 10.1016/j.molmed.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 8.Firestein GS, Zvaifler NJ. How important are T cells in chronic rheumatoid synovitis? Arthritis Rheum. 1990;33:768–73. doi: 10.1002/art.1780330602. [DOI] [PubMed] [Google Scholar]
- 9.Roelofs MF, et al. Type I interferons might form the link between toll-like receptor (TLR) 3/7 and TLR4-mediated synovial inflammation in rheumatoid arthritis (RA) Ann Rheum Dis. 2009;68:1486–93. doi: 10.1136/ard.2007.086421. [DOI] [PubMed] [Google Scholar]
- 10.Zhang HG, et al. Regulation of tumor necrosis factor alpha-mediated apoptosis of rheumatoid arthritis synovial fibroblasts by the protein kinase Akt. Arthritis Rheum. 2001;44:1555–67. doi: 10.1002/1529-0131(200107)44:7<1555::AID-ART279>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 11.Nasu K, et al. Adenoviral transfer of cyclin-dependent kinase inhibitor genes suppresses collagen-induced arthritis in mice. J Immunol. 2000;165:7246–52. doi: 10.4049/jimmunol.165.12.7246. [DOI] [PubMed] [Google Scholar]
- 12.Nonomura Y, et al. Hypoxia-induced abrogation of contact-dependent inhibition of rheumatoid arthritis synovial fibroblast proliferation. J Rheumatol. 2009;36:698–705. doi: 10.3899/jrheum.080188. [DOI] [PubMed] [Google Scholar]
- 13.Ranieri VM, et al. Drotrecogin alfa (activated) in adults with septic shock. N Engl J Med. 2012;366:2055–64. doi: 10.1056/NEJMoa1202290. [DOI] [PubMed] [Google Scholar]
- 14.Jackson CJ, Xue M. Activated protein C: an anticoagulant that does more than stop clots. Int J Biochem Cell Biol. 2008;40:2692–7. doi: 10.1016/j.biocel.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 15.Esmon CT. New mechanisms for vascular control of inflammation mediated by natural anticoagulant proteins. J Exp Med. 2002;196:561–4. doi: 10.1084/jem.20021088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruf W. Protease-activated receptor signaling in the regulation of inflammation. Crit. Care Med. 2004;32(Suppl 5):S287–92. doi: 10.1097/01.ccm.0000126364.46191.12. [DOI] [PubMed] [Google Scholar]
- 17.Julovi SM, et al. Protease activated receptor-2 mediates activated protein C-induced cutaneous wound healing via inhibition of p38. Am J Pathol. 2011;179:2233–42. doi: 10.1016/j.ajpath.2011.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uchiba M, et al. Activated protein C induces endothelial cell proliferation by mitogen-activated protein kinase activation in vitro and angiogenesis in vivo. Circ Res. 2004;95:34–41. doi: 10.1161/01.RES.0000133680.87668.FA. [DOI] [PubMed] [Google Scholar]
- 19.Xue M, et al. Activated protein C mediates a healing phenotype in cultured tenocytes. J Cell Mol Med. 2009;13:749–57. doi: 10.1111/j.1582-4934.2008.00359.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buisson-Legendre N, Smith S, March L, Jackson C. Elevation of activated protein C in synovial joints in rheumatoid arthritis and its correlation with matrix metalloproteinase 2. Arthritis Rheum. 2004;50:2151–6. doi: 10.1002/art.20313. [DOI] [PubMed] [Google Scholar]
- 21.Furmaniak-Kazmierczak E, et al. Studies of thrombin-induced proteoglycan release in the degradation of human and bovine cartilage. J Clin Invest. 1994;94:472–80. doi: 10.1172/JCI117358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arnett FC, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–24. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 23.Del Rey MJ, et al. Transcriptome analysis reveals specific changes in osteoarthritis synovial fibroblasts. Ann Rheum Dis. 2012;71:275–80. doi: 10.1136/annrheumdis-2011-200281. [DOI] [PubMed] [Google Scholar]
- 24.Iyer VR, et al. The transcriptional program in the response of human fibroblasts to serum. Science. 1999;283:83–7. doi: 10.1126/science.283.5398.83. [DOI] [PubMed] [Google Scholar]
