


Abstract
HIV-1 reverse transcriptase (RT) connection subdomain mutations at positions 348, 369 and 376 have been associated with resistance to non-nucleoside RT inhibitors (NNRTIs). N348I may interfere with the initiation of (+)-strand DNA synthesis by reducing polypurine tract (PPT) removal in the presence of nevirapine. The effect of NNRTIs on the RNase H-mediated cleavage of PPT-containing template-primers has been studied with wild-type HIV-1 RT and mutants N348I, T369I, T369V, T376S and N348I/T369I. In the presence of NNRTIs, all RTs were able to stimulate PPT cleavage after primer elongation. The enhancing effects of nevirapine and efavirenz were reduced in RTs carrying mutation N348I, and specially N348I/T369I. However, those mutations had no effect on rilpivirine-mediated cleavage. Prior to elongation, the PPT remains resilient to cleavage, although efavirenz and rilpivirine facilitate RNase H-mediated trimming of its 3′-end. The integrity of the 3′-end is essential for the initiation of (+)-strand DNA synthesis. In the presence of dNTPs, rilpivirine was the most effective inhibitor of (+)-strand DNA synthesis blocking nucleotide incorporation and preventing usage of available PPT primers. The N348I/T369I RT showed reduced ability to generate short RNA products revealing a cleavage window defect. Its lower RNase H activity could be attributed to enhanced rigidity compared to the wild-type enzyme.
INTRODUCTION
The human immunodeficiency virus type 1 (HIV-1) reverse transcriptase (RT) is a major target of antiretroviral therapy (1–3). The HIV-1 RT is a DNA polymerase that can use either DNA or RNA strands as templates for DNA synthesis. The RT also possesses an RNase H activity responsible for the degradation of RNA in RNA/DNA complexes. RT DNA polymerase and RNase H activities are needed to convert the viral genomic RNA into double-stranded DNA that integrates into the genome of the host cell. During reverse transcription, the first DNA strand (i.e. (–)-strand DNA) is synthesized by extending the 3′-end of a specific tRNA using the viral RNA as template. The RT RNase H activity degrades the RNA template, with the exception of two short polypurine tracts (PPTs), located at the center and at the 3′ end of the viral RNA genome. These PPTs are used as major initiation sites for (+)-strand DNA synthesis. The tRNA and PPT primers are later removed by the RNase H activity of the RT (for reviews see (4,5)).
Inhibitors of the DNA polymerization activity constitute the backbone of current therapies against HIV infection and AIDS. These drugs can be classified into nucleoside/nucleotide RT inhibitors (NRTIs) and non-nucleoside RT inhibitors (NNRTIs). There are five NNRTIs approved for clinical use: nevirapine, delavirdine, efavirenz, etravirine and rilpivirine, although delavirdine is rarely used due to its inferior antiviral efficacy and inconvenient dosing schedule (3,6). The HIV-1 RT is a heterodimer composed of subunits of 66 and 51 kDa (p66 and p51, respectively). The NNRTI binding site is a hydrophobic pocket in the palm subdomain of p66, ∼10 Å away from the polymerase active site. Leu100, Lys101, Lys103, Val106, Thr107, Val108, Val179, Tyr181, Tyr188, Val189, Gly190, Phe227, Trp229, Leu234, Pro236 and Tyr318 in p66 and Glu138 in p51 delineate the NNRTI binding pocket ((7,8); reviewed in (9)). NNRTIs have a low genetic barrier and single amino acid substitutions affecting residues at their binding pocket confer high-level resistance to nevirapine, delavirdine and efavirenz (e.g. K103N or Y181C) (reviewed in (3)). Kinetic studies have shown that NNRTIs slow the rate of the chemical reaction of nucleotide incorporation (10,11). Based on structural studies, it has been proposed that NNRTIs block reverse transcription by (i) changing the mobility of the RT thumb subdomain, (ii) distorting the catalytic triad formed by the side chains of Asp110, Asp185 and Asp186 in p66 or (iii) by repositioning the primer grip in a non-active conformation (reviewed in (2,9)).
Apart from their inhibitory effects on DNA polymerization, it has been shown that nevirapine and efavirenz stimulate the RNase H activity of HIV-1 RT ((12–14); reviewed in (15)). The HIV-1 RT can bind nucleic acid substrates in one of the two orientations. Single-molecule spectroscopy studies using PPT-containing RNA/DNA hybrids mimicking the initiation of (+)-strand DNA synthesis showed that the 3′ end of the PPT could be located at the DNA polymerase catalytic site (polymerase-dependent mode) or at the RNase H active site (16,17). Nevirapine and efavirenz promote binding in a polymerase-independent mode, thereby facilitating the cleavage and subsequent removal of the PPT (16,17). On the other hand, the presence of dNTPs favors binding in a polymerase-dependent mode, thereby facilitating the addition of nucleotides at the 3′ end of the PPT and the initiation of (+)-strand DNA synthesis. This process is particularly sensitive to NNRTI inhibition (18), while premature removal of the PPT impairs reverse transcription (19). Several studies have shown that NNRTIs, such as nevirapine or efavirenz, can modulate RNase H activity through long-range interactions that depend on the structure of the RNA/DNA hybrid ((14,20–22); reviewed in (15)). However, little is known about the effects of recently approved diarylpyrimidines such as etravirine or rilpivirine.
Several studies have demonstrated that mutations in the connection subdomain and the RNase H domain of HIV-1 RT can have an impact on resistance to RT inhibitors ((23–26); reviewed in (27)). Specifically, mutations away from the NNRTI binding pocket such as N348I, T369I or V and A376S confer low-to-moderate levels of resistance to nevirapine and delavirdine in phenotypic assays carried out with recombinant HIV-1 (28,29). Furthermore, HIV-1 clones carrying RT mutations N348I and T369I show high-level resistance to both drugs and moderate resistance to efavirenz, although the presence of both mutations confers a severe fitness defect (28,29). The increased prevalence of N348I and A376S in treated patients has been related to previous exposure to nevirapine while the presence of those mutations has been associated with an increased risk of virological failure (30,31). Both amino acid substitutions confer increased affinity for the template-primer (31,32), while N348I reduces the association rate of the RT and nevirapine (32). However, Asn348 and Ala376 are away from the NNRTI binding pocket and their effects on NNRTI resistance can be explained only by assuming long-range interactions along the nucleic acid binding cleft. N348I has a detrimental effect on the RNase H activity of the RT (30,33). During (+)-strand DNA synthesis, nevirapine and efavirenz facilitate the premature removal of the PPT by favoring its binding in a polymerase-independent orientation (16), although it has been shown that the presence of N348I could attenuate the effects of nevirapine (19).
In this work, we have extended these analyses to recently approved inhibitors (etravirine and rilpivirine) as well as other RT connection subdomain mutations that have been associated with resistance to NNRTIs. Our study provides mechanistic insight into how the double-mutant N348I/T369I in HIV-1 RT could affect PPT removal and RNase H cleavage specificity in the absence or presence of NNRTIs.
MATERIALS AND METHODS
Mutagenesis, expression and purification of recombinant RTs
Wild-type (WT) HIV-1BH10 RT and mutant K103N were obtained as previously described (31,34). Other mutants were obtained after site-directed mutagenesis using the standard QuikChangeTM protocol (Stratagene). Briefly, complementary mutagenic primers (Supplementary Table S1) were used to amplify the entire plasmid p66RTB that contained the WT RT-coding sequence of the HIV-1BH10 strain, in a thermocycling reaction carried out with the high-fidelity Pfu DNA polymerase. After mutagenesis, the entire RT-coding regions were sequenced and, if correct, used for RT expression and purification. Mutant and WT RTs were obtained as heterodimers composed of subunits of 66 and 51 kDa (p66 and p51, respectively), with a polyhistidine tag at the C-terminus of p66 (34,35). Enzymes were quantified by active site titration before biochemical studies (36).
