


Abstract
The efficiency of Nucleotide Excision Repair (NER)process is crucial for maintaining genomic integrity because in many organisms, including humans, it represents the only system able to repair a wide range of DNA damage. The aim of the work was to investigate whether the efficiency of the repair of photoproducts induced by UV-light is affected by the circadian phase at which irradiation occurred. NER activity has been analyzed in human quiescent fibroblasts (in the absence of the cell cycle effect), in which circadian rhythmicity has been synchronized with a pulse of dexamethasone. Our results demonstrate that both DNA damage induction and repair efficiency are strictly dependent on the phase of the circadian rhythm at which the cells are UV-exposed. Furthermore, the differences observed between fibroblasts irradiated at different circadian times (CTs) are abolished when the clock is obliterated. In addition, we observe that chromatin structure is regulated by circadian rhythmicity. Maximal chromatin relaxation occurred at the same CT when photoproduct formation and removal were highest. Our data suggest that the circadian clock regulates both the DNA sensitivity to UV damage and the efficiency of NER by controlling chromatin condensation mainly through histone acetylation.
INTRODUCTION
The efficient repair of DNA damage stemming from endogenous cellular byproducts and dangerous environmental exposures, such as the ultraviolet (UV) component of sunlight, is crucial for maintaining genomic stability and preventing cancer initiation and progression. The two major classes of UV-induced DNA lesions are cyclobutane pyrimidine dimers (CPDs) and pyrimidine (6-4) pirimidone photoproducts (6-4PPs), both of which play an important role in skin aging and skin cancer (1,2). Dimer formation triggers a complex cellular process, the DNA damage response (DDR), which includes checkpoint activation, chromatin remodeling, DNA repair and/or apoptosis. In many organisms including humans, the Nucleotide Excision Repair (NER) represents the only system that is able to repair a wide range of DNA adducts (3). The system can be broadly divided into two major sub pathways: the global genome NER (GG-NER), which is responsible for repairing lesions throughout the entire genome, and the transcription-coupled NER (TC-NER), which specifically repairs DNA lesions of active genes recognized by stalled RNA polymerase II (4,5). CPDs, which represent 70–80% of the UV damage, induce a kink of 7–9° in the DNA helix, mainly within nucleosomes, as opposed to 6-4PPs, the 20–30% of photoproducts, which produce a bend of 44° mainly in the inter-nucleosome linker (6). Probably due to the features of the two kinds of photoproducts, the removal of CDPs occurs slowly compared with that of 6-4PPs. Despite differences in the efficiency of recognizing photoproducts, the removal of dimers proceeds in the same way through a dual incision of DNA around the damaged site and the consequent excision of a 27–30 nucleotide oligomer in a process carried out by six excision repair factors: RPA, Xeroderma pigmentosum group A (XPA), XPC, XPG, XPF-ERCC1 and TFIIH (7). In the end, the gap created is filled by a process that requires DNA polymerases δ or ϵ as well as the accessory replication proteins and a correctly balanced pool of dNTPs (8,9).
A growing number of studies have recently demonstrated that the circadian clock is involved in the control of the DDR. It has been reported that circadian clock components, such as BMAL1-CLOCK, PER1, PER2, PER3 and RORα are involved in controlling the cellular response to genotoxic stress (10–13). Beyond the regulatory connection between the clock and UV-induced DNA damage repair, it has been reported that NER displays a circadian rhythm in mice, possibly through oscillations in the expression of XPA protein, the DNA damage recognition protein for this pathway (2). Since XPA is involved in the first step of NER and represents the rate-limiting factor, a time-dependent variation in its relative abundance resulted, at least at the in vivo level, in an impaired DNA repair capability when UV exposure occurred in anti-phase with its expression (2,10,14).
It has become increasingly clear that chromatin remodeling is one of the processes through which the circadian clock regulates gene transcription (15). Histone acetylation is a marker for transcription activation which is achieved by remodeling the chromatin to make it more accessible to the transcription machinery (16). Histone methylation, on the other hand, acts as a signal for recruitment of chromatin remodeling factors, which can either activate or repress transcription (15). The key molecule for this epigenetic control of gene expression is CLOCK, a central component of the circadian pacemaker, recently found to have histone acetyltransferase (HAT) activity, essential for circadian clock-controlled gene expression. CLOCK is able to acetylate the lysines 9 and 14 of histone H3, stimulating the opening of the chromatin and promoting gene transcription. BMAL1, the heterodimerization partner of CLOCK, seems to be involved in enhancing the HAT function (17). The histone deacetyalse sirtuin 1 (SIRT1) was, moreover, found to regulate circadian rhythms by counteracting the HAT activity of CLOCK (18). SIRT1 operates, thus, as a rheostat of the circadian machinery, modulating the amplitude and ‘tightness’ of CLOCK-mediated acetylation and the consequent transcription cycles in metabolically active tissues. Some investigators have recently reported that the most pervasive circadian regulation observed at the genome scale are rhythms in H3K4me3, H3K9ac, H3K27ac and RNAPII recruitment and transcription initiation, which occur at thousands of expressed genes regardless of whether mRNA metabolism was detectable (19). This demonstrates that gene expression per se exhibits circadian rhythms with temporally separated activation and repression phases.
In this study, we have analyzed the NER capacity in cultures of human primary fibroblasts, in which circadian rhythms have been synchronized by applying a pulse of dexamethasone. Our aim was to investigate the role of circadian clock in regulating the repair efficiency of DNA damage induced by UV-light in quiescent cells, without the influence of cell cycle progression. Our results indicate that both DNA sensitivity to UV-induced damage and the efficiency of photoproduct removal are strictly dependent on the circadian time (CT) at which the cells were exposed to UV-light. In addition, we found that chromatin structure was regulated by circadian rhythmicity and the maximum chromatin relaxation occurred at the same CT at which photoproduct removal was most efficient, showing that circadian clock regulates NER activity by controlling levels of chromatin condensation.
MATERIALS AND METHODS
Cell cultures and circadian clock synchronization
Normal human skin fibroblasts, C63 cells, were cultured as previously described (20). Quiescent cultures were obtained by growing the cells to confluence, changing the medium to Dulbecco's modified Eagle's medium (DMEM)-0.1% , and keeping the cells in this condition for further 7–10 days. Cultures received fresh medium every 3–4 days. Quiescence was analyzed by flow cytometry after propidium iodide staining as previously described (21). Other normal human skin fibroblasts, derived from a healthy newborn donor, were cultured in the same conditions of C63 cells. Mouse dermal fibroblasts (MDF) wild-type and homozygous mutant BMAL1m/m and REV-ERBαm/m MDFs were present in the laboratory of U.A. (22). MDFs were grown in the same conditions of C63 cells. Circadian rhythmicity of clock gene expression was obtained by addition of DMEM-0.1% Fetal bovine serum (FBS) supplemented with 100 nM dexamethasone (Sigma-Aldrich). After 2 h the medium was removed and cells were washed two times with fresh medium w/o serum. After the dexamethasone shock, quiescent C63 cells were incubated again with DMEM-0.1% FBS.
Cell treatments
After dexamethasone pulse, the cells were irradiated with a 254-nm UVS-11 mineral light lamp at a fluency rate of 1.5 J/m2/s. Before irradiation, the culture medium was removed and fresh medium was added after irradiation. When indicated, sodium butyrate (5 mM, Sigma-Aldrich) was added immediately after synchronization and maintained throughout the experiments. In experiments performed to analyze chromatin condensation, as positive controls were used cells treated with 300 mM H2O2 for 30 min or with 5 mM NaBu for 24 h. Cells were fixed, treated with 0.1N HCl, stained with AO at pH 2.6 and their fluorescence measured by flow cytometry (see below).
