


Abstract
Nucleotides are required for a wide variety of biological processes and are constantly synthesized denovo in all cells. When cells proliferate, increased nucleotide synthesis is necessary for DNA replication and for RNA production to support protein synthesis at different stages of the cell cycle, during which these events are regulated at multiple levels. Therefore the synthesis of the precursor nucleotides is also strongly regulated at multiple levels. Nucleotide synthesis is an energy intensive process that uses multiple metabolic pathways across different cell compartments and several sources of carbon and nitrogen. The processes are regulated at the transcription level by a set of master transcription factors but also at the enzyme level by allosteric regulation and feedback inhibition. Here we review the cellular demands of nucleotide biosynthesis, their metabolic pathways and mechanisms of regulation during the cell cycle. The use of stable isotope tracers for delineating the biosynthetic routes of the multiple intersecting pathways and how these are quantitatively controlled under different conditions is also highlighted. Moreover, the importance of nucleotide synthesis for cell viability is discussed and how this may lead to potential new approaches to drug development in diseases such as cancer.
INTRODUCTION
A large fraction of the genome is now known to be transcribed into a wide range of RNAs whose functions are still being ascertained (1–4). Even in quiescent cells, there is considerable turnover of RNA involved in cell maintenance, repair and regulation. Proliferating cells must up-regulate RNA and DNA biosynthesis as an essential component of cell division, which can modulate, at least in part, the rate of the overall cell cycle (5,6). This requires increased expression of the genes associated with nucleotide synthesis in late G1 phase (5–15).
Nucleotide synthesis is regulated by several critical transcription factors (cf. Table 1), MYC and Rb/E2F in particular, which if mutated or overexpressed are associated with transformation and uncontrolled proliferation leading to cancer (16–23). MYC directly regulates the expression of genes that encode the enzymes in the nucleotide biosynthetic pathways and in the feeder pathways for the production of the precursors of all nucleotides (15,24–26), as well as coordinates RNA and protein biosynthesis (27,28). MYC also influences expression of specific microRNAs that regulate enzymes required for cell proliferation (22,29–31).
Table 1. Genes and enzymes involved in nucleotide biosynthesis.
| Gene name | Enzyme | Chromosome location | Reaction (Figure No.) | Predicted and known regulators |
|---|---|---|---|---|
| 1. Purines | ||||
| PRPS | Phosphoribosyl Pyrophosphate Synthetase | Xq22.3 | (Supplementary Figure S1) | GR Sox5 p53 FOXD1 Nkx2-5 FOXO3b POU2F1 POU2F1a FOXO4 GR-alpha |
| PPAT | phosphoribosyl pyrophosphate amidotransferase | 4q12 | 1 (3) | Bach1, GATA-1, Bach2, C/EBPa, CHOP-10, Brachyury,Roaz, Arnt, USF-1; MYC |
| GART | Trifunctional enzyme. Phosphoribosylglycinamide Formyltransferase Phosphoribosylglycinamide Formyltransferase, Phosphoribosylglycinamide Synthetase; Phosphoribosylaminoimidazole Synthetase | 21q22.11 | 2,3,5 (3) | SRY, HOXA9, HOXA9B, Meis-1, CUTL1, Stat5a, FOX O3/O3a/ O3b, MYC |
| PFAS | Formylglycinamide ribotide amidotransferase | 17p13.1 | 4 (3) | AML1a,MAX,MAX1;MYC |
| PAICS | Bifunctional Phosphoribosyl aminoimidazole Carboxylase | 4q12 | 6,7 (3) | AREB6, p53, CP2, STAT3, MyoD, MYC |
| ADSL | Adenylosuccinate Lyase | 22q13.2 | 8 (3) | MEF-2,RORa2, |
| 12 (S2) | ARP-1,POU2F_1,1a,F2,F2B; MYC | |||
| ATIC | Bifunctional 5-Aminoimidazole-4-Carboxamide Ribonucleotide Formyltransferase/IMP Cyclohydrolase | 2q35 | 9,10 (3) | FOXO1/a, C/EBPb,SREBP-1a,b,c, NFkB, AP-1, c-jun; MYC |
| IMPDH1 | IMP dehydrogenase | 7q31.3-q32 | 13 (3) | c-Fos USF1 AP-1 NRF-2 USF-1 c-Jun MYC |
| GMPS | GMP synthetase | 3q25.31 | 13 (3) | E2F-4 E2F-3a E2F-5 E2F-1 E2F p53 E2F-2 C/EBPalpha |
| ADSS | Adenylosuccinate synthetase | 1q44 | 11 (3) | USF1 Pax-5 NRSF form 1 USF2 CUTL1 NRSF form 2 Roaz FOXC1 STAT3 IRF-7A |
| ADSL | Adenylosuccinate Lyase | 22q13.2 | 12 (S2) | |
| 8 (3) | ||||
| AK1 | Adenylate kinase | 9q34.1 | c-Fos p53 AP-2alpha isoform 3 AP-1 AP-2alpha isoform 2 AP-2alpha isoform 4 c-Jun AP-2alpha AP-2alphaA | |
| NME | Nucleoside Diphosphate Kinase | 17q21.3 | c-Fos AP-1 ATF-2 c-Jun | |
| 2. Pyrimidines | ||||
| CAD, trifunctional | Carbamoyl-Phosphate Synthetase 2 | 2p22-p21 | 1 (Figure 2) | PPAR-gamma1 AP-1 ATF-2 MyoD c-Jun PPAR-gamma2 CUTL1 ; MYC, Hif1a, ER, SP-1 |
| Aspartate Transcarbamylase | 2 (Figure 2) | |||
| Dihydroorotase | 3 (Figure 2) | |||
| DHODH | Dihydroorotate Dehydrogenase | 16q22 | 4 (Figure 2) | AhR AML1a p300 CUTL1 NF-kappaB POU3F2 Evi-1 Arnt GATA-2 NF-kappaB1 ; MYC |
| UMPS | Bifunctional Uridine Monophosphate Synthetase: orotate phosphoribosyltransferase | 3q13 | 5 (Figure 2) | POU2F1a ER-alpha AML1a HTF AREB6 E2F E2F-1 POU2F1 POU2F1a ; MYC |
| UMPS | OMP decarboxylase | 3q21.2 | 6 (Figure 2) | ER-alpha AML1a HTF AREB6 E2F E2F-1 POU2F1 POU2F1a |
| NME | Nucleoside Diphosphate Kinase | 17q21.3 | c-Fos AP-1 ATF-2 c-Jun ; MYC | |
| CTPS | CTP Synthase 1,2 | 1p34.1 | ||
| 3. ribose/PPP | ||||
| G6PD | Glucose-6-phosphate dehydrogenase | Xq28 | (Figure 1) | TBP p53 ATF-2 c-Jun |
| H6PD | D-glucono-1,5-lactone lactone hydrolase (also G6PDH activity) | 1p36 | (Figure 1) | ER-alpha Spz1 NF-1 GCNF RORalpha2 Max GCNF-1 Ik-1 c-Myc |
| PGD | 6-phosphogluconate dehydrogenase (deficiency not associated8 (3) with disease) | 1p36.22 | (Figure 1) | Bach1 Sox5 NF-1/L NF-1 HOXA5 NF-AT C/EBPalpha |
| RPIA | ribose 5-phosphate isomerase A | 2p11.2 | (Figure 1) | NMyc GR AML1a NCX/Ncx MyoD GR-alpha LCR-F1 |
| RPE | ribulose 5-phosphate epimerase | 2q32-q33.3 | (Figure 1) | Sp1 AP-1 ATF-2 |
| TALDO1 | Transaldolase 1 | 11p15.5-p15.4 | (Figure 1) | Pax-5 POU3F1 CUTL1 HNF-3beta YY1 AREB6 SRY FOXO4 FOXJ2 |
| TKT | transketolase | 3p14.3 | (Figure 1) | NF-1 Sp1 p53 HFH-1 LUN-1 Egr-4 C/EBPalpha |
| 4. Feeder pathways | ||||
| GOT1 | Glutamic-Oxaloacetic Transaminase8 (3) (cytoplasmic) | 10q24.1-q25.1 | STAT1 STAT1beta Egr-4 STAT1alpha AREB6 Egr-2 PPAR-gamma1 FOXO1a PPAR-gamma2 FOXO1 | |
| GOT2 | Glutamic-Oxaloacetic Transaminase (mitochondrial) | 16q21 | MIF-1 NF-kappaB GATA-2 AREB6 SRY POU2F1a NF-kappaB2 FOXJ2 (long isoform) FOXJ2 NF-kappaB1 | |
| GLUD1 | Glutamate dehydrogenase 1 | 10q23.3 | HOXA9B HOXA9 ER-alpha Elk-1 Pax-2 Pax-2a FOXJ2 (long isoform) ZIC2/Zic2 Meis-1a Meis-1 | |
| PHGDH | 3-Phosphoglycerate Dehydrogenase | 1p12 | 2 (Figure 4) | GR Max1 IRF-1 CUTL1 PPAR-alpha Max NRF-2 GR-alpha c-Myc |
| PSAT1 | Phosphoserine Aminotransferase 1 | 9q21.2 | 3 (Figure 4) | POU2F2 (Oct-2.1) Oct-B1 oct-B3 oct-B2 POU2F2 POU2F2C POU2F1 POU2F1a c-Jun POU2F2B |
| PSPH | Phosphoserine Phosphatase | 7p11.2 | 4 (Figure 4) | AREB6 GR CREB p53 deltaCREB SEF-1 (1) GR-alpha Nkx2-5 |
| SHMT1 | Serine Hydroxymethyltransferase 1 (cyto) | 17p11.2 | 5 (Figure 4) | AhR CHOP-10 CBF-B CBF-A NF-YA c-Myb CP1A NF-Y CBF(2) C/EBPalpha |
| SHMT2 | Serine Hydroxymethyltransferase 2 (mito) | 12q12-q14 | 5 (Figure 4) | E2F-3a E2F-1 Sp1 E2F-2 GATA-1 |
| TYMS | Thymidylate synthetase | 18p11.32 | AML1a p53 ATF-2 Egr-1 RREB-1 FAC1 POU2F1 POU2F1a ARP-1 MRF-2 MYC | |
| DHFR | Dihydrofolate reductase | 5q11.2-13.2 | USF1 Sp1 p53 USF-1:USF-2 C/EBPalpha Pax-3 POU2F1 POU2F1a USF-1 | |
| MTHFD1 | Trifunctional: Methylenetetrahydrofolate Dehydrogenase (NADP+ Dependent) 1, Methenyltetrahydrofolate Cyclohydrolase, Formyltetrahydrofolate Synthetase | 14q24 | RFX1 NF-YA NF-YC STAT5A CBF-C NF-YB HOXA5 CP1A CP1C NF-Y | |
| RRM1 | Ribonucleotide reductase subunit 1 | 11p15.5 | E2F-4 E2F-3a E2F-5 E2F E2F-1 p53 HOXA5 Lmo2 E2F-2 ; MYC | |
| RRM2 | Ribonucleotide reductase subunit 2 | 2p25-p24 | CREB AP-2alpha isoform 3 Sp1 AP-1 deltaCREB AP-2alpha isoform 4 AP-2alpha isoform 2 AP-2alpha AP-2alphaA | |
The gene nomenclature and regulation (including allosteric regulators) refer to mammalian systems. Predicted transcription factors are from http://www.genecards.org/, which recognize consensus binding sites in the promoters of given genes. Factors in bold represent those experimentally verified by at least one method. MYC binding to gene promoters is mainly from Liu (15); MYC regulation of TS, IMPDH2, PRPS2 is from (24) and E2F from (17). MYC regulation of nucleotide biosynthesis was confirmed by Kim et al. (28).
Despite its functional importance, nucleotide metabolism and cell cycle control have received much less attention than genomics and functional genomics, although there have been multiple metabolic targets derived from the relevant processes for human disease therapy, such as the antimetabolites MTX (32,33), gemcitabine (34), purine analogues (35), suicide inhibitors like 5-FU (36), a range of antiviral nucleotide analogues (37,38) and traditional RNA-seeking antibiotics (39).
Although numerous recent reviews have dealt with metabolic adaptations in proliferating cells (40–50), there has been little emphasis on nucleotide biosynthesis and its regulation. Here we review the regulation of energy and metabolic pathways needed for nucleotide biosynthesis in proliferating mammalian cells.
CELLULAR CONTENT OF NUCLEOTIDES AND NUCLEIC ACIDS
RNA and DNA content of mammalian cells
The DNA content of cells in an organism is fixed, and does not depend on the cell size. In contrast, other cellular components depend on cell volume, as the concentration is regulated. Thus, the protein concentration of mammalian cells is about 200 mg/ml (20% solution), which can occupy ∼16% of the cell volume, not counting the shells of ‘bound’ water in macromolecules. However, cell volumes vary widely - by more than an order of magnitude even for a given organism (see below). This means that the macromolecular content per cell with the exception of DNA varies over a factor of 10-fold or more from one cell type to another.
Quiescent mammalian cells in G0 or G1 are typically diploid, and contain the minimum amount of both DNA and RNA. In order to pass into S phase, the genes for DNA biosynthesis must first be upregulated. Furthermore, actively proliferating cells must double other macromolecular content as they enter M phase and divide into two daughter cells. As the major macromolecular component of cells is protein, protein biosynthesis must also be greatly upregulated during S phase. This will require an increase in the number of ribosomes and thus rRNA, as well as the concomitant energy production needed to meet the enhanced demand for the highly endergonic nucleotide biosynthesis (7,13,51). However, it has been argued that the utilization for macromolecule biosynthesis is a small fraction of the total cellular adenosine triphosphate (ATP) consumption (52,53). Nevertheless, the ATP concentrations (and probably the more relevant ATP/ADP.Pi ratio which is the thermodynamic potential (54,55)) cannot fall below critical levels to maintain cell viability. The ATP requirements show a hierarchy of proliferation > maintenance and repair > membrane potential (46,56–57), i.e. proliferation ceases first, while maintaining membrane potentials goes last. However in mouse Ehrlich Ascites cells, no such energy hierarchy was detected (58).
The content of mammalian cell biomass has been documented for different conditions (59,60). However, it is important to specify the cell type and conditions, whether it be cells in G0/G1 or proliferating cells, as well as the cell size. For example, all proliferating cells double their biomass as they complete a cell cycle. However, in the diploid state, the amount of DNA per cell is independent of the cell size, as it is fixed by the genome C-value (61). For human diploid cells, this is about 6.5 pg per cell based on 6 billion base pairs. However, cell volumes vary greatly, at least by an order of magnitude from small hematopoietic cells of 0.2–0.5 pl to large hepatocytes of volume >5 pl (60,62–63). As other macromolecules are maintained at about the same concentration per cell, the biomass content scales with cell size, as has been emphasized recently by Vaquez etal. (51).
Biomass accounts for around 30% of the cell volume (i.e. 70% water v/v). Given the relative abundances of the various macromolecules, their effective partial specific volumes and the density of mammalian cells, (64–66), (67) the biomass content of a cell is ca. 0.35–0.4 g/ml. In resting cells, biomass comprises ∼50–60% protein, 15% lipid, 5% carbohydrates, 5% small metabolites and ions, and the remainder the nucleic acids (i.e. ca. 15–20%). In a small cell, the DNA content may be up to 3% of the biomass, but in a large cell it is a much smaller fraction (cf. 0.2%). The remaining nucleic acid content is RNA, of which 85% is rRNA and <5% is mRNA. Mammalian cells have been said to have an RNA content of 3% of the biomass (68). However, this seems to be rather variable depending on cell type and size (58). Note that this is considerably less than the >20% estimated for Escherichia coli (52).
The RNA content of a cell does not necessarily represent the amount of RNA synthesis however, because the RNAs are synthesized as larger precursors that are then processed to the mature size, with recycling of the released mononucleotides. For example the ratio of intron to exon length in the human genome is about 28 (69) and the length of mature rRNAs are about 50% of the pre rRNA transcript (70). This requires at least three-fold more RNA synthesis than the amount of mature RNA present in the cell.
Increased protein biosynthetic rate scales with cell size and progression into S-phase. To make protein faster, the cell must make more ribosomes, which requires more rRNA and therefore an increase in the rate of production of rNTPs, which suggests that RNA and protein biosynthesis are coordinately regulated. It has been shown that MYC and its downstream target translation initiation factor eIF4E achieve this via controlling the cis-regulatory element in the 5′ UTR of phosphoribosylpyrophosphate synthetase (PRPS2) (27), which catalyzes the first committed step in nucleotide biosynthesis (cf. Figures 1 and 3A).
Figure 1.