- 25.Hiramitsu T, et al. Intercellular adhesion molecule-1 mediates the inhibitory effects of hyaluronan on interleukin-1beta-induced matrix metalloproteinase production in rheumatoid synovial fibroblasts via down-regulation of NF-kappaB and p38. Rheumatology (Oxford) 2006;45:824–32. doi: 10.1093/rheumatology/kel026. [DOI] [PubMed] [Google Scholar]
- 26.Neumann E, et al. Cell culture and passaging alters gene expression pattern and proliferation rate in rheumatoid arthritis synovial fibroblasts. Arthritis Res Ther. 2010;12:R83. doi: 10.1186/ar3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosengren S, Boyle DL, Firestein GS. Acquisition, culture, and phenotyping of synovial fibroblasts. Methods Mol Med. 2007;135:365–75. doi: 10.1007/978-1-59745-401-8_24. [DOI] [PubMed] [Google Scholar]
- 28.Lu Y, Xie L, Chen J. A novel procedure for absolute real-time quantification of gene expression patterns. Plant Methods. 2012;8:9. doi: 10.1186/1746-4811-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo X, et al. Fractalkine stimulates cell growth and increases its expression via NF-kappaB pathway in RA-FLS. Int J Rheum Dis. 2012;15:322–9. doi: 10.1111/j.1756-185X.2012.01721.x. [DOI] [PubMed] [Google Scholar]
- 30.Laragione T, Gulko PS. Liver X receptor regulates rheumatoid arthritis fibroblast-like synoviocyte invasiveness, matrix metalloproteinase 2 activation, interleukin-6 and CXCL10. Mol Med. 2012;18:1009–17. doi: 10.2119/molmed.2012.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Julovi SM, Ito H, Hiramitsu T, Yasuda T, Nakamura T. Hyaluronan inhibits IL-1beta-stimulated collagenase production via down-regulation of phosphorylated p38 in SW-1353 human chondrosarcoma cells. Mod Rheumatol. 2008;18:263–70. doi: 10.1007/s10165-008-0067-7. [DOI] [PubMed] [Google Scholar]
- 32.Lefevre S, et al. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat Med. 2009;15:1414–20. doi: 10.1038/nm.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Firestein GS, Yeo M, Zvaifler NJ. Apoptosis in rheumatoid arthritis synovium. J Clin Invest. 1995;96:1631–8. doi: 10.1172/JCI118202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Imamura F, et al. Monoclonal expansion of synoviocytes in rheumatoid arthritis. Arthritis Rheum. 1998;41:1979–86. doi: 10.1002/1529-0131(199811)41:11<1979::AID-ART13>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 35.Meinecke I, et al. Modification of nuclear PML protein by SUMO-1 regulates Fas-induced apoptosis in rheumatoid arthritis synovial fibroblasts. Proc Natl Acad Sci U S A. 2007;104:5073–8. doi: 10.1073/pnas.0608773104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muller-Ladner U, et al. Synovial fibroblasts of patients with rheumatoid arthritis attach to and invade normal human cartilage when engrafted into SCID mice. Am J Pathol. 1996;149:1607–15. [PMC free article] [PubMed] [Google Scholar]
- 37.Egles C, et al. Denatured collagen modulates the phenotype of normal and wounded human skin equivalents. J Invest Dermatol. 2008;128:1830–7. doi: 10.1038/sj.jid.5701240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nonomura Y, Kohsaka H, Nagasaka K, Miyasaka N. Gene transfer of a cell cycle modulator exerts anti-inflammatory effects in the treatment of arthritis. J Immunol. 2003;171:4913–9. doi: 10.4049/jimmunol.171.9.4913. [DOI] [PubMed] [Google Scholar]
- 39.Woods JM, et al. A cell-cycle independent role for p21 in regulating synovial fibroblast migration in rheumatoid arthritis. Arthritis Res Ther. 2006;8:R113. doi: 10.1186/ar1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mavers M, et al. Cyclin-dependent kinase inhibitor p21, via its C-terminal domain, is essential for resolution of murine inflammatory arthritis. Arthritis Rheum. 2012;64:141–52. doi: 10.1002/art.33311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ohori M, Takeuchi M, Maruki R, Nakajima H, Miyake H. FR180204, a novel and selective inhibitor of extracellular signal-regulated kinase, ameliorates collagen-induced arthritis in mice. Naunyn Schmiedebergs Arch Pharmacol. 2007;374:311–6. doi: 10.1007/s00210-006-0117-7. [DOI] [PubMed] [Google Scholar]