RNA extraction, reverse transcription and real-time PCR
Total RNA was extracted from cells using TRIzol reagent (Life Technologies) according to the manufacturer's instructions. The quality and quantity of RNA were assessed by NanoDrop 1000 spectrophotometer (Thermo Scientific) and by denaturing agarose gel electrophoresis with ethidium bromide staining. Total RNA (1 μg) was transcribed to cDNAs using the ImProm-II™ Reverse Transcription System (Promega) and random hexamers (Life Technologies). Transcript levels were detected in 25 ng of cDNA by Sybr Green GoTaq® qPCR Master Mix (Promega). Reactions were performed in quadruplicate in 96-well optical plates with a 7500 real-time PCR system (Applied Biosystems). GAPDH and RPL32 were used as endogenous control for normalizations. Primers list is reported in Supplementary Information.
Cell extracts and western blot
Whole cell extracts were obtained by lysing the cells in Radioimmunoprecipitation assay (RIPA) buffer. The cytosolic and nuclear fraction were obtained as described in (23). Briefly the cells were passed through a needle (26G × 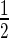 ’) in hypotonic extraction buffer supplemented with protease inhibitor cocktail (Sigma-Aldrich P8340) and phosphatase inhibitor (PhosSTOP, Roche 04906845001). The cytosolic fraction was separated by centrifugation and nuclei were lysed in RIPA buffer containing protease and phosphatase inhibitor cocktail. Histone enriched fraction was obtained by incubating the nuclear pellet with 0.2 N HCl overnight on ice. The western blot procedure and the antibody list are reported in Supplementary Information.
’) in hypotonic extraction buffer supplemented with protease inhibitor cocktail (Sigma-Aldrich P8340) and phosphatase inhibitor (PhosSTOP, Roche 04906845001). The cytosolic fraction was separated by centrifugation and nuclei were lysed in RIPA buffer containing protease and phosphatase inhibitor cocktail. Histone enriched fraction was obtained by incubating the nuclear pellet with 0.2 N HCl overnight on ice. The western blot procedure and the antibody list are reported in Supplementary Information.
Analysis of γ-H2AX
Flow cytometer analysis of γ-H2AX were performed as previously described (21,23). Data were collected from 25 000 cells/sample by FACSCanto™ II flow cytometer (Becton Dickinson) and analyzed with FACSDiva™ software (Becton Dickinson).
Analysis of 6-4PPs and CPDs after UV irradiation
Dot blot and were performed to determine the relative amounts of 6-4PPs and CPDs in total genomic DNA from cells collected at different times after UV irradiation. Dot-Blot analysis was performed as previously described (9). For Enzyme-Linked Immunosorbent Assay (ELISA) assay the detailed procedure is described in Supplementary Information.
Flourometric analysis of DNA unwinding (FADU)
Flourometric analysis of DNA unwinding (FADU) was performed according to Birnboim et al. (24) as previously described (9).
Unscheduled DNA synthesis (UDS)
Immediately after irradiation, the medium of quiescent C63 cells was substituted with pre-warmed DMEM-0.1% FBS supplemented with 0.3 μM of [3H]-TdR (specific activity 20 000 CPM). At fixed time-points cells were harvested, centrifuged at 4°C and washed twice with cold phosphate buffered saline (PBS). Pellets (0.6 × 106 cells) were thus dissolved in 150 μl of lysis buffer (10 mM Tris–HCl pH 7.4, 10 mM NaCl, 5% sodium dodecyl sulphate and 10 mM ethylenediaminetetraacetic acid (EDTA)). Lysates were spotted in glass microfiber GF/C filters and dried 10 min under infrared lamp. Filters were washed five times in 5% trichloroacetic acid, two times in 100% ethanol and dried again under infrared lamp. Filters were placed inside scintillation tubes with 4 ml of scintillation liquid (ultima gold/F, Perkin Elmer). Radioactivity of samples was reported as count per minute/106 cells (CPM/106 cells).
Transfection with siRNAs
Confluent C63 cells were transfected with 30 nM of Stealth RNAi™ siRNAs anti-Bmal1 (HSS100704 and HSS180959, Life Technologies) or siRNA negative control (Life Technologies) in DMEM-0.1% FBS without antibiotics using RNAiMAX (Life Technologies). After 3 days medium was diluted 1:1 with fresh DMEM-0.1% FBS and cells were incubated for additional 3 days in presence of 15 nM siRNA. Fibroblasts were thus transfected a second time with 30 nM of siRNA in DMEM-0.1% FBS without antibiotics (25). After 24 h cells medium was exchanged with DMEM-0.1% FBS supplemented with 100 nm dexamethasone for 2 h, washed twice and incubated in the previous medium containing siRNA until UV irradiation.
Micrococcal nuclease assay
The cells were harvested and washed twice with PBS supplemented with 5 mM Sodium butyrate (NaBu). Nuclei were isolated from cytoplasm by passing cells through a fine needle in hypotonic extraction buffer, supplemented with protease inhibitor cocktail and 5 mM NaBu, followed by sucrose cushion centrifugation (1.2 M sucrose, 5 mM NaBu, 0.1 mM phenylmethylsulfonyl fluoride (PMSF), 0.5 mM dithiothreitol (DTT), 60 mM KCl, 15 mM NaCl, 5 mM MgCl2, 0.1 mM EDTA, 15 mM Tris–HCl pH 7.5) at 3500 g for 1 h at 4°C. Nuclei were, thus, resuspended in 500 μl of MNase digestion buffer (0.3 M sucrose, 5 mM NaBu, 0.2 mM PMSF, 4 mM MgCl2, 1 mM CaCl2, 50 mM Tris–HCl pH 7.5) containing 15 U of microccocal nuclease (New England Biolabs) and incubated at 37°C. After 0, 4, 5, 6 and 8 min 90 μl of treated nuclei were transferred on ice in a new tube containing 4 μl of 0.5 M EDTA. Samples were centrifuged and the digested DNA were purified by phenol–chloroform extraction. DNA was dissolved in TE buffer (10 mM Tris–HCl, 1 mM EDTA, pH 7.5) and quantified by using NanoDrop 1000 spectrophotometer. Hundred nanograms of DNA was electrophoresed on 2% agarose Tris-borate-EDTA gel with 0.5 μg/ml EtBr and analyzed by using a Gel Doc XR system (Bio-Rad).
Acridine orange assay
The cells were harvested, washed twice in PBS supplemented with 1 mM MgCl2, and fixed in 70% ethanol overnight at 4°C. Cells were thus centrifuged, washed with PBS and incubated with 100 μg/ml RNase A (Sigma-Aldrich) for 1 h at 37°C. Subsequently samples were centrifuged and cells were treated with 250 μl of 0.1 M HCl for 30 s followed by 1 ml of 0.1 M phosphate-citrate buffer (pH 2.6) containing 6 μg/ml of Acridine/Orange at room temperature (26,27). Green and red mean fluorescence intensity were measured in 25 000 cells using a FACSCanto™ II flow cytometer (Becton Dickinson). The fraction of dsDNA was calculated as FdsDNA = Green/(Green + Red) (28).
Statistical analysis
Statistical analysis was performed using the Prism 5 software (Graphpad). Figures were prepared using Corel Draw X6 (Corel corporation).