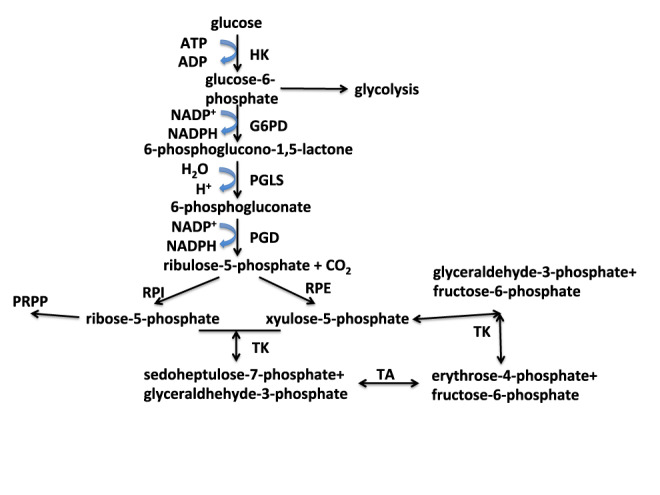
De novo nucleotide biosynthesis: generation of activated ribose. 5-phosphoribose-1-pyrophosphate (PRPP) is the activated form of ribose used for nucleotide biosynthesis and is derived from ribose-5-phosphate from the pentose phosphate pathway (PPP).Ribose-5-phosphate is produced via both oxidative and non-oxidative branches of the PPP. The oxidative branch also generates two NADPH. The oxidative branch comprises the reactions catalyzed by G6PD, PGLS and PGD. The non-oxidative branch interconverts five carbon sugars with four and six carbon sugars using the transaldolase (TA) and transketolase (TK) reactions. HK: hexokinase; G6PD: glucose-6-phosphate dehydrogenase; PGLS: 6-phosphogluconolactonase; PGD: 6-phosphogluconate dehydrogenase; RPI: ribulose-5-phosphate isomerase; RPE: PGLS 3-epimerase; TK: transketolase; TA: transaldolase.
Cellular concentrations of rNTPs and ATP requirement for nucleic acid metabolism
To sustain energy and precursors for RNA biosynthesis, the ATP concentration in cells is generally maintained above 1 mM (71) with a high ATP/ADP.Pi ratio (72), so that there is sufficient free energy available to drive endergonic reactions. There are limits to the maintenance of these levels, below which different aspects of the cell functions cease, eventually leading to necrotic cell death (57,73–74). Some cells maintain very high concentrations of ATP (>5 mM) for specialized mechanical work, such as skeletal muscle (75) and cardiac myocytes (76). Similarly, other rNTPs are maintained at sufficient concentrations to activate and drive anabolic processes (see above) while supplying the materials for nucleic acid synthesis. Thus in cells, it is typically observed that the nucleotide concentrations are in the order ATP > UTP > GTP > CTP with the CTP level kept at ∼1 μmol/g (77,78). Nucleotide sugars are also needed to activate metabolites for various anabolic processes including UDP-glucose and other UDP-hexoses (for carbohydrates synthesis), CDP-choline (for lipid synthesis), GDP sugars (e.g. GDP mannose for glycosyltransferases), NAD(P)+,FAD/FMN (for mediating redox reactions) and ADP ribosylation for a wide range of regulatory functions (79,80). Collectively these compounds are typically present at >1 mM. For a cell of 1 pl volume, the amount of nucleotides is of the order 4–10 fmol. To maintain these pools at constant concentrations as a cell divides, these nucleotides must be synthesized.
For a cell to divide, the entire biomass including metabolites (which is cell size dependent) is doubled as it progresses from the beginning of G1 through the cell cycle and cytokinesis. As described above, a cell with a volume of 0.2–5 pl has a biomass content of 0.08–2 ng, comprising roughly 0.05–1.2 ng protein, 0.01–0.3 ng RNA, 6.5 pg DNA, 0.01–0.3 ng lipid and 0.004–0.1 ng each of carbohydrates and metabolites. The nucleic acids for cell doubling alone amount to 50–950 fmol nucleotides, which use eight ATP equivalents on average for denovo synthesis or 0.4–8 pmol ATP. However, as the RNAs are synthesized as larger precursor molecules and energy is required for recycling the released nucleoside monophosphate (NMPs), the actual ATP usage for RNA synthesis must be larger. For nucleic acid biosynthesis, which is energetically costly, the nucleotide synthesis consumes 0.5–9 pmol exogenous carbon, which is comparable in number to the ATP hydrolysis. Thus, to make nucleic acids for cell proliferation purposes, cells have to upregulate both energy metabolism and the nucleotide biosynthetic pathways. As expected nucleotide biosynthesis is greatly stimulated as cells enter rapid growth (7,11,16,81). Progression through the cell cycle is tightly regulated by numerous transcription factors, and is associated with changes in volume, energy and anabolic metabolism as cells progress through S-phase (15–16,49,51,62,81–87).
NUCLEIC ACID SYNTHESIS: ENERGETICS AND NUTRIENT REQUIREMENTS
To maintain homeostasis, dividing cells need to replenish nucleotides at the same rate as cell division. Thus, the progression of the cell cycle must be tightly linked to the ability of the cell to acquire nutrients, generate metabolic energy and to drive anabolism, including nucleotide/nucleic acid biosynthesis. Although there are salvage pathways and cells can take up nucleotides (88–90), most proliferating cells synthesize nucleotides and nucleic acids denovo, mainly from glucose, glutamine and CO2. The metabolic demands of nucleic acid synthesis have been reviewed recently (47).
Bioenergetics of nucleotide biosynthesis
The different parts of the nucleotides derive from various carbon and nitrogen sources in the cell (cf. Figures 1–3 and below), and the assembly of the mature rNTPs has a high metabolic demand. Supplementary Table S1 summarizes the numbers of nucleoside triphosphate (NTP) equivalents (number of phosphates released from ATP and GTP) needed to make one molecule each of the four rNTPs, according to Figures 1–3. Thus starting from glucose, three ATP equivalents are needed to make the activated ribose-5′-phosphoribosepyrophosphate (PRPP), which is produced by the reaction of 5′-phosphoribose with ATP, driven by the release of the good leaving group 5′-AMP (Supplementary Figure S1). The pyrimidine rings are synthesized first as uracil from aspartate, CO2 (or bicarbonate) and glutamine (Figure 2), which requires two ATP. One of the biosynthetic steps, i.e. orotate dehydrogenase reaction, occurs in the mitochondria (91), while the remainder reside in the cytoplasm. The amination of U to C consumes an additional ATP; denovo synthesis of 5′-UMP and 5′-CMP therefore require four and five molecules of ATP respectively. Aspartate provides three of the four carbon atoms of pyrimidines, which derive largely from glutamine and to a lesser extent from glucose (92) (and see below).
Figure 3.
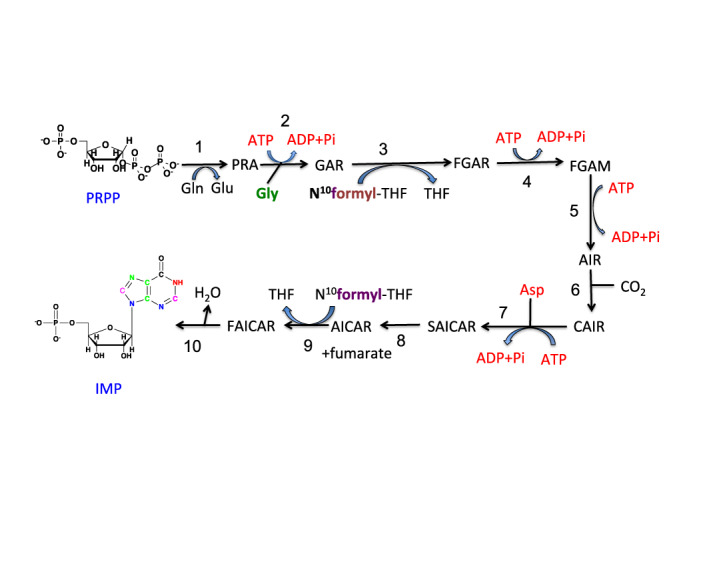
Purine biosynthesis: synthesis of IMP. Various atoms of the purine ring originate from different sources, i.e. N3, N9 derive from the amido group of Gln (blue), N7, C5, C4 derive from Gly (green), C6 from CO2 (black), N1 from the amino group of Asp (red) and C2, C8 from N10formyl-tetrahydrofolate. Enzyme names: (1) glutamine phosphoribosylpyrophosphate amidotransferase (PPAT); (2) glycinamide ribotide synthase (GART); (3) glycinamide ribotide transformylase (GART); (4) formylglycinamide synthase (PFAS); (5) aminoimidazole ribotide synthase (GART); (6) aminoimidazole ribotide carboxylase (PAICS); (7) succinylaminoimidazolecarboxamide ribotide synthase (PAICS); (8) adenylosuccinate lyase (ADSL); (9) aminoimidazole carboxamide ribotide transformylase (ATIC); (10) IMP cyclohydrolase (ATIC). IMP is the common precursor of AMP and GMP. The pathway from IMP to GMP and AMP are shown in Supplementary Figure S2.
Figure 2.
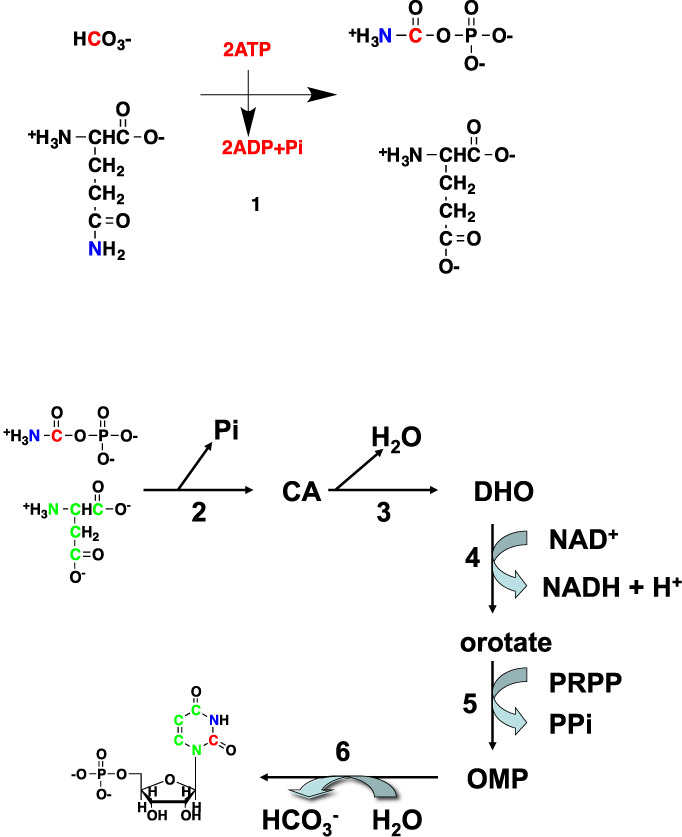
Pyrimidine biosynthesis. CA: carbamoyl aspartate; DHO: dihydroorotate; OMP: orotate monophosphate. Enzyme names: (1) carbamoyl phosphate synthase II (CPSII); (2) aspartate transcarbamoylase (ATCase); (3) carbamoyl aspartate dehydratase = dihydroorotase [CAD encodes enzymes 1 + 2 + 3]; (4) dihydroorotate dehydrogenase; (5) orotate phosphoribosyltransferase; (6) orotidine-5-phosphate decarboxylase (OMP decarboxylase). The activities of 5 and 6 reside in a single bifunctional polypeptide encoded by the UMPS gene. Atom colors denotes origins: red from CO2, green from aspartate and ultimately glucose or Gln, blue from Gln.
Unlike pyrimidines, the synthesis of purine nucleotides is entirely cytoplasmic, with the nucleobase being built directly on the activated PRPP (again, using three ATPs starting from glucose) (Figure 3A). The five carbon atoms of the purine ring derive from CO2, glycine and the one-carbon unit N10-formyl-TetraHydroFolate (THF), which is derived from the serine-glycine pathway via N5,N10-methylene-THF (Figure 4). The sources of serine and glycine may be exogenous, and/or via denovo synthesis from predominantly glucose (93–96). The production of the common intermediate in purine biosynthesis, Inosine Monophosphate (IMP), uses seven ATPs (Figure 3A; Supplementary Table S1). An additional GTP or ATP is used to convert IMP into AMP or GMP, respectively (Figure 3B). The NMPs are then converted to the triphosphate nucleotides by the action of nucleotide kinases, which use two additional ATP molecules for a total of 10. Hence for a genome of 50% GC content and 6 × 109 nucleotides, DNA replication alone would consume around 6 × 1010 ATPs per cycle or close to 0.1 pmol ATP/cell and around 15 fmol glucose equivalents in terms of carbon. The energy demand for RNA synthesis may be 10-fold higher during a cell cycle, thus accounting for about 1 pmol ATP/cell.
Figure 4.
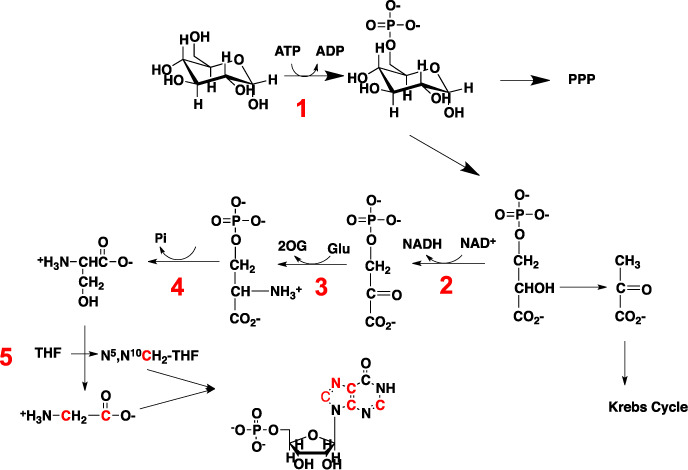
Glycine, serine and aspartate pathways. Synthesis of glycine and N5,N10-methylene tetrahydrofolate (N5,N10-CH2-THF) from glucose via the One-Carbon pathway. N5,N10-CH2-THF is further converted to N10-formyl-THF for incorporation into purine rings. Enzymes: 1: Hexokinase (HK); 2: 3-phosphoglycerate dehydrogenase (PHGDH); 3: phosphoserine aminotransferase (PSAT); 4: phosphoserine phosphatase (PSPH); 5: Serine Hydroxymethyltransferase (SHMT).
The production of DNA further requires the reduction of rNDPs to dNDPs via ribonucletide reductase, which is NADPH-dependent (97), therefore requiring the supply of around 0.1 pmol NADPH/cell per division (and see below). The source of this NADPH is variable according to cell type (98), but the oxidative branch of the pentose phosphate pathway (PPP) is an efficient cytoplasmic source, which also links glucose metabolism to ribose production. As in general the DNA content of cells is lower than that of RNA, and compared with rNTPs, dNTPs have fewer functions other than nucleic acid synthesis, the concentrations of the dNTPs relatively low in cells, typically in the micromolar range in cells in G1, and rising ca. 5–10-fold in late G1 or during S phase (99,100).
In addition to NADPH, as alluded to earlier, the nucleotide biosynthesis pathways have feeder pathways that provide for the carbon and nitrogen precursors, including the amino acids aspartate, glutamine, serine and glycine as well as CO2. These feeder pathways are glycolysis, the PPP (Figure 1), the serine-glycine pathways (Figure 4), the Krebs cycle without or with anaplerotic inputs (Figure 5) and glutamine amidotransferase reactions (Figures 2 and 3).
Figure 5.