- 42.Thiel MJ, et al. Central role of the MEK/ERK MAP kinase pathway in a mouse model of rheumatoid arthritis: potential proinflammatory mechanisms. Arthritis Rheum. 2007;56:3347–57. doi: 10.1002/art.22869. [DOI] [PubMed] [Google Scholar]
- 43.Fukushima A, Boyle DL, Corr M, Firestein GS. Kinetic analysis of synovial signalling and gene expression in animal models of arthritis. Ann Rheum Dis. 2010;69:918–23. doi: 10.1136/ard.2009.112201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ahn NG, Nahreini TS, Tolwinski NS, Resing KA. Pharmacologic inhibitors of MKK1 and MKK2. Methods Enzymol. 2001;332:417–31. doi: 10.1016/s0076-6879(01)32219-x. [DOI] [PubMed] [Google Scholar]
- 46.Soares HP, Ni Y, Kisfalvi K, Sinnett-Smith J, Rozengurt E. Different patterns of Akt and ERK feedback activation in response to rapamycin, active-site mTOR inhibitors and metformin in pancreatic cancer cells. PLoS One. 2013;8:e57289. doi: 10.1371/journal.pone.0057289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carracedo A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065–74. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meloche S, Pouyssegur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene. 2007;26:3227–39. doi: 10.1038/sj.onc.1210414. [DOI] [PubMed] [Google Scholar]
- 49.Woods D, et al. Raf-induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21Cip1. Mol Cell Biol. 1997;17:5598–611. doi: 10.1128/mcb.17.9.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kunisch E, et al. Predominant activation of MAP kinases and pro-destructive/pro-inflammatory features by TNF alpha in early-passage synovial fibroblasts via TNF receptor-1: failure of p38 inhibition to suppress matrix metalloproteinase-1 in rheumatoid arthritis. Ann Rheum Dis. 2007;66:1043–51. doi: 10.1136/ard.2006.062521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pillinger MH, et al. Cyclooxygenase-2-derived E prostaglandins down-regulate matrix metalloproteinase-1 expression in fibroblast-like synoviocytes via inhibition of extracellular signal-regulated kinase activation. J Immunol. 2003;171:6080–9. doi: 10.4049/jimmunol.171.11.6080. [DOI] [PubMed] [Google Scholar]
- 52.Barchowsky A, Frleta D, Vincenti MP. Integration of the NF-kappaB and mitogen-activated protein kinase/AP-1 pathways at the collage-nase-1 promoter: divergence of IL-1 and TNF-dependent signal transduction in rabbit primary synovial fibroblasts. Cytokine. 2000;2000;79 doi: 10.1006/cyto.2000.0743. [DOI] [PubMed] [Google Scholar]
- 53.Binder NB, et al. Tumor necrosis factor-inhibiting therapy preferentially targets bone destruction but not synovial inflammation in a tumor necrosis factor-driven model of rheumatoid arthritis. Arthritis Rheum. 2013;65:608–17. doi: 10.1002/art.37797. [DOI] [PubMed] [Google Scholar]
- 54.Luo SF, et al. Involvement of MAPKs and NF-kappaB in tumor necrosis factor alpha-induced vascular cell adhesion molecule 1 expression in human rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2010;62:105–16. doi: 10.1002/art.25060. [DOI] [PubMed] [Google Scholar]
- 55.Cha HS, et al. A novel spleen tyrosine kinase inhibitor blocks c-Jun N-terminal kinase-mediated gene expression in synoviocytes. J Pharmacol Exp Ther. 2006;317:571–8. doi: 10.1124/jpet.105.097436. [DOI] [PubMed] [Google Scholar]
- 56.Smolen JS, et al. Evidence of radiographic benefit of treatment with infliximab plus methotrexate in rheumatoid arthritis patients who had no clinical improvement: a detailed subanalysis of data from the anti-tumor necrosis factor trial in rheumatoid arthritis with concomitant therapy study. Arthritis Rheum. 2005;52:1020–30. doi: 10.1002/art.20982. [DOI] [PubMed] [Google Scholar]