RESULTS
Photoproduct removal in dexamethasone-shocked fibroblasts
We investigated the role of circadian clock in regulating the repair of UV-induced DNA damage in the C63 human skin fibroblast cell line. Since previous experimental evidence in proliferating cells (29, 30) suggested a correlation between cell cycle phases and DNA repair efficiency, we decided to perform the experiments with quiescent cells in order to separate the cell cycle from the circadian cycle. C63 cells were grown to confluency and cultured in 0.1% FBS for an additional 7 days to reach quiescence. This was confirmed by flow cytometry analyses (Supplementary Figure S1). We then treated the cells with a 2 h pulse of dexamethasone in order to induce the rhythmic expression of clock genes. This method, which has already been used to synchronize clock gene expression in different types of cycling cells (22,31,32), here has been successfully applied to quiescent cells, as proved by the time-course of Bmal1 and Per2 gene expression (Figure 1). Per2 mRNA had two troughs at the CTs 16 and 40 (hours after dexamethasone pulse) and a peak at CT28, with an overall period of about 24 h. Bmal1 mRNA exhibited two transcriptional oscillations in anti-phase with Per2, with a trough at CT 24–28 and a peak at CT 36–40. The rhythmicity in qRT-PCR was confirmed by western blot analysis, where BMAL1 and PER2 protein levels were quantified in the CT 12–44 window (Figure 1A–C). Using specific antibodies we investigated whether the capacity of NER to remove the two main UV photoproducts, the CPDs and the 6-4 pyrimidone photoproducts (6-4PPs), differed in relation to the rhythmic expression of BMAL1 and PER2 genes. The dot-blot assay performed on quiescent fibroblasts, irradiated with UV light (12 J/m2) every 4 h at CT 12–36, showed that the efficiency of 6-4PP removal analyzed 1 h after irradiation oscillated in phase with BMAL1 (Figure 1D), with a peak at CT16 and a trough at CT28, in agreement with results obtained in vivo by other authors (2,7,10).
Figure 1.
Circadian rhythmicity and DNA repair efficiency. (A) Relative levels of Per2 and Bmal1 mRNAs in C63 cells were analyzed every 4 h after dexamethasone pulse during an interval of 32 h. (B) Representative western blot depicting the amounts of PER2 and BMAL1 proteins. Ponceau staining served as a loading control. (C) Relative levels of BMAL1 and PER2 proteins measured every 4 h after dexamethasone pulse. (D) Removal of 6-4PP photoproducts was determined by Dot-blot assay every 4 h after dexamethasone pulse. All the values in (A) and (C) are the mean ± SEM of three (Per2) or four (Bmal1) independent experiments; values in (D) are the mean ± SEM of three independent experiments.
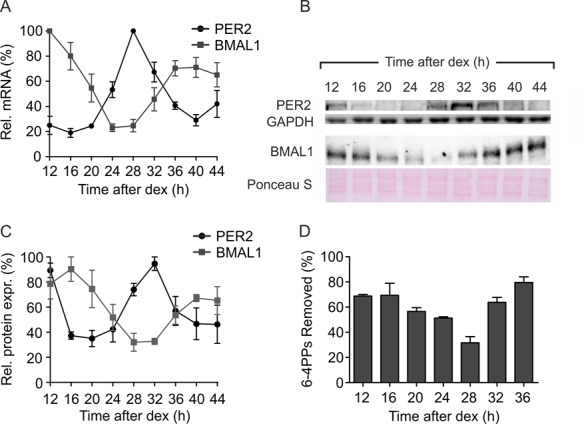
On the basis of these results, we selected two CTs separated by a 12 h interval, CT16 and CT28, so that UV irradiation was administered at two opposite extremes of BMAL1 and PER2 protein expression. When the cells were irradiated at these two CTs, the removal of 6-4PPs was almost completed within 3 h in both groups, with a significantly faster kinetics in CT16 cells compared to CT28 cells (Figure 2A and B). The difference was marked at 0.5 h after irradiation (5% versus 44% of 6-4PPs removed, CT28 versus CT16) and persisted up to 1 h after irradiation (32 versus 69% of 6-4PPs removed, CT28 versus CT16). Previous data (2,9,33,34) demonstrated that CPDs, which represent the majority of UV photoproducts, were removed more slowly than 6-4PPs. This is in agreement with our results showing that in C63 cells CPDs are very inefficiently repaired and 40% of CPDs persisted 24 h after irradiation. Due to the slow repair kinetics of CPDs in C63 cells no significant difference between CT16 and CT28 irradiated cells were observed (Supplementary Figure S2). We then quantified the phosphorylation on S139 of histone H2AX (γ-H2AX) by flow cytometry during a period of 24 h following irradiation. In CT16 cells the signal of γ-H2AX started to increase at 0.5 h, reached a peak at 4 h and decreased until 24 h, at which time the fluorescence intensity was similar to that of non-irradiated cells. The kinetics of γ-H2AX fluorescence signal in CT28-irradiated fibroblasts followed the same parabolic trend, but with significantly higher values, that may be due to the longer persistence of DNA damages when the cells were irradiated at this CT (Figure 2C).
Figure 2.
DNA repair in C63 cells irradiated with UV light (12 J/m2) at CT16 and CT28. (A) Percentage of 6-4PP photoproducts removed within 3 h of post-irradiation incubation. Insert: representative Dot-blot of 6-4PPs remaining at different times after irradiation. (B) Fluorescence intensity of γ-H2AX during a 24 h of post-irradiation incubation. (C) Percentage of dsDNA during 24 h of post-irradiation incubation was determined by FADU assay, which measures the fluorescence of EtBr bound to alkali-treated DNA. (D) DNA resynthesis has been determined by unscheduled DNA synthesis (UDS) assay. The results are plotted as mean ± SEM obtained in three (A, C and D) or four (B) independent experiments. The P-value refers to a 2-way ANOVA. ***P < 0.001, **P < 0.01, *P < 0.05, CT16 versus CT28, Bonferroni post-hoc test.
To better understand the difference in repair kinetics of CT16 and CT28 irradiated cells we analyzed the efficiency of the second step of the NER process that consists in restoring the integrity of DNA molecule filling the 27–30 nucleotide gap created by the dimer removal. Prior to the ligation step, alkali treatment of the nicked DNA produces single-stranded regions at the sites of the initial damage. The amount of double-stranded DNA (dsDNA) provides, thus, a measure of the ongoing, although incomplete, repair process (35,36). This parameter can be quantified by FADU from the fluorescence of DNA-bound EtBr. In CT16 and CT28 irradiated cells, fluorescence intensity dropped immediately after irradiation as a consequence of the rapid loss of dsDNA caused by the initial step of the repair process. Interesting differences in dsDNA recovery were noted between the two groups of cells. First, the cells irradiated at CT28 contained less dsDNA at all time-points with respect to the CT16 cells (Figure 2C), suggesting that slower repair kinetics reflected a higher residual damage. Second, after the initial drop in dsDNA percentage, the CT16 group began a rapid recovery, as opposed to the CT28 one in which the quantity of dsDNA was unaltered for up to 1 h after irradiation, due to the slower kinetics of 6-4PPs removal and/or defective DNA re-synthesis (Figure 2C). Finally, after 6 h from irradiation CT28 cells contained only 68% of dsDNA, as opposed to 88% in CT16 cells (Figure 2C). The DNA resynthesis step of NER was analyzed by the unscheduled DNA synthesis (UDS) assay, which showed a significantly higher incorporation of [3H]-TdR in CT16-irradiated fibroblasts within the first 30 min after irradiation with respect to CT28 cells (Figure 2D). However, the incorporation of [3H]-TdR increased over time together with the progression of DNA repair and no differences between the two groups were detected at later times (45 and 60 min from irradiation).