Nucleotides, template-primers and NNRTIs
Stock solutions (100 mM) of deoxyribonucleoside triphosphates (dNTPs) were obtained from GE Healthcare. [γ-32P]ATP (10 mCi/ml; 3000 Ci/mmol) was provided by Perkin Elmer. Synthetic oligonucleotides were obtained from Life Technologies and Sigma. Template-primers used in nucleotide incorporation and RNase H activity assays are given in Supplementary Table S2. RT inhibitors were obtained from the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH (where etravirine and rilpivirine were supplied to the Program by Tibotec Pharmaceuticals, Inc.).
Nucleotide incorporation and RT inhibition assays
Assays were carried out with template-primer D38/25PGA (Supplementary Table S2). The primer was labeled at its 5′ end with 1 μCi of [γ-32P]ATP and 5 U of T4 polynucleotide kinase (Promega) in 70-mM Tris-HCl pH 7.6 buffer containing 10-mM MgCl2, 5-mM dithiothreitol (DTT) and 10-μM adenosine triphosphate (ATP). Template and primer were annealed in 150-mM NaCl and 150-mM magnesium aspartate, the template/primer molar ratio was adjusted to 1:1 and the final concentration was 300 nM. Nucleotide incorporation assays in the absence or presence of NNRTIs were carried out in a total volume of 12 μl. A preincubation mix (5 μl) of purified RT (14.4–36 nM) and labeled template-primer (72 nM) was maintained at 37ºC during 10 min in 120-mM Hepes pH 7.0 buffer, containing 24-mM NaCl, 24-mM MgCl2, 208-mM potassium acetate, 1.6-mM DTT and 8% (w/v) polyethyleneglycol 6000, and then mixed with 3 μl of 4% dimethyl sulfoxide (DMSO) or appropriate concentrations of the corresponding NNRTI (in 4% DMSO). Then, the sample was incubated for 10 min at 37ºC. Four microliter of a solution containing 100-μM 2′-deoxythymidine 5′-triphosphate (dTTP), diluted in 130-mM potassium acetate, 1-mM DTT and 5% (w/v) polyethyleneglycol 6000 was added and the reaction was then stopped after 20 s by mixing an aliquot of 4 μl with an equal volume of stop solution (10-mM ethylenediaminetetraacetic acid in 90% formamide containing 3-mg/ml xylene cyanol (FF) and 3-mg/ml bromophenol blue) and heating at 90ºC for 10 min. Samples were analyzed by denaturing polyacrylamide gel electrophoresis and quantified by phosphorimaging. NNRTI inhibition was determined from the relative amounts of dTTP incorporated in the presence of different NNRTI concentrations, compared with nucleotide incorporation in the absence of drug. The IC50 values for each inhibitor were obtained from the inflection points in the corresponding dose-response curves.
RNase H activity assays
Template-primers used in these assays are listed in Supplementary Table S2 and include RNA/DNA complexes, as well as a complex containing a DNA strand and a chimeric RNA–DNA primer (e.g. T57d/PPT17r8d). Assays were carried out in 50-mM Tris-HCl pH 8.0 buffer, containing 50-mM NaCl and 5-mM MgCl2 (37). Template-primer concentrations were usually in the range 25–50 nM, and RT concentrations varied between 25 and 125 nM, depending on the assays. A 2- to 5-fold excess of enzyme over template-primer was used in all assays. Reactions were initiated by adding MgCl2. Aliquots were removed at appropriate times (usually between 20 s and 5 min) and quenched with an equal volume of stop solution. In assays carried out under single-turnover conditions, RTs and template-primers were preincubated for 5 min at 37ºC and reactions were then initiated by adding MgCl2 and sodium heparin to final concentrations of 5 mM and 3 mg/ml, respectively. Products were analyzed by denaturing polyacrylamide gel electrophoresis and quantified by phosphorimaging. The amount of hydrolyzed product generated over time was fitted to a single-exponential decay equation: [P] = A × e−kobs × t, where kobs is an apparent kinetic constant of the RNase H cleavage reaction.
HIV drug susceptibility assays
Recombinant HIV-1 variants were obtained after co-transfecting MT-4 cells with an RT-deleted HXB2-D clone previously linearized with BstEII and a polymerase chain reaction product containing the nucleotide sequence encoding for the 66-kDa subunit of the RT (38). The RT-coding region of recovered viruses was fully sequenced in order to check for reversions or undesired mutations. MT-4 cells and the deleted HXB2-D clone were obtained from the AIDS Reagent Program (Medical Research Council). MT-4 cells were used to propagate the recombinant virus. Their susceptibility to RT inhibitors was determined as previously described, using a multiplicity of infection of 0.003 (37).
RESULTS
Previous reports have shown that mutations affecting residues away from the NNRTI binding pocket confer low-to-moderate resistance to nevirapine and delavirdine, and in a lesser extent to efavirenz ((28,29); reviewed in (27)). Phenotypic assays carried out in MT-4 cells with recombinant HIV-1 bearing mutations N348I, T369I, T369V, T376S and the double mutant N348I/T369I were consistent with previous estimates and revealed that the double mutant shows significant resistance to nevirapine (Table 1). On the other hand, none of the tested mutations had a significant impact on resistance to efavirenz, etravirine or rilpivirine. These results were broadly consistent with the IC50 values obtained with recombinant RTs in nucleotide incorporation reactions using heteropolymeric DNA/DNA template-primers, although the N348I/T369I RT showed low-level resistance to rilpivirine in these assays (Supplementary Table S3).
Table 1. Susceptibility of HIV-1 constructs to RT inhibitorsa.
| RT | IC50 (nM) | ||||
|---|---|---|---|---|---|
| AZT | Nevirapine | Efavirenz | Etravirine | Rilpivirine | |
| WT | 4.3 ± 0.6 | 15.0 ± 1.7 | 1.1 ± 0.2 | 1.0 ± 0.1 | 0.11 ± 0.01 |
| K103N | 14 ± 2 (3.2) | 1831 ± 226 (>100) | 44.5 ± 15.0 (42) | 1.2 ± 0.4 (1.3) | 0.13 ± 0.02 (1.2) |
| N348I | 16.3 ± 1.5 (3.8) | 33.0 ± 4.6 (2.2) | 1.2 ± 0.3 (1.1) | 1.2 ± 0.1 (1.2) | 0.26 ± 0.10 (2.3) |
| N348I/T369I | 5.0 ± 1.0 (1.1) | 102.3 ± 2.5 (6.8) | 1.4 ± 0.3 (1.3) | 1.1 ± 0.1 (1.1) | 0.08 ± 0.03 (0.7) |
| T369I | 7.0 ± 1.0 (1.6) | 28.3 ± 1.5 (1.9) | 1.1 ± 0.1 (1.0) | 1.0 ± 0.1 (1.0) | 0.12 ± 0.03 (1.0) |
| T369V | 18.7 ± 3.1 (4.3) | 27.3 ± 11.8 (1.8) | 1.0 ± 0.1 (1.0) | 1.1 ± 0.1 (1.1) | 0.27 ± 0.03 (2.5) |
| T376S | 7.3 ± 1.5 (1.7) | 26.0 ± 7.2 (1.7) | 1.5 ± 0.2 (1.4) | 1.4 ± 0.2 (1.4) | 0.19 ± 0.05 (1.7) |
aThe IC50 values shown are averages ± standard deviations of at least three tests, with each one performed six times. The fold increase in IC50 relative to wild-type HXB2 virus control carrying the RT sequence of BH10 is shown between parentheses.
Effect of RT connection subdomain mutations on PPT removal
The selection of N348I and other mutations under therapy with NNRTIs, particularly with nevirapine, could be related to effects in other steps of the reverse transcription process. It has been previously shown that initiation of (+)-strand DNA synthesis is particularly sensitive to NNRTI inhibition (18), while PPT removal is a critical step at the initiation of (+)-strand DNA synthesis.