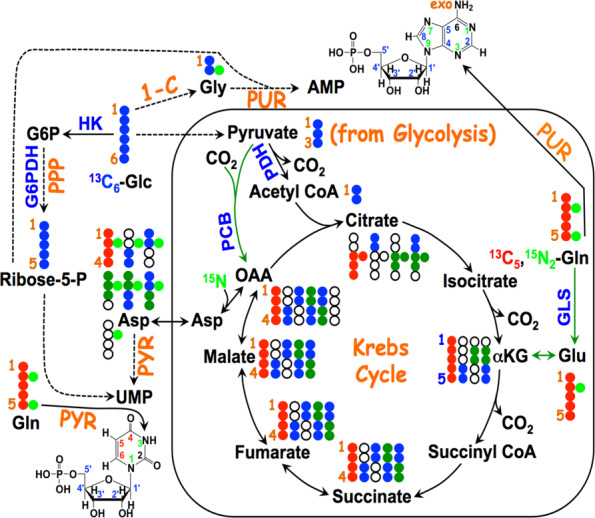
Atom resolved tracing from glucose and glutamine into ribonucleotides. The 13C labels from 13C6-Glc ( ) are incorporated into the ribose unit (via PPP), uracil ring (via the Krebs cycle–pyrimidine synthesis path or PYR) of UMP or adenine ring (via the one-carbon or 1-C to purine synthesis path or PUR) of AMP (structures shown). The 13C (
) are incorporated into the ribose unit (via PPP), uracil ring (via the Krebs cycle–pyrimidine synthesis path or PYR) of UMP or adenine ring (via the one-carbon or 1-C to purine synthesis path or PUR) of AMP (structures shown). The 13C ( ) and 15N labels (
) and 15N labels ( ) from 13C5,15N2-Gln are expected to go into the uracil ring (via the anaplerotic glutaminolysis or GLS-Krebs cycle-PYR path) of UMP and the adenine ring (via the PUR path) of AMP. The color of the label for atomic positions in the UMP and AMP structures is matched with that of 13C or 15N label derived from the glucose or glutamine tracer, except for C4-C6 of UMP where glucose or Gln-derived 13C is not delineated. Three examples of labeled uracil ring delineate contribution of 13C from 13C6-Glc or 13C5,15N2-Gln after one Krebs cycle turn without or with pyruvate carboxylation. The 13C labeling patterns of the Krebs cycle intermediates and Asp account for the 13C scrambling in succinate due to its symmetry and anaplerotic input (green arrows and
) from 13C5,15N2-Gln are expected to go into the uracil ring (via the anaplerotic glutaminolysis or GLS-Krebs cycle-PYR path) of UMP and the adenine ring (via the PUR path) of AMP. The color of the label for atomic positions in the UMP and AMP structures is matched with that of 13C or 15N label derived from the glucose or glutamine tracer, except for C4-C6 of UMP where glucose or Gln-derived 13C is not delineated. Three examples of labeled uracil ring delineate contribution of 13C from 13C6-Glc or 13C5,15N2-Gln after one Krebs cycle turn without or with pyruvate carboxylation. The 13C labeling patterns of the Krebs cycle intermediates and Asp account for the 13C scrambling in succinate due to its symmetry and anaplerotic input (green arrows and  ) from pyruvate carboxylation into the Krebs cycle after the first turn. Open circles: 12C; HK: hexokinase; G6PDH: glucose-6-phosphate dehydrogenase; PDH: pyruvate dehydrogenase; GLS: glutaminase; PCB: pyruvate carboxylase; OAA: oxaloacetate; αKG: α-ketoglutarate; exo: exocyclic.
) from pyruvate carboxylation into the Krebs cycle after the first turn. Open circles: 12C; HK: hexokinase; G6PDH: glucose-6-phosphate dehydrogenase; PDH: pyruvate dehydrogenase; GLS: glutaminase; PCB: pyruvate carboxylase; OAA: oxaloacetate; αKG: α-ketoglutarate; exo: exocyclic.
The metabolic energy needed to drive nucleotide biosynthesis is therefore substantial, and is expected to be derived from a combination of glycolysis and oxidative phosphorylation. The ATP yields for oxidation of different substrates assuming perfectly coupled mitochondria are given in Supplementary Table S2. Different cell types use different strategies that are also dependent on the tissue environment including the nutrient supply, which is a point of focus for understanding metabolic reprogramming in cancers (25,44,47,101–107). Although the oxygen concentration in tissues (e.g. solid tumors) can become quite low, reaching <1% or 10 μM (108,109), it is still sufficient to saturate cytochrome c oxidase, which has a low μM Km for oxygen (110,111). However, other metabolic reprogramming occurs at oxygen levels lower than about 2–4%, due to the activation of HIF1α (112,113), which leads to accelerated glycolysis but inhibition of the Krebs cycle activity. HIF1α is constitutively degraded by the proteasome under normoxia (21% oxygen, ca. 210 μM in air-saturated water at 37°C) due to the regulation by HIF proline hydroxylase. However, when oxygen levels drop to below 2–4%, this enzyme activity is greatly attenuated (114), leading to HIF1α activation (112,115–116).
Moreover, the cellular respiration rate depends on cell type, the number of mitochondria present, the nutrient being oxidized (cf. Supplementary Table S2) and the overall metabolic demand. The oxygen consumption rate of some cancer cells has been measured as 4 fmol/min/cell under normoxia (21% oxygen), to 2 fmol/min/cell under hypoxia (1% O2) (117–120). Such cells can thus generate ca. 5 fmol ATP/min under 1% oxygen by oxidative phosphorylation. When compared with the nucleotide synthesis requirement during a cell cycle of ca. 1 pmol ATP, it would need about 3 h to produce this amount of ATP from oxidative phosphorylation to support nucleotide synthesis alone. However, hypoxic cells also ferment glucose to lactate, producing 2 ATP/mol glucose and in cancer cells glycolysis is typically accelerated many fold (42,121–122), which may produce ATP at a rate comparable to the more efficient mitochondrial oxidation of fuels including fatty acids (104,123), Gln (25,44–45,81,106,119,124–127) and ketone bodies (46) (Supplementary Table S2). It is notable that in the interstitial fluid of solid tumors, the glucose levels are very low with depletion of lipids compared with the blood supply in the tumor (128). This could reflect the high energy demand of tumor cells, which oxidize nutrients at a high rate.
Enzymes and gene locations involved in nucleotide biosynthesis and ‘feeder’ pathways
Figures 1–4 and Table 1 lists the enzymes and genes directly involved in the denovo biosynthesis of nucleotides as well as the relevant ‘feeder pathways’ that supply the carbon, nitrogen and phosphorus, as well as metabolic energy. The genes are distributed over nine chromosomes for the purines and five for the pyrimidines, plus several others for the ‘feeder pathways’. The expression of the genes is thought to be controlled by several transcription factors, though relatively few have been directly demonstrated experimentally.
A notable feature of the enzymes involved in nucleobase biosynthesis is the multifunctional nature, including PPAT, GART and MTHFD1 for purines and carbamoyl phosphate synthase II (CPSII) aspartate transcarbamoylase (ATCase) dihydroorotase trifunctional enzyme (CAD) and UMPS for pyrimidines. These enzymes are thought to be rate limiting in nucleotide biosynthesis, though in practice this concept may not be helpful in understanding regulation (129–132). This is because a coordinate activation of multiple enzymes is necessary for enhancing flux through the pathway (131,133).
Coordinate regulation of expression and flux control
The rate of cell growth is ultimately limited by the supply of materials and energy, and therefore by the flux through the relevant metabolic pathways. Nucleic acid biosynthesis, like any other complex intersecting metabolic networks requires coordination. The flux through even a simple linear pathway depends on the supply of the initial metabolite, the concentration and activities of all of the enzymes in the pathway, and the presence of any feedback or other regulatory controls. It is not well appreciated that the control of flux generally does not reside in any single enzyme or even the same enzymes under all conditions (129,131–134). Fundamentally, at steady state, the flux is the same at every point in the pathway. The concept of a rate-limiting enzyme is therefore at best conditional. In metabolic control analysis (MCA), the goal is to decipher how much control each enzyme in a pathway exerts on the net flux (which can be non-zero only where the system is not at equilibrium). A rate-limiting step in a multistep process is one that controls the net flux, such that changes in any of the other steps in the process have no influence on the rate. Glycolysis for example, is often considered to have three rate-limiting enzymes, HK, PFK-1 and PK. A single rate limiting enzyme is one that completely controls the pathways flux and its flux control coefficient (FCC) is unity. FCC is defined as ∂lnJ/∂lnvi where J is the net flux and vi is the enzymatic activity of the ith enzyme. If there are three equally rate limiting steps, then the flux control is shared equally among the three enzymes. As the sum of the FCC is unity, then each enzyme now has an FCC of one-third (131). In the context of MCA, this is equivalent to having the FCC of 0.33 for each of these enzymes, which is a form of distributed flux control. This means that changing the concentration of any one of these enzymes by 10-fold will change the flux by only a factor of 2, compared with a 10-fold change in flux if these enzymes were simultaneously changed 10-fold. In poorly vascularized tumors, however, the flux control resides mainly at the glucose uptake and HK steps (46,135).
Distributed control is a general feature of metabolic pathways. Even if there is one enzyme that is rate limiting, increasing its activity indefinitely will only transfer the rate limiting step to other enzymes in the pathway, thereby limiting the effect of up regulating a given enzyme activity (by expression or other enzyme-level activity control).
In pyrimidine biosynthesis, the rate limiting step has been described as CPSII (the first enzyme of CAD) (11) and dihydroorotate dehydrogenase (DHODH) (136), the fourth step in the pathway (Figure 2), again implying distributed flux control. This and the fact that several enzymes in both purine and pyrimidine biosynthesis have activities residing on a single polypeptide chain indicates a level of coordinate, stoichiometric expression and the possibility of channeling intermediates from one enzyme to another (see next section).
A means to coordinate expression of functionally linked enzymes that are coded for on different chromosomes is to make use of a common or a set of transcription factors. Although the promoters of the nucleotide genes have binding sites for a variety of transcription factors, there are many in common, one that stands out is MYC (along with related factors that interact with MYC (e.g. MAX and E2F), which have been independently verified by validation experiments such as MYC promoter occupancy assays (Table 1). The regulation of the expression of many genes by MYC is also linked to signaling pathways that respond to growth sensing cues, such as the EGFR/MAP kinase pathway (5,137–139), Hif1/2α (140–142) or estrogen receptor/Sp1 (137,143) for CAD regulation. Although not yet completely understood, cross-talk among these gene regulatory networks is likely to add additional complexities to homeostatic control.
In addition to transcriptional-level regulation, direct modulation of various enzyme activities of the pathways is important, as described below.
Substrate level control
Several of the enzymes involved in nucleotide biosynthesis are regulated at the enzyme level by allosteric interactions, primarily by feedback inhibition. For example, the activities of the enzymes PPAT and CAD multifunctional enzymes are strongly inhibited by relevant pathway products (11,139). Purine biosynthesis is inhibited by AMP and GMP and Pi that act on PRPP synthetase (Supplementary Figure S1) (7) and by AXP and GXP (adenosine and guanosine mono, di or trihosphates) at two sites on the PRPP amidotransferase (Figure 3) (144–146). It is also evident that PRPP amidotransferase activity may be stimulated by the substrate PRPP (147). The branch point at IMP to AMP and GMP (Supplementary Figure S2) is further regulated by allosteric interactions, in which AMP and GMP inhibit the adenylosuccinate synthetase and IMP dehydrogenases, respectively (148–150). The enzyme activities therefore depend not only on the concentrations of the immediate substrates, but also on the end products. Substrate availability in eukaryotic cells can be an important means of controlling flux through pathways, either by physical compartmentation (important in pyrimidine biosynthesis) or by diffusion limits between different pools in large cells (151).
A potentially powerful means of overcome kinetic/diffusion limitations is to channel intermediates in multienzyme complexes (152,153). The observation that several of the enzymes in mammals are expressed as multifunctional enzymes (Figures 2 and 3, Table 1), and that these activities do not always catalyze sequential reactions has lead to the hypothesis that there might be a purinosome (91), in which intermediates are channeled rather than freely equilibrating with the bulk cytoplasm. Whereas the evidence for specific stoichiometric associations invivo is weak, kinetic experiments invitro are consistent with channeling, implying the possibility of weak and transient complex formation (154). Putative purinosome bodies have been detected invivo (91), but they may also be artifacts of the constructs used for visualization (155).
The pyrimidine synthesis pathway is controlled by feedback inhibition by UTP acting on the CPSII domain of the CAD trifunctional enzyme (Figure 2, Table 1) (5,11), which is also activated by PRPP, thereby integrating regulation of purine and pyrimidine biosynthesis (11). As for purine biosynthesis, mammalian pyrimidine biosynthesis is characterized by multifunctional enzymes (Table 1), in contrast to the homologues of E.coli, where CPS and ATCase are two non-associated polypeptides and are differentially regulated at the metabolite level (11). For efficient capturing of the CAD product dihydroorotate (DHO) by the mitochondrial DHODH, and the observation of the association of both CAD and mitochondria with the cytoskeleton, it has been suggested that CAD could be translocated via the cytoskeleton to the DHODH site (11). One complication is that up to 30% of the CAD may translocate to the nucleus during the S phase of the cell cycle, when demand for pyrimidine biosynthesis is maximal. This would compromise the DHODH efficiency and favor an alternative moonlighting function for nuclear CAD (11). However, DHO could still be readily diffuse from the nucleus into the mitochondria as the latter are in close proximity to the nuclear membrane (156).
Synthesis of the deoxyNTPs
DNA synthesis requires a source of the four dNTPs, which derive from the ribonucleotides at the level of rNDPs by reduction at the 2′ position of the ribose subunit. The synthesis of dNTPs occurs cytoplasmically using the enzyme ribonucleotide reductase (RNR) (157), though the possibility of RNR activity inside mitochondria has been reported (158). This enzyme acts upon the rNDP and reduces the 2′-OH of ribose to the deoxy state, using NADPH as the electron source via thioredoxin and thioredoxin reductase. The resulting dNDPs are then converted to the dNTPs via dinucleotide kinase and ATP.
The DNA-unique nucleotide, dNTP is synthesized by methylation of dUMP by the action of thymidylate synthase using N5,N10-methylene tetrahydrofolate as the methyl carbon donor. dUMP is itself formed by a specific phosphatase activity on dUTP or by deamination of dCMP formed from hydrolysis of DNA (99). The dTMP is then converted to dTTP by the sequential action of two kinases.
The R1 subunit of RNR and TS are also regulated by MYC (Table 1). RNR expression is cell cycle dependent and as expected it rises during S phase (159,160). There is an alternative form of the R2 protein called p53R2 that is constitutively expressed at low levels but is upregulated in TP53(+) cells in the presence of DNA damage, and seems to be involved in DNA repair (161–163). The supply of dNTPs are moreover regulated at the substrate level by the supply of NADPH and allosteric interactions of RNR with ATP, dATP and the other dNTPs. RNR has two allosteric sites, both on the RRM encoded subunit. One binds either ATP (activating) or dATP (inhibitory). The ratio of the rATP to dATP therefore regulates net flux to dNDP according to demand—a high ratio of ATP/dATP favors a higher reductive flux and DNA synthesis, whereas a low ratio signals low demand for dNDP. The other allosteric site regulates substrate specificity (164). Thus binding of (d)ATP induces reduction of CDP or UDP, dGTP induces reduction of ADP and dTTP induces reduction of GDP. Together, these activities determine the supply of dNTPs during S phase (net DNA synthesis) and for repair synthesis.
STABLE ISOTOPE TRACING OF NUCLEOTIDE SYNTHESIS
Although the metabolic pathways for nucleotide metabolism are well established, the detailed regulation at the enzyme (such as substrate channeling) and transcriptional levels under different conditions is less well understood. In order to define such regulation and the contribution of different nutrient sources for these complex intersecting pathways, it is necessary to measure the pathways in cells and tissues, using tracer technology such that individual atoms can be traced from the source to the product. In principle, radioisotopes can be used to trace precursors through metabolic pathways into final products (165,166). In practice stable (non-radioactive) isotopes offer more versatility and have the advantage of being fully compatible with live cells or tissues and making it possible to follow the fate of individual atoms from a precursor into detected products (167). The main analytical tools are mass spectrometry and nuclear magnetic resonance (NMR). Mass spectrometry distinguishes 13C or 15N from the more abundant isotopes 12C and 14N by virtue of the mass difference, which is nominally one mass unit. At sufficiently high resolution, such as FT-MS, differences in neutron mass can readily be detected. Thus the mass of a nucleotide containing one 13C atom versus one 15N atom differ by 0.00634 amu which is readily resolved at a resolution of 400,000 (168). This means that simultaneous dual labeling with both 15N and 13C can be used to determine the incorporation of the number and type of atoms (isotopologues) into nucleotides. NMR in contrast depends on the difference in magnetic properties of the nuclei of 13C versus 12C for example. 12C is magnetically silent, whereas 13C can either be directly detected or indirectly by its influence on the directly attached proton (92,169–170) which has the advantage of greatly enhanced sensitivity. NMR detects both the position (isotopomers) and the amount of incorporation of the labeled atoms. The analysis of the isotopomers and isotopologues of intermediates and products provides very detailed information about the biosynthetic routes to the products under study (25,167). Thus use of 13C and/or 15N labeled precursors coupled with labeled product analysis by MS and NMR offers the optimal approach for this purpose, as these techniques can directly detect the number and positions of 13C or 15N in the intermediates and products of nucleotide biosynthesis, thereby robustly defining the flow of atoms through the intersecting pathways. Other stable isotopes can also be used to measure nucleotide synthesis and incorporation into RNA and DNA. Hellerstein's group (14,171) has used both 13C glucose and 2H glucose with GC-MS detection of purine nucleotide released from DNA to follow DNA synthesis in cells and tissues. The overall turnover of nucleic acids in tissues can also be determined by measuring the incorporation of deuterons from D2O into DNA (172). Recently 14C-1 and 14C-6 glucose was used in comparison with [1–2H]-glucose or [3–2H]-glucose with LC MS to detect the transfer of a deuteron to NADP+ in the oxidative branch of the PPP (166).