The circadian clock modulates UV sensitivity and UV-damage repair
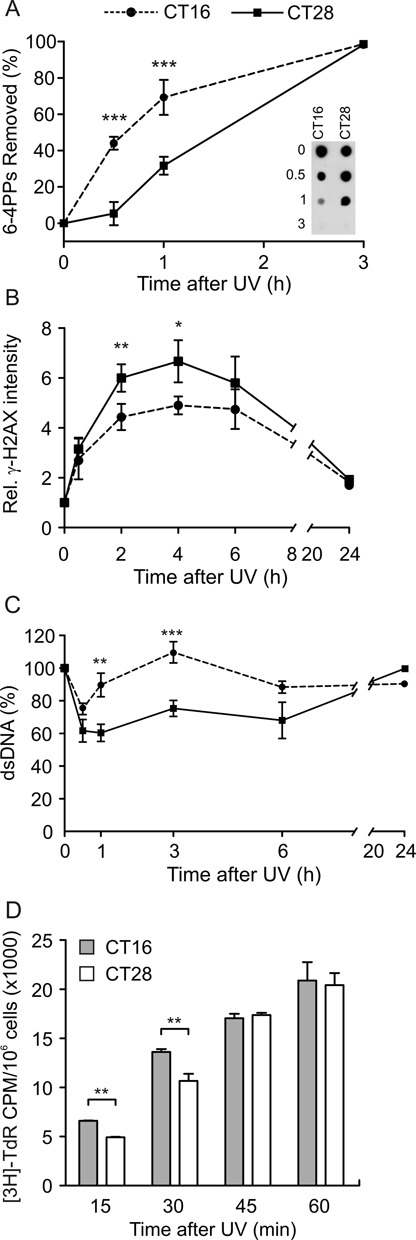
We next investigated whether, the sensitivity to UV light was regulated by the circadian rhythmicity. To this end, the cells were transfected with siRNAs to interfere with Bmal1 gene expression. In those experimental conditions, fibroblasts were found to become arrhythmic, even after the dexamethasone shock, as demonstrated by the low and similar level of Per2 and Rev-Erb a gene expression at both CT16 and CT28 (Supplementary Figure S3). Fibroblasts transfected with siBmal1 or siCTRL were irradiated at CTs 16 and 28 and photoproduct formation was analyzed immediately after UV exposure. As expected, in fibroblasts transfected with siCTRL, which retained an intact circadian rhythmicity, the difference in the amount of 6-4PPs and CPDs formed at the two CTs of irradiation persisted. In contrast, it was totally abrogated in cells transfected with siBmal1 (Figure 3A). Interestingly, by analyzing the removal of 6-4PP photoproducts at early times after irradiation (0.5 and 1 h), we found that fibroblasts irradiated at CT28 (CT28 siCTRL) exhibited a delay in the damage removal (Figure 3B). Half an hour after irradiation, no removal activity was observed in those cells in contrast to the 47% of 6-4PPs removed in cells irradiated at CT16 (CT16 siCTRL). By silencing Bmal1 expression, the efficiency of DNA repair was markedly reduced in cells irradiated at CT16 (CT16 siBmal1) and 30 min after irradiation only 23% of dimers have been removed (Figure 3B). In that case, the efficiency of DNA repair in CT16 siBmal1 fibroblasts was almost the same as that observed in CT28 siBMAL1 cells. The results obtained by silencing BMAL1 expression suggested that the circadian clock plays an important role in regulating DNA sensitivity to UV light and the following DNA repair process.
Figure 3.
Photoproduct formation and repair in C63 cells transfected with siBmal1. (A) The quantity of 6-4PP and CPD photoproducts formed immediately after UV irradiation at CT16 and CT28 was determined in fibroblasts transfected with siBMAL1 and siCTRL. (B) Kinetics of [6-4]PP removal in transfected fibroblasts irradiated at CT16 and CT28. The results reported in (A) and (B) were obtained in three independent experiments and plotted as mean ± SEM. The P-value refers to t-test. *P < 0.05, CT16 versus CT28.
Influence of circadian times on XPA expression and localization
Some investigators recently reported that the repair of UVB-induced DNA damage in mice is defective at night, due to the decreased levels of XPA-mediated excision repair (2). The same authors, however, later reported that cells derived from mice mutant for different circadian clock genes are indistinguishable from the wild-type in their response to UV irradiation (14). To evaluate if the difference observed in the removal of photoproducts could be attributed to a different expression of the XPA protein, we measured the amount of XPA protein in quiescent C63 cells after dexamethasone pulse. Western blot analyses performed on total and nuclear cell fractions showed that in the time-interval 12–44 h there were no appreciable variations either in total protein amount and in nuclear content (Figure 4A and B). Similar results were obtained in quiescent skin fibroblasts derived from a different healthy donor (Supplementary Figure S4). The level of XPA mRNA in fibroblasts transfected with siBMAL1 or siCTRL measured by qRT-PCR did not vary between CT16 and CT28 cells, showing that the circadian rhythmicity did not affect the XPA gene transcription (Figure 4C). To confirm this result, we investigated the effect of circadian clock mutations on XPA transcription. MDF cultures, derived from wild-type and BMAL1 or REV-ERBα mutant newborn mice, were analyzed for the relative amount of XPA transcript. The mRNA expression was found unchanged in both BMAL1m/m and REV-ERBαm/m MDFs in comparison with the transcripts collected from the wild-type cells, demonstrating that the circadian clock does not control XPA gene expression in cultured cells (Figure 4D).
Figure 4.
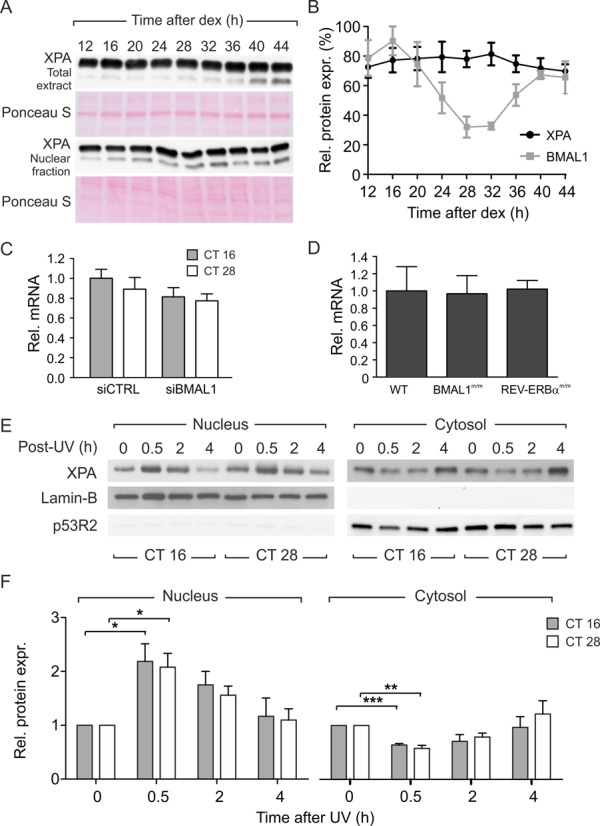
XPA gene expression and protein localization. (A) Representative western blot depicting the amounts of XPA protein in total and nuclear extracts. Ponceau staining served as a loading control. (B) The protein amount of XPA was analyzed in quiescent C63 cells every 4 h after dexamethasone pulse. For comparison we showed oscillation of BMAL1 content measured in the same condition. (C) Relative level of XPA mRNA in CT16 and CT28 fibroblasts transfected with siCTRL or siBmal1. (D) Relative level of XPA mRNA in primary dermal fibroblasts derived from wild-type mice, and from Bmal1 and Rev-erbα mutant mice. (E) Representative western blot of XPA amount in cytosol and nucleus of C63 cells at CT16 and CT28 before and after UV irradiation. Nuclear marker was Lamin B and cytosolic marker p53R2 (23). (F) Quantification of relative XPA amount in cytosol and nucleus. The results reported in (B) were obtained from four (BMAL1) and six (XPA) independent experiments; the results in (C), (D) and (F) from three independent experiments. All the results were plotted as mean ± SEM. The P-value refers to a 2-way ANOVA. ***P < 0.001, *P < 0.05, Bonferroni post-hoc test.