Template-primers mimicking the initial steps of (+)-strand DNA synthesis were used to study the effects of RT mutations on PPT removal. In one of them (T57d/PPT17r8d) the shorter strand was a chimeric RNA–DNA oligonucleotide that contained the PPT sequence located at the 3′ end of the HIV-1 genome (17 ribonucleotides), with an 8-nt extension of DNA. As shown in Figure 1A, RT connection subdomain mutations T369I, T369V and T376S had a minor effect on the RT's ability to cleave the PPT17r8d oligonucleotide at position –17 (G↓A site) that corresponds to the RNA(PPT)–DNA junction. However, as in the case of mutant T369I shorter PPT products of 16 and 15 nucleotides were also observed in reactions carried out with mutants T369V and T376S (data not shown). N348I, particularly in combination with T369I, showed very low efficiency in the RNase H cleavage reaction. Interestingly, when the template-primer used was a complex containing a 29-nt DNA and the 17-nt PPT RNA, we did not observe any secondary cleavage and the PPT remained intact (Figure 1B). This complex (PPT29DNA/PPT17r) was used because it mimics the first step in (+)-strand DNA synthesis. Its premature degradation by the RT RNase H activity would abort reverse transcription, since the integrity of the 3′ end of the PPT is essential in this process (39,40).
Figure 1.

RNase H activity of WT and mutant HIV-1 RTs during (+)-strand DNA synthesis. (A) Representative gel showing the cleavage of the PPT17r8d strand in the template-primer T57d/PPT17r8d complex (sequence given below) obtained with the WT RT and mutants N348I, T369I and N348I/T369I. The T57d/PPT17r8d complex mimics a (+)-strand DNA synthesis intermediate where eight dNTPs have been added to the 17-nucleotide PPT-RNA primer. The template-primer (25 nM) was incubated in the presence of RTs at 125-nM concentration. Aliquots were removed at 20 s, 40 s, and 1, 2, 3, 4 and 5 min. Arrows indicate the positions of the observed cleavages. Time courses of the RNase H cleavage reactions obtained with all tested RTs are shown on the right (data were obtained from three independent experiments). (B) RNase H activity of WT, N348I/T369I and N348I mutant enzymes on a PPT29DNA/PPT17r complex. This complex mimics the first intermediate in the (+)-strand DNA synthesis reaction, containing the RNA primer before the incorporation of dNTPs.
Effects of NNRTIs on PPT removal by HIV-1 RTs bearing mutations in the connection subdomain
Previous studies have shown an increase in PPT cleavage efficiency in reactions carried out with the WT RT in the presence of nevirapine and efavirenz (19). In addition, those authors demonstrated that N348I alone counteracts the effect of nevirapine, and to a lesser degree, efavirenz. We have now extended those observations to other RT connection subdomain mutants as well as to the recently approved inhibitor rilpivirine. In all cases, NNRTI addition resulted in a more efficient PPT cleavage of the chimeric RNA–DNA substrate (Figure 2A). The largest effects were observed at saturating concentrations (i.e. in the micromolar range), and nevirapine and efavirenz were stronger promoters of the RNase H activity than rilpivirine. N348I alone or in combination with T369I suppressed in part the enhancing effect of nevirapine and efavirenz on the RNase H activity of the enzyme. In contrast, in reactions carried out in the presence of rilpivirine, the endonucleolytic activity of mutants N348I and N348I/T369I was similar to that observed for the WT enzyme. The double mutant showed increased cleavage efficiency in the presence of NNRTIs, but the differences between rilpivirine and the other drugs were much smaller. Interestingly, differences in PPT cleavage efficiencies between the WT and the double-mutant RT in the absence of inhibitor were largely reduced in the presence of NNRTIs (Figure 2B). Thus, although the RNase H activity of the N348I/T369I RT is very low, addition of NNRTIs enhances its RNase H activity to similar levels as those observed with the WT RT.
Figure 2.

Effect of HIV-1 RT connection subdomain mutations on the RNase H cleavage of a PPT-containing template-primer in the presence of NNRTIs. (A) Cleavage of the chimeric PPT(RNA)-DNA oligonucleotide in the T57d/PPT17r8d complex in the presence of increasing concentrations of nevirapine (0, 0.08, 0.16, 0.32, 0.63, 1.25, 2.5 and 5 μM). Template-primer and RT concentrations in these assays were 25 and 125 nM, respectively. Aliquots were withdrawn after a 20-s incubation. C stands for control (uncleaved PPT17r8d primer). Graphical representations of the percentage of PPT17r8d primer cleavage by WT and mutant RTs in the absence of drug or in the presence of 5-μM nevirapine, 12.5-μM efavirenz or 5-μM rilpivirine are shown below. (B) Time courses of cleavage reactions carried out in the presence of nevirapine, efavirenz and rilpivirine. WT, N348I and/or N348I/T369I RTs (50 nM) were mixed with 25-nM template-primer (T57d/PPT17r8d), and RNase H cleavage was monitored in the absence or presence of drug. Aliquots were removed at 20 s, 40 s and 1, 2, 3 and 4 min. Cleavage products corresponding to bands of 17, 16 and 15 nucleotides are indicated in the gel. NVP, nevirapine; EFV, efavirenz; RPV, rilpivirine.
The increased efficiency of PPT removal mediated by NNRTIs is not observed in the presence of mutations that abolish drug binding
The HIV-1 RT mutation K103N confers high-level resistance to nevirapine and efavirenz due to the inability of those drugs to bind the mutant RT. RNase H assays carried out with this enzyme and the T57d/PPT17r8d complex demonstrated that the inhibitor concentration had no impact on the PPT cleavage efficiency of the RT (Supplementary Figure S1A). However, NNRTI concentration influenced PPT removal in reactions carried out with both WT and the double-mutant N348I/T369I, with highest efficiencies at 0.6–5-μM nevirapine (Figure 2A and Supplementary Figure S1A). In reactions carried out in the presence of dNTPs with the complex PPT29DNA/PPT17r that mimics the initial step of (+)-strand DNA synthesis, the mutant K103N RT was able to extend the RNA primer with similar efficiencies in the presence or absence of inhibitor (Supplementary Figure S1B). On the other hand, the WT RT was able to produce significant amounts of a fully extended product of 29 nucleotides in the absence of nevirapine, although some cleavage of the PPT tract was observed in the corresponding extension reactions. In the presence of the drug, there was enhanced PPT degradation before nucleotide incorporation and premature PPT removal and this resulted in the inhibition of (+)-strand DNA synthesis (Supplementary Figure S1B). These results are consistent with an increased binding of the RNA/DNA complex in an RNase H-competent mode that would facilitate polymerase-independent cleavage of the PPT.
The combination of N348I and T369I affects the RNase H cleavage window of the HIV-1 RT
We analyzed whether the reduced efficiency of PPT cleavage shown by mutant RTs N348I and N348I/T369I could be related to the positioning of the susceptible G↓A cleavage site in the RNA template relative to the DNA polymerase active site. For this purpose, we used RNA/DNA template-primers with recessed DNA 3′ ends containing the PPT sequence with the G↓A cleavage site in the RNA located at a distance of 16, 17 or 18 nucleotides from the 3′ end of the DNA primer.