- 57.Landewe R, van der Heijde D, Klareskog L, van Vollenhoven R, Fatenejad S. Disconnect between inflammation and joint destruction after treatment with etanercept plus methotrexate: results from the trial of etanercept and methotrexate with radiographic and patient outcomes. Arthritis Rheum. 2006;54:3119–25. doi: 10.1002/art.22143. [DOI] [PubMed] [Google Scholar]
- 58.Emery P, et al. Less radiographic progression with adalimumab plus methotrexate versus methotrexate monotherapy across the spectrum of clinical response in early rheumatoid arthritis. J Rheumatol. 2009;36:1429–41. doi: 10.3899/jrheum.081018. [DOI] [PubMed] [Google Scholar]
- 59.Julovi SM, Ito H, Nishitani K, Jackson CJ, Nakamura T. Hyaluronan inhibits matrix metal-loproteinase-13 in human arthritic chondrocytes via CD44 and P38. J Orthop Res. 2011;29:258–64. doi: 10.1002/jor.21216. [DOI] [PubMed] [Google Scholar]
- 60.Feldmann M, Brennan FM, Maini RN. Role of cytokines in rheumatoid arthritis. Annu Rev Immunol. 1996;14:397–440. doi: 10.1146/annurev.immunol.14.1.397. [DOI] [PubMed] [Google Scholar]
- 61.Schett G, et al. Activation, differential localization, and regulation of the stress-activated protein kinases, extracellular signal-regulated kinase, c-JUN N-terminal kinase, and p38 mitogen-activated protein kinase, in synovial tissue and cells in rheumatoid arthritis. Arthritis Rheum. 2000;43:2501–12. doi: 10.1002/1529-0131(200011)43:11<2501::AID-ANR18>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 62.Makarov SS. NF-kappa B in rheumatoid arthritis: a pivotal regulator of inflammation, hyperplasia, and tissue destruction. Arthritis Res. 2001;3:200–6. doi: 10.1186/ar300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xue M, March L, Sambrook PN, Fukudome K, Jackson CJ. Endothelial protein C receptor is overexpressed in rheumatoid arthritic (RA) synovium and mediates the anti-inflammatory effects of activated protein C in RA monocytes. Ann Rheum Dis. 2007;66:1574–80. doi: 10.1136/ard.2006.068239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang X, Huang Y, Navarro MT, Hisoire G, Caulfield JP. A proof-of-concept and drug-drug interaction study of pamapimod, a novel p38 MAP kinase inhibitor, with methotrexate in patients with rheumatoid arthritis. J Clin Pharmacol. 2010;50:1031–8. doi: 10.1177/0091270009357433. [DOI] [PubMed] [Google Scholar]
- 65.Genovese MC, et al. A 24-week, randomized, double-blind, placebo-controlled, parallel group study of the efficacy of oral SCIO-469, a p38 mitogen-activated protein kinase inhibitor, in patients with active rheumatoid arthritis. J Rheumatol. 2011;38:846–54. doi: 10.3899/jrheum.100602. [DOI] [PubMed] [Google Scholar]
- 66.Coulthard LR, et al. Genetic variants within the MAP kinase signalling network and anti-TNF treatment response in rheumatoid arthritis patients. Ann Rheum Dis. 2011;70:98–103. doi: 10.1136/ard.2010.133249. [DOI] [PubMed] [Google Scholar]
- 67.de Launay D, et al. Selective involvement of ERK and JNK mitogen-activated protein kinases in early rheumatoid arthritis (1987 ACR criteria compared to 2010 ACR/EULAR criteria): a prospective study aimed at identification of diagnostic and prognostic biomarkers as well as therapeutic targets. Ann Rheum Dis. 2012;71:415–23. doi: 10.1136/ard.2010.143529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–7. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 69.Xue M, et al. Protease-activated receptor 2, rather than protease-activated receptor 1, contributes to the aggressive properties of synovial fibroblasts in rheumatoid arthritis. Arthritis Rheum. 2012;64:88–98. doi: 10.1002/art.33323. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.