Because some authors demonstrated that XPA import after irradiation into the nucleus varied between stressed and unstressed cells and in relation to the phase of cell cycle (29, 30), we tested whether the CTs affected the cellular localization of XPA. Cytosolic and nuclear lysates were collected at different time-points (0–4 h) after irradiation and analyzed by western blotting for XPA protein. As previously shown (Figure 4A and B), the amount of XPA protein did not differ at any CTs either in total or nuclear extracts before irradiation. Early after irradiation (0.5 h), XPA amount significantly increased in the nuclear fractions and decreased over time (2 and 4 h) reaching the initial values, with almost identical profiles in both CT16 and CT28 cells. In the cytosolic fractions of both cell groups the pattern of XPA amount was opposite (Figure 4E and F). To exclude the involvement of a circadian control on XPA localization we analyzed XPA amount in nuclear and cytosolic extracts from siBmal1 transfected cells irradiated at the two CTs. In transfected fibroblasts, in which the circadian clock was obliterated, the trend of nuclear and cytosolic XPA amount was very similar to that previously observed in non-transfected cells (Supplementary Figure S4). Thus, our results clearly show that the nuclear amount of XPA significantly increases early after irradiation and decreases 4 h later, independently on the circadian phase at which irradiation occurred.
The circadian clock modulates chromatin accessibility
Looking for the cause of the different repair efficiency observed in quiescent C63 cells irradiated at CTs 16 and 28, we examined whether the oscillations of circadian proteins may regulate the extent of chromatin condensation. Previous experimental data (28,37–39) showed that differences in chromatin condensation and DNA damage repair could depend on the different level of histone acetylation. We investigated whether the acetylation at lysine 9 of histone H3 (H3K9ac) varied between CTs 16 and 28 after dexamethasone pulse. Our data showed that the amount of H3K9ac was significantly higher in CT16 compared with CT28 cells and the difference disappeared when the cells were transfected with siBmal1 (Figure 5A and B). After UV irradiation, the level of H3K9ac decreased in both CT16 and CT28 cells, in agreement with previous data showing a decrease of H3K9ac in cells treated with the radiomimetic agent phleomycin (40). In contrast, no significant differences after irradiation were detected in siBmal1-transfected cells. We expected that inhibition of histone deacetylase by sodium butyrate (NaBu) would abolish the differences in photoproduct formation and DNA repair efficiency observed in cells irradiated at CTs 16 and 28 (Figure 5C). To test our hypothesis, we incubated quiescent dexamethasone synchronized C63 cells with NaBu. This treatment did not disrupt circadian oscillations leaving PER2 and BMAL1 transcription levels unaffected (Supplementary Figure S4). In accordance with our hypothesis, NaBu-treatment lead to similar levels of 6-4PPs in both CT16 and CT28 UV-irradiated cells (Figure 5C). Although in general the levels were higher than in untreated cells. The rate of the removal of 6-4PPs in CT28 cells was markedly enhanced when the cells were incubated with the inhibitor of histone deacetylase NaBu, reaching the values detected for CT16 cells treated or untreated with NaBu (Figure 5D). These data strongly suggested that the circadian clock affects DNA damage formation and repair efficiency by regulating the level of histone acetylation.
Figure 5.
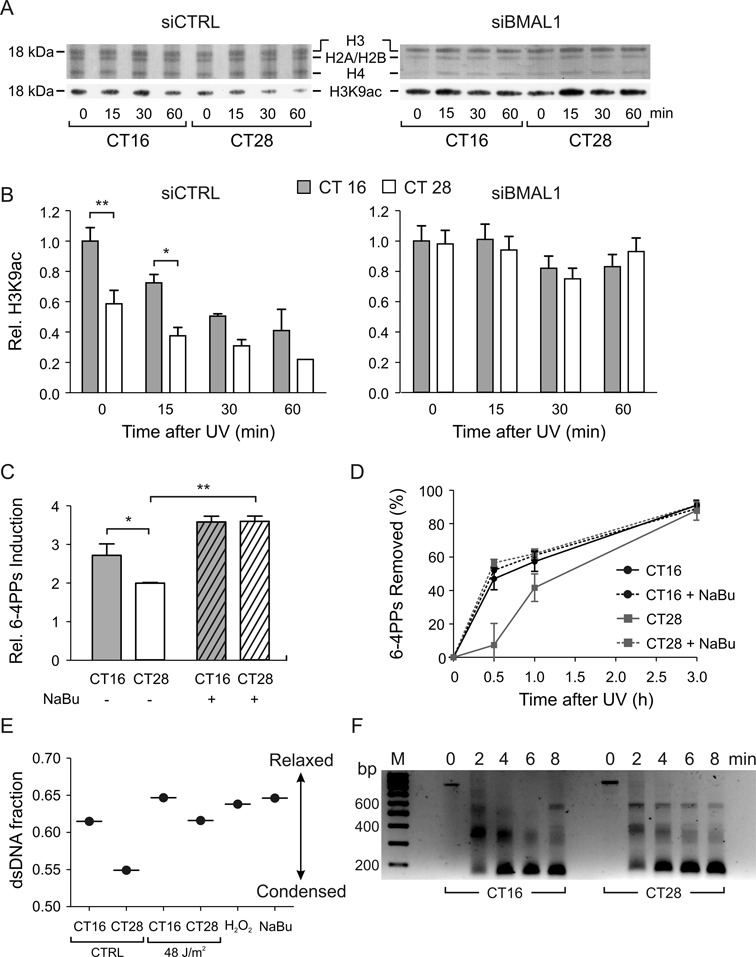
Analysis of chromatin conformation in C63 cells at CT16 and CT28. (A) Representative western blot of histones H3, H2A/H2B, H4 and H3K9ac of C63 cells transfected with siCTRL or siBmal1 and UV-irradiated at CT16 and CT28. (B) The relative content of H3K9ac, normalized over total H3 amount, was analyzed before and after irradiation. The P-value refers to a 2-way ANOVA. **P < 0.01, CT16 versus CT28, Bonferroni post-hoc test. (C) The amount of 6-4PPs formed immediately after irradiation was determined in CT16 and CT28 cells NaBu-treated or untreated. The P-value refers to a 2-way ANOVA, Bonferroni post-hoc test. *P < 0.05, CT16 versus CT28. **P < 0.01, CT28 NaBu treated versus CT28 NaBu untreated. (D) Removal of 6-4PP photoproducts in CT16 and CT28 cells NaBu-treated or untreated. (E) Chromatin relaxation was assayed by partial DNA denaturation and AO staining at CTs 16 and 28 before and after UV irradiation and after treatment with H2O2 or NaBu. Red and green fluorescence was measured by flow cytometry. (F) Micrococcal nuclease digestion of chromatin from CT16 and CT28 cells. Representative ethidium bromide-stained gel shows increased digestion of chromatin to three- di-and mono-nucleosomes (600, 400, 200 bp, respectively) in CT16 compared with CT28 cells. The results plotted as mean ± SEM were obtained from three (B, C, D) independent experiments. In (E) results are plotted as mean of two independent experiments.
We then evaluated the extent of chromatin condensation in CT16 and CT28 cells analyzing the propensity of DNA in situ to undergo denaturation induced by heat or acid. The level of DNA denaturation can be assayed in chromatin by the metachromatic fluorochrome acridine orange (AO). After binding of AO to double-stranded (ds) DNA green fluorescence can be observed, while interaction of AO with the denatured, single-stranded (ss) DNA results in red fluorescence. It was observed (41) that the DNA in condensed chromatin, as that of mitotic, apoptotic and quiescent G0 cells, was more easily denatured by heat or acid treatment than the DNA of cells having less condensed chromatin. Therefore, the ratio of green to red fluorescence of AO-stained cells provides information on the relative proportion of denatured ssDNA (reflecting the more condensed chromatin) versus dsDNA (reflecting the less condensed chromatin) (27). To this end, we investigated the susceptibility of DNA to denaturation at CT16 and CT28 by the HCl/AO assay described by Darzynkiewicz et al. (26). To have a positive control of chromatin relaxation, we treated the cells with the histone deacetylase inhibitor, NaBu, which increases both chromatin accessibility and NER efficiency (34). We found that chromatin condensation significantly differed between the two groups of cells, and the fraction of dsDNA, i.e. the less condensed chromatin, was higher in CT16 cells than in CT28 cells. After UV irradiation, dsDNA fraction increased in both CT16 and CT28 cells approaching the values of NaBu- and H2O2-treated cells (Figure 5E). The different level of chromatin condensation in CT16 and CT28 cells was confirmed by assaying their DNA to the micrococcal nuclease digestion. By increasing the time of incubation with micrococcal nuclease, we detected a higher degree of digested chromatin to three-, di- and mono-nucleosomes in CT16 compared with CT28 cells (Figure 5F).