As shown in Figure 3A, the PPT29RNA in the 29/28mer hybrid was efficiently cleaved at position –16 in reactions carried out with the WT RT and the single mutants T369I, T369V and T376S. Overall cleavage efficiencies of N348I RT were similar to those determined for the WT enzyme (Supplementary Table S4), although the N348I mutant produced a smaller amount of secondary products. A slight reduction in cleavage efficiency was observed with the double-mutant N348I/T369I. In reactions carried out with this RT, cleavage at position –16 occurred at a very slow rate, while larger bands corresponding to cuts at positions 18 and 19 nucleotides away from the 3′ end of the primer were also observed. Interestingly, when the cleavage window was one-nucleotide longer, all tested RTs showed increased endonucleolytic activity (Figure 3B and Supplementary Table S4). However, cleavages at positions –17 and –18 were predominant in reactions carried out with the N348I/T369I RT, while all other RTs rendered a single product of 21 nucleotides (corresponding to RNAs cleaved at position –17) (Figure 3B). In assays carried out with the 29/30mer (i.e. PPT29RNA/PPT30DNA), all RTs showed identical cleavage patterns (Figure 3C) with similar RNase H catalytic rates, except for the single-mutant T369V that showed an increased kobs in comparison with the other enzymes (Supplementary Table S4). Taken together, the analyses with template-primers of different lengths indicate that the combination of N348I and T369I has an impact on the RNase H cleavage window and reduces the RT's ability to produce shorter RNA products.
Figure 3.
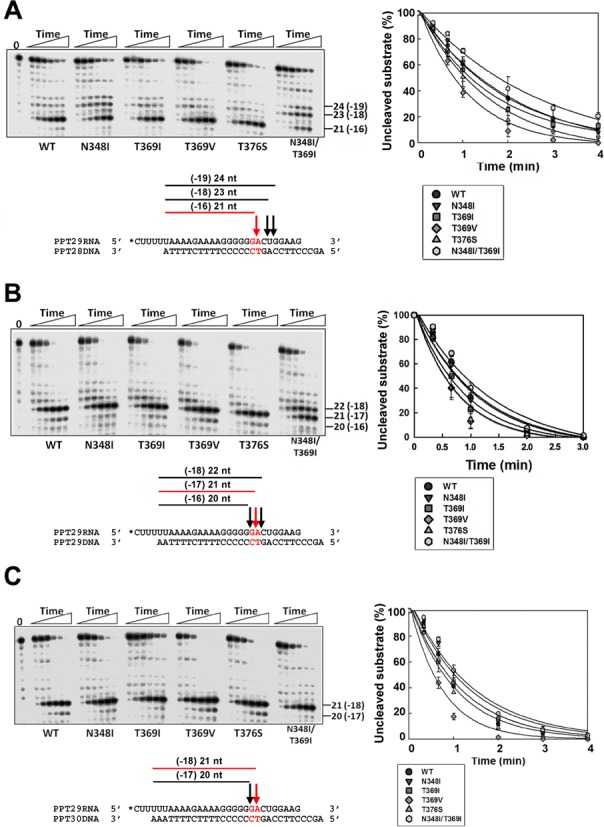
Effect of HIV-1 RT connection subdomain mutations on the RNase H cleavage window. Time courses of the RNase H cleavage reaction were obtained under multiple-turnover conditions with template-primers that mimic (−) strand DNA synthesis intermediates with PPT-containing templates: (A) PPT29RNA/PPT28DNA, (B) PPT29RNA/PPT29DNA and (C) PPT29RNA/PPT30DNA. Reactions were carried out in the presence of RT and template-primers at concentrations of 50 and 25 nM, respectively. Aliquots were removed at 20 s, 40 s and 1, 2, 3 and 4 min. Template-primer nucleotide sequences and relevant cleavage sites are shown below the corresponding gels. Time courses of RNase H reactions obtained for each enzyme are shown on the right. Represented values were determined from three independent experiments [means ± standard deviations (error bars)]. The calculated kobs values are given in Supplementary Table S4.
Data shown in Figure 3 were consistent with the results of RNase H cleavage assays carried out under single-turnover conditions (Supplementary Figure S2). RNA oligonucleotides of 21 bases resulting from cleavage of the G↓A bond at position –16 of template-primer PPT29RNA/PPT28DNA represented 42.8% of all products in reactions catalyzed by the WT RT. However, the efficiency of this cleavage was almost negligible in reactions carried out with mutants N348I and N348I/T369I. On the other hand, all RTs showed the same specificity with the 29/30mer and rendered the 21-nucleotide product with yields above 90%. Interestingly, cleavage patterns obtained with the N348I RT and template-primer PPT29RNA/PPT29DNA revealed its higher tendency to produce shorter oligonucleotides in comparison with the double mutant. Thus, the 21-base oligonucleotide represented 25.4% of all cleavage products, while this value was reduced to 13.9% in reactions catalyzed by the N348I/T369I RT.
Additional experiments also showed that the differences between WT, N348I and N348I/T369I RTs were reduced when the assays were carried out in the presence of nevirapine or efavirenz (Supplementary Figure S3). Short cleavage products representing cuts at positions –17, –16 or even –15 were obtained with all three enzymes in the presence of NNRTIs. Although the double mutant showed a slower cleavage rate in comparison with the WT enzyme, these differences were only detectable for the shorter cleavage products (i.e. those resulting from cuts at position –15).
RNA templates containing the PPT sequence are resilient to cleavage. In order to test whether the observed differences in cleavage specificity between the mutant N348I/T369I and the other studied RTs could be attributed to the specific PPT sequence, we compared the RNase H cleavage patterns of WT and mutant RTs N348I and N348I/T369I using two heteropolymeric RNA/DNA template-primers. First, with the 31/21mer (31Trna/21P) we found that in the absence of the drug, the major cleavage site for the WT RT is at position –16 (Supplementary Figure S4). As described above for the PPT29RNA/PPT28DNA complex, the N348I RT shows reduced cleavage efficiency at this position, while the activity of the double mutant is considerably reduced. However, when nevirapine is included in the reaction, the three RTs were efficient in generating a 26-nt RNA (i.e. resulting from cleavage at position –16), as well as one-nucleotide shorter products. However, shorter products resulting from cleavage at position –13 appeared only in reactions catalyzed by the WT enzyme. On the other hand, experiments carried out with the D30RNA/25PGA hybrid revealed that primary cleavage of D30RNA by WT RT occurs at position –18, while secondary cuts are observed at positions –16, –15 and –14 (Supplementary Figure S4). Interestingly, in the absence of drugs, differences in the cleavage rates at position –18 between the three tested RTs were very small. However, secondary cleavages at positions –15 and –14 were almost negligible in reactions carried out with the double mutant in the presence of nevirapine. These results are consistent with an impaired ability to cleave shorter RNAs by the double-mutant N348I/T369I RT.
PPT stability and initiation of (+)-strand DNA synthesis in the presence of NNRTIs
Nucleotide incorporation at the 3′ end of the PPT can be inhibited in the presence of NNRTIs. In addition, the RNase H activity of the RT can also affect the integrity of the PPT (Supplementary Figure S1B). Experiments carried out with the PPT17r/PPT29DNA template-primer showed that the stability of the PPT might be different depending on the NNRTI used in the assays. Thus, rilpivirine and efavirenz are more potent than nevirapine and etravirine in stimulating PPT degradation (Figure 4A). Cleavage of the PPT in these assays can be detected only if the template-primer binds in a polymerase-independent mode, although hybrids were designed to favor a polymerase-dependent binding mode. Both WT and N348I/T369I RTs showed similar PPT cleavage patterns in the presence of NNRTIs. However, at higher concentrations of nevirapine (i.e. 1—5 μM), degradation of the PPT by the WT enzyme was increased relatively to the double mutant. Since the efficiency of the extension of trimmed PPTs is very low, these results imply that the initiation of (+)-strand DNA synthesis might be impaired in the presence of NNRTIs.
Figure 4.