Our group has developed the stable isotope resolved metabolomics or SIRM approach (173–175) for this purpose, which has been applied to mapping nucleotide biosynthesis in cultured cells or tissues insitu (19,92,101,168,173,176–177). Figure 5 tracks the expected incorporation of labeled atoms from 13C6-glucose (, for 13C) and 13C5,15N2-glutamine ( for 13C and for 15N) into nucleotide products via purine (PUR) and pyrimidine (PYR) synthesis as well as feeder pathways including glycolysis, the Krebs cycle in the absence or presence of anaplerotic pyruvate carboxylation or glutaminolysis, PPP and one-carbon pathway. Based on SIRM profiling of the various labeled intermediates and products, these intersecting pathways for nucleotide biosynthesis can be rigorously reconstructed and quantified even in human subjects insitu (154). Examples of stable isotope tracer studies on nucleotide biosynthesis are described below.
Tracking ribose synthesis via the pentose phosphate pathway
The ribose unit of the nucleotides derives from the PPP, and for most cells and tissues, this means that as few tissues are gluconeogenic the carbon originates from glucose either exogenous or via G-1P by phosphorolysis of glycogen. Two-dimensional (2D) NMR can be applied to crude cell or tissue extracts and discriminates the ribose subunit of purine and pyrimidine nucleotides as well as those of NAD+. The TOCSY experiment detects the scalar interactions between the ribose H1′–H2′–H3′–H4′ which appear as discrete cross peaks. When the carbon atom is 12C, the cross peaks are single, whereas if °C is present, it splits the attached proton resonance into two peaks separated by the one-bond coupling constant of 130–160 Hz. The patterns of thee so-called satellite peaks are characteristic according to which atoms are enriched in 13C, which gives rise to the isotopomer distribution that can be quantified (167,178). Complementary isotope edited experiments can be used to determine the complete isotopomer distribution in complex systems as needed (177,179). This is straightforward to demonstrate in the free nucleotide pool in cells by 2D 1H NMR analysis, as shown in Supplementary Figure S3. Here the ribose subunits of the free nucleotide pools are highly enriched with 13C when the cells are exposed to 13C glucose, but not at all when exposed to 13C Gln. The production of 13C5-ribose-containing nucleotides from 13C6-glucose, as determined by FT-ICR-MS analysis (513C isotopologue in Supplementary Figure S4) is also consistent with the PPP activity The use of glucose as the primary carbon source for ribose biosynthesis applies to many different cell types (92,168,175,179–180).
However, it is difficult to delineate whether the 13C5-ribose labeling pattern results from oxidative, non-oxidative or both branches of the PPP. The oxidative branch (Figure 1) generates ribose-5-phosphate from glucose-6-phosphate (G6P) via decarboxylation of the C1 of G6P while producing two molecules of NADPH. This reaction is especially important in cells that have a high demand for NADPH, including erythrocytes (to remove H2O2 in a highly oxidizing environment (181)) and proliferating cells that are actively making fatty acids (166). The alternative non-oxidative branch reversibly converts ribose-5-phosphate and erythrose-4-phosphate into fructose-6-phosphate and glyceraldehyde-3-phosphate, and viceversa (182), which serves a dual purpose, namely to direct glucose carbon back into glycolysis and for nucleotide ribose biosynthesis. This branch does not generate NADPH. Boros etal. have established a simple GC-MS based stable isotope tracing method that can discriminate between these two branches of pathways, while estimating the net lactic fermentation flux (12,183–186). This method utilizes 13C2-1,2 glucose as tracer, which undergoes decarboxylation to become exclusively 13C-1-ribose via the oxidative branch of the PPP, whereas via extensive scrambling in the non-oxidative branch, ribose is labeled at the C1 and C2 positions. This scrambling arises from the reversibility of the reactions catalyzed by transaldolase and transketolase (cf. Figure 1A), which in fact may be exacerbated by the exchanges in the individual half reactions (187). Chemical exchange in reactions that are near equilibrium equilibrate label across the reaction, such that there can be label reaching a metabolite even where there is net flux in the opposite direction. This is a very general problem, that must be accounted for in any atom-resolved tracer experiments. The ratios of the different labeled ribose species provides an estimate of the relative flow through the oxidative and non-oxidative pathways subject to the caveats noted by Kleijn et al. (187). 14C-1,6 glucose has also been used to discriminate between the oxidative and non-oxidative branches, which relies on radiometric detection of CO2 released by the oxidation at C1′ (166). NMR analysis of free nucleotides can determine positional enrichment in the ribose subunits of intact nucleotides (92) without the need for fragmentation, which may provide a more direct discrimination of between the oxidative and non-oxidative branches and the relative flux through the two branches (188) (and A.N. Lane, unpublished).
It appears that both pathways may operate leading to net nucleotide ribose biosynthesis. Depending on the cell type, and possibly the growth conditions, the oxidative branch can account for very little to most of the flow of glucose into ribose (12,101,182–183,185–186,189–192). Such direct readout of the two pathway activities is required to determine the functional consequences of relevant gene or even enzyme expression in the two pathways (Table 1), as post-translational and substrate level regulations can further alter the metabolic outcome. Despite its complexity and importance to cell proliferation and maintenance, the flux through the PPP is only 1–7% of the net glucose flux to pyruvate (183,188,193–194).
Tracking pyrimidine ring biosynthesis
The pyrimidine ring is synthesized independent of ribose-5-P, with all but one reaction occurring in the cytoplasm. The one exception is the DHODH reaction, which resides on the mitochondrial membrane and is intimately coupled to the electron transport chain (ETC). Mitochondria with a defective ETC cannot make UMP. As Figures 2 and 5 show, four of the ring atoms of uracil derived from aspartate, one from CO2 and the N3 from Gln via the cytoplasmic CPSII enzyme. Aspartate is a non-essential amino acid present in blood plasma at about 20 μM (195) and can be transported into the cell by Na+-dependent anionic transporters (196) or synthesized denovo by transamination of oxalacetate (OAA) in either the mitochondria or the cytoplasm. This reaction in fact is an important component of the malate/aspartate shuttle for transferring electrons from NADH in the cytoplasm into mitochondria. In addition to be the entry point of the Krebs cycle, OAA is the product of the anaplerotic reaction catalyzed by pyruvate carboxylase (PC), which is important in some cancers (179,197–198).
If aspartate and thus OAA is used for pyrimidine biosynthesis, anaplerosis is needed to replenish OAA to sustain the functioning of the Krebs cycle. In addition to PC, another common anaplerotic reaction in cancer cells and other proliferating cells (175,194) is glutaminolysis, in which glutamine is first hydrolyzed to glutamate and ammonia followed by either transamination with OAA to produce Asp + α-ketoglutarate (αKG) (199) or oxidative deamination via GDH to αKG + ammonium ions. αKG can also be converted to OAA by the normal functioning of the Krebs cycle (86,125–127,200–204) (Figure 5). The relative importance of these two anaplerotic reactions probably depends on cell type and growth conditions to maintain energy production, nitrogen balance and anabolic metabolism (175,179,194,198,205). The main difference between PC and glutaminolytic anaplerotic pathways is the production of ammonium ions by the latter, which in excess is toxic to cells and must be dealt with. Moreover, glutaminolysis competes with other Gln deamidation reactions as the direct nitrogen source for many anabolic processes including nucleotide synthesis (45,206–207). These considerations would probably in part govern the choice of anaplerotic pathways that proliferating cells adopt to meet their growth demand.
Again, tracer technologies can readily discriminate between glutamine and glucose as carbon sources and the different feeder pathways leading to uracil ring biosynthesis. [U-13C]-glucose enters the Krebs cycle via pyruvate either as 13C2-acetyl CoA (via PDH) or 13C3-OAA (via PC), which in turn gives rise to distinct labeling patterns in citrate, Asp and ultimately the uracil ring. For example, PC- () or PDH-derived 13C labeled Asp () leads to the synthesis of 13C3–4,5,6-UMP or 13C1–6-UMP, respectively, after one Krebs cycle turn, as shown in Figure 5. Although [U-13C]-Gln also produces 13C3–4,5,6-UMP () via the first Krebs cycle turn, the 13C labeling patterns of Glu, αKG and citrate derived from [U-13C]-Gln are distinct from those derived from [U-13C]-glucose (Figure 5) (25,81,92). Quantitative analysis of the isotope distribution in these intermediates and products provides a means of determining the relative contribution of Gln and glucose to uracil biosynthesis (92). It is generally found that in proliferating cells, whether transformed or primary cells that are stimulated to proliferate, Gln is the preferred carbon source for pyrimidine ring over glucose (92), but the precise balance is cell-dependent (Lane & Fan, unpublished data), and possibly also condition dependent.
Cells in culture synthesize a large fraction of the aspartate pool that is used for pyrimidine biosynthesis from glutamine and glucose (81,92,101,179,208), even in media such as RPMI that contain aspartate (0.15 mM) at concentrations much higher than that present in blood (ca. 20 μM). Also, in cells treated with 13C5,15N2-Gln, it is found by high resolution FT-ICR-MS that the aspartate pool is enriched mainly in the 13C415N and 13C414N isotopologues, despite the presence of high levels of exogenous aspartate (168). Both labeled Asp species are produced as a consequence of glutaminolysis and transamination. 13C5,15N2 glutamine produces 13C5,15N1 glutamate and then 13C5-αKG via glutaminase and aminotransferase activity. αKG enters into the Krebs cycle to produce 13C4-OAA, which is transaminated to form 13C415N1-Asp or 13C414N1-Asp in the mitochondria (Figure 5). The aspartate-malate shuttle then transfers the labeled Asp to the cytoplasm to supply both carbon (C4–C6) and nitrogen (N1) for pyrimidine biosynthesis. Thus, tracking the 13C and/or 15N fate from labeled precursors to the various intermediates and pyrimidine products enable a complete reconstruction of the feeder and pyrimidine synthesis pathways, including the delineation of different compartmental events.
However, there are few examples of this type of analysis in the literature, further studies are needed to determine whether other significant carbon sources are used by some cells (e.g. threonine in stem cells (209)) or under different growth conditions.
Tracking purine ring biosynthesis
The denovo synthesis of the purine ring is considerably more complex than that of the pyrimidine ring, as the former is built up atom by atom on the phosphorylated ribose unit in the cytoplasm (Figure 3). The carbon sources are CO2, glycine and N10-formyltetrahydrofolate (Figure 4). Overall, glycine contributes up to four of the five carbon atoms in the purine ring, including indirectly via N10-formyl THF. Glycine is a non-essential amino acid that is present in human blood at ca. 0.25 mM (210) (HMDB: url-http://www.hmdb.ca/). It can also be synthesized from glucose via glycolytically produced 3-phosphoglycerate (3-PGA) (Figure 4), via the choline-betaine pathway (211) or from other sources such as threonine in some organisms, though not humans (212). It appears that the glycine precursor for purine biosynthesis is synthesized denovo from serine via glycolysis or mitochondrially via the glycine cleavage system (213) (Figure 4), and there may even be a net efflux of glycine in proliferating cells (214,215). In fact, exogenous serine, or endogenously synthesized serine, but not exogenous glycine is the major source of the one carbon units for purine biosynthesis (216). The nitrogen in purines derives from glycine (N7), glutamine (N3, N9) and Asp (N1) (Figure 3).
Incorporation of ring nitrogen atoms (N3 and N9) from the amido nitrogen of glutamine is readily observed in the free nucleotide pool using 15N-enriched Gln tracer coupled with 2D 1H(15N}-HSQC NMR analysis. The N3 and N9 of AXP and GXP can be detected in crude cell or tissue extracts using the 2-bond scalar coupling of N9 to C8H or N3 to C2H (cf. AXP data in Figure 6). This 15N incorporation pattern is evident in different cell types (19,92,177). In addition, using high resolution FT-ICR-MS, the incorporation of 13C and 15N from [U-13C,15N]-Gln into nucleotide can be simultaneously tracked by utilizing the difference in the effective neutron mass in 15N versus 13C. This capability makes it practical to reconstruct both carbon and nitrogen pathways without the need for separate experiments (168). Supplementary Figure S4 illustrates FT-ICR-MS analysis of the 13C labeling pattern of AMP derived from [U-13C]-glucose (168). Together with the NMR analysis for 13C labeled positions, we can ascertain that 13C5-AMP represents AMP with fully 13C labeled ribose subunit, while 13C6-9-AMP have fully 13C labeled ribose plus 1–4 13C labels in the adenine ring (and see also (92,168,177). These data indicate that PPP, glycolysis, Ser-Gly-N10-formyl THF variably contribute to denovo synthesis of AMP. Thus, NMR and MS are synergistic in defining the source and detailed labeling patterns for nucleotides, from which the feeder and nucleotide synthesis pathways can be reconstructed and their modulation by transcription factors or environmental conditions can be elucidated.
Figure 6.
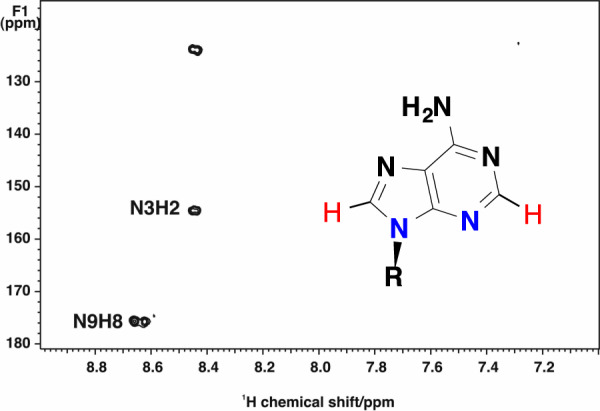
15N incorporation from [U-13C,15N]-glutamine into purines detected by HSQC. A549 cells were grown the presence of [U-13C,15N]-glutamine for 24 h. 1H{15N} HSQC NMR spectra were recorded at 18.8 T using an INEPT delay optimized for two bond couplings in aromatic systems. The two ring 15N atoms derived from the amido N of Gln (blue) are indirectly detected by their coupled protons (red) as cross-peaks of N3 to H2 and N9 to H8 in the adenine ring of AXP. Reproduced with kind permission from Springer Science+Business Media from J. Biomolec NMR (Springer) J. Biomol NMR. 2011 April; 49(3–4): 267–280. doi:10.1007/s10858-011-9484-6 (figure 9).
Recently a tracer approach for following both NAD(P)H production from deuteriated substrates was used to determine the synthesis of glycine in cancer cells and it was shown that the serine/glycine pathway was a net producer of both mitochondrial glycine and NADPH (213,217).
Salvage pathways
RNA in particular is constantly turned over in cells, both during the production of mature RNAs from longer precursors and to regulate the amounts of, e.g. mRNA. The breakdown of polymeric RNA and DNA results in release of NMPs, which can be recycled by the action of the nucleotide kinases. This process contributes to the nucleotide pools but cannot be used for net synthesis. Cells may also transport nucleobases from the external environment and add the appropriate sugar.
Addition of the nucleobase to ribose is achieved either via the PRPP step for purines, or catalyzed by specific pyrimidine phosphorylase to add the base to ribose-1-phosphate (218). Cytidine and deoxycytidine can be salvaged by the agency of cytidine deaminase, producing uridine or deoxy uridine, and then feed into the uracil pathway (219). This in essence randomizes the UTP/CTP pool and must be accounted for where isotope labeling experiments are performed. Cytidine can also be salvaged more directly via uridine-cytidine kinase producing (d)CMP (220). The relative importance of salvage versus denovo synthesis likely depend on the growth conditions and on the specific tissue. Recently it has ben argued that salvage pathways are very important in colorectal cancers for example, albeit without the assistance of isotope tracers (221).
Tracking nucleotide incorporation into nucleic acids.
The free nucleotide pools in cells are straightforward to measure as they are relatively abundant, and are readily labeled using 13C glucose or 13C,15N glutamine (92,168,177,179). It is also practical to extract both DNA and RNA from 13C and/or 15N labeled cells and determine incorporation of exogenously supplied labeled tracers into nucleic acids (via free nucleotides) after hydrolysis to the NMPs (183,185). Although not all of the free nucleotide pool enters nucleic acids, it appears that there is a major common pool of labeled free nucleotides that is used for RNA synthesis (92).