DISCUSSION
The role of the circadian clock in DDR has recently attracted increasing attention because of its possible implications in cancer development (2,42). While the relationships between the circadian clock, cell cycle control and apoptosis induction have been clarified to some extent (43, 44), the effect of the circadian clock on DDR pathways is still unclear (14,45).
Our research aimed to analyze the cell response to UV irradiation in clock-synchronized quiescent human skin fibroblasts in the attempt to evaluate if and how the efficiency of DNA repair varies within a 24 h time-interval of circadian oscillation. Our results showed significant differences in DNA repair when UV irradiation was performed at two different CTs separated by 12 h, CT16 and CT28, corresponding to the maximum and minimum, respectively, of BMAL1 expression. At the same CTs PER2 expression oscillated in anti-phase. In cells irradiated at CT16 the removal of 6-4PPs was very fast, whereas photoproduct removal in cells irradiated at CT28 was slower, although both cell groups were able to remove all 6-4PPs within the first 3 h from irradiation. The kinetics of CPD removal was scarcely informative because of the very slow rate of recognition and removal of this kind of photoproducts. Indeed, no significant differences were noted between the two-irradiation groups in the time interval in which circadian proteins were induced to oscillate by dexamethasone pulse while it is well known that the phosphorylation of histone H2AX on S139 (γ-H2AX) is an important step in DNA double-strand break repair, the functional role of this event after UV irradiation is less apparent. The presence of γ-H2AX is correlated to persistent DNA gaps created by the excision of photoproducts, and its disappearance signals that the gaps have been filled by dNTP polymerization (9,46). Analysis of the γ-H2AX quantity and the state of winding of the DNA double helix within the first hours after irradiation revealed that greater residual damage persists in CT28 irradiated cells because of the slower DNA repair activity. UDS assay confirmed this result showing that within the first 30 min from irradiation the incorporation of [3H]-TdR was higher in CT16 cells with respect to CT28 cells. At the later times, the re-synthesis step of DNA repair did not differ between the two irradiation groups, probably because of the contribution of the gap filling activity after CPD removal, that started slower than 6-4PPs and was very similar between CT16 and CT28 cells.
All our data on the removal of 6-4PPs, the percentage of dsDNA and the rate of DNA re-synthesis during the repair process demonstrated that the cells irradiated at BMAL1 peak expression (CT16) were prone to initiate the repair process. In contrast, in cells irradiated at BMAL1 trough expression (CT28), the repair process started slowly. These data are in agreement with previous results, obtained in different mouse tissues, showing that NER activity exhibits a robust circadian oscillation (7,10,47). Nevertheless, while in those experimental systems the oscillation has been ascribed to the circadian rhythmicity of XPA expression in antiphase with Cry1 expression (2,7,10,30,47) in our cultures of quiescent fibroblasts we did not observe any rhythmicity neither in the basal expression or in the nuclear content of XPA. Moreover, after UV irradiation, XPA amounts rapidly increased into the nucleus of both CT16 and CT28 cells, without any influence of the circadian phase. Discrepancies in the amount and nature of circadianly oscillating genes have been already reported between ex vivo cells and intact mice. In particular, in ex vivo hepatocytes and in some cell lines only few clock genes have been shown to cycle compared to the thousands of clock-controlled genes exhibiting a circadian rhythmicity in the liver and other mouse organs and tissues (48).
Interestingly, when we analyzed the induction of CPDs and 6-4PPs immediately after irradiation, we found that a significantly higher amount of both photoproducts was formed in fibroblasts irradiated at CT16 compared to CT28. It appears that the cells, which were more prone to start the repair process, were also more sensitive to UV damage. Geyfman et al. (13) recently reported that the mouse epidermis had different susceptibilities to UVB-induced DNA damage at different times during a 24 h period of the circadian clock. The authors correlated the higher amount of photoproducts formed at Zeitgeber Time (ZT) 20, with respect to ZT 8, to the culmination of S phase at ZT 20, making DNA more sensitive to UV damage. Moreover, they demonstrated that BMAL1 is responsible for the time-of-day-dependent UV sensitivity, with maximal photoproduct formation when BMAL1 expression was at its minimum. Our experiments were conducted in quiescent fibroblasts blocked in G1/G0 phase; therefore we cannot attribute the different susceptibilities to UV light to events dependent on cell cycle phases. In contrast to the results obtained in mouse epidermis (13), the formation of photoproducts was maximal when BMAL1 expression peaked in cultures of quiescent fibroblasts. Moreover, all the different features that characterize the more efficient photo-damage repair in CT16 cells were abolished when BMAL1 expression was inhibited and the cells became arrhythmic. As demonstrated by the higher amount of photoproducts formed in cells with relaxed as opposed to more condensed chromatin, chromatin conformation also plays an important role in the susceptibility to UV-induced damage. Our results show that the amount of H3K9ac was significantly higher in cells at CT16 compared to those at CT28, and this could be related to the circadian rhythmicity in H3K9 acetylation, RNAPII recruitment, chromatin remodeling, as well as gene expression per se, as recently reported to a genome scale in mammals (19). We speculate that a similar scenario could be envisaged also in our case, with a proportional reduction in H3K9 and H3K27 methylation at CT16 (required for increasing the chromatin accessibility) followed by an accumulation at CT28 (promoting chromatin transition to a non-permissive conformation). Nevertheless other histone modifications could be reasonably involved in this process as reported in Arabidopsis thaliana where the peak-to-trough circadian oscillation is paralleled by a sequential accumulation of H3K56ac and H3K9ac followed by H3K4me3 and H3K4me2 (49). In particular, the phase of H3K56ac and H3K9ac matched that of the expression of LHY, CCA1, PRR9, PRR7, TOC1 and LUX genes showing temporally coinciding peaks. Although the mechanism underlying this process remains largely unknown, H3K4me3 could be actively involved in the modulation of the transition from activation to repression, probably acting as a substrate for histone demethylases. Following UV irradiation, the scenario of histone modifications becomes even more complicated to portray due to contrasting results in the literature. Nevertheless, as previously reported in a study performed on human cell cultures (50), we also observed in both CT16 and CT28 cells a reduction in H3K9ac after UV irradiation, probably an indirect effect of transcriptional inhibition, concurrently to a rapid phosphorylation of H2AX.
Taken together, our results lead to two major conclusions. First of all, we observed that formation of DNA damage and repair efficiency were strictly dependent on the CT at which the cells were exposed to UV light. Second, although the integrity of the circadian clock is not essential to carry out DNA repair, we found that its disruption by siBMAL1 transfection affected the efficiency of photoproduct removal. In addition, our work suggests that circadian rhythmicity in chromatin conformation regulates the activation and efficiency of NER in quiescent human fibroblasts. Besides the influence of chromatin condensation, it is possible that other factors modulated by the circadian clock affected the progress and conclusion of the repair process. This is suggested by our data of FADU and γ-H2AX kinetics, which indicated that a higher amount of DNA gaps persisted in CT28 cells up to 4–6 h from irradiation. Since we didn't observed differences in dNTPs incorporation long afterward UV irradiation, we hypothesize that this effect could be related to a different efficiency in DNA ligation. It has been recently observed that the DNA repair synthesis and ligation impairment slow-down the rate of UV photoproduct removal in human quiescent fibroblasts, with an accumulation of RPA-bound small, excised, damage-containing DNA (sedDNA) oligonucleotides (51). On the basis of our findings and those reported by Kemp et al. (51), we can also speculate on a synergic effect between circadian chromatin remodeling and DNA ligation efficiency, which eventually results in a different sensitivity and effectiveness of UV-induced DNA repair during the CT progression.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online.