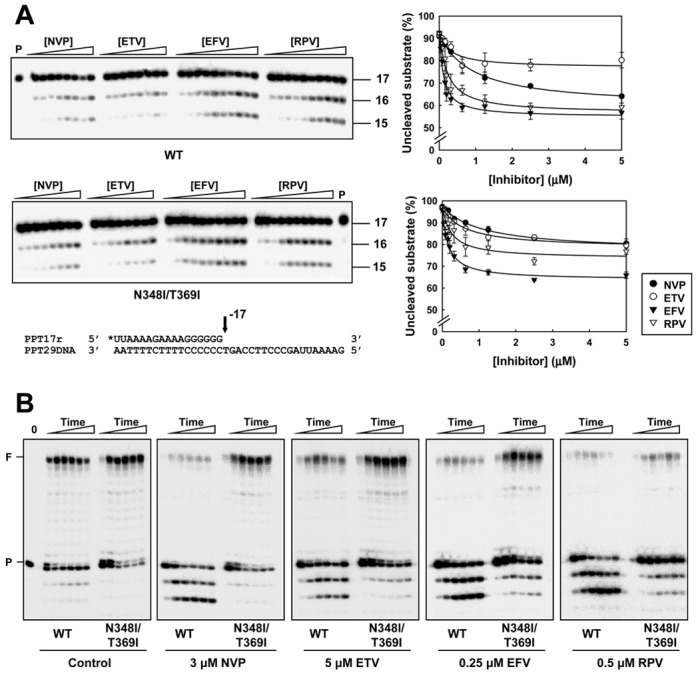
Effects of NNRTIs on PPT susceptibility to RNase H cleavage by WT and mutant N348I/T369I RTs. (A) Cleavage of the PPT primer in the presence of nevirapine, etravirine, efavirenz and rilpivirine. Reactions were carried out in the presence of 25-nM template-primer (PPT29DNA/PPT17r) and 125-nM RT. The RT and the inhibitor were preincubated at room temperature during 5 min. In reactions carried out with WT RT aliquots were taken 20 s after adding the template-primer, while in the case of the N348I/T369I RT time points were collected 60 s after adding the PPT29DNA/PPT17r complex. Nevirapine (NVP) and etravirine (ETV) concentrations in the assays were 0, 0.08, 0.16, 0.31, 0.625, 1.25, 2.5 and 5 μM. For efavirenz (EFV) and rilpivirine (RPV) the concentrations used were 0, 0.04, 0.08, 0.16, 0.20, 0.31, 0.625, 1.25, 2.5 and 5 μM. Graphical representations of the results obtained with WT and mutant N348I/T369I RTs are shown on the right. Represented values were determined from three independent experiments [means ± standard deviations (error bars)]. P stands for uncleaved PPT oligonucleotide. (B) PPT primer extension assays carried out in the presence of NNRTIs. Reactions were carried out in the presence of 25-nM RT, 25-nM template-primer (PPT29DNA/PPT17r) and all four dNTPs (each at 200-μM concentration). Aliquots were withdrawn at 0.5, 1, 2, 3, 4 and 5 min.
In addition, significant inhibition of (+)-strand DNA synthesis due to cleavage of the PPT primer in reactions catalyzed by the WT RT was observed with rilpivirine and efavirenz at concentrations that were 10 times lower than those needed for nevirapine and etravirine (Figure 4B). Unlike the WT RT, the combination of mutations N348I and T369I facilitates the extension of the PPT primer to produce a full-length product of 29 nucleotides in the presence of nevirapine (Figure 4B). These compensatory effects were also observed with efavirenz and etravirine, although the differences in the amount of extended product obtained with WT and mutant RTs were reduced. On the other hand, in the presence of rilpivirine, we observed a significant inhibition of PPT extension with both WT and N348I/T369I RTs. Although WT RT increased PPT degradation under these conditions, both RTs were very inefficient in extending the primer and rendered low amounts of fully extended product. Interestingly, these inhibitory effects were not observed with a mutant RT that contained the rilpivirine resistance-associated mutation E138K combined with the two substitutions in the connection subdomain (i.e. E138K/N348I/T369I) (Supplementary Figure S5). Taking into account the effects on PPT degradation and primer elongation, our results reveal that rilpivirine is the most efficient and potent inhibitor of (+)-strand DNA synthesis initiation, among the NNRTIs analyzed in this study.
DISCUSSION
HIV-1 RT connection subdomain mutations are selected during therapy with NNRTIs, particularly with nevirapine (30,31). A recent search in the Stanford HIV Drug Resistance Database (http://hivdb.stanford.edu/; accessed on 22 September 2014) showed that the prevalence of N348I and T369I in untreated patients infected with HIV-1 group M subtype B was lower than 0.1%, while in patients treated with nevirapine, these figures went up to 11.1 and 1.5% for N348I and T369I, respectively (P < 0.001 in both cases). Significant increases of prevalence were also observed for patients treated with efavirenz where N348I and T369I were present in 8.7% and 2.2% of all isolates, respectively (P < 0.001). Similar analyses carried out with T369V and A376S only reveal a significant increase of prevalence in the case of T369V and nevirapine (8.8% in nevirapine-treated patients versus 3.0% in untreated individuals). The studied amino acid substitutions had a relatively small impact on NNRTI resistance (Table 1), but the combination of N348I and T369I was previously described as conferring a serious defect in viral replication capacity (28).
In agreement with previous reports, our results demonstrate that RT functions can be modulated by RT connection subdomain mutations despite the distant location of Asn348 and Thr369, relative to the DNA polymerase and RNase H active sites, and the NNRTI binding pocket. Several mechanisms have been proposed to explain how RT connection subdomain mutations could influence NNRTI resistance ((19,30,32,33); reviewed in (27)). Thus, mutations that affect template-primer binding could indirectly modulate NNRTI binding (e.g. A376S) (31). Reduced RNase H activity mediated by N348I and other mutations in its vicinity increases the stability of the RT/template-primer/NNRTI complex and therefore allows more time for dissociation of the NNRTI from the RT/template-primer binary complex (26). The correct positioning of the template-primer in the nucleic acid binding cleft may also have an influence on the dynamics of NNRTI binding. This is particularly important during the initiation of (+)-strand DNA synthesis where the PPT is used as a primer for DNA elongation and needs to be removed by the RNase H activity of the RT. The initiation of (+)-strand DNA synthesis is characterized by slow polymerization where high dNTP levels are needed for the incorporation of the first two nucleotides, followed by rapid dissociation of the RT and frequent kinetic pauses during primer extension (18,41).
Our data confirm that initiation of (+)-strand DNA synthesis during reverse transcription is significantly affected by N348I (19), but we also show that other individual mutations in the RT connection subdomain such as T369I or V or T376S do not seem to affect PPT removal. Nevertheless, we also found that T369I enhances the effects of N348I RT by producing a further reduction of its RNase H activity. The lower replication capacity of HIV-1 bearing RT mutations N348I/T369I (28) could be attributed to the reduced RNase H cleavage efficiency of the N348I/T369I RT observed in our studies with PPT-containing template-primers as well as conventional heteropolymeric hybrids. Nevertheless, nevirapine and efavirenz are strong enhancers of the PPT removal reaction (Figure 2A), while N348I and in a larger extent N348I/T369I counteract these effects. The results obtained with the WT RT and the single-mutant N348I RT in the presence of nevirapine and efavirenz are similar to those previously reported by Biondi et al. (19), although in their assays the suppressive effect of N348I was observed only with nevirapine. Differences between their results and ours could be attributed to the NNRTI concentration (a higher amount of efavirenz is needed to achieve maximum levels of PPT cleavage) or to amino acid sequence differences found in the RTs used in those experiments. Our results also show that rilpivirine has a small stimulatory effect on PPT removal during (+)-strand DNA synthesis in comparison with nevirapine and efavirenz, although this effect cannot be suppressed by RT connection subdomain mutations. In addition, recent studies have shown that N348I produces a moderate increase (around 2-fold) in the levels of rilpivirine resistance conferred by E138K, although the reduced DNA polymerase and RNase H activities of the N348I mutant restrict the emergence of the rilpivirine resistance mutation due to the diminished replication capacity of the virus (42).