Tracking nucleotide synthesis invivo
The amount of denovo nucleotide biosynthesis correlates well with cell proliferation rates. On the other hand, tissues typically have a low mitotic index, even in tumors (which can be highly heterogeneous and may contain a rather low fraction of cancer cells) (222), so that the amount of denovo nucleotide biosynthesis is small. Nevertheless, using [U-13C]-glucose tracing in SCID mice, 13C incorporation into free nucleotide ribose was observed in some tissues (176), as well as in lung tumors resected from human subjects (175,223). The feeder pathways for nucleotide synthesis haves also been traced invivo in human glioblastomas using [U-13C]-glucose as tracer (194,198). 13C2-glycine was detected by 13C NMR in [U-13C]-glucose labeled glioblastoma tissues, demonstrating synthesis from glucose via the 3PGA-serine pathways (cf. Figure 4). This is despite the presence of 0.25 mM glycine in blood (210). In addition to the free nucleotide pool which is relatively well sampled by good extraction procedures (92,168,224), it is also possible to determine the enrichment in DNA and RNA (12,92,171–172,183,225). With modern instrumentation, the mass isotope ratios are generally good, at around 1% accuracy for medium resolution mass spectrometers, to <0.5% for high-resolution FT-MS instruments. Isotope accuracy in NMR is limited mainly by spectral quality rather than any instrumental issue, as the detectors are fundamentally linear over a dynamic range of ca. 220. The major issues of accuracy and precision are related to experimental design and sample preparation. NMR methods do not require any fragmentation of the nucleotides, whereas GC-MS does. The experimental design must take into consideration multiple inputs to the nucleotide synthesis (see above), as well as turnover rates, which in tissues is complicated by the heterogeneity of cells, the efficiency of extraction and the generation of the nucleotides from the different kinds of polymeric material. Nevertheless, with careful consideration of the kinetics and of compartmentation, adequate models can be built that account for the observations without an excess of parameters, as shown for example for the synthesis of UDP-N-acetylglucosamine (173).
Thus, stable isotope tracers coupled with state-of-the-science NMR and MS analysis for labeling patterns of relevant metabolites provide an unprecedented opportunity for tracking nucleotide biosynthesis invivo, even in human subjects, under different tissue contexts and pathological conditions. These advances are expected to greatly accelerate our understanding in the regulation of human nucleotide metabolism, the perturbation of which is crucial to the pathogenesis of many human diseases, including cancer.
CONCLUSIONS AND FUTURE DIRECTIONS
Nucleotides are used in a wide variety of metabolic function sin all cells, as coenzymes, for regulation, for activating substrates, for anabolic purposes as well as providing the subunits of the nucleic acids. Maintenance of nucleotide levels is therefore fundamental to cellular function. Proliferating cells, whether normal division during embryogenesis or in controlled proliferation in the hematopoietic system and stem cell compartments for example, as well as dysregulated cell division in cancer all need to maintain the supply of nucleotides. Unsurprisingly then the supply of nucleotides is strongly regulated and tied to the cell cycle. This is achieved both at the gene expression level by a variety of transcription factors as well as the substrates level regulation of the large number of enzymes needed to synthesize the nucleotides.
Although the basic nucleotide synthesis pathways are known and their energy demand can be estimated, the specific requirement for nutrient precursors/energy and the regulatory networks for modulating nucleotide biosynthesis in dividing cells remain unclear, particularly in terms of dependence on cell type and pathological conditions. This is in part due to the lack of powerful tools for elucidating the actual paths from nutrient precursors through various feeder pathways to denovo synthesized nucleotides. Furthermore, the amount of RNA synthesis greatly exceeds the amount of polymeric RNA present in a cell, owing to the extensive turnover. Characterization of gene and protein expression in these paths can provide useful clues, but such studies need to be validated by functional pathway analysis.
Stable isotope tracers in conjunction with state-of-the-science metabolomics methodologies is especially well suited for such functional studies not only in model cells and animals but also directly in human subjects. The relatively few stable isotope tracer-based studies reported have already uncovered important new insights into nucleotide biosynthesis, such as preference for endogenously synthesized precursors such as glycine and aspartate over those externally supplied, and how resources are reallocated according to the environmental conditions, especially in pathological conditions such as cancers (‘metabolic reprogramming’ (22,44)). Such information cannot be obtained without tracer methods. These considerations point to the need to be able to assess the nucleotide synthesis according to tissue pathology and nature, for which these new technologies can now be applied with some ease (176,194,197–198,226–227). It is notable that many drugs are targeted at nucleotide synthesis at the level of the availability of nucleotides or act as chain terminators (32–39,228–229).
In the future we expect further systems biochemical advances in systems biochemical approaches in which the detailed energy and anabolic pathways are integrated with the ‘feeder’ pathways and the specific gene expression networks that are linked to the cell cycle and thus nucleotide demand for DNA and RNA synthesis. As these are likely to be cell type and environment specific, the modem high throughput ‘omics approaches, such as SIRM described in this review are well suited to define the biochemical details of normal and pathological cell function, which directly informs the optimum modes for therapeutic intervention. As dysregulations of nucleotide metabolism are commonly involved in human disease pathogenesis, such understanding is expected to have important diagnostic and therapeutic benefits in relevant human diseases including cancer and diabetes.
Supplementary Material
FUNDING
NIH [P01CA163223-01A1, R01ES022191-01, 1R21CA133688-02, 1 U24 DK097215-01A1, in part]; Carmen L. Buck endowment (to A.N.L.); Edith Gardner endowment (T.W.M.F.). The open access publication charge for this paper has been waived by Oxford University Press - NAR Editorial Board members are entitled to one free paper per year in recognition of their work on behalf of the journal.
Conflict of interest statement. None declared.
REFERENCES
- 1.Hangauer M.J., Vaughn I.Q., McManus M.T. Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs. PLoS Genet. 2013;9:e1003569. doi: 10.1371/journal.pgen.1003569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Djebali S., Davis C.A., Merkel A., Dobin A., Lassmann T., Mortazavi A., Tanzer A., Lagarde J., Lin W., Schlesinger F., et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Graur D., Zheng Y.C., Price N., Azevedo R.B.R., Zufall R.A., Elhaik E. On the immortality of television sets: “function” in the human genome according to the evolution-free gospel of ENCODE. Genome Biol. Evol. 2013;5:578–590. doi: 10.1093/gbe/evt028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dunham I., Kundaje A., Aldred S.F., Collins P.J., Davis C., Doyle F., Epstein C.B., Frietze S., Harrow J., Kaul R., et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sigoillot F.D., Berkowski J.A., Sigoillot S.M., Kotsis D.H., Guy H.I. Cell cycle-dependent regulation of pyrimidine biosynthesis. J. Biol. Chem. 2003;278:3403–3409. doi: 10.1074/jbc.M211078200. [DOI] [PubMed] [Google Scholar]
- 6.Quéméneur L., Gerland L.-M., Flacher M., Ffrench M., Revillard J.-P., Genestier L. Differential control of cell cycle, proliferation, and survival of primary T lymphocytes by purine and pyrimidine nucleotides. J. Immunol. 2003;170:4986–4995. doi: 10.4049/jimmunol.170.10.4986. [DOI] [PubMed] [Google Scholar]
- 7.Fridman A., Saha A., Chan A., Casteel D.E., Pilz R.B., Boss G.R. Cell cycle regulation of purine synthesis by phosphoribosyl pyrophosphate and inorganic phosphate. Biochem. J. 2013;454:91–99. doi: 10.1042/BJ20130153. [DOI] [PubMed] [Google Scholar]
- 8.Wahl A.F., Geis A.M., Spain B.H., Wong S.W., Korn D., Wang T.S.F. Gene-expression of human DNA polymerase-alpha during cell-proliferation and the cell-cycle. Mol. Cell. Biol. 1988;8:5016–5025. doi: 10.1128/mcb.8.11.5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang W., Fridman A., Blackledge W., Connelly S., Wilson I.A., Pilz R.B., Boss G.R. The phosphatidylinositol 3-kinase/akt cassette regulates purine nucleotide synthesis. J. Biol. Chem. 2009;284:3521–3528. doi: 10.1074/jbc.M806707200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laliberte J., Yee A., Xiong Y., Mitchell B.S. Effects of guanine nucleotide depletion on cell cycle progression in human T lymphocytes. Blood. 1998;91:2896–2904. [PubMed] [Google Scholar]
- 11.Evans D.R., Guy H.I. Mammalian pyrimidine biosynthesis: fresh insights into an ancient pathway. J. Biol. Chem. 2004;279:33035–33038. doi: 10.1074/jbc.R400007200. [DOI] [PubMed] [Google Scholar]
- 12.Boren J., Cascante M., Marin S., Comin-Anduix B., Centelles J.J., Lim S., Bassilian S., Ahmed S., Lee W.N.P., Boros L.G. Gleevec (ST1571) influences metabolic enzyme activities and glucose carbon flow toward nucleic acid and fatty acid synthesis in myeloid tumor cells. J. Biol. Chem. 2001;276:37747–37753. doi: 10.1074/jbc.M105796200. [DOI] [PubMed] [Google Scholar]
- 13.Kondo M., Yamaoka T., Honda S., Miwa Y., Katashima R., Moritani M., Yoshimoto K., Hayashi Y., Itakura M. The rate of cell growth is regulated by purine biosynthesis via ATP production and G(1) to S phase transition. J. Biochem. 2000;128:57–64. doi: 10.1093/oxfordjournals.jbchem.a022730. [DOI] [PubMed] [Google Scholar]
- 14.Macallan D.C., Fullerton C.A., Neese R.A., Haddock K., Park S.S., Hellerstein M.K. Measurement of cell proliferation by labeling of DNA with stable isotope-labeled glucose: studies in vitro, in animals, and in humans. Proc. Natl. Acad. Sci. U.S.A. 1998;95:708–713. doi: 10.1073/pnas.95.2.708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Y.-C., Li F., Handler J., Huang C.R.L., Xiang Y., Neretti N., Sedivy J.M., Zeller K.I., Dang C.V. Global regulation of nucleotide biosynthetic genes by c-Myc. PLoS One. 2008;3:e2722. doi: 10.1371/journal.pone.0002722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang H., Mannava S., Grachtchouk V., Zhuang D., Soengas M.S., Gudkov A.V., Prochownik E.V., Nikiforov M.A. c-Myc depletion inhibits proliferation of human tumor cells at various stages of the cell cycle. Oncogene. 2008;27:1905–1915. doi: 10.1038/sj.onc.1210823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zeller K., Zhao X., Lee C., Chiu KP, Yao F., Yustein J.T., Ooi H.S., Orlov Y.L., Shahab A., Yong H.C., et al. Global mapping of c-Myc binding sites and target gene networks in human B cells. Proc. Natl. Acad. Sci. U.S.A. 2006;103:7834–17839. doi: 10.1073/pnas.0604129103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu S., Balakrishnan A., Bok R.A., Anderton B., Larson P.E.Z., Nelson S.J., Kurhanewicz J., Vigneron D.B., Goga A. (13)C-pyruvate imaging reveals alterations in glycolysis that precede c-Myc-induced tumor formation and regression. Cell Metab. 2011;14:131–142. doi: 10.1016/j.cmet.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuneva M.O., Fan T.W.-M., Allen T.D., Higashi R.M., Ferraris D.V., Tsukamoto T., Matés J.M., Alonso F.J., Wang C., Seo Y., et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 2012;15:157–170. doi: 10.1016/j.cmet.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nie Z., Hu G., Wei G., Cui K., Yamane A., Resch W., Wang R., Green D.R., Tessarollo L., Casellas R., et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell. 2012;151:68–79. doi: 10.1016/j.cell.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin C.Y., Loven J., Rahl P.B., Paranal R.M., Burge C.B., Bradner J.E., Lee T.I., Young R.A. Transcriptional amplification in tumor cells with elevated c-Myc. Cell. 2012;151:56–67. doi: 10.1016/j.cell.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tong X.M., Zhao F.P., Thompson C.B. The molecular determinants of de novo nucleotide biosynthesis in cancer cells. Curr. Opin. Genet. Dev. 2009;19:32–37. doi: 10.1016/j.gde.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bester A.C., Roniger M., Oren Y.S., Im M.M., Sarni D., Chaoat M., Bensimon A., Zamir G., Shewach D.S., Kerem B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell. 2011;145:435–446. doi: 10.1016/j.cell.2011.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mannava S., Grachtchouk V., Wheeler L.J., Im M., Zhuang D.Z., Slavina E.G., Mathews C.K., Shewach D.S., Nikiforov M.A. Direct role of nucleotide metabolism in C-MYC-dependent proliferation of melanoma cells. Cell Cycle. 2008;7:2392–2400. doi: 10.4161/cc.6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Le A., Lane A.N., Hamaker M., Bose S., Barbi J., Tsukamoto T., Rojas C.J., Slusher B.S., Zhang H., Zimmerman L.J., et al. Myc induction of hypoxic glutamine metabolism and a glucose-independent TCA cycle in human B lymphocytes. Cell Metab. 2012;15:110–121. doi: 10.1016/j.cmet.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dang C.V., Le A., Gao P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin. Cancer Res. 2009;15:6479–6483. doi: 10.1158/1078-0432.CCR-09-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cunningham J.T., Moreno M.V., Lodi A., Ronen S.M., Ruggero D. Protein and nucleotide biosynthesis are coupled by a single rate-limiting enzyme, PRPS2, to drive cancer. Cell. 2014;157:1088–1103. doi: 10.1016/j.cell.2014.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim J., Lee J.-H., Iyer V.R. Global identification of Myc target genes reveals its direct role in mitochondrial biogenesis and its E-box usage in vivo. PLoS One. 2008;3:e1798. doi: 10.1371/journal.pone.0001798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao P., Tchernyshyov I., Chang T.C., Lee Y.S., Kita K., Ochi T., Zeller K.I., De Marzo A.M., Van Eyk J.E., Mendell J.T., et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762–765. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O'Donnell K.A., Wentzel E.A., Zeller K.I., Dang C.V., Mendell J.T. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 31.Han H., Sun D., Li W., Shen H., Zhu Y., Li C., Chen Y., Lu L., Li W., Zhang J., et al. A c-Myc-microRNA functional feedback loop affects hepatocarcinogenesis. Hepatology. 2013;57:2378–2389. doi: 10.1002/hep.26302. [DOI] [PubMed] [Google Scholar]
- 32.Johnston A., Gudjonsson J.E., Sigmundsdottir H., Ludviksson B., Valdimarsson H. The anti-inflammatory action of methotrexate is not mediated by lymphocyte apoptosis, but by the suppression of activation and adhesion molecules. Clin. Immunol. 2005;114:154–163. doi: 10.1016/j.clim.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 33.Goodsell D.S. The molecular perspective: methotrexate. Oncologist. 1999;4:340–341. [PubMed] [Google Scholar]