Acknowledgments
We thank Vera Bianchi and Peter Reichard for suggestions and critically reading the manuscript. We are grateful for their help to Elisa Franzolin for cell transfection, Filippo Cendron for western blots, Lisa Marchioretto for MNase assays and Dr Ueli Schibler for RORα mice.
FUNDING
Department of Biology, University of Padova (to L.C.); Epigenomics Flagship Project - EPIGEN (National Research Council of Italy - CNR to R.C.); Fondazione CARIPLO—Scientific Research in Biomedicine 2011 (to R.C.); Telethon Project GGP11011 (to R.C.); Swiss National Science Foundation (to U.A). Funding for open access charge: EPIGEN (National Research Council of Italy - CNR to R.C.).
Conflict of interest statement. None declared.
REFERENCES
- 1.Meeran S.M., Punathil T., Katiyar S.K. IL-12 deficiency exacerbates inflammatory responses in UV-irradiated skin and skin tumors. J. Invest. Dermatol. 2008;128:2716–2727. doi: 10.1038/jid.2008.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gaddameedhi S., Selby C.P., Kaufmann W.K., Smart R.C., Sancar A. Control of skin cancer by the circadian rhythm. Proc. Natl. Acad. Sci. U.S.A. 2011;108:18790–18795. doi: 10.1073/pnas.1115249108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sancar A., Lindsey-Boltz L.A., Kang T.-H., Reardon J.T., Lee J.H., Ozturk N. Circadian clock control of the cellular response to DNA damage. FEBS Lett. 2010;584:2618–2625. doi: 10.1016/j.febslet.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lagerwerf S., Vrouwe M.G., Overmeer R.M., Fousteri M.I., Mullenders L.H.F. DNA damage response and transcription. DNA Repair (Amst). 2011;10:743–750. doi: 10.1016/j.dnarep.2011.04.024. [DOI] [PubMed] [Google Scholar]
- 5.Ray A., Milum K., Battu A., Wani G., Wani A.A. NER initiation factors, DDB2 and XPC, regulate UV radiation response by recruiting ATR and ATM kinases to DNA damage sites. DNA Repair (Amst). 2013;12:273–283. doi: 10.1016/j.dnarep.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim J.K., Patel D., Byong-Seok C. Contrasting structural impacts induced by cis-syn cyclobutane dimer and (6-4) adduct in DNA duplex decamers: implication in mutagenesis and repair activity. Photochem. Photobiol. 1995;62:44–50. doi: 10.1111/j.1751-1097.1995.tb05236.x. [DOI] [PubMed] [Google Scholar]
- 7.Kang T.-H., Lindsey-Boltz L.A., Reardon J.T., Sancar A. Circadian control of XPA and excision repair of cisplatin-DNA damage by cryptochrome and HERC2 ubiquitin ligase. Proc. Natl. Acad. Sci. U.S.A. 2010;107:4890–4895. doi: 10.1073/pnas.0915085107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palomera-Sanchez Z., Bucio-Mendez A., Valadez-Graham V., Reynaud E., Zurita M. Drosophila p53 is required to increase the levels of the dKDM4B demethylase after UV-induced DNA damage to demethylate histone H3 lysine 9. J. Biol. Chem. 2010;285:31370–31379. doi: 10.1074/jbc.M110.128462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pontarin G., Ferraro P., Bee L., Reichard P., Bianchi V. Mammalian ribonucleotide reductase subunit p53R2 is required for mitochondrial DNA replication and DNA repair in quiescent cells. Proc. Natl. Acad. Sci. U.S.A. 2012;109:13302–13307. doi: 10.1073/pnas.1211289109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kang T.-H., Reardon J.T., Kemp M., Sancar A. Circadian oscillation of nucleotide excision repair in mammalian brain. Proc. Natl. Acad. Sci. U.S.A. 2009;106:2864–2867. doi: 10.1073/pnas.0812638106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Im J.S., Jung B.H., Kim S.E., Lee K.H., Lee J.K. Per3, a circadian gene, is required for Chk2 activation in human cells. FEBS Lett. 2010;584:4731–4734. doi: 10.1016/j.febslet.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 12.Kim H., Lee J.M., Lee G., Bhin J., Oh S.K., Kim K., Pyo K.E., Lee J.S., Yim H.Y., Kim K.I., et al. DNA damage-induced RORα Is crucial for p53 stabilization and increased apoptosis. Mol. Cell. 2011;44:797–810. doi: 10.1016/j.molcel.2011.09.023. [DOI] [PubMed] [Google Scholar]
- 13.Geyfman M., Kumar V., Liu Q., Ruiz R., Gordon W., Espitia F., Cam E., Millar S.E., Smyth P., Ihler A., et al. Brain and muscle Arnt-like protein-1 (BMAL1) controls circadian cell proliferation and susceptibility to UVB-induced DNA damage in the epidermis. Proc. Natl. Acad. Sci. U.S.A. 2012;109:11758–11763. doi: 10.1073/pnas.1209592109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gaddameedhi S., Reardon J.T., Ye R., Ozturk N., Sancar A. Effect of circadian clock mutations on DNA damage response in mammalian cells. Cell Cycle. 2012;11:3481–3491. doi: 10.4161/cc.21771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sahar S., Sassone-Corsi P. The epigenetic language of circadian clocks. Handb. Exp. Pharmacol. 2013;217:29–44. doi: 10.1007/978-3-642-25950-0_2. [DOI] [PubMed] [Google Scholar]
- 16.Jenuwein T., Allis C.D. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 17.Doi M., Hirayama J., Sassone-Corsi P. Circadian regulator CLOCK is a histone acetyltransferase. Cell. 2006;125:497–508. doi: 10.1016/j.cell.2006.03.033. [DOI] [PubMed] [Google Scholar]
- 18.Nakahata Y., Kaluzova M., Grimaldi B., Sahar S., Hirayama J., Chen D., Guarente L.P., Sassone-Corsi P. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell. 2008;134:329–340. doi: 10.1016/j.cell.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koike N., Yoo S.-H., Huang H.-C., Kumar V., Lee C., Kim T.-K., Takahashi J.S. Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science. 2012;338:349–354. doi: 10.1126/science.1226339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rampazzo C., Fabris S., Franzolin E., Crovatto K., Frangini M., Bianchi V. Mitochondrial thymidine kinase and the enzymatic network regulating thymidine triphosphate pools in cultured human cells. J. Biol. Chem. 2007;282:34758–34769. doi: 10.1074/jbc.M705923200. [DOI] [PubMed] [Google Scholar]
- 21.Bee L., Fabris S., Cherubini R., Mognato M., Celotti L. The efficiency of homologous recombination and non-homologous end joining systems in repairing double-strand breaks during cell cycle progression. PLoS One. 2013;8:e69061. doi: 10.1371/journal.pone.0069061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmutz I., Ripperger J.A., Baeriswyl-Aebischer S., Albrecht U. The mammalian clock component PERIOD2 coordinates circadian output by interaction with nuclear receptors. Genes Dev. 2010;24:345–357. doi: 10.1101/gad.564110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pontarin G., Fijolek A., Pizzo P., Ferraro P., Rampazzo C., Pozzan T., Thelander L., Reichard P.A., Bianchi V. Ribonucleotide reduction is a cytosolic process in mammalian cells independently of DNA damage. Proc. Natl. Acad. Sci. U.S.A. 2008;105:17801–17806. doi: 10.1073/pnas.0808198105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Birnboim H.C., Jevcak J.J. Fluorometric method for rapid detection of DNA strand breaks in human white blood cells produced by low doses of radiation. Cancer Res. 1981;41:1889–1892. [PubMed] [Google Scholar]