The effects of nevirapine on PPT removal have been explained by its capacity to favor the interaction between the RT and the template-primer in a polymerase-independent binding mode (16). In their report, these authors also mention that similar effects were observed with efavirenz, although evidence of this behavior was not presented. By using a blunt-ended RNA/DNA template-primer with a recessed RNA 3′ end, we show that rilpivirine and efavirenz have a stronger tendency to adopt a polymerase-independent binding mode in comparison with nevirapine and etravirine, and this could facilitate the premature degradation of the PPT (Figure 4). However, the differences between WT and N348I/T369I RT were more pronounced with nevirapine. The stability of the PPT primer at high concentrations of nevirapine was lower in the presence of WT RT than in the presence of the double mutant. Another important conclusion of these experiments is that rilpivirine seems to be effective in promoting the degradation of the PPT and inhibiting its extension in the presence of dNTPs, being almost equally effective against the WT enzyme and the double mutant N348I/T369I. An intriguing observation relates to differences observed between the two closely related diarylpyrimidines. Unlike rilpivirine, etravirine has a minor effect on trimming the PPT primer. Both inhibitors are chemically similar, although they adopt a slightly different conformation in the NNRTI binding site (43–45) where the cyanovinyl moiety of rilpivirine makes extensive contacts with Phe227 and Trp229. These interactions, absent in the etravirine complex, affect the positioning of the primer at the DNA polymerase active site and could facilitate nucleic acid binding in a polymerase-independent orientation.
Our studies with RNA/DNA complexes of different lengths show for the first time that N348I influences the RNase H cleavage window and that this effect is enhanced by the presence of T369I. WT and N348I/T369I RTs had similar efficiencies and cleavage patterns when RNA products of 21 nucleotides were generated (i.e. resulting from cleavage at position –18). However, when the PPT sequence resistant to hydrolysis was made one or two nucleotides shorter, the differences in cleavage patterns between WT and N348I/T369I RTs became evident. In addition, experiments carried out with the 29/28mer hybrid showed that the double mutant had significantly reduced efficiency in the generation of shorter RNAs, in comparison with the WT RT or the single mutants T369I, T369V and T376S. The different cleavage windows finally result in the conservation of the PPT primer in the presence of nevirapine, etravirine and efavirenz, and a higher efficiency of (+)-strand DNA synthesis in reactions catalyzed by the double-mutant N348I/T369I.
A structural interpretation of these differences is difficult since both mutations map away from the DNA polymerase and RNase H active sites. However, Asn348 and Thr369 are surrounded by hydrophobic and aromatic residues in the HIV-1 RT structure and mutations to Ile in both cases could enhance hydrophobicity and make stronger interactions in p66 and p51. An enhanced rigidity of the mutant RT would be consistent with its reduced ability to cleave shorter RNAs. In agreement with this proposal, it has been suggested that a hydrogen bond between Tyr427 and Asn348 could be important in stabilizing the C-terminal region of p51, and this could have an impact on substrate positioning (46). The impact on drug resistance caused by mutations in the connection subdomain has been attributed to conformational changes that influence the orientation and positioning of the template-primer (22). Moreover, the likely increased rigidity caused by mutations such as N348I imposes constraints in the structure of the NNRTI binding pocket. As a result, subtle but significant changes in the DNA polymerase and RNase H active sites may occur, and allosteric effects could be responsible for the differences observed between mutant RTs in their susceptibility to NNRTIs (22,47,48).
Neither Asn348 nor Thr369 interact directly with the RNA/DNA hybrid (22,47,48), although Thr369 locates at α-helix K in p66 and through interactions with α-helix L in p51 (residues 394–404) could have an influence on the conformation of the DNA strand (22). The α-helix L of p51 is part of the so-called ‘RNase H primer grip’ (47). Mutations affecting residues of the RNase H primer grip often reduce polymerization-independent cleavage of the RNA template (49). A recent report has shown that the A400T mutation reduces RNase H activity while producing a small increase in resistance to nevirapine and efavirenz (50). Additional hydrogen bonding between the side chains of Thr400 and Glu396 has been proposed by those authors to explain changes in the conformation of the RNase H primer grip that could account for the altered RNase H activity of mutants affecting Ala400. Unlike mutations analyzed in our study whose prevalence in untreated patients is lower than 7.5%, A400T is a common polymorphism in HIV-1 group M isolates with a prevalence of nearly 56% in untreated patients. Modeling studies have shown little differences between ternary complexes containing the WT or the double-mutant N348I/T369I RT, associated with a DNA/DNA template-primer and nevirapine (51). However, it is clear from our studies that when associated with RNA/DNA complexes, the combination of mutations N348I and T369I has a major impact in the RNase H activity of the RT. These results are also in agreement with the notion that a number of distal connection subdomain mutations contribute to drug resistance by altering the dynamic properties of the RT and contact networks involving amino acid residues in the viral enzyme, identified by using molecular dynamics (51).
In summary, our studies show that in general all NNRTIs favor the premature removal of the PPT at the initiation of (+)-strand DNA synthesis. However, rilpivirine appears to be most effective at inhibiting PPT elongation, as well as decreasing its availability due to RNase H-mediated cleavage. Although N348I and more efficiently N348I/T369I could revert some of the effects of NNRTIs on PPT removal, their efficiency with rilpivirine is almost negligible. The decreased RNase H activity conferred by both mutations is apparently caused by a loss in the ability of the HIV-1 RT to render shorter RNA products, suggesting that those mutations could affect the rigidity of a hypothetical hinge located at the connection subdomain of the RT and linking the two functional domains of the enzyme. Our study provides insight on how connection subdomain mutations could affect the dynamics of coupling between HIV-1 RT activities, a mechanism that is relevant to understand antiretroviral drug resistance, and in a more general perspective the effects of distant mutations on the catalytic activity of multifunctional enzymes.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Footnotes
Present addresses:
Gilberto Betancor, Department of Infectious Diseases, King's College London, Guy's Hospital, London SE1 9RT, UK.
Barbara Marcelli, Department of Molecular Genetics, Groningen Biomolecular Sciences and Biotechnology Institute, University of Groningen, 9747 AG Groningen, The Netherlands.
Cristina Andrés, Laboratorio de Retrovirología e Inmunopatogenia Viral, Institut d'Investigacions Biomèdiques August Pi i Sunyer, 08036 Barcelona, Spain.
FUNDING
The Spanish Ministry of Economy and Competitiveness [BIO2010/15542 and BIO2013-48788-C2-1-R (to L.M.-A.); SAF2013-41421-R (to M.A.M.)]; the Spanish Ministry of Health, Social Services and Equality [EC11-025]; the Fundación Ramón Areces, Madrid, Spain (institutional grant to CBMSO). Funding for open access charge: The Spanish Ministry of Economy and Competitiveness [BIO2013-48788-C2-1-R].
Conflict of interest statement. None declared.