- 34.Plunkett W., Huang P., Xu Y., Heinemann V., Grunewald R., Gandhi V. Gemcitabine: metabolism, mechanisms of action, and self-potentiation. Semin. Oncol. 1995;22:3–10. [PubMed] [Google Scholar]
- 35.Evans W. Pharmacogenetics of thiopurine S-methyltransferase and thiopurine therapy. Ther. Drug Monit. 2004;26:186–191. doi: 10.1097/00007691-200404000-00018. [DOI] [PubMed] [Google Scholar]
- 36.Longley D., Harkin D., Johnston P. 5-fluorouracil: mechanisms of action and clinical strategies. Nat. Rev. Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 37.Jordheim L.P., Durantel D., Zoulim F., Charles Dumontet C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 2013;12:447–464. doi: 10.1038/nrd4010. [DOI] [PubMed] [Google Scholar]
- 38.Li F., Maag H., Alfredson T. Prodrugs of nucleoside analogues for improved oral absorption and tissue targeting. J. Pharm. Sci. 2008;97:1109–1134. doi: 10.1002/jps.21047. [DOI] [PubMed] [Google Scholar]
- 39.Schroeder R., Waldsich C., Wank H. Modulation of RNA function by aminoglycoside antibiotics. EMBO J. 2000;19:1–9. doi: 10.1093/emboj/19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shaw R.J., Cantley L.C. Decoding key nodes in the metabolism of cancer cells: sugar & spice and all things nice. F1000 Biol. Rep. 2012;4:2. doi: 10.3410/B4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cairns R.A., Harris I.S., Mak T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 42.Koppenol W.H., Bounds P.L., Dang C.V. Otto Warburg's contributions to current concepts of cancer metabolism. Nat. Rev. Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- 43.Fan T.W.-M., Lorkiewicz P., Sellers K., Moseley H.N.B., Higashi R.M., Lane A.N. Stable isotope-resolved metabolomics and applications to drug development. Pharmacol. Ther. 2012;133:366–391. doi: 10.1016/j.pharmthera.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.DeBerardinis R.J., Lum J.J., Hatzivassiliou G., Thompson C.B. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 45.DeBerardinis R.J., Cheng T. Q's next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010;29:313–324. doi: 10.1038/onc.2009.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moreno-Sanchez R., Rodriguez-Enriquez S., Marin-Hernandez A., Saavedra E. Energy metabolism in tumor cells. FEBS J. 2007;274:1393–1418. doi: 10.1111/j.1742-4658.2007.05686.x. [DOI] [PubMed] [Google Scholar]
- 47.Vander Heiden M.G., Cantley L.C., Thompson C.B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gillies R.J., Gatenby R.A. Adaptive landscapes and emergent phenotypes: why do cancers have high glycolysis. J. Bioenerg. Biomembr. 2007;39:251–257. doi: 10.1007/s10863-007-9085-y. [DOI] [PubMed] [Google Scholar]
- 49.Morrish F., Neretti N., Sedivy J.M., Hockenbery D.M. The oncogene c-Myc coordinates regulation of metabolic networks to enable rapid cell cycle entry. Cell Cycle. 2008;7:1054–1066. doi: 10.4161/cc.7.8.5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cantor J.R., Sabatini D.M. Cancer cell metabolism: one hallmark, many faces. Cancer Discov. 2012;2:1–18. doi: 10.1158/2159-8290.CD-12-0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dolfi S.C., Chan L.L.-Y., Qiu J., Tedeschi P.M., Bertino J.R., Hirshfield K.M., Oltvai Z.N., Vazquez A. The metabolic demands of cancer cells are coupled to their size and protein synthesis rates. Cancer Metab. 2013;1:20. doi: 10.1186/2049-3002-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lunt S.Y., Vander Heiden M.G. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Ann. Rev. Cell Dev. Biol. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- 53.Schmid D., Burmester G.R., Tripmacher R., Kuhnke A., Buttgereit F. Bioenergetics of human peripheral blood mononuclear cell metabolism in quiescent, activated, and glucocorticoid-treated states. Biosci. Rep. 2000;20:289–302. doi: 10.1023/a:1026445108136. [DOI] [PubMed] [Google Scholar]
- 54.Roberts J.K.M., Lane A.N., Clark R.A., Nieman R.H. Relationships between the rate of synthesis of ATP and the concentrations of reactants and products of ATP hydrolysis in maize root-tips, determined by P-31 nuclear magnetic-resonance. Arch. Biochem. Biophys. 1985;240:712–722. doi: 10.1016/0003-9861(85)90080-3. [DOI] [PubMed] [Google Scholar]
- 55.Slater E.C., Rosing J., Mol A. The phosphorylation potential generated by respiring mitochondria. Biochim. Biophys. Acta. 1973;292:534–553. doi: 10.1016/0005-2728(73)90003-0. [DOI] [PubMed] [Google Scholar]
- 56.Brahimi-Horn M.C., Pouyssegur J. Oxygen Sensing and Hypoxia-Induced Responses. Vol. 43. London: Biochemical Soceity; 2007. pp. 165–178. [Google Scholar]
- 57.Buttgereit F., Brand M.D. A hierarchy of ATP-consuming processes in mammalian-cells. Biochem. J. 1995;312:163–167. doi: 10.1042/bj3120163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schmidt H., Siems W., Muller M., Dumdey R., Rapoport S.M. ATP-producing and consuming processes of Ehrlich mouse ascites tumor cells in proliferating and resting phases. Exp. Cell Res. 1991;194:122–127. doi: 10.1016/0014-4827(91)90140-p. [DOI] [PubMed] [Google Scholar]
- 59.Prescher J.A., Bertozzi C.R. Chemistry in living systems. Nat. Chem. Biol. 2005;1:13–21. doi: 10.1038/nchembio0605-13. [DOI] [PubMed] [Google Scholar]
- 60.Mourant J.R., Yamada Y.R., Carpenter S., Dominique L.R., Freyer J.P. FTIR spectroscopy demonstrates biochemical differences in mammalian cell cultures at different growth stages. Biophys. J. 2003;85:1938–1947. doi: 10.1016/S0006-3495(03)74621-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Greilhuber J., Dolezel J., Lysák M.A., Bennett M.D. The origin, evolution and proposed stabilization of the terms ‘genome size’ and ‘C-value’ to describe nuclear DNA contents. Ann. Bot. 2005;95:255–260. doi: 10.1093/aob/mci019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sharma S., Cabana R., Shariatmadar S., Krishan A. Cellular volume and marker expression in human peripheral blood apheresis stem cells. Cytometry A. 2008;73:160–167. doi: 10.1002/cyto.a.20524. [DOI] [PubMed] [Google Scholar]
- 63.David H. The hepatocyte. Development, differentiation, and ageing. Exp. Pathol. Suppl. 1985;11:1–148. [PubMed] [Google Scholar]
- 64.Bonifacio G.F., Brown T., Conn G.L., Lane A.N. Comparison of the electrophoretic and hydrodynamic properties of DNA and RNA oligonucleotide duplexes. Biophys. J. 1997;73:1532–1538. doi: 10.1016/S0006-3495(97)78185-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kratky O., Leopold H., Stabinger H. The determination of the partial specific volume of proteins by the mechanical oscillator technique. Methods Enzymol. 1973;27:98–110. doi: 10.1016/s0076-6879(73)27007-6. [DOI] [PubMed] [Google Scholar]
- 66.Zucker R.M., Cassen B. The separation of normal human leukocytes by density and classification by size. Blood. 1969;34:591–600. [PubMed] [Google Scholar]
- 67.Durchschlag H. Determination of the partial specific volume of conjugated proteins. Colloid Polymer Sci. 1989;267:1139–1150. [Google Scholar]
- 68.Alberts B., Bray D., Lewis J., Raff M., Roberts K., Watson J. Molecular Biology of the Cell. 3rd edn. NY: Garland Publishing; 1994. [Google Scholar]
- 69.Sakharkar K.K., Chow V.T.K., Kangueane P. Distributions of exons and introns in the human genome. In Silico Biol. 2004;4:387–393. [PubMed] [Google Scholar]
- 70.Nazar R.N. Ribosomal RNA processing and ribosome biogenesis in eukaryotes. IUBMB Life. 2004;56:457–465. doi: 10.1080/15216540400010867. [DOI] [PubMed] [Google Scholar]
- 71.Gadian D.G. NMR and its applications to living systems. 2nd edn. Oxford: Oxford University Press; 1995. [Google Scholar]
- 72.Manfredi G., Yang L., Gajewski C.D., Mattiazzi M. Measurements of ATP in mammalian cells. Methods. 2002;26:317–326. doi: 10.1016/S1046-2023(02)00037-3. [DOI] [PubMed] [Google Scholar]
- 73.Eguchi Y., Shimizu S., Tsujimoto Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997;57:1835–1840. [PubMed] [Google Scholar]
- 74.Zong W.X., Thompson C.B. Necrotic death as a cell fate. Genes Dev. 2006;20:1–15. doi: 10.1101/gad.1376506. [DOI] [PubMed] [Google Scholar]
- 75.MacIntosh B.R., Holash R.J., Renaud J.M. Skeletal muscle fatigue - regulation of excitation-contraction coupling to avoid metabolic catastrophe. J. Cell Sci. 2012;125:2105–2114. doi: 10.1242/jcs.093674. [DOI] [PubMed] [Google Scholar]
- 76.Tatsumia T., Shiraishia J., Keiraa N., Akashia K., Manoa A., Yamanakaa S., Matobaa S., Fushikib S., Flissc H., Nakagawaa M. Intracellular ATP is required for mitochondrial apoptotic pathways in isolated hypoxic rat cardiac myocytes. Cardiovasc. Res. 2003;59:428–440. doi: 10.1016/s0008-6363(03)00391-2. [DOI] [PubMed] [Google Scholar]
- 77.Plagemann P.G.W. Nucleotide pools in novikoff rat hepatoma cells growing in suspension culture. J. Cell Biol. 1972;52:131–146. doi: 10.1083/jcb.52.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Traut T.W. Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 1994;140:1–22. doi: 10.1007/BF00928361. [DOI] [PubMed] [Google Scholar]
- 79.Dolle C., Skoge R.H., VanLinden M.R., Ziegler M. NAD biosynthesis in humans—enzymes, metabolites and therapeutic aspects. Curr. Top. Med. Chem. 2013;13:2907–2917. doi: 10.2174/15680266113136660206. [DOI] [PubMed] [Google Scholar]
- 80.Tallis M., Morra R., Barkauskaite E., Ahel I. Poly(ADP-ribosyl)ation in regulation of chromatin structure and the DNA damage response. Chromosoma. 2014;123:79–90. doi: 10.1007/s00412-013-0442-9. [DOI] [PubMed] [Google Scholar]
- 81.Reynolds M.R., Lane A.N., Kemp S., Liu Y., Hill B., Dean D.C., Clem B.F. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene. 2014;33:556–566. doi: 10.1038/onc.2012.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lin W.Y., Arthur G. Phospholipids are synthesized in the G2/M phase of the cell cycle. Int. J. Biochem. Cell Biol. 2007;39:597–605. doi: 10.1016/j.biocel.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 83.Hordern J., Henderson J. Comparison of purine and pyrimidine metabolism in G1 and S phases of HeLa and Chinese hamster ovary cells. Can. J. Biochem. 1982;60:422–433. doi: 10.1139/o82-050. [DOI] [PubMed] [Google Scholar]
- 84.Yalcin A., Clem B.F., Simmons A., Lane A., Nelson K., Clem A.L., Brock E., Siow D., Wattenberg B., Telang S., et al. Nuclear targeting of 6-Phosphofructo-2-kinase (PFKFB3) increases proliferation via cyclin-dependent kinases. J. Biol. Chem. 2009;284:24223–24232. doi: 10.1074/jbc.M109.016816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bao Y., Mukai K., Hishiki T., Kubo A., Ohmura M., Sugiura Y., Matsuura T., Nagahata Y., Hayakawa N., Yamamoto T., et al. Energy management by enhanced glycolysis in G(1)-phase in human colon cancer cells in vitro and in vivo. Mol. Cancer Res. 2013;11:973–985. doi: 10.1158/1541-7786.MCR-12-0669-T. [DOI] [PubMed] [Google Scholar]
- 86.Moncada S., Higgs E.A., Colombo S.L. Fulfilling the metabolic requirements for cell proliferation. Biochem. J. 2012;446:1–7. doi: 10.1042/BJ20120427. [DOI] [PubMed] [Google Scholar]
- 87.Colombo S.L., Palacios-Callender M., Frakich N., Carcamo S., Kovacs I., Tudzarova S., Moncada S. Molecular basis for the differential use of glucose and glutamine in cell proliferation as revealed by synchronized HeLa cells. Proc. Natl. Acad. Sci. U.S.A. 2011;108:21069–21074. doi: 10.1073/pnas.1117500108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Young J.D., Yao S.Y.M., Baldwin J.M., Cass C.E., Baldwin S.A. The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Mol. Aspects Med. 2013;34:529–547. doi: 10.1016/j.mam.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 89.Ipata P.L., Balestri F. The functional logic of cytosolic 5 ‘-nucleotidases. Curr. Med. Chem. 2013;20:4205–4216. doi: 10.2174/0929867311320340002. [DOI] [PubMed] [Google Scholar]
- 90.Austin W.R., Armijo A.L., Campbell D.O., Singh A.S., Hsieh T., Nathanson D., Herschman H.R., Phelps M.E., Witte O.N., Czernin J., et al. Nucleoside salvage pathway kinases regulate hematopoiesis by linking nucleotide metabolism with replication stress. J. Exp. Med. 2012;209:2215–2228. doi: 10.1084/jem.20121061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhao H., French J., Fang Y., Benkovic S.J. The purinosome, a multi-protein complex involved in the de novo biosynthesis of purines in humans. Chem. Commun. 2013;49:4444–4452. doi: 10.1039/c3cc41437j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fan T.W.-M., Tan J.L., McKinney M.M., Lane A.N. Stable isotope resolved metabolomics analysis of ribonucleotide and RNA metabolism in human lung cancer cells. Metabolomics. 2012;8:517–527. doi: 10.1007/s11306-011-0337-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vazquez A., Tedeschi P.M., Bertino J.R. Overexpression of the mitochondrial folate and glycine-serine pathway: a new determinant of methotrexate selectivity in tumors. Cancer Res. 2013;73:478–482. doi: 10.1158/0008-5472.CAN-12-3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tedeschi P.M., Markert E.K., Gounder M., Lin H., Dvorzhinski D., Dolfi S.C., Chan L.Y., Qiu J., DiPaola R.S., Hirshfield K.M., et al. Contribution of serine, folate and glycine metabolism to the ATP, NADPH and purine requirements of cancer cells. Cell Death Dis. 2013;4:e877. doi: 10.1038/cddis.2013.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Possemato R., Marks K.M., Shaul Y.D., Pacold M.E., Kim D., Birsoy K., Sethumadhavan S., Woo H.-K., Jang H.G., Jha A.K., et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346–350. doi: 10.1038/nature10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Morrish F., Isern N., Sadilek M., Jeffrey M., Hockenbery D.M. c-Myc activates multiple metabolic networks to generate substrates for cell-cycle entry. Oncogene. 2009;28:2485–2491. doi: 10.1038/onc.2009.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cory J.G., Sato A. Regulation of ribonucleotide reductase activity in mammalian cells. Mol. Cell. Biochem. 1983;53:257–266. doi: 10.1007/BF00225258. [DOI] [PubMed] [Google Scholar]
- 98.Frederiks W.M., Vizan P., Bosch K.S., Vreeling-Sindelarova H., Boren J., Cascante M. Elevated activity of the oxidative and non-oxidative pentose phosphate pathway in (pre)neoplastic lesions in rat liver. Int. J. Exp. Pathol. 2008;89:232–240. doi: 10.1111/j.1365-2613.2008.00582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Corey J.G., Sato A. Regulation of ribonucleotide reductase activity in mammalian cells. Mol. Cell. Biochem. 1983;53:257–266. doi: 10.1007/BF00225258. [DOI] [PubMed] [Google Scholar]
- 100.Ferraro P., Franzolin E., Pontarin G., Reichard P., Bianchi V. Quantitation of cellular deoxynucleoside triphosphates. Nucleic Acids Res. 2009;38:e85. doi: 10.1093/nar/gkp1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yang Y., Lane A.N., Ricketts C.J., Carole Sourbier C., Wei M.-H., Shuch B., Pike L., Wu M., Rouault T.A., Boros L.G., et al. Metabolic reprogramming for producing energy and reducing power in fhydratase ncells from hereditary leiomyomatosis renal cell carcinoma. PLos One. 2013;8:e72179. doi: 10.1371/journal.pone.0072179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mullen A.R., Wheaton W.W., Jin E.S., Chen P.-H., Sullivan L.B., Cheng T., Yang Y., Linehan W.M., Chandel N.S., DeBerardinis R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2011;481:385–388. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wise D.R., Ward P.S., Shay J.E.S., Cross J.R., Gruber J.J., Sachdev U.M., Platt J.M., DeMatteo R.G., Simon M.C., Thompson C.B. Hypoxia promotes isocitrate dehydrogenasedependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. PNAS. 2011;108:19611–19616. doi: 10.1073/pnas.1117773108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kamphorst J.J., Cross J.R., Fan J., de Stanchina E., Mathew R., White E.P., Thompson C.B., Rabinowitz J.D. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl. Acad. Sci. U.S.A. 2013;110:8882–8887. doi: 10.1073/pnas.1307237110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.DeBerardinis R.J., Sayed N., Ditsworth D., Thompson C.B. Brick by brick: metabolism and tumor cell growth. Curr. Opin. Genet. Dev. 2008;18:54–61. doi: 10.1016/j.gde.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Frezza C., Zheng L., Folger O., Rajagopalan K.N., MacKenzie E.D., Jerby L., Micaroni M., Chaneton B., Adam J., Hedley A., et al. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature. 2011;477:225–228. doi: 10.1038/nature10363. [DOI] [PubMed] [Google Scholar]