- 25.Franzolin E., Pontarin G., Rampazzo C., Miazzi C., Ferraro P., Palumbo E., Reichard P., Bianchi V. The deoxynucleotide triphosphohydrolase SAMHD1 is a major regulator of DNA precursor pools in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 2013;110:14272–14277. doi: 10.1073/pnas.1312033110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Darzynkiewicz Z., Traganos F., Zhao H., Halicka H.D., Skommer J., Wlodkowic D. Analysis of individual molecular events of DNA damage response by flow- and image-assisted cytometry. Methods Cell. Biol. 2011;103:115–147. doi: 10.1016/B978-0-12-385493-3.00006-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Halicka H.D., Zhao H., Podhorecka M., Traganos F., Darzynkiewicz Z. Cytometric detection of chromatin relaxation, an early reporter of DNA damage response. Cell Cycle. 2009;8:2233–2237. doi: 10.4161/cc.8.14.8984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rubbi C.P., Milner J. p53 is a chromatin accessibility factor for nucleotide excision repair of DNA damage. EMBO J. 2003;22:975–986. doi: 10.1093/emboj/cdg082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Z., Musich P.R., Serrano M.A., Dong Z., Zou Y. XPA-mediated regulation of global nucleotide excision repair by ATR is p53-dependent and occurs primarily in S-phase. PLoS One. 2011;6:e28326. doi: 10.1371/journal.pone.0028326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Z., Musich P.R., Cartwright B.M., Wang H., Zou Y. UV-induced nuclear import of XPA is mediated by importin-α4 in an ATR-dependent manner. PLoS One. 2013;8:e68297. doi: 10.1371/journal.pone.0068297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Balsalobre A., Brown S.A., Marcacci L., Tronche F., Kellendonk C., Reichardt H.M., Schütz G., Schibler U. Resetting of circadian time in peripheral tissues by glucocorticoid signaling. Science. 2000;289:2344–2347. doi: 10.1126/science.289.5488.2344. [DOI] [PubMed] [Google Scholar]
- 32.Nagoshi E., Saini C., Bauer C., Laroche T., Naef F., Schibler U. Circadian gene expression in individual fibroblasts: Cell-autonomous and self-sustained oscillators pass time to daughter cells. Cell. 2004;119:693–705. doi: 10.1016/j.cell.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 33.Kang T.H., Reardon J.T., Sancar A. Regulation of nucleotide excision repair activity by transcriptional and post-transcriptional control of the XPA protein. Nucleic Acids Res. 2011;39:3176–3187. doi: 10.1093/nar/gkq1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nishinaga M., Kurata R., Onishi K., Kuriyama K., Wakasugi M., Matsunaga T. Establishment of a microplate-formatted cell-based immunoassay for rapid analysis of nucleotide excision repair ability in human primary cells. Photochem. Photobiol. 2012;88:356–362. doi: 10.1111/j.1751-1097.2012.01073.x. [DOI] [PubMed] [Google Scholar]
- 35.Erixon K., Ahnström G. Single-strand breaks in DNA during repair of UV-induced damage in normal human and xeroderma pigmentosum cells as determined by alkaline DNA unwinding and hydroxylapatite chromatography. Mutat. Res. Mol. Mech. Mutagen. 1979;59:257–271. doi: 10.1016/0027-5107(79)90164-7. [DOI] [PubMed] [Google Scholar]
- 36.Baumstark-Khan C., Hentschel U., Nikandrova Y., Krug J., Horneck G. Fluorometric analysis of DNA unwinding (FADU) as a method for detecting repair-induced DNA strand breaks in UV-irradiated mammalian cells. Photochem. Photobiol. 2000;72:477–484. doi: 10.1562/0031-8655(2000)072<0477:faoduf>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 37.Ikura T., Tashiro S., Kakino A., Shima H., Jacob N., Amunugama R., Yoder K., Izumi S., Kuraoka I., Tanaka K., et al. DNA damage-dependent acetylation and ubiquitination of H2AX enhances chromatin dynamics. Mol. Cell. Biol. 2007;27:7028–7040. doi: 10.1128/MCB.00579-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun Y., Jiang X., Price B.D. Tip60: connecting chromatin to DNA damage signaling. Cell Cycle. 2010;9:930–936. doi: 10.4161/cc.9.5.10931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palomera-Sanchez Z., Zurita M. Open, repair and close again: chromatin dynamics and the response to UV-induced DNA damage. DNA Repair (Amst). 2011;10:119–125. doi: 10.1016/j.dnarep.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 40.Tjeertes J. V, Miller K.M., Jackson S.P. Screen for DNA-damage-responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. EMBO J. 2009;28:1878–1889. doi: 10.1038/emboj.2009.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dobrucki J., Darzynkiewicz Z. Chromatin condensation and sensitivity of DNA in situ to denaturation during cell cycle and apoptosis–a confocal microscopy study. Micron. 2001;32:645–652. doi: 10.1016/s0968-4328(00)00069-x. [DOI] [PubMed] [Google Scholar]
- 42.Gery S., Koeffler H.P. Circadian rhythms and cancer. Cell Cycle. 2010;9:1097–1103. doi: 10.4161/cc.9.6.11046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gery S., Komatsu N., Baldjyan L., Yu A., Koo D., Koeffler H.P. The circadian gene per1 plays an important role in cell growth and DNA damage control in human cancer cells. Mol. Cell. 2006;22:375–382. doi: 10.1016/j.molcel.2006.03.038. [DOI] [PubMed] [Google Scholar]
- 44.Kowalska E., Ripperger J. A, Hoegger D.C., Bruegger P., Buch T., Birchler T., Mueller A., Albrecht U., Contaldo C., Brown S. NONO couples the circadian clock to the cell cycle. Proc. Natl. Acad. Sci. U.S.A. 2013;110:1592–1599. doi: 10.1073/pnas.1213317110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kondratov R. V. Cell-autonomous circadian DNA damage response: is the case closed. Cell Cycle. 2012;11:3720–3721. doi: 10.4161/cc.22115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cleaver J.E. γ-h2Ax: biomarker of damage or functional participant in DNA repair ‘all that glitters is not gold!’. Photochem. Photobiol. 2011;87:1230–1239. doi: 10.1111/j.1751-1097.2011.00995.x. [DOI] [PubMed] [Google Scholar]
- 47.Kang T.H., Leem S.H. Modulation of ATR-mediated DNA damage check point response by Cryptochrome1. Nucleic Acids Res. 2014;42:4427–4434. doi: 10.1093/nar/gku094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hughes M.E., DiTacchio L., Hayes K.R., Vollmers C., Pulivarthy S., Baggs J.E., Panda S., Hogenesch J.B. Harmonics of circadian gene transcription in mammals. PLoS Genet. 2009;5:e1000442. doi: 10.1371/journal.pgen.1000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Malapeira J., Khaitova L.C., Mas P. Ordered changes in histone modifications at the core of the Arabidopsis circadian clock. Proc. Natl. Acad. Sci. U.S.A. 2012;109:21540–21545. doi: 10.1073/pnas.1217022110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tjeertes J.V., Miller K.M., Jackson S.P. Screen for DNA-damage-responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. EMBO J. 2009;28:131878–131889. doi: 10.1038/emboj.2009.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kemp M.G., Gaddameedhi S., Choi J.H., Hu J., Sancar A. DNA repair synthesis and ligation affect the processing of excised oligonucleotides generated by human nucleotide excision repair. J. Biol. Chem. 2014;289:26574–26583. doi: 10.1074/jbc.M114.597088. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.