REFERENCES
- 1.Menéndez-Arias L. Targeting HIV: antiretroviral therapy and development of drug resistance. Trends Pharmacol. Sci. 2002;23:381–388. doi: 10.1016/s0165-6147(02)02054-0. [DOI] [PubMed] [Google Scholar]
- 2.Sarafianos S.G., Marchand B., Das K., Himmel D.M., Parniak M.A., Hughes S.H., Arnold E. Structure and function of HIV-1 reverse transcriptase: molecular mechanisms of polymerization and inhibition. J. Mol. Biol. 2009;385:693–713. doi: 10.1016/j.jmb.2008.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Menéndez-Arias L. Molecular basis of human immunodeficiency virus type 1 drug resistance: overview and recent developments. Antiviral Res. 2013;98:93–120. doi: 10.1016/j.antiviral.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 4.Herschhorn A., Hizi A. Retroviral reverse transcriptases. Cell. Mol. Life Sci. 2010;67:2717–2747. doi: 10.1007/s00018-010-0346-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matamoros T., Álvarez M., Barrioluengo V., Betancor G., Menéndez-Arias L. Reverse transcriptase and retroviral replication. In: Kušić-Tišma J, editor. DNA Replication and Related Cellular Process. Croatia: InTech, Rijeka; 2011. pp. 111–142. [Google Scholar]
- 6.Sluis-Cremer N. The emerging profile of cross-resistance among the nonnucleoside HIV-1 reverse transcriptase inhibitors. Viruses. 2014;6:2960–2973. doi: 10.3390/v6082960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kohlstaedt L.A., Wang J., Friedman J.M., Rice P.A., Steitz T.A. Crystal structure at 3.5 Å resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science. 1992;256:1783–1790. doi: 10.1126/science.1377403. [DOI] [PubMed] [Google Scholar]
- 8.Ren J., Esnouf R., Garman E., Somers D., Ross C., Kirby I., Keeling J., Darby G., Jones Y., Stuart D., et al. High resolution structures of HIV-1 RT from four RT-inhibitor complexes. Nat. Struct. Biol. 1995;2:293–302. doi: 10.1038/nsb0495-293. [DOI] [PubMed] [Google Scholar]
- 9.Ren J., Stammers D.K. Structural basis for drug resistance mechanisms for non-nucleoside inhibitors of HIV reverse transcriptase. Virus Res. 2008;134:157–170. doi: 10.1016/j.virusres.2007.12.018. [DOI] [PubMed] [Google Scholar]
- 10.Spence R.A., Kati W.M., Anderson K.S., Johnson K.A. Mechanism of inhibition of HIV-1 reverse transcriptase by nonnucleoside inhibitors. Science. 1995;267:988–993. doi: 10.1126/science.7532321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spence R.A., Anderson K.S., Johnson K.A. HIV-1 reverse transcriptase resistance to nonnucleoside inhibitors. Biochemistry. 1996;35:1054–1063. doi: 10.1021/bi952058+. [DOI] [PubMed] [Google Scholar]
- 12.Palaniappan C., Fay P.J., Bambara R.A. Nevirapine alters the cleavage specificity of ribonuclease H of human immunodeficiency virus 1 reverse transcriptase. J. Biol. Chem. 1995;270:4861–4869. doi: 10.1074/jbc.270.9.4861. [DOI] [PubMed] [Google Scholar]
- 13.Hang J.Q., Li Y., Yang Y., Cammack N., Mirzadegan T., Klumpp K. Substrate-dependent inhibition or stimulation of HIV RNase H activity by non-nucleoside reverse transcriptase inhibitors. Biochem. Biophys. Res. Commun. 2007;12:341–350. doi: 10.1016/j.bbrc.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 14.Radzio J., Sluis-Cremer N. Efavirenz accelerates HIV-1 reverse transcriptase ribonuclease H cleavage, leading to diminished zidovudine excision. Mol. Pharmacol. 2008;73:601–606. doi: 10.1124/mol.107.038596. [DOI] [PubMed] [Google Scholar]
- 15.Delviks-Frankenberry K.A., Nikolenko G.N., Pathak V.K. The ‘connection’ between HIV drug resistance and RNase H. Viruses. 2010;2:1476–1503. doi: 10.3390/v2071476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abbondanzieri E.A., Bokinsky G., Rausch J.W., Zhang J.X., Le Grice S.F.J., Zhuang X. Dynamic binding orientations direct activity of HIV reverse transcriptase. Nature. 2008;453:184–189. doi: 10.1038/nature06941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu S., Abbondanzieri E.A., Rausch J.W., Le Grice S.F.J., Zhuang X. Slide into action: dynamic shuttling of HIV reverse transcriptase on nucleic acid substrates. Science. 2008;322:1092–1097. doi: 10.1126/science.1163108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grobler J.A., Dornadula G., Rice M.R., Simcoe A.L., Hazuda D.J., Miller M.D. HIV-1 reverse transcriptase plus-strand initiation exhibits preferential sensitivity to non-nucleoside reverse transcriptase inhibitors in vitro. J. Biol. Chem. 2007;282:8005–8010. doi: 10.1074/jbc.M608274200. [DOI] [PubMed] [Google Scholar]
- 19.Biondi M.J., Beilhartz G.L., McCormick S., Götte M. N348I in HIV-1 reverse transcriptase can counteract the nevirapine-mediated bias toward RNase H cleavage during plus-strand initiation. J. Biol. Chem. 2010;285:26966–26975. doi: 10.1074/jbc.M110.105775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gopalakrishnan V., Benkovic S. Effect of a thiobenzimidazolone derivative on DNA strand transfer catalyzed by HIV-1 reverse transcriptase. J. Biol. Chem. 1994;269:4110–4115. [PubMed] [Google Scholar]
- 21.Shaw-Reid C.A., Feuston B., Munshi V., Getty K., Krueger J., Hazuda D.J., Parniak M.A., Miller M.D., Lewis D. Dissecting the effects of DNA polymerase and ribonuclease H inhibitor combinations on HIV-1 reverse-transcriptase activities. Biochemistry. 2005;44:1595–1606. doi: 10.1021/bi0486740. [DOI] [PubMed] [Google Scholar]
- 22.Lapkouski M., Tian L., Miller J.T., Le Grice S.F.J., Yang W. Complexes of HIV-1 RT, NNRTI and RNA/DNA hybrid reveal a structure compatible with RNA degradation. Nat. Struct. Mol. Biol. 2013;20:230–236. doi: 10.1038/nsmb.2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nikolenko G.N., Palmer S., Maldarelli F., Mellors J.W., Coffin J.M., Pathak V.K. Mechanism for nucleoside analog-mediated abrogation of HIV-1 replication: balance between RNase H activity and nucleotide excision. Proc. Natl. Acad. Sci. U.S.A. 2005;102:2093–2098. doi: 10.1073/pnas.0409823102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Delviks-Frankenberry K.A., Nikolenko G.N., Barr R., Pathak V.K. Mutations in human immunodeficiency virus type 1 RNase H primer grip enhance 3′-azido-3′-deoxythymidine resistance. J. Virol. 2007;81:6837–6845. doi: 10.1128/JVI.02820-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hachiya A., Kodama E.N., Sarafianos S.G., Schuckmann M.M., Sakagami Y., Matsuoka M., Takiguchi M., Gatanaga H., Oka S. Amino acid mutation N348I in the connection subdomain of human immunodeficiency virus type 1 reverse transcriptase confers multiclass resistance to nucleoside and nonnucleoside reverse transcriptase inhibitors. J. Virol. 2008;82:3261–3270. doi: 10.1128/JVI.01154-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nikolenko G.N., Delviks-Frankenberry K.A., Pathak V.K. A novel molecular mechanism of dual resistance to nucleoside and nonnucleoside reverse transcriptase inhibitors. J. Virol. 2010;84:5238–5249. doi: 10.1128/JVI.01545-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Menéndez-Arias L., Betancor G., Matamoros T. HIV-1 reverse transcriptase connection subdomain mutations involved in resistance to approved non-nucleoside inhibitors. Antiviral Res. 2011;92:139–149. doi: 10.1016/j.antiviral.2011.08.020. [DOI] [PubMed] [Google Scholar]