- 107.Tennant D.A., Duran R.V., Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat. Rev. Cancer. 2010;10:267–277. doi: 10.1038/nrc2817. [DOI] [PubMed] [Google Scholar]
- 108.Cardenas-Navia L.I., Mace D., Richardson R.A., Wilson D.F., Shan S., Dewhirst M.W. The pervasive presence of fluctuating oxygenation in tumors. Cancer Res. 2008;68:5812–5819. doi: 10.1158/0008-5472.CAN-07-6387. [DOI] [PubMed] [Google Scholar]
- 109.Schroeder T., Yuan H., Viglianti B.L., Peltz C., Asopa S., Vujaskovic Z., Dewhirst M.W. Spatial heterogeneity and oxygen dependence of glucose consumption in R3230Ac and fibrosarcomas of the Fischer 344 rat. Cancer Res. 2005;65:5163–5171. doi: 10.1158/0008-5472.CAN-04-3900. [DOI] [PubMed] [Google Scholar]
- 110.Chandel N.S., Budinger G.R., Schumacker P.T. Molecular oxygen modulates cytochrome c oxidase function. J. Biol. Chem. 1996;271:18672–18677. doi: 10.1074/jbc.271.31.18672. [DOI] [PubMed] [Google Scholar]
- 111.Kraba K., Kempe H., Wikström M. Explaining the enigmatic KM for oxygen in cytochrome c oxidase: a kinetic model. Biochim. Biophys. Acta. 2011;1807:348–358. doi: 10.1016/j.bbabio.2010.12.015. [DOI] [PubMed] [Google Scholar]
- 112.Kim J.W., Tchernyshyov I., Semenza G.L., Dang C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 113.Semenza G.L. Regulation of mammalian O-2 homeostasis by hypoxia- inducible factor 1. Annu. Rev. Cell Dev. Biol. 1999;15:551–578. doi: 10.1146/annurev.cellbio.15.1.551. [DOI] [PubMed] [Google Scholar]
- 114.Myllyla R., Tuderman L., Kivirikko K.I. Mechanism of the prolyl hydroxylase reaction. Eur. J. Biochem. 1977;80:349–357. doi: 10.1111/j.1432-1033.1977.tb11889.x. [DOI] [PubMed] [Google Scholar]
- 115.Jokilehto T., Jaakkola P.M. The role of HIF prolyl hydroxylases in tumour growth. J. Cell. Mol. Med. 2010;14:758–770. doi: 10.1111/j.1582-4934.2010.01030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bos R., van der Groep P., Greijer A.E., Shvarts A., Meijer S., Pinedo H.M., Semenza G.L., van Diest P.J., van der Wall E. Levels of hypoxia-inducible factor-1 alpha independently predict prognosis in patients with lymph node negative breast carcinoma. Cancer. 2003;97:1573–1581. doi: 10.1002/cncr.11246. [DOI] [PubMed] [Google Scholar]
- 117.Strovas T.J., McQuaide S.C., Anderson J.B., Nandakumar V., Kalyuzhnaya M.G., Burgess L.W., Holl M.R., Meldrum D.R., Lidstrom M.E. Direct measurement of oxygen consumption rates from attached and unattached cells in a reversibly sealed, diffusionally isolated sample chamber. Adv. Biosci. Biotechnol. 2010;1:398–408. doi: 10.4236/abb.2010.15053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Telang S., Lane A.N., Nelson K.K., Arumugam S., Chesney J.A. The oncoprotein H-RasV12 increases mitochondrial metabolism. Mol. Cancer. 2007;6:77. doi: 10.1186/1476-4598-6-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yang Y., Lane A.N., Fan T.W.-M., Rickets C., Wu M., Boros L., Linehan W.M. Understanding how fumarate hydratase (FH) null cells use its central carbon for energy and malignant development. PLoS One. 2013;8:e72179. doi: 10.1371/journal.pone.0072179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wu M., Neilson A., Swift A.L., Moran R., Tamagnine J., Parslow D., Armistead S., Lemire K., Orrell J., Teich J., et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am. J. Physiol. Cell Physiol. 2007;292:C125–C136. doi: 10.1152/ajpcell.00247.2006. [DOI] [PubMed] [Google Scholar]
- 121.Marín-Hernández A., Gallardo-Pérez J.C., Rodríguez-Enríquez S., Encalada R., Moreno-Sánchez R., Saavedra E. Modeling cancer glycolysis. Biochim. Biophys. Acta. 2010;1807:755–767. doi: 10.1016/j.bbabio.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 122.Warburg O. Versuche an überlebendem Carcinomgewebe (Methoden) Biochem. Zeitschr. 1923;142:317–333. [Google Scholar]
- 123.Guppy M., Leedman P., Zu X., Russell V. Contribution by different fuels and metabolic pathways to the total ATP turnover of proliferating MCF-7 breast cancer cells. Biochem. J. 2002;364:309–315. doi: 10.1042/bj3640309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Liu W., Le A., Lane A.N., Fan T.W.-M., Dang C.V., Phang J.M. The reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses to c-MYC. Proc. Natl. Acad. Sci. U.S.A. 2012;109:8983–8988. doi: 10.1073/pnas.1203244109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.DeBerardinis R.J., Mancuso A., Daikhin E., Nissim I., Yudkoff M., Wehrli S., Thompson C.B. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. U.S.A. 2007;104:19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Brennan L., Corless M., Hewage C., Malthouse J.P.G., McClenaghan N.H., Flatt P.R., Newsholme P. C-13 NMR analysis reveals a link between L-glutamine metabolism, D-glucose metabolism and gamma-glutamyl cycle activity in a clonal pancreatic beta-cell line. Diabetologia. 2003;46:1512–1521. doi: 10.1007/s00125-003-1184-7. [DOI] [PubMed] [Google Scholar]
- 127.Newsholme E.A., Crabtree B., Ardawi M.S.M. Glutamine metabolism in lymphocytes: its biochemical, physiological and clinical importance. Q. J. Exp. Physiol. 1985;70:473–489. doi: 10.1113/expphysiol.1985.sp002935. [DOI] [PubMed] [Google Scholar]
- 128.Gullino P.M., Clark S.H., Grantham F.H. The interstial fluid of solid tumors. Cancer Res. 1964;24:780–797. [PubMed] [Google Scholar]
- 129.Fell D. Understanding the Control of Metabolism. London: Portland Press; 1997. [Google Scholar]
- 130.Papin J.A., Price N.D., Wiback S.J., Fell D.A., Palsson B.O. Metabolic pathways in the post-genome era. Trends Biochem. Sci. 2003;28:250–258. doi: 10.1016/S0968-0004(03)00064-1. [DOI] [PubMed] [Google Scholar]
- 131.Poulo J.M., Elston T., Lane A.N., Macdonald J.M., Cascante M. Introduction to Metabolic Control Analysis. In: Fan TW-M, Higashi RM, Lane AN, editors. Handbook of Metabolomics. NY: Humana Press; 2012. [Google Scholar]
- 132.Kohen E., Kohen C., Hirschberg J.G., Wouters A.W., Thorell B., Westerhoff H.V., Charyulu K.K.N. Metabolic control and compartmentation in single living cells. Cell Biochem. Funct. 1983;1:3–16. doi: 10.1002/cbf.290010103. [DOI] [PubMed] [Google Scholar]
- 133.Thomas S., Fell D.A. The role of multiple enzyme activation in metabolic flux control. Adv. Enzyme Reg. 1998;38:65–85. doi: 10.1016/s0065-2571(97)00012-5. [DOI] [PubMed] [Google Scholar]
- 134.Kacser H., Burns J. The control of flux. Symp. Soc. Exp. Biol. 1973;27:65–104. [PubMed] [Google Scholar]
- 135.Marin-Hernandez A., Rodriguez-Enriquez S., Vital-Gonzalez P.A., Flores-Rodriguez F.L., Macias-Silva M., Sosa-Garrocho M., Moreno-Sanchez R. Determining and understanding the control of glycolysis in fast-growth tumor cells—flux control by an over-expressed but strongly product-inhibited hexokinase. FEBS J. 2006;273:1975–1988. doi: 10.1111/j.1742-4658.2006.05214.x. [DOI] [PubMed] [Google Scholar]
- 136.Baumgartner R., Walloschek M., Kralik M., Gotschlich A., Tasler S., Mies J., Leban J. Dual binding mode of a novel series of DHODH inhibitors. J. Med. Chem. 2006;49:1239–1247. doi: 10.1021/jm0506975. [DOI] [PubMed] [Google Scholar]
- 137.Abdelrahim M., Samudio I., Smith R., Burghardt R., Safe S. Small inhibitory RNA duplexes for Sp1 mRNA block basal and estrogen-induced gene expression and cell cycle progression in MCF-7 breast cancer cells. J. Biol. Chem. 2002;277:28815–28822. doi: 10.1074/jbc.M203828200. [DOI] [PubMed] [Google Scholar]
- 138.Sigoillot F.D., Sigoillot S.M., Guy H.I. Breakdown of the regulatory control of pyrimidine biosynthesis in human breast cancer cells. Int. J. Cancer. 2004;109:491–498. doi: 10.1002/ijc.11717. [DOI] [PubMed] [Google Scholar]
- 139.Graves L.M., Guy H.I., Kozlowski P., Huang M., Lazarowski E., Pope R.M., Collins M.A., Dahlstrand E.N., Earp H.S., Evans D.R. Regulation of carbamoyl phosphate synthetase by MAP kinase. Nature. 2000;403:328–332. doi: 10.1038/35002111. [DOI] [PubMed] [Google Scholar]
- 140.Gordan J.D., Lal P., Dondeti V.R., Letrero R., Parekh K.N., Oquendo C.E., Greenberg R.A., Flaherty K.T., Rathmell W.K., Keith B., et al. HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell. 2008;14:435–446. doi: 10.1016/j.ccr.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chen K.F., Lai Y.Y., Sun H.S., Tsai S.J. Transcriptional repression of human cad gene by hypoxia inducible factor-1 alpha. Nucleic Acids Res. 2005;33:5190–5198. doi: 10.1093/nar/gki839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gordan J.D., Bertout J.A., Hu C.J., Diehl J.A., Simon M.C. HIF-2 alpha promotes hypoxic cell proliferation by enhancing c-Myc transcriptional activity. Cancer Cell. 2007;11:335–347. doi: 10.1016/j.ccr.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Khan S., Abdelrahim M., Samudio I., Safe S. Estrogen receptor/Sp1 complexes are required for induction of cad gene expression by 17 beta-estradiol in breast cancer cells. Endocrinology. 2003;144:2325–2335. doi: 10.1210/en.2002-0149. [DOI] [PubMed] [Google Scholar]
- 144.Wyngaarden J.B. Regulation of purine biosynthesis and turnover. Adv. Enzyme Regul. 1976;14:25–42. doi: 10.1016/0065-2571(76)90006-6. [DOI] [PubMed] [Google Scholar]
- 145.Switzer R.L., Sogin D.C. Regulation and mechanism of phosphoribosylpyrophosphate synthetase V.Inhibition by end products and regulation by adenosine diphosphate. J. Biol. Chem. 1973;248:1063–1073. [PubMed] [Google Scholar]
- 146.Smith J.L. Glutamine PRPP amidotransferase: snapshots of an enzyme in action. Curr. Opin. Struct. Biol. 1998;8:686–694. doi: 10.1016/s0959-440x(98)80087-0. [DOI] [PubMed] [Google Scholar]
- 147.Bera A.K., Smith J.L., Zalkin H. Dual role for the glutamine phosphoribosylpyrophosphate amidotransferase ammonia channel. Interdomain signaling and intermediate channeling. J. Biol. Chem. 2000;275:7975–7979. doi: 10.1074/jbc.275.11.7975. [DOI] [PubMed] [Google Scholar]
- 148.Holmes E.W., Pehlke D.M., Kelley W.N. Human IMP dehydrogenase: kinetics and regulatory properties. Biochim. Biophys. Acta. 1974;364:209–217. doi: 10.1016/0005-2744(74)90006-0. [DOI] [PubMed] [Google Scholar]
- 149.Hedstrom L. IMP dehydrogenase: structure, mechanism and inhibition. Chem. Rev. 2009;109:2903–2928. doi: 10.1021/cr900021w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Van Der Weyden M.B., Kelly W.N. Human adenylosuccinate synthetase. Partial purification, kinetic and regulatory properties of the enzyme from placenta. J. Biol. Chem. 1974;249:7782–7289. [PubMed] [Google Scholar]
- 151.Anousis N., Carvalho R.A., Zhao P.Y., Malloy C.R., Sherry A.D. Compartmentation of glycolysis and glycogenolysis in the perfused rat heart. NMR Biomed. 2004;17:51–59. doi: 10.1002/nbm.860. [DOI] [PubMed] [Google Scholar]
- 152.Easterby J.S. The analysis of metabolite channelling in multienzyme complexes and multifunctional proteins. Biochem. J. 1989;264:605–607. doi: 10.1042/bj2640605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lane A.N., Kirschner K. Mechanism of the physiological reaction catalyzed by tryptophan synthase from Escherichia-Coli. Biochemistry. 1991;30:479–484. doi: 10.1021/bi00216a025. [DOI] [PubMed] [Google Scholar]
- 154.Rudolph J., Stubbe J. Investigation of the mechanism of phosphoribosylamine transfer from glutamine phosphoribosylpyrophosphate amidotransferase to glycinamide ribonucleotide synthetase. Biochemistry. 1995;37:2241–2250. doi: 10.1021/bi00007a019. [DOI] [PubMed] [Google Scholar]
- 155.Zhao A., Tsechansky M., Ellington A.D., Marcotte E.M. Revisiting and revising the purinosome. Mol. Biosyst. 2014;10:369–374. doi: 10.1039/c3mb70397e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Al-Mehdi A., Pastukh V., Swiger B., Reed D., Patel M., Bardwell G., Pastukh V., Alexeyev M., Gillespie M.N. Perinuclearmitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci. Signal. 2012;5:ra47. doi: 10.1126/scisignal.2002712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Nordlund N., Reichard P. Ribonucleotide reductases. Annu. Rev. Biochem. 2006;75:681–706. doi: 10.1146/annurev.biochem.75.103004.142443. [DOI] [PubMed] [Google Scholar]
- 158.Chimploy K., Song S.W., Wheeler L.J., Mathews C.K. Ribonucleotide reductase association with mammalian liver mitochondria. J. Biol. Chem. 2013;288:13145–13155. doi: 10.1074/jbc.M113.461111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Engstrom Y., Eriksson S., Jildevik I., Skog S., Thelander L., Tribukait B. Cell cycle-dependent expression of mammalian ribonucleotide reductase. J. Biol. Chem. 1985;260:9114–9116. [PubMed] [Google Scholar]
- 160.Guarino E., Salguero I., Kearsey S.E. Cellular regulation of ribonucleotide reductase in eukaryotes. Semin. Cell Dev. Biol. 2014;30:97–103. doi: 10.1016/j.semcdb.2014.03.030. [DOI] [PubMed] [Google Scholar]
- 161.Lin Z.P., Lee Y., Lin F., Belcourt M.F., Li P.N., Cory J.G., Glazer P.M., Sartorelli A.C. Reduced level of ribonucleotide reductase R2 subunits increases dependence on homologous recombination repair of cisplatin-induced DNA damage. Mol. Pharmacol. 2011;80:1000–1012. doi: 10.1124/mol.111.074708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Tanaka H., Arakawa H., Yamaguchi T., Shiraishi K., Fukuda S., Matsui K., Takei Y., Nakamura Y. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature. 2000;404:42–49. doi: 10.1038/35003506. [DOI] [PubMed] [Google Scholar]
- 163.Nakano K., Balint E., Ashcroft M., Vousden K.H. A ribonucleotide reductase gene is a transcriptional target of p53 and p73. Oncogene. 2000;19:4283–4289. doi: 10.1038/sj.onc.1203774. [DOI] [PubMed] [Google Scholar]
- 164.Nordlund P., Reichard P. Ribonucleotide reductases. Annu. Rev. Biochem. 2006;75:681–706. doi: 10.1146/annurev.biochem.75.103004.142443. [DOI] [PubMed] [Google Scholar]
- 165.Rennie M. An introduction to the use of tracers in nutrition and metabolism. Proc. Nutr. Soc. 1999;58:935–944. doi: 10.1017/s002966519900124x. [DOI] [PubMed] [Google Scholar]
- 166.Fan J., Ye J.B., Kamphorst J.J., Shlomi T., Thompson C.B., Rabinowitz J.D. Quantitative flux analysis reveals folate-dependent NADPH production. Nature. 2014;510:298–302. doi: 10.1038/nature13236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lane A.N., Fan T.W., Higashi R.M. Isotopomer-based metabolomic analysis by NMR and mass spectrometry. Methods Cell Biol. 2008;84:541–588. doi: 10.1016/S0091-679X(07)84018-0. [DOI] [PubMed] [Google Scholar]
- 168.Lorkiewicz P.K., Higashi R.M., Lane A.N., Fan T.W.-M. High information throughput analysis of nucleotides and their isotopically enriched isotopologues by direct-infusion FTICR-MS. Metabolomics. 2012;8:930–939. doi: 10.1007/s11306-011-0388-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lane A.N. Introduction to Metabolomics. In: Fan TW-M, Lane AN, Higashi RM, editors. Handbook of Metabolomics. NY: Humana; 2012. [Google Scholar]
- 170.Higashi R.M., Fan T.W.-M., Lorkiewicz P.K., Moseley H.N.B., Lane A.N. Stable isotope-labeled tracers for metabolic pathway elucidation by GC-MS and FT-MS. Methods Mol. Biol. 2014;1198:147–167. doi: 10.1007/978-1-4939-1258-2_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Neese R.A., Siler S.Q., Cesar D., Antelo F., Lee D., Misell L., Patel K., Tehrani S., Shah P., Hellerstein M.K. Advances in the stable isotope-mass spectrometric measurement of DNA synthesis and cell proliferation. Anal. Biochem. 2001;298:189–195. doi: 10.1006/abio.2001.5375. [DOI] [PubMed] [Google Scholar]
- 172.Robinson M.M., Turner S.M., Hellerstein M.K., Hamilton K.L., Miller B.F. Long-term synthesis rates of skeletal muscle DNA and protein are higher during aerobic training in older humans than in sedentary young subjects but are not altered by protein supplementation. FASEB J. 2011;25:3240–3249. doi: 10.1096/fj.11-186437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Moseley H.N.B., Lane A.N., Belshoff A.C., Higashi R.M., Fan T.W.-M. Non-steady state modeling of UDP-GlcNAc biosynthesis is enabled by stable isotope resolved metabolomics (SIRM) BMC Biol. 2011;9:37. doi: 10.1186/1741-7007-9-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Lane A.N., Fan T.W.-M., Bousamra II, Higashi R.M., Yan J., Miller D.M. Clinical applications of stable isotope-resolved metabolomics (SIRM) in non-small cell lung cancer. Omics. 2011;15:173–182. doi: 10.1089/omi.2010.0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Fan T.W., Lane A.N., Higashi R.M., Farag M.A., Gao H., Bousamra M., Miller D.M. Altered regulation of metabolic pathways in human lung cancer discerned by 13C stable isotope-resolved metabolomics (SIRM) Mol. Cancer. 2009;8:41. doi: 10.1186/1476-4598-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Fan T.W.-M., Lane A.N., Higashi R.M., Yan J. Stable isotope resolved metabolomics of lung cancer in a SCID mouse model. Metabolomics. 2011;7:257–269. doi: 10.1007/s11306-010-0249-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Fan T.W.-M., Lane A.N. NMR-based stable isotope resolved metabolomics in systems biochemistry. J. Biomol. NMR. 2011;49:267–280. doi: 10.1007/s10858-011-9484-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Lane A.N., Fan T.W. Quantification and identification of isotopomer distributions of metabolites in crude cell extracts using 1H TOCSY. Metabolomics. 2007;3:79–86. [Google Scholar]
- 179.Fan T.W.-M., Kucia M., Jankowski K., Higashi R.M., Rataczjak M.Z., Rataczjak J., Lane A.N. Proliferating rhabdomyosarcoma cells shows an energy producing anabolic metabolic phenotype compared with Primary Myocytes. Mol. Cancer. 2008;7:79. doi: 10.1186/1476-4598-7-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Moseley H.N.B., Higashi R.M., Fan T.W.-M., Lane A.N. Proceedings of Bioinformatics 2011. Rome: SciTePress; 2011. pp. 108–115. [Google Scholar]
- 181.Arese P., Turrini F., Schwarzer E. Band 3/complement-mediated recognition and removal of normally senescent and pathological human erythrocytes. Cell Physiol. Biochem. 2005;16:133–146. doi: 10.1159/000089839. [DOI] [PubMed] [Google Scholar]
- 182.Ramos-Montoya A., Lee W.N.P., Bassilian S., Lim S., Trebukhina R.V., Kazhyna M.V., Ciudad C.J., Noe V., Centelles J.J., Cascante M. Pentose phosphate cycle oxidative and nonoxidative balance: a new vulnerable target for overcoming drug resistance in cancer. Int. J. Cancer. 2006;119:2733–2741. doi: 10.1002/ijc.22227. [DOI] [PubMed] [Google Scholar]
- 183.Lee W.-N.P., Boros L.G., Puigjaner J., Bassilian S., Lim S., Cascante M. Mass isotopomer study of the nonoxidative pathways of the pentose cycle with [1,2-13C2]glucose. Am. J. Physiol. Endocrinol. Metab. 1998;274:E843–E851. doi: 10.1152/ajpendo.1998.274.5.E843. [DOI] [PubMed] [Google Scholar]
- 184.Marin S., Lee W.N.P., Bassilian S., Lim S., Boros L.G., Centelles J.J., Fernandez-Novell J.M., Guinovart J.J., Cascante M. Dynamic profiling of the glucose metabolic network in fasted rat hepatocytes using 1,2-C-13(2) glucose. Biochem. J. 2004;381:287–294. doi: 10.1042/BJ20031737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Centelles J.J., Ramos-Montoya A., Lim S., Bassilian S., Boros L.G., Marin S., Cascante M., Lee W.N.P. Metabolic profile and quantification of deoxyribose synthesis pathways in HepG2 cells. Metabolomics. 2007;3:105–111. [Google Scholar]
- 186.Kominsky D.J., Klawitter J., Brown J.L., Boros L.G., Melo J.V., Eckhardt S.G., Serkova N.J. Abnormalities in glucose uptake and metabolism in imatinib-resistant human BCR-ABL-positive cells. Clin. Cancer Res. 2009;15:3442–3450. doi: 10.1158/1078-0432.CCR-08-3291. [DOI] [PubMed] [Google Scholar]
- 187.Kleijn R.J., van Winden W.A., van Gulik W.M., Heijnen J.J. Revisiting the 13C-label distribution of the non-oxidative branch of the pentose phosphate pathway based upon kinetic and genetic evidence. FEBS J. 2005;272:4970–4982. doi: 10.1111/j.1742-4658.2005.04907.x. [DOI] [PubMed] [Google Scholar]
- 188.Miccheli A., Tomassini A., Puccetti C., Valerio M., Peluso G., Tuccillo F., Calvani M., Manetti C., Conti F. Metabolic profiling by C-13-NMR spectroscopy: [1,2-C-13(2)] glucose reveals a heterogeneous metabolism in human leukemia T cells. Biochimie. 2006;88:437–448. doi: 10.1016/j.biochi.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 189.Boros L.G., Puigjaner J., Cascante M., Lee W.N.P., Brandes J.L., Bassilian S., Yusuf F.I., Williams R.D., Muscarella P., Melvin W.S., et al. Oxythiamine and dehydroepiandrosterone inhibit the nonoxidative synthesis of ribose and tumor cell proliferation. Cancer Res. 1997;57:4242–4248. [PubMed] [Google Scholar]
- 190.Boros L.G., Lee W.P., Muscarella P., Fisher W.E., Schirmer W.J., Melvin W.S. Inhibitors of the oxidative and non-oxidative pentose phosphate pathways inhibit pancreatic cancer cell proliferation. Gastroenterology. 1997;112:A540. [Google Scholar]
- 191.Vizan P., Boros L.G., Figueras A., Capella G., Mangues R., Bassilian S., Lim S., Lee W.-N.P., Cascante M. K-ras codon-specific mutations produce distinctive metabolic phenotypes in human fibroblasts. Cancer Res. 2005;65:5512–5515. doi: 10.1158/0008-5472.CAN-05-0074. [DOI] [PubMed] [Google Scholar]
- 192.Ying H., Kimmelman A.C., Lyssiotis A.C., Hua S., Chu G.C., Fletcher-Sananikone E., Locasale J.W., Son J., Zhang H., Coloff J.L., et al. Oncogenic kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–670. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Delgado T.C., Castro M.M., Geraldes C.F., Jones J.G. Quantitation of erythrocyte pentose pathway flux with [2-(13)]Glucose and H-1 NMR analysis of the lactate methyl signal. Magn. Reson. Med. 2004;51:1283–1286. doi: 10.1002/mrm.20096. [DOI] [PubMed] [Google Scholar]
- 194.Marin-Valencia I., Yang C.D., Mashimo T., Cho S., Baek H., Yang X.L., Rajagopalan K.N., Maddie M., Vemireddy V., Zhao Z.Z., et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab. 2012;15:827–837. doi: 10.1016/j.cmet.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Wishart D.S.E.A. HMDB 3.0-The Human Metabolome Database in 2013. Nucleic Acids Res. 2013;41:D801–D807. doi: 10.1093/nar/gks1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Broer S. Amino acid transport across mammalian intestinal and renal epithelia. Phyiol. Rev. 2008;88:249–286. doi: 10.1152/physrev.00018.2006. [DOI] [PubMed] [Google Scholar]
- 197.Sellers K., Fox M.P., Bousamra M., Slone S., Higashi R.M., Miller D.M., Wang Y., Yan J., Yuneva M.O., Deshpande R., et al. Pyruvate carboxylase is critical in non-small cell lung cancer. J. Clin. Invest. 2014. doi:10.1172/JCI72873. [DOI] [PMC free article] [PubMed]
- 198.Maher E.A., Marin-Valencia I., Bachoo R.M., Mashimo T., Raisanen J., Hatanpaa K.J., Jindal A., Jeffrey F.M., Choi C.H., Madden C., et al. Metabolism of U-13C glucose in human brain tumors in vivo. NMR Biomed. 2012;25:1234–1244. doi: 10.1002/nbm.2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Son J., Lyssiotis C.A., Ying H., Wang X., Hua S., Ligorio M., Perera R.M., Ferrone C.R., Mullarky E., Ng S.-C., et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Yang C., Sudderth J., Dang T., Bachoo R.G., McDonald J.G., DeBerardinis R.J. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or akt signaling. Cancer Res. 2009;69:7986–7993. doi: 10.1158/0008-5472.CAN-09-2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Wise D.R., DeBerardinis R.J., Mancuso A., Sayed N., Zhang X.-Y., Pfeiffer H.K., Nissim I., Daikhin E., Yudkoff M., McMahon S.B., et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. U.S.A. 2008;105:18782–18787. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Mazurek S., Eigenbrodt E., Failing K., Steinberg P. Alterations in the glycolytic and glutaminolytic pathways after malignant transformation of rat liver oval cells. J. Cell. Physiol. 1999;181:136–146. doi: 10.1002/(SICI)1097-4652(199910)181:1<136::AID-JCP14>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 203.Kelly A., Li C.H., Gao Z.Y., Stanley C.A., Matschinsky F.M. Glutantinolysis and insulin secretion—from bedside to bench and back. Diabetes. 2002;51:S421–S426. doi: 10.2337/diabetes.51.2007.s421. [DOI] [PubMed] [Google Scholar]
- 204.McKeehan W.L. Glycolysis, glutaminolysis and cell proliferation. Cell Biol. Int. Rep. 1982;6:635–650. doi: 10.1016/0309-1651(82)90125-4. [DOI] [PubMed] [Google Scholar]
- 205.Cheng T., Sudderth J., Yang C., Mullen A.R., Jin E.S., Mates J.M., DeBerardinis R.J. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc. Natl. Acad. Sci. U.S.A. 2011;108:8674–8679. doi: 10.1073/pnas.1016627108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Donadio A.C., Lobo C., Tosina M., Rosa V., Martin-Rufian M., Campos-Sandoval J.A., Mates J.M., Marquez J., Alonso F.J., Segura J.A. Antisense glutaminase inhibition modifies the O-GlcNAc pattern and flux through the hexosamine pathway in breast cancer cells. J. Cell. Biochem. 2008;103:800–811. doi: 10.1002/jcb.21449. [DOI] [PubMed] [Google Scholar]
- 207.Mates J.M., Segura J.A., Alonso F.J., Marquez J. Pathways from glutamine to apoptosis. Front. Biosci. 2006;11:3164–3180. doi: 10.2741/2040. [DOI] [PubMed] [Google Scholar]
- 208.Thornburg J.M., Nelson K.K., Lane A.N., Arumugam S., Simmons A., Eaton J.W., Telang S., Chesney J. Targeting aspartate aminotransferase in breast cancer. Breast Cancer Res. 2008;10:R84. doi: 10.1186/bcr2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Wang J., Alexander P., Wu L., Hammer R., Cleaver O., McKnight S. Dependence of mouse embryonic stem cells on threonine catabolism. Science. 2009;325:435–439. doi: 10.1126/science.1173288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Wishart D.S., Jewison T., Guo A.C., Wilson M., Knox C., Liu Y.F., Djoumbou Y., Mandal R., Aziat F., Dong E., et al. HMDB 3.0-The Human Metabolome Database in 2013. Nucleic Acids Res. 2013;41:D801–D807. doi: 10.1093/nar/gks1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Wang W.W., Wu Z.L., Dai Z.L., Yang Y., Wang J.J., Wu G.Y. Glycine metabolism in animals and humans: implications for nutrition and health. Amino Acids. 2013;45:463–477. doi: 10.1007/s00726-013-1493-1. [DOI] [PubMed] [Google Scholar]
- 212.Edgar A.J. Mice have a transcribed L-threonine aldolase/GLY1 gene, but the human GLY1 gene is a non-processed pseudogene. BMC Genomics. 2005;6:32–44. doi: 10.1186/1471-2164-6-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Lewis C.A., Parker S.J., Fiske B.P., McCloskey D., Gui D.Y., Green C.R., Vokes N.I., Feist A.M., Vander Heiden M.G., Metallo C.M. Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol. Cell. 2014;55:253–263. doi: 10.1016/j.molcel.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Murphy T.A., Dang C.V., Young J.D. Isotopically nonstationary 13C flux analysis of Myc-induced metabolic reprogramming in B-cells. Metab. Eng. 2013;15:206–217. doi: 10.1016/j.ymben.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Dolfi S.C., Chan L.L.-Y., Qiu J., Tedeschi P.M., Bertino J.R., Hirshfield K.M., Oltvai Z.N., Vazquez A. The metabolic demands of cancer cells are coupled to their size and protein synthesis rates. Cancer Metab. 2013;1:20. doi: 10.1186/2049-3002-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Labuschagne C., van den Broek N., Mackay G., Vousden K., Maddocks O.D. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 2014;7:1248–1258. doi: 10.1016/j.celrep.2014.04.045. [DOI] [PubMed] [Google Scholar]
- 217.Guin S., Pollard C., Ru Y.B., Lew C.R., Duex J.E., Dancik G., Owens C., Spencer A., Knight S., Holemon H., et al. Role in tumor growth of a glycogen debranching enzyme lost in glycogen storage disease. J. Natl. Cancer Inst. 2014;106:dju062. doi: 10.1093/jnci/dju062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Pérignon J.L., Bories D.M., Houllier A.M., Thuillier L., Cartier P.H. Metabolism of pyrimidine bases and nucleosides by pyrimidine-nucleoside phosphorylases in cultured human lymphoid cells. Biochim. Biophys. Acta. 1987;928:130–136. doi: 10.1016/0167-4889(87)90113-3. [DOI] [PubMed] [Google Scholar]
- 219.Navaratnam N., Sarwar R. An overview of cytidine deaminases. Int. J. Hematol. 2006;83:195–200. doi: 10.1532/IJH97.06032. [DOI] [PubMed] [Google Scholar]
- 220.Suzuki N.N., Koizumi K., Fukushima M., Matsuda A., Inagaki F. Structural basis for the specificity, catalysis, and regulation of human uridine-cytidine kinase. Structure. 2004;12:751–764. doi: 10.1016/j.str.2004.02.038. [DOI] [PubMed] [Google Scholar]
- 221.Qi Z., Voit E.O. Identification of cancer mechanisms through computational systems modeling. Transl. Cancer Res. 2014;3:233–242. doi: 10.3978/j.issn.2218-676X.2014.05.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Meyer J.S., Cosatto E., Graf H.P. Mitotic index of invasive breast carcinoma. Arch. Pathol. Lab. Med. 2009;133:1826–1833. doi: 10.5858/133.11.1826. [DOI] [PubMed] [Google Scholar]
- 223.Sellers K., Fox M.P., Bousamra M., Slone S., Higashi R.M., Miller D.M., Wang Y., Yan J., Yuneva M.O., Deshpande R., et al. Pyruvate carboxylase is upregulated in NSCLC. J. Clin. Invest. 2014. in press. [DOI] [PMC free article] [PubMed]
- 224.Fan T.W. Considerations of Sample Preparation for Metabolomics Investigation. In: Fan TW, Lane AN, Hiagshi RM, editors. Handbook of Metabolomics. Vol. 17. New York: Humana; 2012. [Google Scholar]
- 225.Hellerstein M.K., Neese R.A. Mass isotopomer distribution analysis at eight years: theoretical, analytic, and experimental considerations. Am. J. Physiol. 1999;276:E1146–E1170. doi: 10.1152/ajpendo.1999.276.6.E1146. [DOI] [PubMed] [Google Scholar]
- 226.Bousamra M., Day J., Fan T.W.-M., Higashi R.M., Kloecker G., Lane A.N., Miller D.M. The Handbook of Metabolomics. Vol. 17. Totoya: Humana; 2012. [Google Scholar]
- 227.Lane A.N., Fan T.W., Higashi R.M., Tan J., Bousamra M., Miller D.M. Prospects for clinical cancer metabolomics using stable isotope tracers. J. Exp. Molec. Pathol. 2009;86:165–173. doi: 10.1016/j.yexmp.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Christopherson R.I., Lyons S.D., Wilson P.K. Inhibitors of de novo nucleotide biosynthesis as drugs. Acc. Chem. Res. 2002;35:961–971. doi: 10.1021/ar0000509. [DOI] [PubMed] [Google Scholar]
- 229.Teschner S., Burst V. Leflunomide: a drug with a potential beyond rheumatology. Immunotherapy. 2010;2:637–650. doi: 10.2217/imt.10.52. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.