- 28.Gupta S., Fransen S., Paxinos E.E., Stawiski E., Huang W., Petropoulos C.J. Combinations of mutations in the connection domain of human immunodeficiency virus type 1 reverse transcriptase: assessing the impact on nucleoside and nonnucleoside reverse transcriptase inhibitor resistance. Antimicrob. Agents Chemother. 2010;54:1973–1980. doi: 10.1128/AAC.00870-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gupta S., Vingerhoets J., Fransen S., Tambuyzer L., Azijn H., Frantzell A., Paredes R., Coakley E., Nijs S., Clotet B., et al. Connection domain mutations in HIV-1 reverse transcriptase do not impact etravirine susceptibility and virologic responses to etravirine-containing regimens. Antimicrob. Agents Chemother. 2011;55:2872–2879. doi: 10.1128/AAC.01695-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yap S.-H., Sheen C.-W., Fahey J., Zanin M., Tyssen D., Lima V.D., Wynhoven B., Kuiper M., Sluis-Cremer N., Harrigan P.R., et al. N348I in the connection domain of HIV-1 reverse transcriptase confers zidovudine and nevirapine resistance. PLoS Med. 2007;4:e335. doi: 10.1371/journal.pmed.0040335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paredes R., Puertas M.C., Bannister W., Kisic M., Cozzi-Lepri A., Pou C., Bellido R., Betancor G., Bogner J., Gargalianos P., et al. A376S in the connection subdomain of HIV-1 reverse transcriptase confers increased risk of virological failure to nevirapine therapy. J. Infect. Dis. 2011;204:741–752. doi: 10.1093/infdis/jir385. [DOI] [PubMed] [Google Scholar]
- 32.Schuckmann M.M., Marchand B., Hachiya A., Kodama E.N., Kirby K.A., Singh K., Sarafianos S.G. The N348I mutation at the connection subdomain of HIV-1 reverse transcriptase decreases binding to nevirapine. J. Biol. Chem. 2010;285:38700–38709. doi: 10.1074/jbc.M110.153783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ehteshami M., Beilhartz G.L., Scarth B.J., Tchesnokov E.P., McCormick S., Wynhoven B., Harrigan P.R., Götte M. Connection domain mutations N348I and A360V in HIV-1 reverse transcriptase enhance resistance to 3′-azido-3′-deoxythymidine through both RNase H-dependent and -independent mechanisms. J. Biol. Chem. 2008;283:22222–22232. doi: 10.1074/jbc.M803521200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matamoros T., Deval J., Guerreiro C., Mulard L., Canard B., Menéndez-Arias L. Suppression of multidrug-resistant HIV-1 reverse transcriptase primer unblocking activity by α-phosphate-modified thymidine analogues. J. Mol. Biol. 2005;349:451–463. doi: 10.1016/j.jmb.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 35.Boretto J., Longhi S., Navarro J.-M., Selmi B., Sire J., Canard B. An integrated system to study multiply substituted human immunodeficiency virus type 1 reverse transcriptase. Anal. Biochem. 2001;292:139–147. doi: 10.1006/abio.2001.5045. [DOI] [PubMed] [Google Scholar]
- 36.Kati W.M., Johnson K.A., Jerva L.F., Anderson K.S. Mechanism and fidelity of HIV reverse transcriptase. J. Biol. Chem. 1992;267:25988–25997. [PubMed] [Google Scholar]
- 37.Betancor G., Puertas M.C., Nevot M., Garriga C., Martínez M.A., Martinez-Picado J., Menéndez-Arias L. Mechanisms involved in the selection of HIV-1 reverse transcriptase thumb subdomain polymorphisms associated with nucleoside analogue therapy failure. Antimicrob. Agents Chemother. 2010;54:4799–4811. doi: 10.1128/AAC.00716-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kellam P., Larder B.A. Recombinant virus assay: a rapid, phenotypic assay for assessment of drug susceptibility of human immunodeficiency virus type 1 isolates. Antimicrob. Agents Chemother. 1994;38:23–30. doi: 10.1128/aac.38.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huber H.E., Richardson C.C. Processing of the primer for plus strand DNA synthesis by human immunodeficiency virus 1 reverse transcriptase. J. Biol. Chem. 1990;265:10565–10573. [PubMed] [Google Scholar]
- 40.Rausch J.W., Le Grice S.F.J. ‘Binding, bending and bonding’: polypurine tract-primed initiation of plus-strand DNA synthesis in human immunodeficiency virus. Int. J. Biochem. Cell Biol. 2004;36:1752–1766. doi: 10.1016/j.biocel.2004.02.016. [DOI] [PubMed] [Google Scholar]
- 41.Götte M., Kameoka M., McLellan N., Cellai L., Wainberg M.A. Analysis of efficiency and fidelity of HIV-1 (+)-strand DNA synthesis reveals a novel rate-limiting step during retroviral reverse transcription. J. Biol. Chem. 2001;276:6711–6719. doi: 10.1074/jbc.M009097200. [DOI] [PubMed] [Google Scholar]
- 42.Xu H.T., Colby-Germinario S.P., Oliveira M., Han Y., Quan Y., Zanichelli V., Wainberg M.A. The connection domain mutation N348I in HIV-1 reverse transcriptase enhances resistance to etravirine and rilpivirine but restricts the emergence of the E138K resistance mutation by diminishing viral replication capacity. J. Virol. 2014;88:1536–1547. doi: 10.1128/JVI.02904-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Das K., Clark A.D., Jr, Lewi P.J., Heeres J., De Jonge M.R., Koymans L.M., Vinkers H.M., Daeyaert F., Ludovici D.W., Kukla M.J., et al. Roles of conformational and positional adaptability in structure-based design of TMC125-R165335 (etravirine) and related non-nucleoside reverse transcriptase inhibitors that are highly potent and effective against wild-type and drug-resistant HIV-1 variants. J. Med. Chem. 2004;47:2550–2560. doi: 10.1021/jm030558s. [DOI] [PubMed] [Google Scholar]
- 44.Das K., Bauman J.D., Clark A.D. Jr, Frenkel Y.V., Lewi P.J., Shatkin A.J., Hughes S.H., Arnold E. High-resolution structures of HIV-1 reverse transcriptase/TMC278 complexes: strategic flexibility explains potency against resistant mutations. Proc. Natl. Acad. Sci. U.S.A. 2008;105:1466–1471. doi: 10.1073/pnas.0711209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lansdon E.B., Brendza K.M., Hung M., Wang R., Mukund S., Jin D., Birkus G., Kutty N., Liu X. Crystal structures of HIV-1 reverse transcriptase with etravirine (TMC125) and rilpivirine (TMC278): implications for drug design. J. Med. Chem. 2010;53:4295–4299. doi: 10.1021/jm1002233. [DOI] [PubMed] [Google Scholar]
- 46.Chung S., Miller J.T., Lapkouski M., Tian L., Yang W., Le Grice S.F.J. Examining the role of the HIV-1 reverse transcriptase p51 subunit in positioning and hydrolysis of RNA/DNA hybrids. J. Biol. Chem. 2013;288:16177–16184. doi: 10.1074/jbc.M113.465641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sarafianos S.G., Das K., Tantillo C., Clark A.D. Jr, Ding J., Whitcomb J.M., Boyer P.L., Hughes S.H., Arnold E. Crystal structure of HIV-1 reverse transcriptase in complex with a polypurine tract RNA:DNA. EMBO J. 2001;20:1449–1461. doi: 10.1093/emboj/20.6.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Das K., Martinez S.E., Bauman J.D., Arnold E. HIV-1 reverse transcriptase complex with DNA and nevirapine reveals non-nucleoside inhibition mechanism. Nat. Struct. Mol. Biol. 2012;19:253–259. doi: 10.1038/nsmb.2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rausch J.W., Lener D., Miller J.T., Julias J.G., Hughes S.H., Le Grice S.F.J. Altering the RNase H primer grip of human immunodeficiency virus reverse transcriptase modifies cleavage specificity. Biochemistry. 2002;41:4856–4865. doi: 10.1021/bi015970t. [DOI] [PubMed] [Google Scholar]
- 50.Wright D.W., Deuzing I.P., Flandre P., van den Eede P., Govaert M., Setiawan L., Coveney P.V., Marcelin A.-G., Calvez V., Boucher C.A.B., et al. A polymorphism at position 400 in the connection subdomain of HIV-1 reverse transcriptase affects sensitivity to NNRTIs and RNase H activity. PLoS One. 2013;8:e74078. doi: 10.1371/journal.pone.0074078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vijayan R.S., Arnold E., Das K. Molecular dynamics study of HIV-1 RT-DNA-nevirapine complexes explains NNRTI inhibition and resistance by connection mutations. Proteins. 2014;82:815–829. doi: 10.1002/prot.24460. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.